

Abstract
There is mounting evidence indicating an important role for complement in the pathogenesis of cerebral ischemia reperfusion injury, or ischemic stroke. The role of the alternative complement pathway in ischemic stroke has not been investigated, and there is conflicting data on the role of the terminal pathway. Here we show that compared to wild type mice, mice deficient in the alternative pathway protein factor B, or mice treated with the alternative pathway inhibitor CR2-fH, have improved outcomes after 60 minutes middle cerebral artery occlusion (MCAO) and 24 hours reperfusion. Factor B-deficient or CR2-fH treated mice were protected in terms of improved neurological function and reduced cerebral infarct, demyelination, P-selectin expression, neutrophil infiltration and microthrombi formation. Mice deficient in both the classical and lectin pathways (C1q/MBL deficient) were also protected from cerebral IRI, and there was no detectable C3d deposition in the ipsilateral brain of these mice. These data demonstrate that alternative pathway is not alone sufficient to initiate complement activation, and indicate the alternative pathway propagates cerebral injury via amplification of the cascade. Deficiency of C6, a component of the terminal cytolytic membrane attack complex (MAC), had no effect on outcome after ischemic stroke, indicating the MAC is not involved in mediating injury in this model. We additionally show that the protective effect of fB deficiency and CR2-fH treatment is sustained in the sub-acute stage of infarct development, adding to the clinical relevance of these findings.
Introduction
Transient ischemic stroke is a pathological process in which a cerebral vessel is occluded creating an ischemic core, and once the blockage is resolved, either naturally or through pharmaceutical intervention, the returned blood flow creates a paradoxical secondary injury (reviewed in (1, 2)). Stroke is a leading cause of death and disability, and the role of complement in cerebral ischemia reperfusion injury (IRI) has received much attention recently.
Complement can be activated by either the classical, lectin or alternative pathways. The classical and lectin pathways can be activated by antibodies, certain polysaccharides and apoptotic cells. The alternative pathway can be activated spontaneously, but also functions to amplify the other pathways. All pathways converge with the formation of a C3 convertase and the cleavage of C3 to produce C3a and C3b. C3a is a soluble bioactive peptide, and C3b becomes covalently bound to the activating surface, where it is further cleaved to produce iC3b and C3d opsonins. C3 cleavage also leads to C5 cleavage to yield C5a and C5b. C5a has multiple inflammatory activities, including the recruitment and activation of leukocytes, whereas C5b initiates the terminal pathway and formation of the cytolytic membrane attack complex (MAC).
Although there is good clinical evidence for a role of complement in ischemic stroke (3), our current understanding of the complement-dependent mechanisms involved in ischemic stroke derives mostly from murine studies and the use of complement deficient and complement inhibited mice. In a seminal study, it was shown that following middle cerebral artery occlusion (MCAO), ischemic neurons upregulate the classical complement pathway molecule C1q, and that a soluble complement inhibitor, sCR1, is protective (4). Furthermore, an sCR1 derivative with sialyl Lewisx glycosylation inhibited both complement activation and selectin-mediated adhesion and demonstrated enhanced therapeutic activity. Subsequent studies demonstrated that genetic deficiency of the central complement protein C3 (5, 6), as well as inhibition of C3 convertases (5), reduced infarct volumes and inflammation, and improved neurological deficit following MCAO. As noted above, the principle complement effector molecules that are produced following activation of any pathway are C3a, C5a, C3 opsonins and the MAC. In the context of rodent ischemic stroke, both C3a and C5a signaling have been shown to contribute to ischemia related inflammation and injury, since both C3a receptor (6, 7) and C5a receptor (8, 9) antagonism reduced cerebral inflammation, infarct volume and neurological deficit. It should be noted, however, that C5 deficiency is not protective following focal cerebral ischemia, a finding that is not consistent with a role for C5a (6).
A recent study demonstrated an important role for natural self-reactive IgM in activating complement after ischemic stroke (10). Complement activation by natural IgM has also been shown to occur in other organ models of IRI, with IgM-mediated activation occuring via the lectin pathway (11–13). A role for the lectin pathway is also indicated in cerebral IRI, since lectin pathway deficiency is protective in murine models (14, 15). Clinical data also suggest a role for the lectin pathway in human ischemic stroke (14, 16). And although IgM also binds C1q and is a powerful activator of the classical pathway, C1q deficiency is not protective following murine focal cerebral ischemia (17, 6). The role of the alternative pathway of complement activation in ischemic stroke has not been investigated, although this pathway is important for propagating inflammation and injury in multiple pathological conditions (18, 19).
A role of the terminal pathway in ischemic cerebral injury has been investigated using neonatal rats that are developmentally deficient in C9. Reconstitution of neonatal rats with C9 exacerbated cerebral injury following carotid artery ligation with hypoxia, indicating an important role for the MAC (20). On the other hand, in a mouse model of neonatal hypoxic-ischemic brain injury involving permanent ischemia, C6 deficiency was not protective, indicating that the MAC does not play a role in cerebral injury in this model (21). In a model of MCAO and transient ischemia, as utilized in the current study, the effect of CD59a deficiency, a membrane-bound inhibitor of MAC formation, was dependent on gender and the period of ischemia (22).
In the current study, we investigated the role of the alternative pathway in propagating secondary inflammation and injury following transient focal cerebral ischemia and reperfusion. In addition, we further investigated the role of the terminal pathway in transient ischemic stroke following MCAO by a direct approach by utilizing mice that are deficient in C6 and that cannot form the MAC. We also investigated a therapeutic strategy for limiting cerebral inflammation and injury using CR2-fH, which targets sites of complement activation via CR2-mediated recognition of C3 opsonins (23). It has been shown previously that CR2-fH specifically inhibits the alternative pathway (23, 24). A human counterpart of CR2-fH termed TT30, also shown to be specific for the alternative pathway (25), is currently in clinical trials.
Material and Methods
Mice
All mice utilized in these studies, including complement deficient mice, were on the C57BL/6 genetic background, male and 8 to 9 weeks old at the time of study. Wild type mice were obtained from The Jackson Laboratory (Bar Harbor, ME). The following complement deficient strains of mice were used: fB −/− (lack alternative pathway) (26); C1q/MBL −/− (lack classical and lectin pathway) (27); C6 −/− (lack ability to form the MAC) (28, 29); CD59a −/− (lack membrane inhibitor of MAC formation) (30). The C6 −/− and CD59a −/− mice were kindly provided by Dr. B. Paul Morgan (University of Wales, Cardiff, UK). Deficient mice were bred and housed at the Medical University of South Carolina. All procedures were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina, in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals.
Middle cerebral artery occlusion (MCAO)
Mice were subjected to 60 minutes MCAO, followed by a reperfusion period of 24 hours, 72 hours or 7 days before mice were sacrificed and brains isolated for analysis, as previously described (5, 10). Briefly, mice were anesthetized, the external carotid artery (ECA) exposed, and the blunted tip of a 6-0 nylon suture inserted through the ECA into the internal carotid artery. The suture was advanced so as to block the middle cerebral artery. After 60 minutes of ischemia, the suture was withdrawn, the ECA stump ligated, and the incision closed. Laser Doppler flowmetry was used to assess cerebral blood flow before, during and after ischemia; mice not achieving reduction in blood flow to 20 percent of pre-ischemia levels were excluded from analysis. Temperature, blood pressure and heart rate were also monitored before, during and 10 minutes following ischemia.
CR2-fH and CR2-Crry preparation and treatment
The recombinant fusion protein CR2-fH, a site-targeted inhibitor of the alternative pathway (24), was prepared as previously described (23). CR2-Crry, an inhibitor of all complement pathways at the C3 activation step, was prepared as previously described (31). Protein purity was assessed by SDS gel electrophoresis and complement inhibitory activity was evaluated by zymosan assay (23). For complement inhibitor treatments, randomized wt C57BL/6 mice received 0.4 mg CR2-fH, 0.25 mg CR2-Crry or 100 μl vehicle control (PBS) intravenously via tail vein 30 minutes post-reperfusion (5). Inhibitor dosing was based on previously published dose-response data for CR2-fH and CR2-Crry in a model of intestinal ischemia reperfusion injury (23).
24-hour survival
Survival to 24 hours following 60 minutes of MCAO was compared among groups. Survival was compared using Fishers Exact 2×2.
Evaluation of infarct volume
Following sacrifice of mice, brains were perfused with PBS, isolated and placed in a rodent brain matrix (EMS, Hatfield, PA). Two millimeter slices were prepared and stained with 2% triphenyltetrazolium chloride (TTC; Sigma-Aldrich, St. Louis, MO). Total infarct area was assessed using Image-J Analysis Software (National Institutes of Health) and calculated by summation of infarcted areas of all slices for each hemisphere. Animals that did not survive to the established endpoint (24 hours, 72 hours or 7 days) were excluded from the determination of infarct volumes.
Neurological deficit scoring
Neurological deficits were determined as described (32) according to the following scoring system: 0, normal function; 1, flexion of torso and contralateral forelimb when lifted; 2, normal posture at rest with circling to contralateral side when held by tail on flat surface; 3, leaning to contralateral side when at rest; 4, no spontaneous movement. Scoring was performed by an observer blinded to experimental groups.
Histopathology
At sacrifice, mice were perfused with PBS. Brains were then removed, fixed overnight in 4% paraformaldehyde in PBS, and either immersed in 20% sucrose for cryosectioning or processed to paraffin for luxol-fast blue/Nissl (American MasterTech, Lodi, CA) or hematoxylin and eosin (H&E) staining for morphological analysis.
Immunohistochemistry
Paraffin processed sections cut at 8 μm were subjected to antigen retrieval by citrate steam for 30 minutes (IHC World, Ellicot City, MD), and primary antibodies were then applied in PBS for 2 hours at room temperature. Primary antibodies used were goat anti-mouse C3d (1:20; R&D Systems, Minneapolis, MN), goat anti-mouse IgM (1:50; Sigma-Aldrich) and rat anti-mouse Ly6G+Ly6C (Gr-1, 1:20; BD Biosciences, San Jose, CA). Brain sections from mice sacrificed at 72 hours were stained with rabbit anti-mouse GFAP (1:1000; Dako, Carpinteria, CA) in PBS for 2 hours at room temperature. Immunohistochemistry to detect P-selectin expression was done on 8 μm cyrosections stained with rat anti-mouse CD62P (1:10; BD Biosciences) overnight at 4°C. Primary antibodies were detected using the appropriate ImmPress kit (Vector Laboratories, Burlingame, CA), developed using NovaRed (Vector Laboratories) and sealed with CytoSeal-60 (Richard-Allan Scientific, Kalamazoo, MI). Primary antibodies were omitted for negative controls. A blinded observer imaged the slides by light microscopy (Nikon Eclipse E600).
Assessment of complement deposition, microthrombi formation and neutrophil infiltration
Brain tissue sampled was located between +2 mm to −2 mm relative to the bregma, with injury confirmed with luxol fast blue/Nissl or counterstaining. C3d deposition within the penumbra was scored as described previously (33): 0, no observable staining; 1, mild staining; 2, moderate staining; 3, severe staining. To assess microthrombi formation, H&E sections were scored as described previously (5): 0, no observable thrombi; 1, modest red blood cell (RBC) attachment; 2, focal RBC attachment and fibrin; 3, 50% occlusion by RBC and fibrin; 4 total occlusion. To quantify neutrophil infiltration in the penumbra, 10 random fields, at 40x magnification, from Gr-1 stained sections were counted. All assessments were performed by an observer blinded to experimental groups.
Evaluation of apoptosis
Paraffin brain sections from mice sacrificed at 72 hours were evaluated for apoptosis by TACS 2 TdT-DAB in situ apoptosis detection kit (Trevigen Inc., Gaithersburg, MD). The staining protocol was followed according to the manufacturer’s instructions. Negative and positive controls were included with each staining batch. Apoptotic cells in the peri-infarct region of the penumbra were counted from ten random fields at 40x magnification by light microscopy. The observer was blinded to the experimental groups.
Statistical analysis
Statistical analyses were performed using Prism 4 software (GraphPad, La Jolla, CA). Parametric data (infarct volumes) were analyzed for statistical significance with one-way analysis of variance test. Non-parametric data (neurological deficits, C3d deposition scores, microthrombi scores and cell counts) were analyzed for statistical significance with Kruskal-Wallis test. Comparisons between two groups were conducted using Student’s t-test (parametric) or Mann-Whitney test (non-parametric). Differences were considered significant when p<0.05.
Results
Alternative pathway deficiency and inhibition protects against cerebral ischemia and reperfusion injury
Neurological deficit was assessed 24 hours following 60 minutes of cerebral ischemia. Compared to wildtype (wt) controls, mice deficient in either factor B (fB; no alternative pathway) or C1q/MBL (only alternative pathway) displayed significant improvements in median neurological deficit (Figure 1a). There was no significant difference in neurological deficits between fB and C1q/MBL deficient mice. To investigate the role of the alternative pathway in a more clinically relevant paradigm, wt mice were treated with either CR2-fH, a targeted inhibitor of the alternative pathway, or CR2-Crry, a similarly targeted inhibitor that blocks all complement activation pathways. Treatments were given at 30 minutes post reperfusion. Both inhibitors significantly improved median neurological deficit compared to wt control mice, and there was no significant difference either between CR2-fH and CR2-Crry treated mice, or between inhibited mice and fB or C1q/MBL deficient mice (Fig. 1a). Focal cerebral infarct volumes were also analyzed 24 hours after reperfusion. Compared to wt controls, mean infarct volumes were significantly decreased in both fB and C1q/MBL deficient mice, although infarct volumes were significantly smaller in C1q/MBL deficient mice compared to fB deficient mice (Fig. 1b). Furthermore, consistent with the effect of CR2-fH and CR2-Crry on neurological deficit, both inhibitors significantly reduced mean infarct volumes compared to wt controls, and although there was a trend toward improved outcome with CR2-Crry compared to CR2-fH, the difference was not significant (Fig. 1b).
Figure 1.

Effect of complement deficiency and CR2-fH or CR2-Crry treatment on neurological deficit and infarct volume after 60 minutes MCAO and 24 hours reperfusion. (A) Neurological deficit and (B) Infarct volume in C1q/MBL −/− mice, fB−/− mice and wt mice treated with CR2-fH or CR2-Crry 30 minutes after reperfusion. Horizontal bar indicates median neurological score and mean infarct volume, n = 12–15 for neurological deficit and n = 8–11 for infarct volume; *p<0.05, **p<0.01, ***p<0.001.
The terminal complement pathway does not play a role in cerebral injury after 60 minutes transient ischemia
The terminal complement pathway and MAC formation is common to all activation pathways, and has been implicated as a key mediator of IRI in various organs. However, a previous study demonstrated that deficiency of CD59a, an endogenous membrane-bound inhibitor of MAC formation, exacerbated cerebral IRI after a 30 minute, but not after a 60-minute ischemic period (22). To more directly investigate the role of the terminal pathway and the MAC in cerebral IRI, we determined the effect of C6 deficiency, a component protein of the MAC, in the more severe model of cerebral IRI incorporating 60 minutes of MCAO. C6 deficiency was not protective in terms of either neurological function or infarct volume when compared to wt control mice (Fig. 2), indicating that the terminal pathway does not contribute to IRI in this model. We also confirmed the previous data demonstrating that CD59a deficiency was without effect on cerebral injury after 60 minutes of MCAO (Fig. 2). Subsequent studies focused on the role of the alternative pathway in this model of ischemic stroke.
Figure 2.

Effect of terminal pathway deficiency and terminal pathway inhibitor deficiency on neurological deficit and infarct volume after 60 minutes MCAO and 24 hours reperfusion. (A) Neurological deficit in C6 −/− and CD59a−/− mice. Horizontal bar represents median; n=10–11. No statistical difference between groups. (B) Infarct volume in C6 −/− and CD59a−/− mice. Horizontal bar indicates mean, n = 6–8. No statistical difference between groups.
Neuronal tissue integrity
Neuronal integrity, as demonstrated by myelin staining with luxol-fast blue/Nissl staining, was improved in C1q/MBL −/− mice, fB −/− mice and CR2-fH treated mice compared to wt controls. There was no apparent improvement in C6 −/− mice (Fig. 3). Thus, improved neuronal integrity correlates with improvements in infarct volumes and neurological deficits for the different groups of mice shown in figure 1. Taken together, these data indicate that the alternative pathway alone is not sufficient to cause injury following cerebral IR, but that it plays a key role in propagating injury following complement activation by either the classical or lectin pathway, and published data has shown dependence on the lectin pathway (14, 15).
Figure 3.
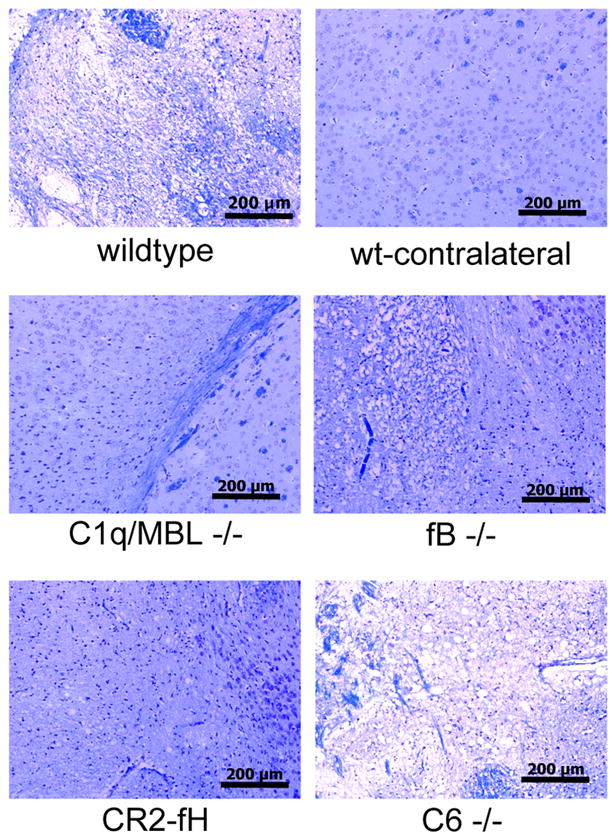
Neuronal tissue integrity in complement deficient and CR2-fH treated mice after 60 minutes MCAO and 24 hours reperfusion. Brain sections from the ipsilateral hemisphere were stained with luxol fast blue/Nissl. Section from contralateral hemisphere shown as undamaged control. Representative images shown, n = 4 per group; 10x magnification, scale bar = 200 μm.
24-hour survival and physiological measurements
C1q/MBL deficiency significantly improved the survival to 24 hours (18/19) compared to wt (12/18, p=0.03 by Fishers Exact 2×2). Mice deficient in fB or treated with CR2-fH appeared to have increased survival rates to 24 hours compared to wt mice (15/18 and 17/20, respectively), but the difference did not reach significance. There was also no significant difference between 24 hour survival rates of wt mice and mice deficient in C6 or CD59a (11/16 and 8/11, respectively).
To confirm that cerebral blood flow was interrupted by the MCAO procedure, blood flow was measured by laser Doppler, and there were no significant differences in cerebral blood flow between any of the groups before, during or 10 minutes after ischemia (not shown). Changes in blood pressure and body temperature can significantly influence the outcome post stroke, and we therefore measured blood pressure, heart rate and temperature before, during and after ischemia. There were no differences between the groups (not shown). Also, as expected, there was no damage observed in the contralateral hemisphere in any group.
Complement deposition is reduced in complement deficient and inhibited mice following MCAO
Penumbral deposition of the C3 activation product, C3d, was examined on sections of brain isolated 24 hours post-reperfusion, at which time complement deposition is maximal in the 60-minute MCAO model (34). C3d staining was seen in the penumbra throughout the ipsilateral hemisphere in wt mice, but was virtually absent in C1q/MBL mice (Fig. 4). Mice deficient in fB or treated with CR2-fH had significantly reduced C3d staining compared to wt mice, and the staining that was observed was associated predominantly with the vasculature in the penumbra (Fig. 4). Brain sections from C6 deficient mice and wt mice had similar levels of C3d staining, and contralateral tissue did not show any C3d staining for any of the experimental groups (not shown). These data support the above outcome measurements in terms of the conclusion that the alternative pathway amplifies complement activation and drives cerebral injury subsequent to classical/lectin pathway initiation.
Figure 4.

C3d deposition in penumbral tissue in complement deficient and CR2-fH treated mice after 60 minutes MCAO and 24 hours reperfusion. (A) Representative immunohistochemistry images taken at 40x magnification. Scale bar = 50 μm. There was no observable C3d staining in the contralateral hemispheres in any group. (B) Quantification of C3d staining. Horizontal bar represents median, n = 5; *p<0.05, **p<0.01.
Reduced complement deposition is not due to reduced IgM binding in ischemic brain
Natural self-reactive IgM antibodies that recognize neoepitopes exposed on ischemic tissue have been shown to play an important role in activating complement and propagating IRI, including cerebral IRI (10). We therefore wanted to confirm that the effect of the different complement deficiencies on cerebral IRI was not due to differences in the binding of IgM post-reperfusion. Immunohistochemical analysis of brain sections isolated 24 hours after reperfusion revealed similar levels of IgM staining in all groups, and IgM staining was absent in sections isolated from the contralateral hemisphere (Fig. 5).
Figure 5.

IgM deposition in penumbral tissue in complement deficient and CR2-fH treated mice after 60 minutes MCAO and 24 hours reperfusion. There were no discernable differences in the levels or distribution of IgM staining between any groups. No IgM staining was observed in the contralateral hemispheres in any group. Representative images, n = 3; 40x magnification, scale bar = 50 μm.
Microthrombi formation is reduced in alternative pathway deficient and inhibited mice
Secondary thrombus formation in the penumbra can perpetuate further ischemic injury and is a significant complication following ischemic stroke. We previously demonstrated fibrinogen deposition and microthrombi formation in the cerebrovasculature following MCAO, and we therefore investigated whether the alternative pathway contributes to this post-ischemic complication. Brain sections were analyzed for the presence of red blood cell accumulation and fibrin deposition as previously described (5). There were significantly lower numbers of microthrombi and partially occluded vessels in fB deficient and CR2-fH treated mice compared to wt mice (Fig. 6). Microthrombi formation appeared to be even lower in C1q/MBL deficient mice compared to fB deficient and CR2-fH treated mice, although the difference did not reach significance.
Figure 6.

Microthrombi formation in penumbral tissue in complement deficient and CR2-fH treated mice after 60 minutes MCAO and 24 hours reperfusion. Microthrombi formation was assessed in H&E stained brain sections. Horizontal bars represent median microthrombi scores, n = 5–7; *p<0.05.
P-selectin expression and neutrophil infiltration is reduced in alternative pathway deficient and inhibited mice
Infiltrating leukocytes are important mediators of post-ischemic inflammation and injury, and complement activation products are involved in their recruitment, either directly or indirectly via up-regulation of inflammatory mediators such as the adhesion molecule P-selectin. The expression of P-selectin and the infiltration of neutrophils are directly linked to cerebral inflammation and injury following ischemic stroke (4, 35), and we therefore examined how modulation of the alternative pathway affects these two parameters. Compared to wt mice, fB deficient and CR2-fH treated mice both had significantly reduced P-selectin expression and neutrophil infiltration in the cerebral vasculature 24 hours after MCAO (Fig. 7). Furthermore, P-selectin expression was undetectable in C1q/MBL deficient mice, and neutrophil infiltration was significantly lower in C1q/MBL deficient mice compared to fB deficient and CR2-fH treated mice. There was no detectable expression of P-selectin in the contralateral hemisphere from any experimental group. Thus, blocking or inhibiting the alternative pathway reduces cerebral inflammation, and this correlates with the above data showing reduced cerebral injury, reduced complement deposition and reduced microthrombi formation. The enhanced effect of C1q/MBL deficiency on the above measured parameters compared to fB deficiency is indicative of the alternative pathway exerting its effect in propagating pathogenesis after ischemic stroke via its function in the complement cascade amplification loop.
Figure 7.

P-selectin expression and neutrophil infiltration in penumbral tissue in complement deficient and CR2-fH treated mice after 60 minutes MCAO and 24 hours reperfusion. (A) P-selectin expression as detected by immunohistochemistry. Expression is associated with the vasculature. Representative images, n = 3, 10x magnification, scale bar = 200 μm. No staining was detected in contralateral hemisphere of any group. (B) Neutrophil infiltration as detected by immunohistochemical staining of GR1+ cells. Horizontal bar represents median, n = 4–5; **p<0.01, ***p<0.001.
CR2-fH treatment is protective at 72 hours following MCAO
To better address the clinical relevance of our findings, we investigated the effect of a single post-reperfusion injection of CR2-fH, as well as fB deficiency, on infarct maturation and longer-term outcome after MCAO. Compared to wt mice, CR2-fH treated or fB deficient mice had significantly reduced median neurological deficits at 3 days after reperfusion (Fig. 8a). Furthermore, the median neurological deficit in CR2-fH treated and fB deficient mice was lower at 72 hours post-reperfusion than it was at 24 hours post-reperfusion. Infarct volumes also remained significantly smaller at 72 hours post reperfusion in CR2-fH treated and fB deficient mice compared to wt mice (Fig. 8b). Since astrocyte responses can be modulated by complement activation products (36), we also determined how alternative pathway inhibition with CR2-fH effects astrogliosis, a hallmark of brain injury (37). As determined by GFAP expression, CR2-fH treatment dampened the reactive astrocyte presence associated with the injury border seen in wt mice at 3 days post-reperfusion (Fig. 8c). Reduction in the GFAP-reactivity was also observed in fB deficient mice. There were no differences between groups in GFAP reactivity in the corresponding contralateral sections (not shown). To determine if CR2-fH treatment impacts delayed cell death, we determined apoptosis in the peri-infarct region of the penumbra at 72 hours following MCAO. As shown in Figure 8d, there were fewer apoptotic cells in the penumbral region of brains from mice treated with CR2-fH compared to wt control mice. The number of apoptotic cells in brains from fB deficient mice was similar to that seen in CR2-fH treated mice. There were no observed differences in the contralateral hemispheres between groups (not shown).
Figure 8.

Effect of fB deficiency and CR2-fH treatment on outcomes after 60 minutes MCAO and 72 hours reperfusion. (A) Neurological deficit. Horizontal bar represents median, n = 5–6; *p<0.05. (B) Infarct volume. Horizontal bar represents mean, n = 5–6; ***p<0.001. (C) Astrocyte GFAP reactivity as assessed by GFAP immunohistochemistry. Representative images shown, n = 3. Magnifications as follows: 4x, scale bar = 500 μm; 10x, scale bar = 200 μm. Contralateral GFAP staining was not different between groups. (D) Apoptosis as evaluated by counting 10 random 40x fields from the peri-infarct region of the penumbra. Horizontal bar represents median, n=3–4; *p<0.05.
CR2-fH mediated protection from cerebral injury persists to 7 days post reperfusion
Whereas C1-INH (38) and C3aR antagonist (39) have been shown to be neuroprotective in stroke models, with improved outcome maintained for 7 days after ischemia, lectin pathway deficiency has been shown to provide only acute (24 hour) protection after MCAO (40). To determine if CR2-fH-mediated protection persists into the subacute phase of stroke, neurological deficit and infarct volume was assessed at 7 days after MCAO. Compared to vehicle treated controls, mice receiving a single dose of CR2-fH at 30 minutes post reperfusion had significantly reduced mean infarct volumes, and displayed a strong trend toward neurological improvement (p=0.07) 7 days following MCAO (Fig. 9). These data indicate that CR2-fH treatment provides sustained neuroprotection, rather than just delaying cerebral ischemic injury.
Figure 9.

Effect of CR2-fH treatment on neurological deficit and infarct volume after 60 minutes of MCAO and 7 days reperfusion. (A) Neurological deficit. Horizontal bar represents median, n = 7; p=0.07. (B) Infarct volume. Horizontal bar indicates mean, n = 7; **p<0.01.
Discussion
This study demonstrates an important role for the alternative complement pathway in the pathogenesis of murine ischemic stroke. Although the alternative pathway can be spontaneously activated, the current data demonstrate that a functioning alternative pathway is not alone sufficient to initiate complement activation and propagate murine cerebral IRI. Rather, the alternative pathway serves to amplify complement activation initiated by the classical and/or lectin pathway. Previous data demonstrating that lectin pathway (14, 15) but not classical pathway (17, 6) deficient mice are protected from cerebral IRI, indicating that it is alternative pathway mediated amplification of the lectin pathway that drives injury in this model of ischemic stroke. Further, we have previously shown an important role for self-reactive natural IgM in propagating cerebral IRI (10), and others have shown that IgM mediates IRI via activation of the lectin pathway, at least following ischemia and reperfusion of the intestine and heart (11–13).
In this study, we also show that mice deficient in C6, a component protein of the MAC, are not protected from cerebral injury after 60 minutes MCAO and 24 hours reperfusion. This finding indicates that the terminal complement pathway and the MAC does not play a role in murine cerebral IRI after 60 minutes MCAO, and is in accord with a previous study that demonstrated mice deficient in CD59a, an inhibitor of the MAC, do not display exacerbated cerebral injury in the same model (22). It is interesting that in contrast to this finding, the MAC is implicated as an important mediator of IRI in various other organs and tissues (41, 42), as well as in other neuroinflammatory conditions such as spinal cord injury (33) and traumatic brain injury (43). Nevertheless, it should be noted that C6 deficiency is protective in a neonatal model of hypoxic-ischemic brain injury involving permanent ischemia, implicating a role for the MAC in a different stroke model (21). Furthermore, CD59a deficient mice show worse outcomes after 30 minutes MCAO, as opposed to 60 minutes MCAO, indicating that the terminal pathway may contribute to cerebral IRI under conditions of less severe injury/ischemia (22). Also, although outcomes were not different between wt and CD59a deficient mice after 60 minutes MCAO, the level of apoptosis was increased in CD59a deficient mice, and the authors suggested that MAC formation following severe ischemic stroke may be redundant and secondary to injury propagated by resident and infiltrating inflammatory cells. In this context, while our data indicate that the MAC is not essential for the progression of complement-dependent cerebral injury after 60 mins MCAO, we demonstrated that the protective effect of upstream blockade of the alternative pathway was associated with reduced levels of apoptosis and inflammation (discussed below). Further studies are needed to fully address the role of the MAC in ischemic stroke, which in experimental models appears to be dependent on the model and/or severity of injury.
The alternative pathway plays an important role in driving inflammation and propagating injury in many pathological conditions (reviewed in (18)), including neuroinflammatory conditions such as traumatic spinal cord injury (33), traumatic brain injury (44) and laser-induced choroidal neovascularization (45). Hallmarks of cerebral IRI (and inflammation in general) are the expression of endothelial adhesion molecules and the infiltration of leukocytes (35, 46, 47), both of which can be directly and indirectly modulated by complement activation products such as C3a and C5a. We demonstrated previously that C3 deficiency or CR2-Crry treatment (inhibits all complement pathways at C3 activation) protects against cerebral IRI and decreases the level of P-selectin expression and neutrophil infiltration and microthrombi formation after 60 minutes MCAO (5). Here we demonstrate that fB deficiency or specific inhibition of the alternative pathway has a similar effect, and we show no significant difference between CR2-fH and CR2-Crry treatment outcomes in terms of neurological deficit and infarct volume at 24 hours post reperfusion. In addition, the reduction in infarct volume and improvement in neurological score seen in CR2-fH treated mice at 24 hours post-reperfusion, was further improved at 72 hours, with improvements persisting for 7 days after reperfusion. Due to the fact that complement has many important physiological functions, specific inhibition of the alternative pathway will likely be a beneficial therapeutic approach compared to inhibition of all complement pathways, and is less likely to have undesirable side effects. In this regard, complement activation products have been shown to have protective roles against certain types neuronal injury (reviewed in (3)), and the classical and lectin pathways are known to play important roles in host defense against some pathogens, as well as provide some important regulatory and homeostatic functions (48, 49). For example, the classical and lectin pathways play a role in the removal of apoptotic and injured cells, important for the resolution of inflammation and tissue repair (50).
Recombinant tissue-type plasminogen activator (tPA) is the only approved pharmacological agent for the treatment of ischemic stroke, and in general it must be administered within 3 hours of symptom onset. tPA promotes reperfusion by enhancing the dissolution of blood clots, but there is the risk of uncontrollable intracranial hemorrhage, and for this reason physicians are often reluctant to use this drug. There is currently no intervention that can reverse the effects of tPA on acute hemorrhage. It is noteworthy that thrombolytic therapy has been shown to activate complement (51), and complement inhibition may thus have the potential to synergize with thrombolytic therapy, possibly even extending the window for tPA treatment. In this context, a human counterpart of CR2-fH, named TT30, is currently in phase 1 clinical trial for nocturnal paroxysmal hemoglobinuria (http://clinicaltrials.gov/ct2/results?term=tt30). TT30 has demonstrated neuroprotection in murine laser-induced choroidal neovascularization (a model of age-related macular degeneration) (52), and has been shown to prevent alternative pathway activity, C3 opsonin accumulation and MAC formation on red blood cells, without interfering with the classical and lectin activation pathways (53).
Acknowledgments
The authors would like to thank Dr. B. Paul Morgan for providing breeding pairs of C6 −/− and CD59a −/− mice.
This work was supported by grants from the National Institutes of Health (HL082485, HL86576) and the Department of Veterans Affairs (Merit Award NURC-051010F).
Abbreviations
- C3aR
C3a receptor
- C5aR
C5a receptor
- fB
factor B
- fH
factor H
- GFAP
glial fibrillary acidic protein
- IRI
ischemia-reperfusion injury
- MCAO
Middle cerebral artery occlusion
- MBL
mannose-binding lectin
- MAC
membrane attack complex
- wt
wildtype
References
- 1.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 2.Schaller B, Graf R. Cerebral ischemia and reperfusion: the pathophysiologic concept as a basis for clinical therapy. J Cereb Blood Flow Metab. 2004;24:351–371. doi: 10.1097/00004647-200404000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Brennan FH, Anderson AJ, Taylor SM, Woodruff TM, Ruitenberg MJ. Complement activation in the injured central nervous system: another dual-edged sword? J Neuroinflammation. 2012;9:137. doi: 10.1186/1742-2094-9-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang J, Kim LJ, Mealey R, Marsh HC, Jr, Zhang Y, Tenner AJ, Connolly ES, Jr, Pinsky DJ. Neuronal protection in stroke by an sLex-glycosylated complement inhibitory protein. Science. 1999;285:595–599. doi: 10.1126/science.285.5427.595. [DOI] [PubMed] [Google Scholar]
- 5.Atkinson C, Zhu H, Qiao F, Varela JC, Yu J, Song H, Kindy MS, Tomlinson S. Complement-dependent P-selectin expression and injury following ischemic stroke. J Immunol. 2006;177:7266–7274. doi: 10.4049/jimmunol.177.10.7266. [DOI] [PubMed] [Google Scholar]
- 6.Mocco J, Mack WJ, Ducruet AF, Sosunov SA, Sughrue ME, Hassid BG, Nair MN, Laufer I, Komotar RJ, Holland MCH, Pinsky DJ, Connolly JES. Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circ Res. 2006;99:209–217. doi: 10.1161/01.RES.0000232544.90675.42. [DOI] [PubMed] [Google Scholar]
- 7.Ducruet AF, Hassid BG, Mack WJ, Sosunov SA, Otten ML, Fusco DJ, Hickman ZL, Kim GH, Komotar RJ, Mocco J, Connolly ES. C3a receptor modulation of granulocyte infiltration after murine focal cerebral ischemia is reperfusion dependent. J Cereb Blood Flow Metab. 2008;28:1048–1058. doi: 10.1038/sj.jcbfm.9600608. [DOI] [PubMed] [Google Scholar]
- 8.Arumugam TV, Tang SC, Lathia JD, Cheng A, Mughal MR, Chigurupati S, Magnus T, Chan SL, Jo DG, Ouyang X, Fairlie DP, Granger DN, Vortmeyer A, Basta M, Mattson MP. Intravenous immunoglobulin (IVIG) protects the brain against experimental stroke by preventing complement-mediated neuronal cell death. Proc Natl Acad Sci U S A. 2007;104:14104–14109. doi: 10.1073/pnas.0700506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim GH, Mocco J, Hahn DK, Kellner CP, Komotar RJ, Ducruet AF, Mack WJ, Connolly ES., Jr Protective effect of C5a receptor inhibition after murine reperfused stroke. Neurosurgery. 2008;63:122–125. doi: 10.1227/01.NEU.0000335079.70222.8D. discussion 125-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elvington A, Atkinson C, Kulik L, Zhu H, Yu J, Kindy MS, Holers VM, Tomlinson S. Pathogenic natural antibodies propagate cerebral injury following ischemic stroke in mice. J Immunol. 2012;188:1460–1468. doi: 10.4049/jimmunol.1102132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McMullen ME, Hart ML, Walsh MC, Buras J, Takahashi K, Stahl GL. Mannose-binding lectin binds IgM to activate the lectin complement pathway in vitro and in vivo. Immunobiology. 2006;211:759–766. doi: 10.1016/j.imbio.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 12.Zhang M, Takahashi K, Alicot EM, Vorup-Jensen T, Kessler B, Thiel S, Jensenius JC, Ezekowitz RA, Moore FD, Carroll MC. Activation of the lectin pathway by natural IgM in a model of ischemia/reperfusion injury. J Immunol. 2006;177:4727–4734. doi: 10.4049/jimmunol.177.7.4727. [DOI] [PubMed] [Google Scholar]
- 13.Busche MN, Pavlov V, Takahashi K, Stahl GL. Myocardial ischemia and reperfusion injury is dependent on both IgM and mannose-binding lectin. Am J Physiol Heart Circ Physiol. 2009;297:H1853–1859. doi: 10.1152/ajpheart.00049.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cervera A, Planas AM, Justicia C, Urra X, Jensenius JC, Torres F, Lozano F, Chamorro A. Genetically-defined deficiency of mannose-binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLoS ONE. 2010;5:e8433. doi: 10.1371/journal.pone.0008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morrison H, Frye J, Davis-Gorman G, Funk J, McDonagh P, Stahl GL, Ritter L. The contribution of mannose binding lectin to reperfusion injury after ischemic stroke. Curr Neurovasc Res. 2011;8:52–63. doi: 10.2174/156720211794520260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osthoff M, Katan M, Fluri F, Schuetz P, Bingisser R, Kappos L, Steck AJ, Engelter ST, Mueller B, Christ-Crain M, Trendelenburg M. Mannose-binding lectin deficiency is associated with smaller infarction size and favorable outcome in ischemic stroke patients. PLoS ONE. 2011;6:e21338. doi: 10.1371/journal.pone.0021338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Simoni MG, Rossi E, Storini C, Pizzimenti S, Echart C, Bergamaschini L. The powerful neuroprotective action of C1-inhibitor on brain ischemia-reperfusion injury does not require C1q. Am J Pathol. 2004;164:1857–1863. doi: 10.1016/S0002-9440(10)63744-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thurman JM, V, Holers M. The central role of the alternative complement pathway in human disease. J Immunol. 2006;176:1305–1310. doi: 10.4049/jimmunol.176.3.1305. [DOI] [PubMed] [Google Scholar]
- 19.Holers VM. The spectrum of complement alternative pathway-mediated diseases. Immunol Rev. 2008;223:300–316. doi: 10.1111/j.1600-065X.2008.00641.x. [DOI] [PubMed] [Google Scholar]
- 20.Imm MD, Feldhoff PW, Feldhoff RC, Lassiter HA. The administration of complement component C9 augments post-ischemic cerebral infarction volume in neonatal rats. Neurosci Lett. 2002;325:175–178. doi: 10.1016/s0304-3940(02)00271-9. [DOI] [PubMed] [Google Scholar]
- 21.Ten VS, Yao J, Ratner V, Sosunov S, Fraser DA, Botto M, Sivasankar B, Morgan BP, Silverstein S, Stark R, Polin R, Vannucci SJ, Pinsky D, Starkov AA. Complement component c1q mediates mitochondria-driven oxidative stress in neonatal hypoxic-ischemic brain injury. J Neurosci. 2010;30:2077–2087. doi: 10.1523/JNEUROSCI.5249-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harhausen D, Khojasteh U, Stahel PF, Morgan BP, Nietfeld W, Dirnagl U, Trendelenburg G. Membrane attack complex inhibitor CD59a protects against focal cerebral ischemia in mice. J Neuroinflammation. 2010;7:15. doi: 10.1186/1742-2094-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Y, Qiao F, Atkinson C, Holers VM, Tomlinson S. A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. J Immunol. 2008;181:8068–8076. doi: 10.4049/jimmunol.181.11.8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banda NK, Levitt B, Glogowska MJ, Thurman JM, Takahashi K, Stahl GL, Tomlinson S, Arend WP, Holers VM. Targeted inhibition of the complement alternative pathway with complement receptor 2 and factor H attenuates collagen antibody-induced arthritis in mice. J Immunol. 2009;183:5928–5937. doi: 10.4049/jimmunol.0901826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fridkis-Hareli M, Storek M, Mazsaroff I, Risitano AM, Lundberg AS, Horvath CJ, Holers VM. Design and development of TT30, a novel C3d-targeted C3/C5 convertase inhibitor for treatment of human complement alternative pathway-mediated diseases. Blood. 2011;118:4705–4713. doi: 10.1182/blood-2011-06-359646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsumoto M, Fukuda W, Circolo A, Goellner J, Strauss-Schoenberger J, Wang X, Fujita S, Hidvegi T, Chaplin DD, Colten HR. Abrogation of the alternative complement pathway by targeted deletion of murine factor B. Proc Natl Acad Sci U S A. 1997;94:8720–8725. doi: 10.1073/pnas.94.16.8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banda NK, Takahashi K, Wood AK, Holers VM, Arend WP. Pathogenic complement activation in collagen antibody-induced arthritis in mice requires amplification by the alternative pathway. J Immunol. 2007;179:4101–4109. doi: 10.4049/jimmunol.179.6.4101. [DOI] [PubMed] [Google Scholar]
- 28.Bhole D, Stahl GL. Molecular basis for complement component 6 (C6) deficiency in rats and mice. Immunobiology. 2004;209:559–568. doi: 10.1016/j.imbio.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Lewis RD, Jackson CL, Morgan BP, Hughes TR. The membrane attack complex of complement drives the progression of atherosclerosis in apolipoprotein E knockout mice. Mol Immunol. 2010;47:1098–1105. doi: 10.1016/j.molimm.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holt DS, Botto M, Bygrave AE, Hanna SM, Walport MJ, Morgan BP. Targeted deletion of the CD59 gene causes spontaneous intravascular hemolysis and hemoglobinuria. Blood. 2001;98:442–449. doi: 10.1182/blood.v98.2.442. [DOI] [PubMed] [Google Scholar]
- 31.Atkinson C, Song H, Lu B, Qiao F, Burns TA, Holers VM, Tsokos GC, Tomlinson S. Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection. J Clin Invest. 2005;115:2444–2453. doi: 10.1172/JCI25208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hata R, Mies G, Wiessner C, Fritze K, Hesselbarth D, Brinker G, Hossmann KA. A reproducible model of middle cerebral artery occlusion in mice: hemodynamic, biochemical, and magnetic resonance imaging. J Cereb Blood Flow Metab. 1998;18:367–375. doi: 10.1097/00004647-199804000-00004. [DOI] [PubMed] [Google Scholar]
- 33.Qiao F, Atkinson C, Kindy MS, Shunmugavel A, Morgan BP, Song H, Tomlinson S. The alternative and terminal pathways of complement mediate post-traumatic spinal cord inflammation and injury. Am J Pathol. 2010;177:3061–3070. doi: 10.2353/ajpath.2010.100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mack WJ, Sughrue ME, Ducruet AF, Mocco J, Sosunov SA, Hassid BG, Silverberg JZ, Ten VS, Pinsky DJ, Connolly ES., Jr Temporal pattern of C1q deposition after transient focal cerebral ischemia. J Neurosci Res. 2006;83:883–889. doi: 10.1002/jnr.20775. [DOI] [PubMed] [Google Scholar]
- 35.Soriano SG, Coxon A, Wang YF, Frosch MP, Lipton SA, Hickey PR, Mayadas TN. Mice deficient in Mac-1 (CD11b/CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke. 1999;30:134–139. doi: 10.1161/01.str.30.1.134. [DOI] [PubMed] [Google Scholar]
- 36.Jauneau AC, Ischenko A, Chan P, Fontaine M. Complement component anaphylatoxins upregulate chemokine expression by human astrocytes. FEBS Lett. 2003;537:17–22. doi: 10.1016/s0014-5793(03)00060-7. [DOI] [PubMed] [Google Scholar]
- 37.Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- 38.Heydenreich N, Nolte MW, Gob E, Langhauser F, Hofmeister M, Kraft P, Albert-Weissenberger C, Brede M, Varallyay C, Gobel K, Meuth SG, Nieswandt B, Dickneite G, Stoll G, Kleinschnitz C. C1-inhibitor protects from brain ischemia-reperfusion injury by combined antiinflammatory and antithrombotic mechanisms. Stroke. 2012;43:2457–2467. doi: 10.1161/STROKEAHA.112.660340. [DOI] [PubMed] [Google Scholar]
- 39.Ducruet AF, Zacharia BE, Sosunov SA, Gigante PR, Yeh ML, Gorski JW, Otten ML, Hwang RY, Derosa PA, Hickman ZL, Sergot P, Connolly ES., Jr Complement Inhibition Promotes Endogenous Neurogenesis and Sustained Anti-Inflammatory Neuroprotection following Reperfused Stroke. PLoS ONE. 2012;7:e38664. doi: 10.1371/journal.pone.0038664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ducruet AF, Sosunov SA, Zacharia BE, Gorski J, Yeh ML, DeRosa P, Cohen G, Gigante PR, Connolly JES. The neuroprotective effect of genetic mannose-binding lectin deficiency is not sustained in the subacute phase of stroke. Transl Stroke Res. 2011;2:588–599. doi: 10.1007/s12975-011-0104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou W, Farrar CA, Abe K, Pratt JR, Marsh JE, Wang Y, Stahl GL, Sacks SH. Predominant role for C5b-9 in renal ischemia/reperfusion injury. J Clin Invest. 2000;105:1363–1371. doi: 10.1172/JCI8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Hu W, Xing W, You T, Xu J, Qin X, Peng Z. The protective role of CD59 and pathogenic role of complement in hepatic ischemia and reperfusion injury. Am J Pathol. 2011;179:2876–2884. doi: 10.1016/j.ajpath.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stahel PF, Flierl MA, Morgan BP, Persigehl I, Stoll C, Conrad C, Touban BM, Smith WR, Beauchamp K, Schmidt OI, Ertel W, Leinhase I. Absence of the complement regulatory molecule CD59a leads to exacerbated neuropathology after traumatic brain injury in mice. J Neuroinflammation. 2009;6:2. doi: 10.1186/1742-2094-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leinhase I, V, Holers M, Thurman JM, Harhausen D, Schmidt OI, Pietzcker M, Taha ME, Rittirsch D, Huber-Lang M, Smith WR, Ward PA, Stahel PF. Reduced neuronal cell death after experimental brain injury in mice lacking a functional alternative pathway of complement activation. BMC Neurosci. 2006;7:55. doi: 10.1186/1471-2202-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rohrer B, Coughlin B, Kunchithapautham K, Long Q, Tomlinson S, Takahashi K, Holers VM. The alternative pathway is required, but not alone sufficient, for retinal pathology in mouse laser-induced choroidal neovascularization. Mol Immunol. 2011;48:e1–8. doi: 10.1016/j.molimm.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ishikawa M, Cooper D, Arumugam TV, Zhang JH, Nanda A, Granger DN. Platelet-leukocyte-endothelial cell interactions after middle cerebral artery occlusion and reperfusion. J Cereb Blood Flow Metab. 2004;24:907–915. doi: 10.1097/01.WCB.0000132690.96836.7F. [DOI] [PubMed] [Google Scholar]
- 47.Jin AY, Tuor UI, Rushforth D, Kaur J, Muller RN, Petterson JL, Boutry S, Barber PA. Reduced blood brain barrier breakdown in P-selectin deficient mice following transient ischemic stroke: a future therapeutic target for treatment of stroke. BMC Neurosci. 2010;11:12. doi: 10.1186/1471-2202-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu JH, Teh BK, Wang L, Wang YN, Tan YS, Lai MC, Reid KB. The classical and regulatory functions of C1q in immunity and autoimmunity. Cell Mol Immunol. 2008;5:9–21. doi: 10.1038/cmi.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takahashi K. Mannose-binding lectin and the balance between immune protection and complication. Expert Rev Anti Infect Ther. 2011;9:1179–1190. doi: 10.1586/eri.11.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bohlson SS, Fraser DA, Tenner AJ. Complement proteins C1q and MBL are pattern recognition molecules that signal immediate and long-term protective immune functions. Mol Immunol. 2007;44:33–43. doi: 10.1016/j.molimm.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 51.Oikonomopoulou K, Ricklin D, Ward PA, Lambris JD. Interactions between coagulation and complement--their role in inflammation. Semin Immunopathol. 2012;34:151–165. doi: 10.1007/s00281-011-0280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rohrer B, Coughlin B, Bandyopadhyay M, Holers VM. Systemic Human CR2-Targeted Complement Alternative Pathway Inhibitor Ameliorates Mouse Laser-Induced Choroidal Neovascularization. J Ocul Pharmacol Ther. 2012;28:402–409. doi: 10.1089/jop.2011.0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Risitano AM, Notaro R, Pascariello C, Sica M, del Vecchio L, Horvath CJ, Fridkis-Hareli M, Selleri C, Lindorfer MA, Taylor RP, Luzzatto L, Holers VM. The complement receptor 2/factor H fusion protein TT30 protects paroxysmal nocturnal hemoglobinuria erythrocytes from complement-mediated hemolysis and C3 fragment. Blood. 2012;119:6307–6316. doi: 10.1182/blood-2011-12-398792. [DOI] [PubMed] [Google Scholar]