

Germline small RNAs called Piwi-interacting RNAs (piRNAs) silence transposable elements to provide genome protection. Tudor domain-containing 1 (TDRD1) is an interaction partner of mouse Piwi proteins and is implicated in piRNA biogenesis. The authors provide biochemical support for a model where only Tudor domains 2–4 of TDRD1 are effective in binding methylated ligands from a mouse Piwi protein MILI. Crystal structure of the third Tudor domain, in complex with a methylated MILI peptide, reveals how a symmetrically dimethylated arginine is recognized by the aromatic cage within the Tudor domain, while additional contacts with nonmethylated residues stabilize the interactions. SAXS analysis of the four tandem Tudor domains reveals an elongated, flexible structure that supports a molecular scaffold role for TDRD1, where it allows assembly of complexes containing Piwi proteins and piRNA biogenesis factors.
Keywords: piRNA, Piwi protein, extended Tudor domain, methylated arginine, X-ray crystallography
Abstract
Piwi-interacting RNAs (piRNAs) are small noncoding RNAs expressed in the germline of animals. They associate with Argonaute proteins of the Piwi subfamily, forming ribonucleoprotein complexes that are involved in maintaining genome integrity. The N-terminal region of some Piwi proteins contains symmetrically dimethylated arginines. This modification is thought to enable recruitment of Tudor domain-containing proteins (TDRDs), which might serve as platforms mediating interactions between various proteins in the piRNA pathway. We measured the binding affinity of the four individual extended Tudor domains (TDs) of murine TDRD1 protein for three different methylarginine-containing peptides from murine Piwi protein MILI. The results show a preference of TD2 and TD3 for consecutive MILI peptides, whereas TD4 and TD1 have, respectively, lower and very weak affinity for any peptide. The affinity of TD1 for methylarginine peptides can be restored by a single-point mutation back to the consensus aromatic cage sequence. These observations were confirmed by pull-down experiments with endogenous Piwi and Piwi-associated proteins. The crystal structure of TD3 bound to a methylated MILI peptide shows an unexpected orientation of the bound peptide, with additional contacts of nonmethylated residues being made outside of the aromatic cage, consistent with solution NMR titration experiments. Finally, the molecular envelope of the four tandem Tudor domains of TDRD1, derived from small angle scattering data, reveals a flexible, elongated shape for the protein. Overall, the results show that TDRD1 can accommodate different peptides from different proteins, and can therefore act as a scaffold protein for complex assembly in the piRNA pathway.
INTRODUCTION
Small RNAs associate with Argonaute proteins to form the RNA-induced silencing complexes (RISC) that mediate transcriptional or post-transcriptional silencing of their nucleic acid targets (Filipowicz et al. 2008). Based on their sequence conservation Argonautes can be partitioned into the Ago and Piwi clades (Carmell et al. 2002). Ago members associate with ∼21 nucleotide microRNAs (miRNAs) or small interfering RNAs (siRNAs) and are responsible for control of gene expression in all cell types. The Piwis, on the other hand, are restricted to the animal gonads, where they associate with ∼24–31 nucleotide Piwi-interacting RNAs (piRNAs). Together, they are implicated in controlling transposon mobility in worms, flies, fishes, amphibians, and mammals (Ghildiyal and Zamore 2009; Malone et al. 2009; Siomi et al. 2011). Loss of Piwi proteins or other factors acting in the pathway leads to activation of transposons, resulting in DNA damage, and finally causing sterility of the individual.
Through a combination of genetic and computational studies, it is now clear that long single-stranded RNAs transcribed from genomic loci called piRNA clusters are major sources of piRNAs (Aravin et al. 2006; Girard et al. 2006; Brennecke et al. 2007). Currently, it is thought that piRNA biogenesis can occur through distinct primary or secondary processing pathways (Senti and Brennecke 2010). Primary processing describes the generation of piRNAs from single-stranded RNA precursors by the action of an unknown nuclease(s), since it has been shown that unlike miRNAs and siRNAs, which have double-stranded precursors, piRNA production does not depend on Dicer or Drosha (Vagin et al. 2006; Houwing et al. 2007). Secondary processing is an elegant mechanism to harness transposon cleavage fragments for new piRNA production. Here, primary piRNA-guided Piwi endonuclease (slicer) action defines the 5′ end of sense secondary piRNAs, which can, in turn, generate the initiating antisense piRNA by cleaving cluster transcripts via the so-called ping-pong amplification cycle (Brennecke et al. 2007; Gunawardane et al. 2007). These piRNAs of opposing polarity usually enter distinct Piwi proteins, which in the case of flies is Aubergine (Aub) and Ago3. The ping-pong cycle in flies is essential to maintaining an overall antisense silencing potential to prevalent transposon populations (Li et al. 2009). In mice, Piwi proteins MILI and MIWI incorporate primary piRNAs, while MIWI2 depends on a functional engagement with MILI to generate its RNA partners via secondary biogenesis. Only piRNA-loaded MIWI2 is licensed for nuclear entry and transposon silencing by DNA methylation (Aravin et al. 2008; Kuramochi-Miyagawa et al. 2008).
Argonaute proteins can be structurally divided into the N-terminal region (N domain), PAZ, MID, and Piwi domains (Sashital and Doudna 2010). In all Argonautes, the small RNA 3′ and 5′ ends are anchored in the PAZ and MID domains, respectively. Also, in several Argonautes, the Piwi domain, which adopts an RNase H fold, harbors the slicer endonuclease activity (Song et al. 2003; Wang et al. 2009b). However, the Ago and Piwi clade members differ in their N domains. Piwi proteins uniquely carry several arginine–glycine (RG) and arginine–alanine (RA) dipeptides in their N domains, and several of these arginines have been shown to be post-translationally modified by methylation (Kirino et al. 2009; Reuter et al. 2009; Vagin et al. 2009). Although these are predominantly symmetrically dimethylated marks, some monomethylated arginines have also been found (Vagin et al. 2009). The symmetrically dimethylarginine (sDMA) marks, catalyzed by the methyltransferase PRMT5, act as affinity tags for members of the Tudor domain protein family (Kirino et al. 2009; Nishida et al. 2009; Reuter et al. 2009; Vagin et al. 2009; Wang et al. 2009a; Patil and Kai 2010; Handler et al. 2011).
The Tudor protein family is defined by the presence of the Tudor domain, which belongs to the “royal family” of modules that can specifically recognize methylated ligands to enable protein–protein interactions (Maurer-Stroh et al. 2003). They are implicated in RNA splicing, chromatin remodeling, and germline development. The prototypic Tudor domain is a ∼60 amino acid β-barrel that builds an aromatic cage capable of accommodating either methylated lysines or arginines (Kim et al. 2006). Structural studies have revealed that there are two distinct groups of Tudor domain-containing proteins, both of which can recognize ligands with methylated arginines. The group defined by the Spinal Motor Neuron (SMN) proteins contains the prototypic Tudor domain, sometimes in multiple copies, and recognizes only the methylated side chain, but not its peptide context (Selenko et al. 2001; Tripsianes et al. 2011). Only recently have the NMR structures of the minimal, prototypical Tudor domains of SMN and SPF30 (survival of motor neuron-related splicing factor 30) been determined in complex with single symmetrically and asymmetrically dimethylated arginine residues. The second group comprises so-called Tudor domain-containing (TDRD) proteins and is represented by the Drosophila TUDOR (Liu et al. 2010a) or the human Staphylococcal nuclease domain-containing 1 (SND1) (Liu et al. 2010b). These proteins contain one or more extended Tudor domains (eTud), typically of ∼180 residues, in which the prototypic Tudor module is fused to a staphylococcal nuclease (SN) domain. The conserved aromatic cage of the Tudor domain is responsible for specifically binding the symmetrically dimethylated arginine side chain, but flanking residues make additional contacts with the SN domain, modulating the affinity of different methylated arginine peptides to the eTud domain (Liu et al. 2010a,b; Chen et al. 2011). The various eTud-peptide structures determined show that there is considerable plasticity in exactly how the flanking regions of the methylated arginine peptide are bound. Furthermore, it has recently been shown that there are subtle, yet systematic, conformational differences between the mode of binding of methylated arginine by the conserved aromatic cage in eTud compared with prototypical Tudor domains (Tripsianes et al. 2011).
Several TDRD proteins were detected in fly, fish, and mouse Piwi complexes, with the Piwi proteins often displaying distinct specificities for different TDRDs (Nishida et al. 2009; Reuter et al. 2009; Vagin et al. 2009; Wang et al. 2009a; Patil and Kai 2010; Handler et al. 2011). Consistent with the involvement of Tudor domains, many such interactions were shown to depend on the presence of sDMAs on Piwi proteins. Both Piwi proteins and TDRDs are colocalized in several cytoplasmic perinuclear granules called the nuage, which are signature features of germ cells (Arkov and Ramos 2010). The TDRD proteins carry varying numbers of Tudor domains and are frequently associated with other domains that impart additional functions such as helicase activity or RNA binding (Handler et al. 2011). All of these suggest the potential to generate an intricate and dynamic network of interactions and assemblies that carry unique functions. The importance of these proteins for germline development is highlighted by the sterility observed in TDRD mutants, which is often accompanied by transposon derepression and disrupted nuage (Siomi et al. 2011).
Tudor domain-containing 1 (TDRD1) is a multidomain protein with an N-terminal MYND (myeloid translocation protein 8, Nervy, and DEAF-1) zinc finger domain, followed by four tandem extended Tudor domains, denoted TD1–4 (Fig. 1A; Chuma et al. 2003). Its expression tightly overlaps with that of Piwi proteins during mouse spermatogenesis, and it is reported to interact with all three Piwi proteins. In embryonic germ cells, TDRD1 associates with MILI and MIWI2, proteins that participate in secondary piRNA biogenesis (Vagin et al. 2009). Indeed, loss of Tdrd1 results in impaired biogenesis of MIWI2-bound piRNAs, reduced transposon DNA methylation, and LINE1 retrotransposon derepression (Reuter et al. 2009; Vagin et al. 2009). Such male mice are infertile. The fish ortholog of TDRD1 is also reported to associate with ping-pong Piwi partners (Huang et al. 2011). Zebrafish lacking Tdrd1 progressively lose germ cells and display transposon derepression, indicating a conserved role for TDRD1 in piRNA biogenesis.
FIGURE 1.

Binding affinity of sDMA containing peptides of MILI to individual TDRD1 eTud domains. (A) Schematic representation of mouse TDRD1 and the N terminus of MILI. TDRD1 has an N-terminal MYND domain and four tandem extended Tudor domains. The N terminus of MILI contains multiple arginine residues being mono- and symmetrically dimethylated (R highlighted in red). The three methylated peptides used in this study are indicated. (B) KDs derived from ITC measurements for three sDMA-containing peptides of MILI binding to the four individual eTud domains of mouse TDRD1. Error bars indicate the standard deviation of two independent measurements. (C) Representative ITC experiments and fits to the data for the four single eTud domains (TD1, TD2, TD3, and TD4) of TDRD1 with the R74me2 peptide of MILI. The data were fitted to a single-site binding model. (D) ITC data for the double eTud domains TD2–3 and TD3–4 with the R74me2 peptide and with the R4574me2s doubly methylated peptide of MILI. The sequence of R4574me2s is indicated below the corresponding curves. The model for fitting the tandem TD2–3 and TD3–4 domains with the R74me2 ligand assumes two different binding sites as the peptide binds differently to each individual Tudor domain. For the R4574me2s peptide binding, a single-site binding model was used (one double methylated peptide to one tandem domain).
Given that multiple sDMA residues in various sequence contexts decorate the N terminus of MILI, distinct specificities of the individual Tudor domains might drive complex formation. Here we used recombinant mouse TDRD1 Tudor domains, individually or in tandem, to examine their specificities for different sDMA-containing MILI peptides. Crystallographic studies of TD3 in its free and MILI sDMA-containing peptide-bound form reveal a unique orientation of the peptide as a whole and also of the sDMA in the aromatic cage. Interestingly, biochemical studies demonstrate that only TD2–4 of TDRD1 are functional in sDMA binding, while in TD1 a single amino acid substitution in the aromatic cage severely impairs sDMA binding. NMR experiments on the TD2 and TD3 domains confirm the crystallographic and thermodynamic findings of the interactions in solution. Finally, SAXS measurements on a construct containing all four tandem Tudor domains reveal a flexible elongated entity, lending support to a molecular scaffolding role for TDRD1 in the piRNA pathway.
RESULTS
Recognition of sDMA-peptides by single Tudor domains of TDRD1
The presence of multiple methylation marks at the N terminus of MILI prompted us to question how they interact with each eTud domain of TDRD1. To address this issue we first expressed and purified a construct spanning all four eTud domains of mouse TDRD1 (residues 232–1094). By partial proteolysis combined with bioinformatics analysis based on known eTud structure (Liu et al. 2010b), we identified well-behaved and soluble constructs of each of the four TDRD1 eTud domains separately (see Materials and Methods). We then measured by Isothermal Titration Calorimetry (ITC) the affinity of each single domain to three different MILI peptides previously shown to contain in vivo a symmetrical-dimethylated arginine (Vagin et al. 2009). The peptides tested were at successive positions and denoted R45me2 (38-GRAGPAGRme2GLVFR), R74me2 (69-LVSMFRme2GMGLDT), and R83me2 (76-MGLDTAFRme2PPSKR) (Fig. 1A). It is noteworthy that, whereas previous structural studies have used peptides in which the methylated arginines have been flanked on both sides by glycine or alanine residues (Liu et al. 2010a,b), this is only true of the R45me2 MILI peptide.
The ITC results are summarized in Figure 1B with representative experimental data and fits shown in Figure 1C. The R45me2 peptide had the highest affinity for TD2 (KD 55 μM) and TD3 (172 μM) domains, with TD4 having a slightly lower affinity (252 μM) and the TD1 domain showing poor binding (685 μM). The R74me2 peptide, in which the Rme2 is only flanked on one side by glycine, showed high affinity only for the TD3 domain (35 μM), with the other domains showing substantially weaker binding. Indeed, when we titrated the R74me2 against the tandem domain constructs TD2–3 and TD3–4 the measured KD of the major binding site in a two-site model is similar to that of the single TD3 domain (respectively, 45 and 51 μM), with a binding to a second site being six- to 10-fold weaker (Fig. 1D). The R83me2 peptide, which has an unusual Phe–Rme2–Pro tripeptide, showed low affinity to all single Tudor domains: TD3 domain had the lowest KD (245 μM), with TD2 (536 μM) and TD4 (831 μM) following. TD1 domain, as for the rest of the peptides, showed a much higher KD value (2.3 mM) (Fig. 1B). These results clearly indicate that the central TD2 and TD3 domains of TDRD1 have the highest affinities for the tested sDMA containing peptides of MILI, with TD4, and especially TD1, having significantly lower affinity.
TD2 and TD3 showed the highest affinity for consecutive singly arginine-methylated MILI peptides, respectively, R45me2 and R74me2. This prompted us to test the binding of tandem TD2–3 and TD3–4 domains to a doubly methylated peptide 43–77 of MILI (43-AGR45me2…FR74me2GMG-77, denoted R4574me2s), which encompasses both the original peptides, for indications of cooperativity. Satisfactory fits were achieved with one binding site (Fig. 1D) and gave a KD of 48.5 μM for TD2–3. This is comparable to the single TD2–R45me2 and TD3–R74me2 affinities (respectively, 55 and 35 μM), showing that there is no enhancement of affinity. For TD3-4, titration of R4574me2s showed a KD of 29 μΜ, which is comparable to the TD3–R74me2 interaction (35 μΜ) and consistent with, at most, weak cooperativity from TD4 binding (252 μΜ for R45me2). Closer scrutiny of the thermodynamic binding parameters shows a favorable additive effect of the binding enthalpies from the single Tudor domains to the double-domain constructs, but that this is compensated for by unfavorable entropic contributions. Taken together, these results suggest that the central TD2 and TD3 domains bind methylarginine peptides essentially independently.
Crystal structure of the TD3–R45me2 peptide complex
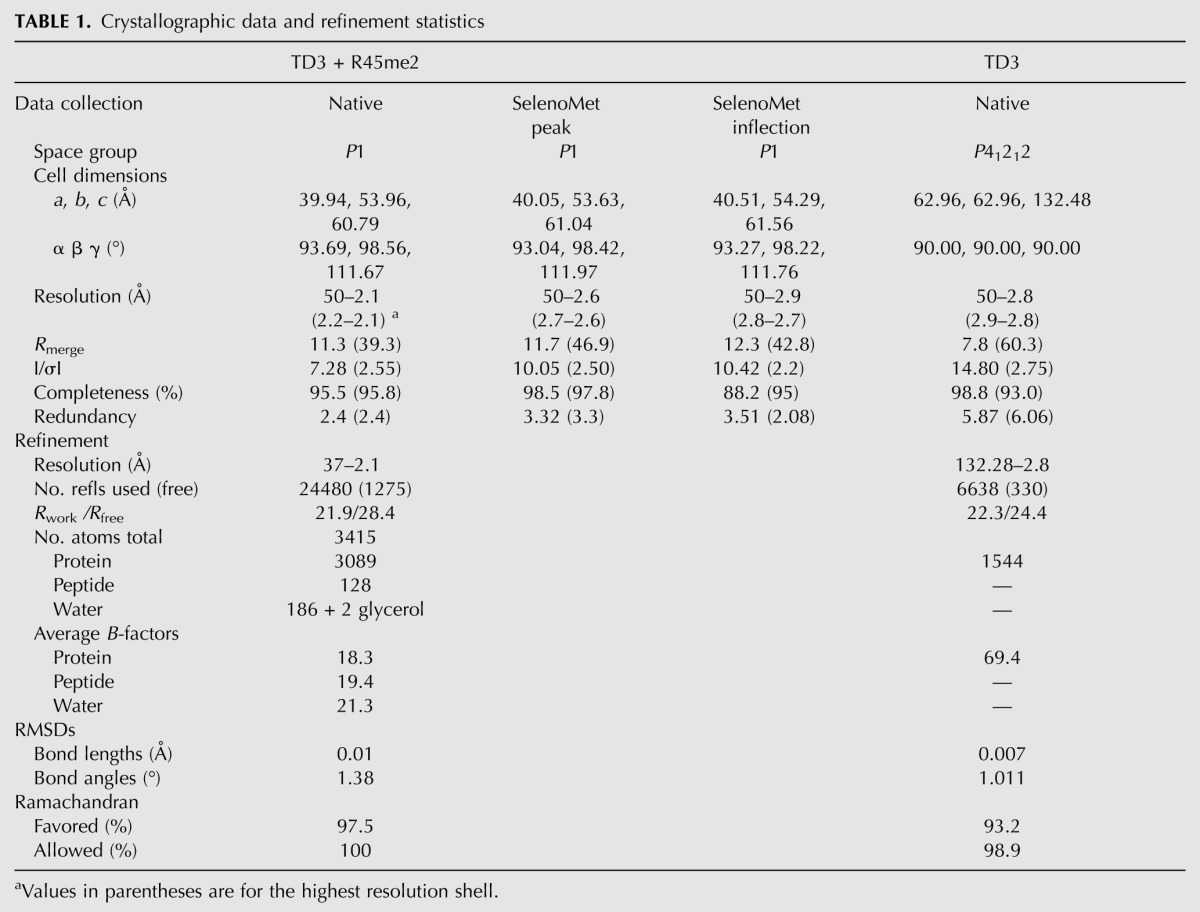
In order to better understand the recognition of MILI-derived sDMA-peptides by the Tudor domains of TDRD1, we tried to co-crystallize each of the domains with the different peptides. Only TD3 (residues 692–892), in the presence of the R45me2 peptide and without (residues 692–917), gave good quality crystals. Since molecular replacement with the known eTud structures did not work, the structure of the TD3–R45me2 peptide complex was solved de novo using selenomethionine-labeled protein and the MAD method and refined at 2.1 Å resolution (Table 1). Subsequently, the unbound TD3 domain was solved, in a different space group, by molecular replacement and refined at 2.8 Å resolution. Unexpectedly, the structure reveals a unique orientation of the bound peptide compared with the reported eTud–sDMA–peptide structures of fly TUDOR–Aub and human SND1–MIWI complexes (Liu et al. 2010a,b).
TABLE 1.
Crystallographic data and refinement statistics
The TDRD1 TD3 domain folds into a bilobal α/β-extended Tudor (eTud) structure with 196/201 residues visible (697–892). It comprises a Tudor core domain (residues 697–702, 731–818) linked by three connections to a SN (staphylococcal nuclease)-like domain (703–730, 819–892) (Fig. 2A). The peptide is bound in a cleft between these two lobes and makes contact with both. The SN-like domain of TD3 is comprised of two β strands (β1 and β2) exposed to the peptide-binding cleft, and three β strands (β8–10) and two α helices (α2 and α3) at the C-terminal part of the structure. The first two β-strands are connected to the Tudor core domain via a long α helix (α1). The Tudor core domain is a barrel comprised of four β-strands (β3–6), each of which contributes key residues to build the hydrophobic/aromatic cage that binds the methylated arginine side chain (Fig. 3A).
FIGURE 2.

TDRD1 TD3 structure in complex with R45me2 peptide. (A) Cartoon representation of TD3 (blue) in complex with the MILI R45me2 peptide (yellow). Secondary structure elements are labeled in white. The aromatic cage residues (orange) and the sDMA (yellow) are represented as sticks. (B) Superposition of the structures of TD3 (blue), SND1 (magenta), and eTud11 (cyan). The C-terminal helix of SND1 is absent from the other two structures. The different orientations of the distal end of the helix α2 could play a determining role in defining the orientation of the peptide. (C) The N-terminal extension (chocolate color) of TD3 makes a third connection between the Tudor core (left) and SN-like (right) subdomains against which it packs via large hydrophobic residues such as W699, W701, and F704 as shown. (D) Structure-based sequence alignment between TD3 (45r4), eTud11 (PDB code 3NTI), and SND1 (PDB code 3OMC). Residues with similarity above 70% are displayed in red.
FIGURE 3.

TD3 binds the R45me2 peptide in a different orientation from that previously observed in eTud domains–peptide complexes. (A) The sDMA in the aromatic cage of TD3 colored as in Figure 2A. The dipeptide GD stabilizing the cage and E798 interacting with N796 are also shown. (B) Details of the interactions between the R45me2 peptide and TD3. Colors are as in A with additionally residues of the SN-like domain interacting with the peptide being in magenta. Blue dashed lines indicate putative hydrogen bonds. (C) Structural comparison of the aromatic cages of TD3 (blue) and eTud11 (green) in complex with their corresponding peptides highlighting the different direction of entry of the two methylarginine groups. All other such structures exhibit the same conformation as eTud11/R13me2. (D) Diagram showing the orientation of various sDMA-containing peptides with reference to the TDRD1 TD3 structure (blue) obtained by superposition of eTud–peptide complexes of known structure. R14me2 (PDB 3OMG, green) and R4me2 (PDB 3OMC, cyan) were co-crystallized with the SND1 protein, while R15me2 (PDB 3NTI, magenta) and R13me2 (PDB 3NTH, wheat) were crystallized with eTud11 from Drosophila TUDOR. All of the above-mentioned peptides enter the aromatic pocket from the “bottom,” while the R45me2 peptide (yellow) enters from the “top” of the structure. The observed peptide residues in each structure are shown at the left with the sDMA highlighted in red. N- and C-termini of the peptides are also indicated. (E) Conserved residues in murine TDRD eTud domains. In a stick-model representation, the residues conserved in most of the eTud domains of TDRD proteins are shown (green). The absolutely conserved R775 is involved in extensive interactions with residues of the Tudor domain, thus stabilizing the aromatic cage. Blue dashed lines indicate hydrogen bonds.
As expected, the overall structure of TD3 resembles the known structures of the 11th extended Tudor (eTud11) domain of Drosophila TUDOR (PDB 3NTI) and human SND1 (PDB 2OMC) with RMSDs of, respectively, 1.72 Å and 1.61 Å, for, respectively, 153 and 173 matched residues (Fig. 2B). The TD3 SN-like domain superposes well with staphylococcal nuclease (PDB 3T16, RMSD 1.70 Å for 84 residues aligned), but, like eTud11, lacks the long C-terminal helix, which is, however, found in the SND1 structure. Despite the overall similarity there are some notable differences between TD3 and the other known eTud domains. Firstly, TD3 has an N-terminal extension (residues 697–707) beyond the N-terminal end of the eTud11 and SND1 structures, which crosses back from the SN-like domain to become an integral part of the Tudor core domain (Fig. 2C). Notably, aromatic residues W699 and W701 form hydrophobic interactions with F817 (Tudor core) and F704 with F727 and M820 (SN domain). K729 interacting with both E708 and the carbonyl oxygen of E703 additionally stabilizes the conformation of this unique extension. The extension, which is found identically positioned in the apo-structure, probably does not directly affect peptide binding since it is underneath the binding cleft. However, it presumably rigidifies the two-lobe structure since it provides a third connection between them. It also explains why constructs modeled on the eTud11 and SND1 structures were not well behaved since they lacked this extension, which is clearly important for stable folding of TD3. Furthermore, it defines a different direction for the linker to the TD2 domain. A second significant difference is the orientation of the helix α2. Compared with the eTud11 and SND1 structures, the C-terminal end of this helix is tilted away from the peptide-binding site (Fig. 2B). This is important in disfavoring contacts to peptides in the previously observed positions and favoring the quite different peptide path observed in the current structure (see below).
Recognition of the sDMA by the aromatic cage
The Tudor core domain of TD3 binds the sDMA of R45me2 via its aromatic cage. The residues forming the cage are highly conserved in all Tudor domains that are known to bind sDMAs. The methylated side chain enters the cage through an opening from the top of the structure and is involved in extensive cation-π and hydrophobic interactions with the aromatic residues F767, Y774, Y794, and F791, respectively, which constitute the cage (Fig. 3A). Y774 forms the back of the cage, with Y794 and F767 sandwiching the methylated side chain from each side. The hydroxyl group of Y794 hydrogen bonds to the of helix α1 residue N740. F791 and the highly conserved asparagine N796 form the ceiling of the binding pocket. The dipeptide, 769-GD, reinforces the cage with G769 peptide stacking on F767 and D770 hydrogen bonding to Y774 (Fig. 3A).
By superimposing the TD3–R45me2 structure with the eTud11–R13me2 or SND1–peptide structures, a remarkable observation is that the methylarginine groups do not overlay, but the methylated guanidine group is rotated 120° about the CZ carbon (Fig. 3C). This is because the side chain enters the cage from a completely different direction. Thus, one methyl group of TD3–R45me2 (CQ2, which points toward F791) coincides with CQ1 in the other structures, whereas CQ1 of TD3–R45me2 coincides with the CD in the other structures (and, reciprocally, the CD of TD3–R45me2 coincides with CQ2 in the other structures) (Fig. 3C). A consequence of this is that the conserved asparagine N796 (which is held in position by an interaction with E798) makes a hydrogen bond to the Nɛ atom of the arginine rather than to Nη1/2 as in other Tudor domain structures. Nevertheless, N796 remains critical for methylarginine binding, as reported for related structures, since the single mutant N796A is enough to significantly weaken the interaction with the R45me2 peptide (KD ∼1.4 mM) (Fig. 4A).
FIGURE 4.

“Active” and “inactive” extended Tudor domains of TDRD1. (A) KDs derived from ITC measurements for the binding of isolated symmetrically dimethylated arginine (Rme2) and R45me2 peptide to individual wild-type (wt) TDRD1 TDs and N325Y, Y774N, and N796A mutants. Error bars represent SD values from two experiments. (B) Representative ITC experiments and fits to the data. The four single eTud domains (TD1, TD2, TD3, and TD4) of TDRD1 with the isolated symmetrically dimethylated arginine (Rme2). (C) Multiple sequence alignment of the four individual Tudor domains of TDRD1. The first red arrow highlights the position of N325 of TD1, and the second arrow the Y774 of the aromatic cage of TD3. Mutations on both residues are critical for binding sDMAs (see A). (D) Binding of TD2 and TD3 domains of TDRD1 to sDMA-containing peptides monitored by NMR. Each panel shows an overlay of the 1H,15N HSQC spectra of the respective domain when free (black), when saturated with an excess of naked sDMA (red), and when saturated with MILI derived sDMA-containing peptides (green or cyan). For well-resolved peaks, chemical-shift perturbations arising from sDMA contacts are annotated with red arrows and those induced by interactions with flanking residues with black arrows in orange background. Notice that other peaks are affected by both sDMA and flanking residues but are not labeled. (E) Pull-down assays of endogenous murine proteins MILI, MIWI, and mouse Vasa homolog (MVH) by individual and multiple TDs of TDRD1. His-tagged constructs with four Tudor domains (TD1–4) and single Tudor domains (TD1, TD1 N325Y mutant TD2, TD3, TD4) were used. Size markers in kiladaltons are indicated.
Recently, solution NMR structures of the core Tudor domains of SMN and SPF30 bound to isolated sDMA or aDMA (asymmetrically dimethylarginine) residues have been described (Tripsianes et al. 2011). Interestingly, the orientation of the aliphatic part of sDMA bound to SMN (PDB 4A4E for SMN) is found to differ from that of aDMA (PDB 4A4G). sDMA is bound to SMN in a similar orientation to that observed in the previous eTud–sDMA peptide complexes (Liu et al. 2010b), except that the two methyl-groups are in the anti–anti conformation in SMN compared with the anti–syn conformation in the eTud complexes. It has been proposed that the latter difference is due to the systematic occurrence in the aromatic cage of single Tudor domain proteins of a tryptophan in place of the phenylalanine most commonly found in eTud domains (F767 in TD3) (Tripsianes et al. 2011). However, the aDMA side chain is bound in SMN in a rotated configuration, which is much closer to that observed for sDMA bound to TD3. In the TD3 complex, the sDMA nevertheless maintains the anti–syn conformation characteristic of eTud domains (Tripsianes et al. 2011). The significance of this correspondence with aDMA binding to core Tudor domains is not clear.
Orientation of the R45me2 peptide in the TD3 domain structure
The MILI R45me2 peptide in the crystal structure reveals seven ordered residues, 44-GRme2GLVFR-50, with A43 poorly ordered. As indicated above, it binds to TD3 in a unique orientation when compared with other known eTud/sDMA–peptide complexes. The C terminus of the peptide is at the “top” of the TD3 domain, whereas in the other structures, the peptide is bound at the “bottom” of the eTud domain (Fig. 3D). However, as in the other known structures, apart from the sDMA, the peptide makes additional contacts with residues in the SN domain as well as the Tudor core domain (Fig. 3B).
The peptide plane of G44p (p denotes a peptide residue), which precedes the methylated arginine, is stabilized by hydrophobic contact with the tip of F767, part of the aromatic cage. This interaction is a consequence of the rotated position of the methylated arginine in the cage and requires the presence of G769 (mentioned above) to avoid steric clash. In SND1 and eTud11 this glycine is substituted by a valine or glutamate, respectively, which would hinder peptide binding (Fig. 2D). Residues C-terminal to the R45me2 form an extended chain and interact principally with residues 716–718 from the distal part of strand β1 and residues 836–843 from the proximal part of helix α2 of the SN-like domain. G46p and L47p make hydrophobic interactions with M716 (a hydrophilic E or Q in other structures) and M717, respectively, both from strand β1, as well as Gln843 on helix α2 (Fig. 3B). The amino group of V48p hydrogen bonds with the carbonyl oxygen of M717, and its side chain makes hydrophobic interactions with M716 and Y718 (Fig. 3B). Next, the benzene ring of F49p, which is stacked on L47p, makes van der Waals contacts with K836, which is on helix α2. The last ordered amino acid of the peptide is R50p, which points out of the structure into the cleft between the SN-like domain and the Tudor core domain. Its amino group makes a hydrogen bond with the carbonyl oxygen of Y718 and its carbonyl group makes a water-mediated hydrogen bond with T839, which is also on helix α2. Finally, the side chain of R50p stacks on Y718 (strand β1) and makes a salt bridge with the carboxyl group of E722 (strand β2) (Fig. 3B). It should be pointed out that there are additional interactions of the peptide with a crystallographic related TD3 molecule (denoted *); these involve two main-chain interactions with the main chain of I781* and a salt bridge of Glu722* with R50p. For reasons given immediately below we do not think that these interactions strongly influence the mode of peptide binding observed.
A significant difference between the TD3 structure and those of eTud11 and SND1 is the orientation of helix α2 of the SN-like domain (Fig. 2B). For SND1 and eTud11 that accommodate their peptides from the bottom (Fig. 3D), the peptide is contacted by residues from the C-terminal (distal) end of this helix (e.g., Asn823 in SND1 equivalent to G848 in TD3) (Fig. 2D) as well as proximal parts of strand β1 (e.g., F686 in SND1, equivalent to V713 in TD3). In TD3 the helix is tilted such that its N-terminal (proximal) end contacts the peptide together with the distal part of strand β1 (as described above), but its distal end is shifted away from the binding cleft and does not contact the peptide (Figs. 2B, 3B,D). This appears to be an important structural adaptation together with, for instance, the presence of M716 and G769 (mentioned above), which permits and accommodates the unusual orientation of the peptide in the TD3 structure. In this respect, we also note that the bound R45me2 peptide is of the form Rme2GΦxΦx, where Φ is a largish hydrophobic residue (M, L, F), which is quite different from bound peptides in other known complex structures. Interestingly, the R74me2 peptide 74-Rme2GMGLD, which binds the best to TD3 (Fig. 1B), also has this pattern. This suggests that TD3 and the path the bound peptide takes are adapted to accommodate these hydrophobic residues. Indeed, some other nontested modified peptides of MILI conserve at least the first hydrophobic position, e.g., 95-Rme2GVLG, 100-Rme2GLSA, 163-Rme2GMDK. Thus, these peptides may all bind to TD3 in a similar way to that observed for the R45me2 peptide, although it is possible that other peptides without these special features might bind in a different manner.
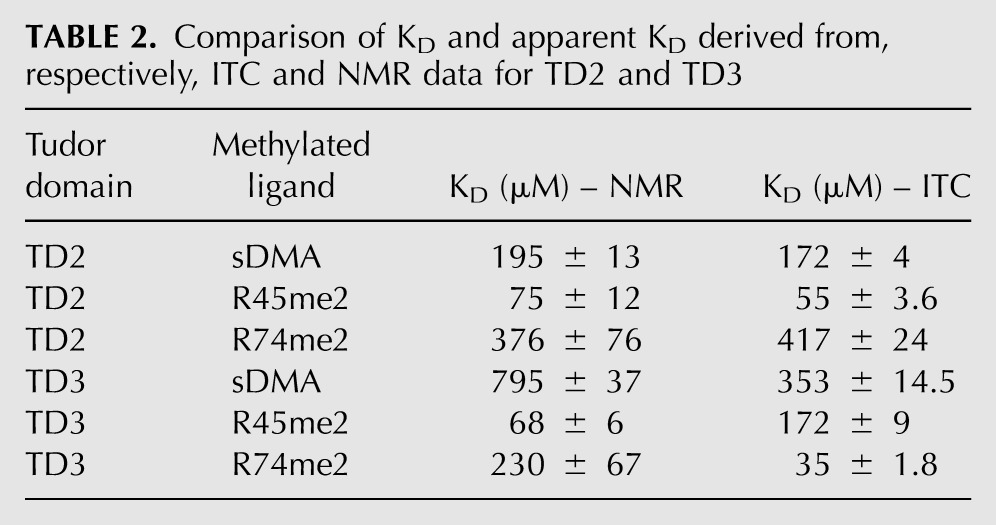
To test the hypothesis of a similar binding mode for R45me2 and R74me2 to TD3, we recorded solution NMR 1H,15N HSQC spectra to monitor the binding of sDMA and MILI sDMA-containing peptides to TD2 and TD3. Each domain binds the respective ligands (sDMA, R45me2, and R74me2) in the fast-exchange regime on the NMR chemical-shift time scale. In all cases the chemical-shift trajectories upon ligand titration are linear and occur at the same rate, indicating a single binding event (Supplemental Fig. 2). The apparent dissociation constants derived from the NMR titrations are generally in good agreement with those measured by ITC, except for an inversion of the relative affinities of TD3 for the two peptides (Table 2). By comparing the saturation points of each titration we can dissect chemical-shift perturbations originating solely by sDMA binding or, additionally, from contacts with the flanking residues present in the peptides (Fig. 4D). This analysis shows that for both TD2 and TD3 domains, the perturbations due to flanking residue interactions are the same irrespective of which peptide is bound (R45me2 or R74me2), indicating that a given eTud domain accommodates both peptides in the same orientation. For TD3 this is presumably the orientation observed in the crystal structure, although confirmation of this would require a full assignment of the NMR spectrum.
TABLE 2.
Comparison of KD and apparent KD derived from, respectively, ITC and NMR data for TD2 and TD3
Structure of unliganded TDRD1 TD3
We solved the structure at 2.8 Å resolution of the TDRD1 TD3 domain in the absence of bound peptide. The construct used was slightly longer (residues 692–917 instead of 692–892) and crystallized in a different space group. The structures are extremely similar (RMSD 0.65 Å for all 191 common visible residues), with small perturbations likely due to crystal contacts. The extra C-terminal residues are not observed, further suggesting that TD3 does not have the last C-terminal helix characteristic of SN domains, and is observed in SND1 but not eTud11. The peptide-binding site is unchanged except for a slight expansion of the aromatic cage, largely due to movement of the 767-FGD loop (Supplemental Fig. 1B). Thus, there is only a small element of induced fit in the peptide binding.
Conserved residues in eTud domains
The interactions of the R45me2 peptide, apart from that of the sDMA itself, clearly depend on the extended nature of TD3 and, indeed, it has previously been noted that the Tudor domains of the TDRD protein family are all found in the extended version (Liu et al. 2010a,b; Vourekas et al. 2010). We therefore used a multiple-sequence alignment of eTud domains from mouse TDRD family proteins to identify possible sequence signatures for such extended domains apart from the aromatic residues forming the cage (Supplemental Fig. 1A). Firstly, we identified highly conserved residues that seem important for establishing the eTud double-domain fold, and these include, in TD3, V714, 723-FY, L736, L743, R775, and 815-LP, whose location is indicated in Figure 3E. V714 and F723 are conserved residues from, respectively, strands β1 and β2 of the SN-like domain and form part of the hydrophobic core of the domain. Adjacent residue Y724 (W in several domains) makes van der Waals contacts across the interdomain interface with G795, a highly conserved feature of the aromatic cage. L736 and L743 are conserved hydrophobic residues found in the amphipathic helix α1 that connects the two domains and pack against the underside of the Tudor core domain, contributing to its hydrophobic core. Perhaps the most striking signature of the mouse eTud domains is the correlated conservation of R775, D793, and 815-LP (Supplemental Fig. 1A). The absolutely conserved R775 (adjacent to aromatic cage residue Y774) found in all eTud domains, but not in single Tudor domains, plays a key role in pinning together different parts of the structure and stabilizing formation of the aromatic cage. The side chain is largely buried, the aliphatic side chain makes hydrophobic contacts with L739, I808, and F817, and the guanidinium group makes a strong salt bridge with conserved D793 (adjacent to aromatic cage residue Y794), as well as three hydrogen bonds to the carbonyl oxygens of F812, L815, and P816 (Fig. 3E). The LP dipeptide is conserved in SND1, but not in eTud11. These considerations suggest that most of the Tudor domains of the mouse TDRD subfamily are, indeed, extended Tudor domains.
Secondly, we examined whether there are any conserved residues outside of the aromatic cage that are involved in extended peptide binding. Equivalent residues E2374 in eTud11 and E708 in SND1 (Fig. 2D), found in the connecting helix α1, interact via hydrogen bonds with the nonmethylated R11 in the R13me2 peptide and R6 in the R4me2 peptide, respectively (Liu et al. 2010a,b). In TD3, the equivalent residue D737 does not interact with the peptide. Furthermore, Q2365 in eTud11 and Q699 in SND1, at equivalent positions on strand β2, interact with R13 and A7 from, respectively, the R15me2 and R4me2 peptides. This residue is conserved as a glutamine in many mouse eTud domains, but is H726 in TD3 (Supplemental Fig. 1A). It does not bind the peptide, but preserves an important hydrogen-bond stabilizing residue, D765, one of the aromatic cage loop residues. The fact that these residues do not have conserved functions in TD3 with regard to peptide binding is not surprising given the very different mode of binding of the R45me2 peptide. Indeed, we have highlighted above adaptations in TD3 that appear to favor the mode of peptide interaction observed.
“Active” and “inactive” eTudor domains
Above, we have argued that all of the mouse TDRD domains are likely to have the eTud fold, but how many are actually active in binding sDMA-containing peptides? Our binding data clearly suggest that TD2, TD3, and TD4 can bind methylated arginine peptides of MILI, albeit with differing affinities. However, TD1 showed only weak binding to all of the peptides tested. To investigate this further we used ITC to measure the affinity of each separate TDRD1 eTud domain with free dimethylated (Rme2), monomethylated (MMA), or unmethylated arginines (Runmethylated) (Fig. 4A,B; Supplemental Fig. 3A). The single residues used were N-terminally acetylated and C-terminally amidated to mimic a peptide context. The isolated Rme2 bound to TD2, TD3, and TD4 with respective affinities of 172, 353, and 275.5 μM, consistent with the results for the peptides and confirming that the non-sDMA parts of the various peptides tested contribute significantly to the affinity (Fig. 4A). The TD1 domain showed the weakest binding with ∼830 μM affinity (Fig. 4A), again suggesting that it may not be involved in methylation-dependent interactions. As expected, the monomethylated (MMA) and unmethylated arginine (R-unmethylated) showed very low and undetectable binding, respectively, to each eTud domain (Supplemental Fig. 3A). This holds true also in a peptide context, since the unmethylated R74 peptide displayed no binding to the TD3 domain, thus highlighting the indispensable role of the symmetrically dimethylated arginine for these interactions (data not shown).
Inspection of the TD1 sequence in comparison to the other three TDRD1 domains shows that N325 is found in place of a usually conserved tyrosine (Y774 in TD3) in the aromatic cage (Fig. 4C). An asparagine at this position is found in mammalian (human, mouse, and bovine) TDRD1, but zebrafish and medaka TDRD1 maintain the tyrosine. To test whether this substitution was responsible for the observed low-binding affinity, we made the mutation N325Y in TD1. ITC measurements with an isolated Rme2 or with the R45me2 peptide show that this single substitution restores binding comparable to the best of the other eTud domains, with the affinity for Rme2 being 117 μM and for the R45me2 peptide 47 μM (Fig. 4A). On the other hand, when the reverse substitution, Y774N, was made in the TD3 domain, binding to the single Rme2 was very weak (2 mM), although the affinity to the R45me2 peptide was reduced from wild type, but not dramatically (270 μM), showing that the additional affinity for the non-sDMA residues could maintain the binding (Fig. 4A). This was not the case, however, for the TD3 mutation Y774A, which failed to bind to either Rme2 or R45me2 (data not shown).
The results above show that substitutions of the conserved asparagine (e.g., N796A in TD3) or an aromatic cage tyrosine (e.g., N325 in TD1, Y774A in TD3) drastically reduce sDMA binding, rendering the domain effectively “inactive.” Indeed, the signature motif of “active” Tudor domains appears to be FXnW(F)YRXnF(Y)XDY(F)GN, consistent with the fact that all reported crystal structures have these residues. On this basis we predict that at least half of the mouse TDRD protein eTud domains have very low or lack binding affinity for sDMA due to the presence of one or more significant deviations from the “active” motif (Supplemental Fig. 1A).
Interaction of endogenous murine proteins by TDRD1 eTud domains
In order to further investigate whether TD1 is “active” or not in the binding of methylated marks on piRNA pathway proteins, we performed pull-down assays from the lysate of adult mouse testes. We tested the binding of the four tandem eTud domains of TDRD1 (TD1–4), as well as each single eTud domain, to endogenous murine proteins MILI and MIWI, both of which are reported to contain sDMAs. Indeed, the presence of arginine modifications on these proteins was confirmed by Western blotting (Supplemental Fig. 3B). Our results show that both proteins can bind TD1 TD4 and single domains TD2, TD3, and TD4, presumably via the methylation marks, but only very weakly to TD1 (Fig. 4E). The germline-specific RNA helicase mouse Vasa homolog (MVH) is a secondary piRNA biogenesis factor (Kuramochi-Miyagawa et al. 2010) that is reported to carry both symmetrical and asymmetrical dimethylarginine modifications (Kirino et al. 2010); the presence of these marks was again verified by Western blotting (Supplemental Fig. 3B). MVH associates with TDRD1 and TDRD6 and is a component of MILI and MIWI complexes (Leroy et al. 1989; Tanaka et al. 2000; Kirino et al. 2010). As shown in Figure 4E, while MVH was retained on beads coated with TD2, TD3, or TD4, it failed to interact with TD1. Furthermore, when pull-down assays were performed with the “reactivated” TD1 domain mutant N325Y and MILI, we observed an increase of the binding signal, although still less than for TD2 and TD3 (Fig. 4E). Thus, we conclude that the TD1 domain of TDRD1 is unlikely to be involved in binding components of the piRNA pathway that are known to be arginine methylated, but it cannot be ruled out that it binds a peptide from some other associated protein.
SAXS analysis of the tandem Tudor domains of TDRD1
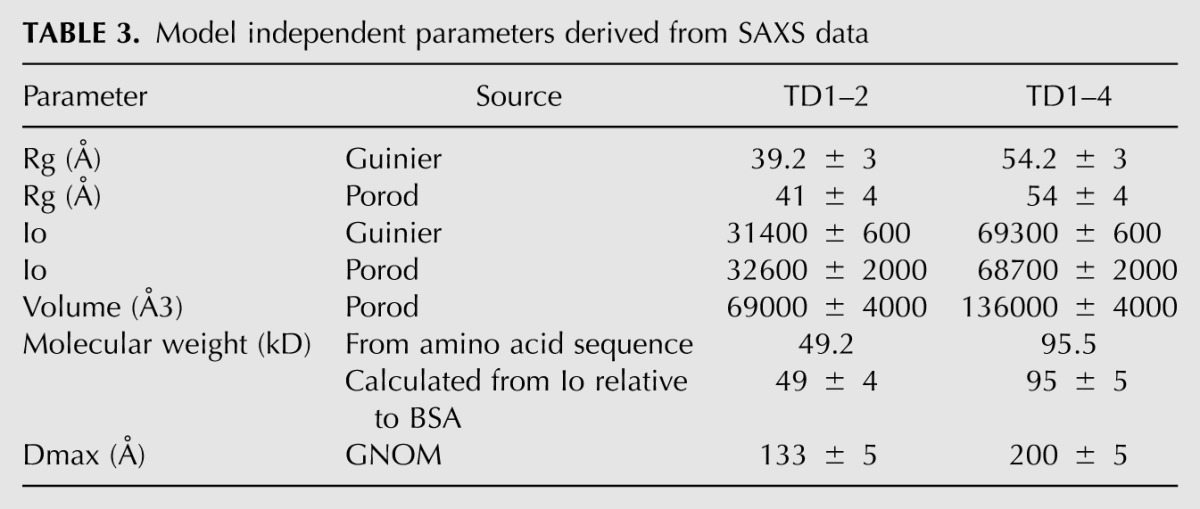
We used small-angle X-ray scattering (SAXS) to determine the overall conformation of the tandem Tudor domains of TDRD1 in solution (Fig. 5). SAXS data were measured on both the four-Tudor domain construct (TD1–4) and the tandem TD1 and TD2 domains (TD1–2). Ab initio modeling was done with DAMMIF, and 40 individual models (for both TD1–4 and TD1–2) were averaged using the program DAMAVER. The output of DAMAVER was refined using it as a starting model for the program DAMMIN to yield the final calculated envelope (see Materials and Methods). The envelope together with model independent parameters such as radius of gyration and Dmax (Table 3) show that TD1–4 has an elongated shape with four distinct lobes that are comparable in size to individual eTud domains and arranged end-to-end (Fig. 5A). The envelope derived from the TD1–2 data is consistent with a double-domain structure similar to either the first two or last two domains of TD1–4 (Fig. 5B).
FIGURE 5.

Overall architecture of the TD1–4 domains of TDRD1 as determined from small-angle scattering data. (A) TD1–4 ab initio models showing “most representative (filtered)” (gray) and “average” envelope (light blue) as given by DAMAVER. The rigid body model was produced using CORAL (Petoukhov and Svergun 2005). TD1 (salmon), TD2 (light red), and TD4 (deep blue) models were based on Tudor-SN (pdb code 2WAC). TD3 (light blue) was fitted using the crystal structure determined here. Flexible linkers connecting the eTud domains, positioned by CORAL, are depicted as spheres (olive green). (B) Ab initio model (orange) derived from the TD1-2 SAXS data. This is consistent with either half of the TD1–4 model. (C) SAXS data (black dots with error bars) and fits for TD1–4 and TD1–2 (solid lines). (Top) TD1–4 data. Fit of the ab initio model shown in A (gray) and the multidomain model created by CORAL (purple). (Bottom) TD1–2 data. Fit of the ab initio model shown in B (orange). Fits of the tandem TD1–2 and TD3–4 parts of the CORAL model are, respectively, shown in red and blue.
TABLE 3.
Model independent parameters derived from SAXS data
To better characterize the molecular envelope of TD1–4, we performed rigid body modeling using the program CORAL (Petoukhov and Svergun 2005). This program translates and rotates the atomic models of individual domains against the SAXS data, with the missing flexible interdomain linkers being modeled as random chains. Models for TD1, TD2, and TD4 were derived by homology modeling from the crystal structure of the D. melanogaster Tudor-SN protein (PDB code: 2WAC), while for TD3 the crystal structure reported here was used. Rigid body modeling simultaneously to both the TD1–4 and TD1–2 data gave χ2 of 2.17 for the TD1–4 data and χ2 of 1.96 and 1.82 for the TD1–2 data (respectively, modeled as TD1–2 or TD3–4) (Fig. 5A,C). The fit was not significantly improved, nor the four-domain model changed when only the TD1–4 data were used (χ2 = 2.05). Although the rigid body fits are of good quality, they are some systematic deviations that are likely due to flexibility of the interdomain linkages. Similarly, the CORAL model fits well within the independently derived ab initio TD1–4 average envelope, although there are some deviations, again consistent with flexibility, with respect to the most representative (filtered) envelope (Fig. 5A). These analyses show that the four-tandem eTud domains of TDRD1 are in an extended conformation in solution, with each domain being potentially accessible for peptide binding.
DISCUSSION
Transposon silencing in mice is initiated by de novo DNA methylation of transposon promoters in the embryonic germline. Repeat-derived piRNAs associating with the nuclear Piwi protein MIWI2 are implicated in specifying this event (Aravin et al. 2008; Kuramochi-Miyagawa et al. 2008). Primary biogenesis feeds many of the piRNAs entering MILI, and some of these can initiate generation of secondary piRNAs entering MIWI2 via the ping-pong pathway. Briefly, this involves the transfer of a target RNA endonucleolytic cleavage fragment from MILI RNPs to MIWI2, where it would mature as a new secondary piRNA. Two additional factors, TDRD1 and MVH, are also implicated in production of MIWI2-bound piRNAs. Infact, Mili, MiliDAH (MILI slicer inactive), Tdrd1, and Mvh mouse mutants all display a failure to load MIWI2, leading to it being mislocalized to the cytoplasm (Aravin et al. 2008; Kuramochi-Miyagawa et al. 2008; Reuter et al. 2009; Vagin et al. 2009; De Fazio et al. 2011). This leads to transposon derepression and hypomethylation of promoter elements in the mutants.
The occurrence of four-tandem extended Tudor domains in TDRD1 and a variety of arginine methylation marks on Piwi and Piwi-associated proteins poses the question of how interactions between the two could help mediate the formation of functional complexes in the piRNA pathway. One prevailing model proposes a molecular scaffold role for proteins like TDRD1, whereby each of the four Tudor domains might specify a unique interaction with a distinct binding partner, such as MILI and MIWI2, facilitating, for example, their interaction for ping-pong piRNA biogenesis. In this respect we note that although MIWI2 is detected in TDRD1 complexes (Vagin et al. 2009), it is unclear how it interacts, as arginine modification is not reported for MIWI2.
To provide further insight into the role of the tandem Tudor domains of TDRD1 we have measured the binding of each of the four domains separately to three different symmetrically dimethylated peptides of MILI and to isolated symmetrically dimethylated arginine. Our data show that whereas TD1 binds very poorly to all peptides, TD2, TD3, and TD4 bind with moderate mi□□o□□□□□micromolar affinity to the three peptides of MILI in vitro. More specifically, we find that whereas favorable flanking regions of a methylated peptide can enhance specificity only moderately upon binding of a single sDMA (e.g., factor of 1–3 for R45me2 or Rme2 binding to the four domains) (Fig. 4A), unfavorable flanking residues can decrease affinity, relative to Rme2. Consequently, different sDMA-containing peptides can modulate the affinity by, at most, an order of magnitude to a given domain, depending on the compatibility of these flanking residues with the binding site (Fig. 1B). Thus, there is some limited preference of a given domain for a given peptide, although it must be borne in mind that we have not tested all relevant methylated peptides. In particular, we suggest that TD3 preferentially recognizes peptides with hydrophobic residues just C-terminal to the sDMA. Furthermore, our ITC measurements revealed an apparent preference for binding of isolated TD2 and TD3 to consecutive MILI peptides, R45me2 and R74me2, respectively. Therefore, we examined whether TDRD1 can integrate inputs from multiple Tudor domains by measuring affinity of a fused peptide combining both of these methylation marks to the tandem TD2–TD3 domains. Somewhat surprisingly, no significantly enhanced affinity was observed, suggesting that binding to each domain is largely independent, probably due to peptide and interdomain flexibility. However, it should be borne in mind that this conclusion is based only on one combination of dually methylated peptide and tandem domains. Our pull-down experiments clearly show that domains TD2, TD3, and TD4 are individually each capable of interacting with endogenous, arginine-methylated MILI, MIWI, and MVH. Taking these results together, we conclude that a 1:1 exclusive interaction of a Tudor domain with a particular sDMA-peptide is unlikely. We favor the hypothesis that TDRD1 can act as a general scaffold capable of engaging one partner through a set of composite contacts or binding multiple partners simultaneously. These various options may be appropriate at different times to create unique complexes or interaction networks dynamically, depending on partner availability.
The crystal structure of the TD3–R45me2 complex unexpectedly reveals a completely different orientation of the peptide compared with other such structures, even with the methylated arginine entering the aromatic cage from a different direction. This is perhaps an adaptation to preferentially bind peptides with large hydrophobic residues just downstream from the methylated arginine as present in the R45me2 and R74me2 peptides and highlights the potential of the extended Tudor domain architecture to adopt multiple binding modes.
The elongated conformation of the SAXS model of TDRD1, with each Tudor domain accessible to receive its interacting partner, provides the strongest evidence to date of the molecular scaffold model. This is further supported by our binding studies with the various Tudor domain constructs and full-length Piwi proteins (MILI and MIWI) and biogenesis factors like MVH present in mouse testes lysates. Thus, it is likely that TDRD1 provides an environment in which the interacting proteins can engage in intermolecular interactions, driving biogenesis of piRNAs.
Our observation that TD1 of mouse TDRD1 is essentially unable to bind methylated arginine peptides is in concordance with previous predictions that many eTud domains in TDRD proteins lack the consensus aromatic cage residues (Liu et al. 2010a; Tripsianes et al. 2011). A single amino acid substitution (N325 instead of Y) within the aromatic cage of TD1 renders it ineffective. However, fishes (separated from humans by 450 million years) have conserved a tyrosine at the same position. As we demonstrated, changing N325 back to Y could restore methylated ligand-binding potential to the mouse TD1, suggesting that the “active” aromatic cage is the major determinant of eTud domain binding and, to a lesser extent, the flanking residues. The functional relevance of such “inactive” domains remains to be determined, but it is possible that they allow methylation-independent interaction with Piwi proteins or other piRNA pathway-associated factors. Indeed, the fly Tudor domain protein Yb lacks any “active” Tudor domains, but is still detected in Piwi complexes and is essential for primary piRNA biogenesis (Saito et al. 2010; Handler et al. 2011). All of these observations point to an evolving role for Tudor domains, some of which might be adopting new roles that go beyond recognition of methylated ligands.
MATERIALS AND METHODS
Protein expression and purification
TD1–4 (four tandem eTud domains of TDRD1)
The four tandem eTud domains of mouse TDRD1 (denoted TD1–4, residues 232–1094) were cloned into pETM11 vector to generate N-terminal His-tagged fusion protein. The recombinant protein was grown in Escherichia coli BL21 (DE3) cells at 37°C and induced with 1 mM IPTG at 20°C for 14 h. Purification was by affinity chromatography on nickel Sepharose fast-flow resin followed by TEV protease cleavage to remove the tag. A further purification step was made by size-exclusion chromatography. Finally, the protein was concentrated as required in buffer containing 20 mM Tris-HCl (pH 7.6), 0.15 M NaCl, and 1 mM DTT.
Identification of TDRD1 extended Tudor-domain pair and individual domain constructs
To experimentally define domain boundaries, multidomain constructs were iteratively subjected to limited proteolysis by trypsin with 1:1000 trypsin:protein ratio. Stable fragments were identified by N-terminal sequencing and mass spectrometry. Partial proteolysis of the TD1–4 construct led first to a stable fragment encompassing the first two Tudor domains (TD1–2, 232–677). Further limited proteolysis by trypsin on the TD1–2 construct gave rise to single domains TD1 (232–476) and TD2 (463–677). Digestion of a construct encompassing TD3–4 (692–1050) yielded a stable fragment containing TD3 (692–892) that was eventually crystallized with the R45me2 peptide. The apo-TD3 structure was obtained by crystallizing a longer construct (Apo-TD3, 692–917), which along with TD4 (916–1113) was designed taking into account secondary structure predictions. Once all single domains had been defined, these were used to design tandem domains 2 and 3 (TD2–3, 463–917) and 3 and 4 (TD3–4, 692–1113).
All of these constructs were cloned, expressed, and purified as described above for the TD1–4 construct. TD2, TD3, apo-TD3, and TD4 were concentrated to 15 mg/mL in a buffer containing 20 mM Tris-HCl (pH 7.6), 150 mM NaCl, and 1 mM DTT for crystallization trials.
Isothermal titration calorimetry (ITC) measurements
ITC measurements were performed in duplicates at 25°C, using an ITC200 Micro-calorimeter (MicroCal, Inc). Experiments included 26 injections of 1.5 μL of peptide solution (3–10 mM) into the sample cell containing 60–90 μM of proteins, in 25 mM Tris-HCl (pH 7.6), 150 mM NaCl, and 5 mM β-mercaptoethanol buffer. All peptides (Peptide Specialty Laboratories GmbH) used for ITC experiments were dissolved or dialyzed into the same buffer as the protein and, if necessary, the pH adjusted to correspond with that of the protein solution. Phenylalanine-containing peptide concentrations were estimated with absorbance spectroscopy using the extinction molar coefficient at 257 nm (195 Μ-1 cm-1). Otherwise, peptide concentrations were estimated from the mass. Control experiments were performed under identical conditions to determine the heat signals that arise from injecting the peptide into the buffer. Binding isotherms were fit by nonlinear regression using Origin Software version 7.0 (MicroCal, Inc). The initial data point was routinely deleted. The ITC data were fit to a one-site binding model, or in the case of tandem Tudor domains, to the sequential binding site model, using software provided by MicroCal (Wiseman et al. 1989; Turnbull and Daranas 2003).
Crystallization trials
Extensive crystallization trials were performed of the various single- and multiple-domain constructs with and without the three MILI sDMA-containing peptides using the EMBL High Throughput Crystallization Facility. Typically, 576 conditions were screened for each protein in 96-well sitting-drop vapor diffusion format. Only apo-TD3 and TD3 bound to the R45me2 peptide ultimately gave good quality crystals.
TD3–R45me2 co-crystallization and structure determination
The TD3 (692–892) complex with R45me2 peptide was prepared by mixing protein and peptide in a 1:5 molar ratio, and co-crystals grew at 20°C in 0.2 M ammonium acetate, 0.05 M sodium cacodylate (pH 6.5), 30% PEG8000, 0.01 M magnesium acetate tetrahydrate. Crystals of the SeMet-labeled protein in complex with the peptide were grown at 20°C in slightly different conditions, 0.2 M NaCl, 0.1 M Bis-Tris (pH 7.0), and 29% PEG 3350.
Crystals belong to the space group of P1 with two complexes per asymmetric unit. A complete native data set was collected to a resolution of 2.1 Å on beamline ID14-EH4 at the ESRF (Grenoble) and processed with XDS (Kabsch 1993). The structure was solved by a SeMet MAD experiment. Data sets with a resolution of 2.7–2.8 Å were collected at the inflection point and peak wavelengths of the Se K-edge. Six SeMet sites were identified using SHARP (de La Fortelle 1997) and phases improved using RESOLVE. The initial model was built manually using COOT (Emsley and Cowtan 2004) and finally automatically built by ARP-wARP (Perrakis et al. 1999). The structure was refined using Refmac5 (with TLS refinement) (Murshudov et al. 1997) to a final R-factor of 21.9% and Rfree of 28.4%, with all residues in allowed (97.7% in favored) regions of the Ramachandran plot, as analyzed by MOLPROBITY (Davis et al. 2004).
Apo-TD3 crystallization and structure determination
Unliganded TD3 (692–917) crystals were grown at 20°C in 10% PEG 1000 and 10% PEG 8000. They belong to space group P41212 and diffract to 2.8 Å resolution. A complete data set was collected on beamline ID14–EH4, processed with XDS, and the structure solved by molecular replacement with PHASER (McCoy et al. 2005) using the TD3/R45me2 complex structure as a search model. The structure was refined with Refmac5 (including TLS) to a final R-factor of 22.7% and Rfree of 25.5%. A total of 99.8% (95.8%) of residues are in allowed (favored) regions of the Ramachandran plot according to MOLPROBITY.
NMR spectroscopy
15N-labeled samples of TD2 and TD3 were prepared by growing cells in minimal medium supplemented with 15NH4Cl (1 g liter−1) as the sole nitrogen source and expressing and purifying as described above. In the final step of size exclusion chromatography, protein samples were exchanged into a buffer containing 20 mM sodium phosphate (pH 6.5), 50 mM NaCl, and 1 mM DTT.
1H,15N HSQC titrations of 15N-labeled TD2 or TD3 with successive addition of unlabeled ligands were performed on samples containing 200 μM protein to a final concentration ratio of 1:30 or 1:100 excess of the ligand (20 mM sodium phosphate at pH 6.5, 50 mM NaCl, and 7% (v/v) 2H2O added for the lock). Spectra were recorded at 298 K using an AV900 Bruker NMR spectrometer equipped with cryogenic triple resonance gradient probes.
SAXS data collection and analysis
Multidomain fragments containing the four Tudor domains (TD1–4) and Tudor domains 1 and 2 (TD1–2) were expressed and purified as described above. In the last purification step, Superdex 200 size exclusion column was used and 5 mM DTT was added before SAXS measurement. Data were collected at the ESRF BioSAXS station, ID14–EH3, which operates at a fixed energy (13.32 keV, λ = 0.931 Å) (Pernot et al. 2010). A total of 30 μL of protein solution was loaded into a 2-mm quartz capillary mounted in vacuum using an automated robotic system. The system enables the sample to flow through the beam during exposure to minimize the effect of radiation damage. A range of protein concentrations (10, 4.5, and 1.5 mg/mL) were measured to assess and account for interparticle effects. Two-dimensional scattering images were collected using a Vantec 2000 detector (Bruker) placed 1.83 m from the sample. For each sample, 10 frames of 30-sec duration were collected. Individual frames were processed automatically and independently using the software BsxCUBE, yielding individual radially averaged curves of normalized intensity versus scattering angle s = 4πSINθ/λ. Time frames are combined, excluding any data points affected by aggregation due to radiation damage, to give the average scattering curve for each measurement. The average scattering from the buffer alone, measured before and after each sample measurement, was used for background subtraction using the program PRIMUS (Konarev et al. 2003). For both the TD1–4 and TD1–2 data, 40 ab initio models were created with DAMMIF (Franke and Svergun 2009), which were averaged and filtered using DAMAVER (Volkov and Svergun 2003). Rigid body models were produced with CORAL (Petoukhov and Svergun 2005), which allows the addition of missing linkers using a database of random self-avoiding chains. The plots of the 1D fits were produced with SAXSview (saxsview.sourceforge.net) and the figures of the 3D models were produced with PYMOL.
DATA DEPOSITION
Atomic coordinates and structure factors for the TD3-R45me2 peptide complex and apo-TD3 have been deposited in the wwPDB with codes 4B9W and 4B9X, respectively.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank the ESRF–EMBL Joint Structural Biology Group for access to ESRF beamlines and the EMBL–ESRF–ILL–IBS Partnership for Structural Biology for access to structural biology instrumentation, notably the high-throughput crystallization platform. Fruitful discussions with Jordi Xiol and Michael Reuter are also acknowledged.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.034181.112.
REFERENCES
- Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, et al. 2006. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442: 203–207 [DOI] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Bourc'his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ 2008. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 31: 785–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkov AL, Ramos A 2010. Building RNA-protein granules: Insight from the germline. Trends Cell Biol 20: 482–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128: 1089–1103 [DOI] [PubMed] [Google Scholar]
- Carmell MA, Xuan Z, Zhang MQ, Hannon GJ 2002. The Argonaute family: Tentacles that reach into RNAi, developmental control, stem cell maintenance, and tumorigenesis. Genes Dev 16: 2733–2742 [DOI] [PubMed] [Google Scholar]
- Chen C, Nott TJ, Jin J, Pawson T 2011. Deciphering arginine methylation: Tudor tells the tale. Nat Rev Mol Cell Biol 12: 629–642 [DOI] [PubMed] [Google Scholar]
- Chuma S, Hiyoshi M, Yamamoto A, Hosokawa M, Takamune K, Nakatsuji N 2003. Mouse Tudor Repeat-1 (MTR-1) is a novel component of chromatoid bodies/nuages in male germ cells and forms a complex with snRNPs. Mech Dev 120: 979–990 [DOI] [PubMed] [Google Scholar]
- Davis IW, Murray LW, Richardson JS, Richardson DC 2004. MOLPROBITY: Structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res 32: W615–W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Fazio S, Bartonicek N, Di Giacomo M, Abreu-Goodger C, Sankar A, Funaya C, Antony C, Moreira PN, Enright AJ, O'Carroll D 2011. The endonuclease activity of Mili fuels piRNA amplification that silences LINE1 elements. Nature 480: 259–263 [DOI] [PubMed] [Google Scholar]
- de La Fortelle E, Bricogne G 1997. Heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. In Methods in Enzymology, Vol 276 (ed. CW Carter Jr, RM Sweet), pp. 472–494. Academic Press, Inc., San Diego, CA [Google Scholar]
- Emsley P, Cowtan K 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N 2008. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat Rev Genet 9: 102–114 [DOI] [PubMed] [Google Scholar]
- Franke D, Svergun DI 2009. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Crystallogr 42: 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal M, Zamore PD 2009. Small silencing RNAs: An expanding universe. Nat Rev Genet 10: 94–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard A, Sachidanandam R, Hannon GJ, Carmell MA 2006. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442: 199–202 [DOI] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, Siomi MC 2007. A slicer-mediated mechanism for repeat-associated siRNA 5′ end formation in Drosophila. Science 315: 1587–1590 [DOI] [PubMed] [Google Scholar]
- Handler D, Olivieri D, Novatchkova M, Gruber FS, Meixner K, Mechtler K, Stark A, Sachidanandam R, Brennecke J 2011. A systematic analysis of Drosophila TUDOR domain-containing proteins identifies Vreteno and the Tdrd12 family as essential primary piRNA pathway factors. EMBO J 30: 3977–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houwing S, Kamminga LM, Berezikov E, Cronembold D, Girard A, van den Elst H, Filippov DV, Blaser H, Raz E, Moens CB, et al. 2007. A role for piwi and piRNAs in germ cell maintenance and transposon silencing in zebrafish. Cell 129: 69–82 [DOI] [PubMed] [Google Scholar]
- Huang H, Gao Q, Peng X, Choi SY, Sarma K, Ren H, Morris AJ, Frohman MA 2011. piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Dev Cell 20: 376–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 26: 795–800 [Google Scholar]
- Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT 2006. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep 7: 397–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirino Y, Kim N, de Planell-Saguer M, Khandros E, Chiorean S, Klein PS, Rigoutsos I, Jongens TA, Mourelatos Z 2009. Arginine methylation of Piwi proteins, catalyzed by dPRMT5, is required for Ago3 and Aub stability. Nat Cell Biol 11: 652–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirino Y, Vourekas A, Kim N, de Lima Alves F, Rappsilber J, Klein PS, Jongens TA, Mourelatos Z 2010. Arginine methylation of Vasa protein is conserved across phyla. J Biol Chem 285: 8148–8154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI 2003. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36: 1277–1282 [Google Scholar]
- Kuramochi-Miyagawa S, Watanabe T, Gotoh K, Totoki Y, Toyoda A, Ikawa M, Asada N, Kojima K, Yamaguchi Y, Ijiri TW, et al. 2008. DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev 22: 908–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramochi-Miyagawa S, Watanabe T, Gotoh K, Takamatsu K, Chuma S, Kojima-Kita K, Shiromoto Y, Asada N, Toyoda A, Fujiyama A, et al. 2010. MVH in piRNA processing and gene silencing of retrotransposons. Genes Dev 24: 887–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy P, Alzari P, Sassoon D, Wolgemuth D, Fellous M 1989. The protein encoded by a murine male germ cell–specific transcript is a putative ATP-dependent RNA helicase. Cell 57: 549–559 [DOI] [PubMed] [Google Scholar]
- Li C, Vagin VV, Lee S, Xu J, Ma S, Xi H, Seitz H, Horwich MD, Syrzycka M, Honda BM, et al. 2009. Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell 137: 509–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wang JY, Huang Y, Li Z, Gong W, Lehmann R, Xu RM 2010a. Structural basis for methylarginine-dependent recognition of Aubergine by Tudor. Genes Dev 24: 1876–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Chen C, Guo Y, Lam R, Bian C, Xu C, Zhao DY, Jin J, MacKenzie F, Pawson T, et al. 2010b. Structural basis for recognition of arginine methylated Piwi proteins by the extended Tudor domain. Proc Natl Acad Sci 107: 18398–18403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Brennecke J, Dus M, Stark A, McCombie WR, Sachidanandam R, Hannon GJ 2009. Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell 137: 522–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer-Stroh S, Dickens NJ, Hughes-Davies L, Kouzarides T, Eisenhaber F, Ponting CP 2003. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem Sci 28: 69–74 [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ 2005. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr 61: 458–464 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Nishida KM, Okada TN, Kawamura T, Mituyama T, Kawamura Y, Inagaki S, Huang H, Chen D, Kodama T, Siomi H, et al. 2009. Functional involvement of Tudor and dPRMT5 in the piRNA processing pathway in Drosophila germlines. EMBO J 28: 3820–3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil VS, Kai T 2010. Repression of retroelements in Drosophila germline via piRNA pathway by the Tudor domain protein Tejas. Curr Biol 20: 724–730 [DOI] [PubMed] [Google Scholar]
- Pernot P, Theveneau P, Giraud T, Fernandes R, Nurizzo D, Spruce D, Surr J, McSweeney S, Round A, Felisaz F, et al. 2010. New beamline dedicated to solution scattering from biological macromolecules at the ESRF. J. Physics, Conference Series 247: doi: 10.1088/1742-6596/247/1/012009 [Google Scholar]
- Perrakis A, Morris R, Lamzin VS 1999. Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Petoukhov MV, Svergun DI 2005. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J 89: 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M, Chuma S, Tanaka T, Franz T, Stark A, Pillai RS 2009. Loss of the Mili-interacting Tudor domain–containing protein-1 activates transposons and alters the Mili-associated small RNA profile. Nat Struct Mol Biol 16: 639–646 [DOI] [PubMed] [Google Scholar]
- Saito K, Ishizu H, Komai M, Kotani H, Kawamura Y, Nishida KM, Siomi H, Siomi MC 2010. Roles for the Yb body components Armitage and Yb in primary piRNA biogenesis in Drosophila. Genes Dev 24: 2493–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sashital DG, Doudna JA 2010. Structural insights into RNA interference. Curr Opin Struct Biol 20: 90–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selenko P, Sprangers R, Stier G, Buhler D, Fischer U, Sattler M 2001. SMN Tudor domain structure and its interaction with the Sm proteins. Nat Struct Biol 8: 27–31 [DOI] [PubMed] [Google Scholar]
- Senti KA, Brennecke J 2010. The piRNA pathway: A fly's perspective on the guardian of the genome. Trends Genet 26: 499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi MC, Sato K, Pezic D, Aravin AA 2011. PIWI-interacting small RNAs: The vanguard of genome defence. Nat Rev Mol Cell Biol 12: 246–258 [DOI] [PubMed] [Google Scholar]
- Song JJ, Liu J, Tolia NH, Schneiderman J, Smith SK, Martienssen RA, Hannon GJ, Joshua-Tor L 2003. The crystal structure of the Argonaute2 PAZ domain reveals an RNA binding motif in RNAi effector complexes. Nat Struct Biol 10: 1026–1032 [DOI] [PubMed] [Google Scholar]
- Tanaka SS, Toyooka Y, Akasu R, Katoh-Fukui Y, Nakahara Y, Suzuki R, Yokoyama M, Noce T 2000. The mouse homolog of Drosophila Vasa is required for the development of male germ cells. Genes Dev 14: 841–853 [PMC free article] [PubMed] [Google Scholar]
- Tripsianes K, Madl T, Machyna M, Fessas D, Englbrecht C, Fischer U, Neugebauer KM, Sattler M 2011. Structural basis for dimethylarginine recognition by the Tudor domains of human SMN and SPF30 proteins. Nat Struct Mol Biol 18: 1414–1420 [DOI] [PubMed] [Google Scholar]
- Turnbull WB, Daranas AH 2003. On the value of c: Can low affinity systems be studied by isothermal titration calorimetry? J Am Chem Soc 125: 14859–14866 [DOI] [PubMed] [Google Scholar]
- Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V, Zamore PD 2006. A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313: 320–324 [DOI] [PubMed] [Google Scholar]
- Vagin VV, Wohlschlegel J, Qu J, Jonsson Z, Huang X, Chuma S, Girard A, Sachidanandam R, Hannon GJ, Aravin AA 2009. Proteomic analysis of murine Piwi proteins reveals a role for arginine methylation in specifying interaction with Tudor family members. Genes Dev 23: 1749–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI 2003. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr 36: 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vourekas A, Kirino Y, Mourelatos Z 2010. Elective affinities: A Tudor-Aubergine tale of germline partnership. Genes Dev 24: 1963–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Saxe JP, Tanaka T, Chuma S, Lin H 2009a. Mili interacts with Tudor Domain Containing Protein 1 in regulating spermatogenesis. Curr Biol 19: 640–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Juranek S, Li H, Sheng G, Wardle GS, Tuschl T, Patel DJ 2009b. Nucleation, propagation and cleavage of target RNAs in Ago silencing complexes. Nature 461: 754–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman T, Williston S, Brandts JF, Lin LN 1989. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem 179: 131–137 [DOI] [PubMed] [Google Scholar]