

Abstract
The proteins MDM2 and MDM4 are key negative regulators of the tumor suppressor protein p53, which are frequently upregulated in cancer cells. They inhibit the transactivation activity of p53 by binding separately or in concert to its transactivation domain. MDM2 is also a ubiquitin ligase that leads to the degradation of p53. Accordingly, MDM2 and MDM4 are important targets for drugs to inhibit their binding to p53. We found from in silico screening and confirmed by experiment that lithocholic acid (LCA) binds to the p53 binding sites of both MDM2 and MDM4 with a fivefold preference for MDM4. LCA is an endogenous steroidal bile acid, variously reported to have both carcinogenic and apoptotic activities. The comparison of LCA effects on apoptosis in HCT116 p53+/+ vs. p53-/- cells shows a predominantly p53-mediated induction of caspase-3/7. The dissociation constants are in the μM region, but only modest inhibition of binding of MDM2 and MDM4 is required to negate their upregulation because they have to compete with transcriptional coactivator p300 for binding to p53. Binding was weakened by structural changes in LCA, and so it may be a natural ligand of MDM2 and MDM4, raising the possibility that MDM proteins may be sensors for specific steroids.
Keywords: HDMX, virtual screening, natural product, cancer pathways, bile acid sensor
The tumor-suppressor protein p53 plays a pivotal role in cancer (1, 2). Often, its function is severely impaired by upregulation of its two key negative regulators, MDM2 and MDM4 (1–3). The N-terminal domains of MDM2 and MDM4 are structurally very similar and both bind to the same sequence in the intrinsically disordered N terminus of p53 (2, 4, 5). The binding cavities within MDM4 and MDM2 are important targets for drug therapy that releases them from p53 (6–10). Nutlins (11), for example, are potent MDM2 inhibitors and are potential therapeutics (12, 13) as well as being invaluable research tools for probing p53 pathways (12, 14), as are spiro-oxindoles, which were found by in silico methods (15, 16). MDM4 has a different specificity for small molecules, binding nutlins, for example, less tightly. Accordingly, MDM4-selective and dual MDM4/MDM2 inhibitors are also being sought (7, 17, 18). We searched for MDM4 inhibitors by structure-based in silico screening of binding (6, 19) and identified LCA as an endogenous inhibitor of both MDM4 and MDM2. LCA is a secondary bile acid formed by bacteria in the gut from its precursor chenodeoxycholic acid (CDCA, Fig S1). It has been variously reported to show both carcinogenic and apoptotic activities (20, 21). LCA is a rare example of a toxic endobiotic that is efficiently detoxicated by conjugation with taurine or glycine, sulfation at C-3 by the sulfotransferase SULT2A1, or metabolism through cytochrome P450 CYP3A enzymes (20). It induces its own metabolism by activating nuclear receptors like the vitamin D receptor (22) (VDR) and the farnesoid X receptor (23) (FXR). Thereby, it inhibits the synthesis of bile acids and promotes the transcription of genes encoding for sulfotransferases and CYP3A enzymes.
Results and Discussion
In Silico Screening.
Because there are varying conformational changes in MDM4 induced by different ligands (4, 24, 25), we applied an ensemble docking approach to four published crystal structures [Protein Data Base ID codes 3FEA (24), 2VYR (25), 3DAB (4), and 3FE7 (24)]. Starting with a virtual database of 3.6 million ready-to-order molecules, we excluded compounds with undesirable properties and docked 87,430 molecules to the ensemble of four MDM4 crystal structures using the program GOLD (26). Subsequently, we applied post-docking filters based on pharmacophore information to eliminate unfavorable binding modes and further narrowed down the list using an expert system (6, 19) (Fig. 1).
Fig. 1.

In silico screening workflow. A database of 3.6 million ready-to-order molecules was filtered to exclude compounds that are either reactive, prone to interfere with fluorescence-based assays or induce protein aggregation (34, 35). A total of 87,430 molecules were then docked to the N-terminal domain of human MDM4 in four crystal structures using GOLD (26, 36, 37) and two independent scoring functions [Goldscore (36) and Chemscore (38)]. Subsequently, post-docking filters based on pharmacophore information of protein–ligand complexes eliminated unfavorable docking poses. Docking scores were normalized and evaluated using an expert system (39).
Biophysical Screening and Characterization.
We screened 295 promising compounds from the final list in a fluorescence anisotropy assay, measuring the displacement of a carboxyfluorescein-labelled p53-N-terminal peptide (LTFEHYWAQLTS-FAM) from the N terminus of MDM4 (residues 16–116) or the N terminus of MDM2 (2–125). The dissociation constants of the labeled peptide were determined by direct titration on a microtiter plate reader (Fig. 2A) to be 12.5 ± 0.3 nM (MDM4) and 5.7 ± 1.2 nM (MDM2), consistent with literature values (27). The stoichiometry of binding of the labeled peptide to MDM4 was found by analytical ultracentrifugation experiments to be 1∶1 (Fig. 2B). Higher DMSO concentrations have a moderately attenuating effect on the binding of the peptide to MDM4 (Fig. 2D) and MDM2 (Fig. 2E). The dissociation constants of LCA were determined by a competition assay (Fig. 2C) to be 15.4 ± 0.6 μM and 66.0 ± 3.3 μM for the MDM4 and MDM2 complexes, respectively. LCA is a dual inhibitor of MDM4/MDM2 with a moderate preference for MDM4.
Fig. 2.

Biophysical characterization of the binding of lithocholic acid to MDM4. (A) Direct titration of 20 nM fluorescent-labeled peptide LTFEHYWAQLTS-FAM with MDM4 (16–116, C17S; circles) or MDM2 (2–125; squares) in the presence of 10% DMSO, 150 mM NaCl, 25 mM potassium phosphate pH 7.2, 5 mM DTT, and 0.2 mg/mL ovalbumin. Each data point represents the mean ± s.d. of 27 (MDM4) or 2 (MDM2) measurements. The KDs for MDM4 and MDM2 are 12.5 ± 0.3 nM and 5.7 ± 1.2 nM, respectively. (B) Analytical ultracentrifugation of 20 μM MDM4 (16–116, C17S) with 5 μM LTFEHYWAQLTS-FAM at a speed of 36,000 rpm and 283 K in the presence of 25 mM sodium phosphate pH 7.2, 150 mM sodium chloride and 5 mM DTT. The theoretical Mr of MDM4 (16–116, C17S; 11,478) bound to LTFEHYWAQLTS-FAM (1851) is 13,329, the calculated Mr of the protein-peptide complex from the experiment is 13,130 ± 170, based on 1∶1 binding stoichiometry. (C) Competitive displacement of the peptide LTFEHYWAQLTS-FAM from the p53-binding site of MDM4 (16–116, C17S; circles) or MDM2 (2–125; squares) by LCA in the presence of 10% DMSO, 150 mM NaCl, 25 mM potassium phosphate pH 7.2, 5 mM DTT, and 0.2 mg/mL ovalbumin, giving dissociation constants of 15.4 ± 0.6 μM and 66.0 ± 3.3 μM, respectively. (D/E) Direct titration of LTFEHYWAQLTS-FAM with (D) MDM4 or (E) MDM2 in 150 mM NaCl, 25 mM potassium phosphate pH 7.2, 5 mM DTT, 0.2 mg/mL ovalbumin and varying amounts of DMSO (5–20%).
We further characterized the binding of LCA to MDM4 by NMR and 1H/15N-HSQC spectra (Fig. 3 A–C). Twenty signals related to the binding cavity shifted significantly as the ligand was increased from 0 to 291 μM in 10 increments, giving a KD of 12.4 ± 1.4 μM (Fig. 3C). MDM4 was progressively stabilized by binding to increasing concentrations of LCA, as found by differential scanning calorimetry measurements (Fig. 3 D and E), the Tm of MDM4 is raised by approximately 1.5 K at 100 μM LCA.
Fig. 3.

(A–C) Concentration-dependent chemical shifts in 15N/1H-HSQC NMR spectra with 70 μM 15N-labeled MDM4, 150 mM NaCl, 25 mM potassium phosphate pH 7.2, and 5 mM DTT. The dissociation constant calculated from these shifts was 12.4 ± 1.4 μM. (D) Differential scanning calorimetry experiments with 70 μM MDM4 and LCA concentrations of 0 μM (black), 5 μM (red), 10 μM (green), 20 μM (cyan), 40 μM (blue), 60 μM (pink), 80 μM (dark red), and 100 μM (orange) in a buffer containing of 5% DMSO, 150 mM NaCl, 25 mM potassium phosphate pH 7.2, and 1 mM TCEP. The value of the apparent Tm is above each curve. (E) The data are fitted to the equation: T = Tm/(1 - (R/ΔSD–N(Tm)) ln(1 + [L]/KD)), where T is the observed melting temperature, Tm that in the absence of ligand L, KD its dissociation constant, and ΔSD–N(Tm) the entropy of denaturation at Tm (6). The resulting KD is found to be 3.8 ± 2.7 μM. (F) Electrostatic potential mapped on the isodensity surface of LCA in the chair-chair-chair-envelope conformation (geometry optimized at TPSS-D/TZVPP level). Blue—negative electrostatic potential to red—positive electrostatic potential. (G) Structure of lithocholic acid.
We investigated whether steroids, in general, and bile acids, in particular, are ligands of MDM4. We measured the binding to MDM2 and MDM4 of more than 50 steroids, including approximately 25 very close structural derivatives of LCA. The most significant binding analogues are summarized in Fig. 4. Modifications of the hydroxyl function in position 3 gave a somewhat greater than sevenfold increase in KD. There were higher losses of affinity on hydroxylations in ring B or C as well as on introduction of double bonds. Conjugation of the bile acids and capping of the side chain gave dramatic loss of binding. LCA was the highest affinity ligand, suggesting it may be a specific MDM4/2 ligand.
Fig. 4.

Specificity of binding of lithocholic acid to MDM4. The affinity of LCA is significantly decreased upon: structural variations of the 3α-hydroxyl function (blue); hydroxylation of the steroid scaffold at positions C-5, C-6, and C-12; introduction of a double bond between C-5 and C-6; truncation of the 4-substituted pentanoic acid moiety; and conjugation of LCA with taurine.
The dissociation constants of LCA do not, at first sight, seem low enough for moderate concentrations to displace tightly bound MDM2 or MDM4 from p53. But this is illusory. If the dissociation constant of MDM4 from p53 is KD(MDM4) and that of LCA KD(LCA), then the effective value of KD(MDM4) in the presence of competing LCA is KD(MDM4)effective = (1 + [LCA]/KD(LCA))xKD(MDM4) (28). If MDM4 is in competition with another protein P for the binding to limiting concentrations of p53, then [P.p53]/[MDM4.p53] = ([P]/KD(P))/([MDM4]/KD(MDM4)effective) (28). For example, when the concentration of LCA = KD(LCA) it raises the effective dissociation constant of MDM4 by a factor of 2, equivalent to lowering its concentration twofold. Because MDM4 and MDM2 are in competition with p300 and other proteins for the binding to p53, the raising of KD(MDM4)effective by LCA at concentrations equal to and above KD(LCA) will be highly important in the balance between transcription of cell-cycle arrest and proapoptotic genes and the MDM2/4 mediated inhibition and degradation of p53. That inhibition would account for the proapoptotic activity of LCA. Single phosphorylations of the N terminus of p53 change the KDs of p300 domains and MDM2 by only small factors (29).
Cellular Studies of Apoptosis Induction.
We examined the effects of LCA on the induction of apoptosis in the human colorectal carcinoma cell line HCT116. Monitoring caspase-3/7 activity through a TriplexGlo assay, we found significantly increased caspase activation for concentrations above 150 μM (Fig. 5A). Concentrations of 250 μM LCA or higher produced a maximal level (> eightfold elevation) of caspase activity. To differentiate between p53 pathway-mediated effects of LCA and complementary ways of apoptosis induction, we treated the isogenic HCT116 p53-/- cell line with identical concentrations of LCA. We observed a comparably small induction of caspase activity with a maximal 2.6-fold increase at 300 μM. These data indicate that the induction of apoptosis by LCA observed in HCT116 p53+/+ cells is predominantly p53-pathway mediated.
Fig. 5.

Effects of LCA (A), Nutlin-3 (B), and HDCA (D) on caspase 3/7 activity in the human colorectal carcinoma cell line HCT116 (gray columns) and the isogenic HCT116 p53-/- cell line (dark gray columns). Comparison of LCA (gray columns) vs. HDCA effects (dark gray columns) on HCT116 p53+/+ is shown in (C). After 24 h treatment time apoptosis is measured by luminescence of a luminogenic substrate cleaved by caspase 3/7 following reaction with luciferase. Data are normalized to a DMSO-only control and are expressed as mean ± SEM (n = 4). Asterisks denote significant differences between treatments as revealed by one-way ANOVA and Bonferroni post hoc test (*** p < 0.001; ** p < 0.01; * p < 0.05).
As a reference compound we used Nutlin-3, which binds tightly to MDM2 and more weakly to MDM4 (30, 31). In HCT116 p53+/+ cells, we found a substantial (> ninefold) increase in caspase-3/7 activity at 50 μM inhibitor concentration (Fig. 5B). However, at lower concentrations, no significant caspase induction was detectable. At concentrations of 50 μM Nutlin-3 or higher, increased caspase activity (up to fourfold) was also observed in HCT116 p53-/- cells, indicating contributions of p53-independent pathways to apoptosis. In conclusion, p53-mediated induction of apoptosis seems to be dominant for both LCA and Nutlin-3.
To investigate whether the observed effects of LCA are due to the inhibition of MDM4, we tested the close structural analog hyodeoxycholic acid (HDCA) that did not bind to MDM4 in vitro (Table S1). The structure of HDCA is identical to LCA, apart from the additional hydroxyl group at C6 and is therefore useful as a control for target-unspecific bile acid effects. Even at high HDCA concentrations (up to 400 μM), no caspase 3/7 response was detectable (Fig. 5C). This correlates well with the lack of affinity of HDCA toward MDM4. Interestingly, HDCA also did not show any p53-independent induction of apoptosis (Fig. 5D), suggesting that increased caspase 3/7 activation by LCA in HCT116 p53-/- cells may not be caused by a target-unspecific bile acid effect.
Conclusions
All those data are consistent with activation of p53 by LCA. The promotion of apoptosis by LCA is explained by its binding to MDM4 and MDM2. Whether or not that function of LCA is a side reaction or a biological response is intriguing and remains to be discovered. There is the possibility that members of the MDM family of proteins are sterol receptors.
Methods
Protein Expression and Purification.
The N terminus of human MDM4 protein (residues 16–116, with the stabilizing mutation C17S) was expressed and purified as described previously (25). Briefly, the N-terminal fusion protein (6xHis/lipoyl domain/TEV protease cleavage site) was overexpressed using Escherichia coli C41 cells in 2xTY medium at 20 °C for 16 h and purified using standard Ni-affinity chromatography protocols. After overnight digestion with TEV protease, the 6xHis/lipoyl domain was removed by a second Ni-affinity chromatography step. Finally, gel filtration chromatography using a Superdex 75 16/60 preparative gel filtration column (GE Healthcare) was done. The molecular weight and the protein purity of > 95% were determined by SDS gel electrophoresis, MALDI-TOF-MS, and ESI–MS. For 1H/15N HSQC experiments the protein expression was carried out in M9 minimal media with 15N-labeled ammonium chloride as the only nitrogen source.
The plasmid encoding the N terminus of MDM2 (residues 2–125) was a gift from Dr. Marina Vaysburd (MRC LMB, Cambridge). The protein was expressed and purified in the same way except for the 6xHis/lipoyl domain tag was substituted with a GST tag, the affinity tag was cleaved with thrombin, and GST affinity chromatography was used.
Peptide Synthesis.
The p53-derived peptide LTFEHYWAQLTS (27) was synthesized by solid-phase peptide synthesis using standard FMOC chemistry, labeled with 5-carboxyfluorescein at the α-amino group of serine, and purified by reversed-phase chromatography. The molecular weight of the labeled peptide was determined by MALDI-TOF MS with α-cyano-4-hydroxycinnamic acid in 50% acetonitrile/water with 0.1% trifluoroacetic acid as a matrix.
Fluorescence Anisotropy Spectroscopy.
The compound screen was carried out as titrations in 96-well plates (Corning 3650) using a Pherastar plate reader (BMG Labtech, Germany) with a 480/520-nm fluorescence polarization module and a Bravo 96-channel pipetting robot (Velocity 11). Buffer conditions for the screen were 25 mM potassium phosphate pH 7.2, 150 mM NaCl, 5 mM DTT, 0.2 mg/mL ovalbumin, and 5% v/v DMSO. For MDM4 and MDM2 screens, protein concentrations of 30 nM and peptide concentrations of 20 nM were used. These concentrations, as well as all buffer conditions were kept constant during the titration, only the compound concentration was varied in 25 steps from 0–1 mM. This was done by aspirating the same volume of the sample prior to addition of an aliquot of compound. All titrations were done at 22 °C. To minimize the errors associated with handling small volumes (< 1 μL), 200 μM stock of compound was used for the first part of the titration, switching to 2 mM for the second part. The compound stock microtiter plates were prepared using an epMotion 5070 pipetting robot (Eppendorf AG).
The direct titration of LTFEHYWAQLTS-FAM with MDM4 and MDM2, respectively, was performed under the same buffer conditions with a final protein concentration of 250 nM, 500 nM, 1,000 nM, and 2,000 nM protein and 20 nM peptide. The content of DMSO was varied to study the influence of DMSO on the dissociation constant (Fig. 2 D and E). The Z factor under final assay conditions was 0.74, indicating that the assay is well suited for discriminating between actives vs. inactives (32). Dissociation constants were calculated from the FP-adapted Cheng-Prusoff equation (33) (http://sw16.im.med.umich.edu/software/calc_ki/index.jsp).
Nuclear Magnetic Resonance Spectroscopy.
Compounds were dissolved in d6-DMSO at 40 mM. NMR samples were freshly prepared by adding dilutions of stock solution to a buffer containing 25 mM potassium phosphate (pH 7.2) 150 mM NaCl, 5 mM DTT, and a final concentration of 5% (v/v) DMSO-d6. 1H/15N-HSQC spectra of MDM4 (70 μM) were acquired at 20 °C on a Bruker Avance-800 spectrometer (800 MHz 1H frequency) using a 5-mm inverse cryogenic probe.
The average weighted 1H/15N chemical shift difference 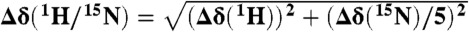 was calculated and considered to be significant if greater than 0.04 ppm. Spectral analysis was performed using Sparky 3.114 and Bruker Topspin 2.0 software. KD values were derived from concentration-dependent chemical shift changes of relevant shifting peaks using the saturation binding equation:
was calculated and considered to be significant if greater than 0.04 ppm. Spectral analysis was performed using Sparky 3.114 and Bruker Topspin 2.0 software. KD values were derived from concentration-dependent chemical shift changes of relevant shifting peaks using the saturation binding equation:
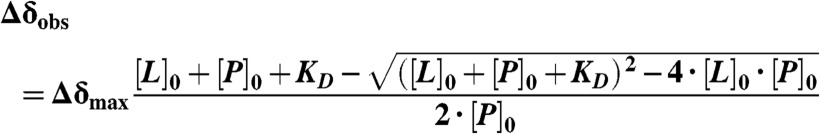 |
where Δδobs is the average weighted chemical shift difference at a particular ligand concentration, Δδmax is the difference between the chemical shifts of the free protein and of the protein in complex, and [L]0 and [P]0 is the initial ligand and protein concentration, respectively.
Analytical Ultracentrifugation.
The sedimentation equilibria were analyzed with a Beckman Optima XL-I ultracentrifuge using a Ti-60 rotor, with samples containing 20 μM MDM4 and 5 μM peptide LTFEHYWAQLTS-FAM in a buffer with 25 mM sodium phosphate pH 7.2, 150 mM NaCl, 5 mM DTT and a density of 1.013 g/cm3 at 36,000 rpm and 10 °C. The calculated Mr of peptide and protein from this experiment is 13,130. The measured was Mr 13,330. Absorption was monitored at 495 nm, specific for the peptide. Equilibration was checked by repetitive scans. For data analysis Ultraspin software was used.
In Silico Modeling.
All quantum chemical calculations were carried out using Turbomole v.6.2 (TURBOMOLE GmbH), and electrostatic potentials were plotted using gOpenMol v. 3.00 (Center for Scientific Computing, Espoo, Finland).
Cell Culture.
Tumor cell lines HCT116 and HCT116 p53-/- (colorectal carcinoma) were maintained in DMEM. Medium was supplemented with 10% fetal calf serum and 10 μg/mL gentamicin. Cells were grown at 37 °C and 5% CO2. Cells were seeded in 96 well plates at 104 cells per well 24 h prior to experiments.
Caspase-3/7 Assay.
Caspase-3/7 activation (a marker of apoptosis) was measured with the ApoTox-Glo Triplex assay (Promega). Twenty-four hours after seeding, medium with double the final concentration of LCA and DMSO was added to an equivalent volume of growth medium. Cells were incubated for 24 h at 37 °C. One-hundred μL of the Caspase-Glo 3/7 reagent was added to the cells, and after 40 min incubation at room temperature, luminescence was recorded using an Orion microplate Luminometer (Berthold Detection Systems). Experiments were performed in quadruplicate.
Supplementary Material
ACKNOWLEDGMENTS.
This work was funded by Medical Research Council Programme Grant G0901534 and the federal state of Baden-Wuerttemberg, Germany.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1215060109/-/DCSupplemental.
References
- 1.Vousden KH, Prives C. Blinded by the light: The growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 2.Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- 3.Toledo F, Wahl GM. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 4.Popowicz GM, Czarna A, Holak TA. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle. 2008;7:2441–2443. doi: 10.4161/cc.6365. [DOI] [PubMed] [Google Scholar]
- 5.Kussie PH, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 6.Boeckler FM, et al. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci USA. 2008;105:10360–10365. doi: 10.1073/pnas.0805326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toledo F, Wahl GM. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int J Biochem Cell Biol. 2007;39:1476–1482. doi: 10.1016/j.biocel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: Drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 9.Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010;20:299–309. doi: 10.1016/j.tcb.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joerger AC, Fersht AR. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harbor Perspect Biol. 2010;2:a000919. doi: 10.1101/cshperspect.a000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vassilev LT, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 12.Tovar C, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc Natl Acad Sci USA. 2006;103:1888–1893. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin Cancer Res. 2008;14:5318–5324. doi: 10.1158/1078-0432.CCR-07-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med. 2007;13:23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 15.Ding K, et al. Structure-based design of potent non-peptide MDM2 inhibitors. J Am Chem Soc. 2005;127:10130–10131. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]
- 16.Ding K, et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J Med Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 17.Popowicz GM, Domling A, Holak TA. The structure-based design of Mdm2/Mdmx-p53 inhibitors gets serious. Angew Chem Int Ed Engl. 2011;50:2680–2688. doi: 10.1002/anie.201003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wade M, Wahl GM. Targeting Mdm2 and Mdmx in cancer therapy: Better living through medicinal chemistry? Mol Cancer Res. 2009;7:1–11. doi: 10.1158/1541-7786.MCR-08-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogel SM, Bauer MR, Boeckler FM. DEKOIS: Demanding evaluation kits for objective in silico screening—A versatile tool for benchmarking docking programs and scoring functions. J Chem Inf Model. 2011;51:2650–2665. doi: 10.1021/ci2001549. [DOI] [PubMed] [Google Scholar]
- 20.Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: Relevance to drug hepatotoxicity. Drug Metab Rev. 2004;36:703–722. doi: 10.1081/dmr-200033475. [DOI] [PubMed] [Google Scholar]
- 21.Katona BW, Anant S, Covey DF, Stenson WF. Characterization of enantiomeric bile acid-induced apoptosis in colon cancer cell lines. J Biol Chem. 2009;284:3354–3364. doi: 10.1074/jbc.M805804200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makishima M, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 23.Parks DJ, et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 24.Kallen J, et al. Crystal structures of human MdmX (HdmX) in complex with p53 peptide analogues reveal surprising conformational changes. J Biol Chem. 2009;284:8812–8821. doi: 10.1074/jbc.M809096200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu GW, Vaysburd M, Allen MD, Settanni G, Fersht AR. Structure of human MDM4 N-terminal domain bound to a single-domain antibody. J Mol Biol. 2009;385:1578–1589. doi: 10.1016/j.jmb.2008.11.043. [DOI] [PubMed] [Google Scholar]
- 26.Hartshorn MJ, et al. Diverse, high-quality test set for the validation of protein-ligand docking performance. J Med Chem. 2007;50:726–741. doi: 10.1021/jm061277y. [DOI] [PubMed] [Google Scholar]
- 27.Czarna A, et al. High affinity interaction of the p53 peptide-analogue with human Mdm2 and Mdmx. Cell Cycle. 2009;8:1176–1184. doi: 10.4161/cc.8.8.8185. [DOI] [PubMed] [Google Scholar]
- 28.Fersht AR. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: W.H. Freeman; 1999. [Google Scholar]
- 29.Teufel DP, Freund SM, Bycroft M, Fersht AR. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc Natl Acad Sci USA. 2007;104:7009–7014. doi: 10.1073/pnas.0702010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shangary S, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laurie NA, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 32.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 33.Nikolovska-Coleska Z, et al. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem. 2004;332:261–273. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 34.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 35.Walters WP, Namchuk M. Designing screens: How to make your hits a hit. Nat Rev Drug Discovery. 2003;2:259–266. doi: 10.1038/nrd1063. [DOI] [PubMed] [Google Scholar]
- 36.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 37.Verdonk ML, et al. Modeling water molecules in protein-ligand docking using GOLD. J Med Chem. 2005;48:6504–6515. doi: 10.1021/jm050543p. [DOI] [PubMed] [Google Scholar]
- 38.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 39.Moitessier N, Englebienne P, Lee D, Lawandi J, Corbeil CR. Towards the development of universal, fast and highly accurate docking/scoring methods: A long way to go. Br J Pharmacol. 2008;153(Suppl 1):S7–26. doi: 10.1038/sj.bjp.0707515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.