

Abstract
Reactive astrogliosis is characterized by a profound change in astrocyte phenotype in response to all CNS injuries and diseases. To better understand the reactive astrocyte state, we used Affymetrix GeneChip arrays to profile gene expression in populations of reactive astrocytes isolated at various time points after induction using two mouse injury models, ischemic stroke and neuroinflammation. We find reactive gliosis consists of a rapid, but quickly attenuated, induction of gene expression after insult and identify induced Lcn2 and Serpina3n as strong markers of reactive astrocytes. Strikingly, reactive astrocyte phenotype strongly depended on the type of inducing injury. Although there is a core set of genes that is upregulated in reactive astrocytes from both injury models, at least 50% of the altered gene expression is specific to a given injury type. Reactive astrocytes in ischemia exhibited a molecular phenotype that suggests that they may be beneficial or protective, whereas reactive astrocytes induced by LPS exhibited a phenotype that suggests that they may be detrimental. These findings demonstrate that, despite well established commonalities, astrocyte reactive gliosis is a highly heterogeneous state in which astrocyte activities are altered to respond to the specific injury. This raises the question of how many subtypes of reactive astrocytes exist. Our findings provide transcriptome databases for two subtypes of reactive astrocytes that will be highly useful in generating new and testable hypotheses of their function, as well as for providing new markers to detect different types of reactive astrocytes in human neurological diseases.
Introduction
Reactive astrogliosis is a universal response of astrocytes to brain injuries and diseases as diverse as trauma, infection, neurodegeneration, and ischemia. The astrocytes' abilities to help support neurons, regulate the blood–brain barrier, remodel the extracellular space, control immune cells, and control synapse formation and function may all be of great import in influencing how the brain fares during and following injury (Pekny and Nilsson, 2005; Sofroniew, 2009).
Reactive astrocytes undergo dramatic morphological changes (Wilhelmsson et al., 2006), and various alterations in gene expression have been observed (Sofroniew, 2009). It has been long debated whether reactive astrocytes are harmful or beneficial. In the past few years, both types of effects have been observed. For instance, reactive astrocytes can inhibit axon regeneration after CNS injury (McKeon et al., 1991; Bradbury et al., 2002; Fitch and Silver, 2008; Alilain et al., 2011) and can produce pro-inflammatory cytokines that exacerbate spinal cord injuries (Brambilla et al., 2005, 2009). Conversely, elegant work involving ablation of reactive astrocytes has demonstrated that reactive astrocytes are crucial for withstanding insult and improving recovery after CNS trauma, after ischemia, and in experimental autoimmune encephalomyelitis (Bush et al., 1999; Faulkner et al., 2004; Voskuhl et al., 2009). Together, these findings demonstrate that reactive astrocytes can play both beneficial and detrimental roles and raise the question of whether there might be different subtypes of reactive astrocytes, elicited depending on the nature of the injury or disease, that differ in their functions.
To more fully characterize the complex molecular changes that occur during reactive astrogliosis, we used Affymetrix GeneChip analysis to profile mRNA from pure populations of quiescent and reactive astrocytes acutely isolated from healthy and injured brains. Gene profiling analysis gives a detailed snapshot of the transcriptional character of a particular cell or tissue state. Though often used to look for differences between healthy and injured tissue, the isolation of individual cell types permits a detailed understanding of which cells exhibit which changes in gene expression (Cahoy et al., 2008; Doyle et al., 2008; Heiman et al., 2008) and increases the resolution of differences between similar cells (Lobo et al., 2006; Sugino et al., 2006). We used expression profiling to investigate the changes that occur during reactive astrogliosis after two complementary injury models: ischemic stroke, which causes extensive cell death, and systemic LPS injection, which causes neuroinflammation (Buttini and Boddeke, 1995; Qin et al., 2007) but leaves brain structure intact. We followed the progression of reactive astrogliosis over the course of the first week and found that it began with a burst of transcription that moderated over the course of a week. Stroke and LPS-induced neuroinflammation are likely to induce reactive gliosis through divergent mechanisms. Nonetheless, there were core changes in gene expression that are shared between the two types of reactive astrocytes. There were, however, as many gene expression differences that differed either qualitatively or quantitatively between these reactive astrocyte populations. These transcriptome datasets give insights into the nature and evolution of the reactive astrocyte response and provide a framework for considering the balance of activities provided by reactive astrocytes during injury and repair.
Materials and Methods
Animals
All experiments were performed on mice from the transgenic mouse line Tg(Aldh1l1-eGFP)OFC789Gsat [GENSAT project (Heintz, 2004)] maintained on a Swiss Webster strain background. Young adult postnatal day 30 (P30) to P35 mice were used for the initial FACS purifications of astrocytes. The in situ hybridization (ISH) and immunohistochemistry experiments were done on mice aged 1–3 months.
Injury models
Neuroinflammation.
A single dose of 5 mg/kg of the endotoxin LPS from Escherichia coli O55:B55 (Sigma-Aldrich) dissolved in normal saline and diluted into endotoxin-free PBS or endotoxin-free PBS control was administered by intraperitoneal injection to a mixture of male and female mice.
Stroke.
Transient ischemia was induced by occluding the middle cerebral artery (MCAO) in young adult male mice as described previously (Han et al., 2009). Occlusion was performed for 1 h, followed by reperfusion. Control animals underwent a sham surgery during which no suture was inserted.
Preparation of live brain cell suspensions
Live single-cell suspensions for each control and experimental condition were made as follows. For the neuroinflammation model, the cortices and corpus callosum from two LPS-injected or saline-injected control animals were combined for each replicate. For the MCAO model, the ipsilateral cortex, corpus callosum, hippocampus, and striatum from three mice that had undergone MCAO and both hemispheres from one mouse that had undergone sham control surgery were used for each replicate. Dissected tissue was treated as described by Cahoy et al. (2008). Briefly, dissected tissue was first diced to 1–3 mm and then digested with 200 U of papain enzyme for 90 min at 34°C in bicarbonate-buffered Earle's balanced salt solution with 0.46% glucose, 26 mm sodium bicarbonate, 0.5 mm EDTA, and 125 U/ml DNase I (Worthington Biochemicals). Digested tissues were dissociated into single-cell suspensions by gentle trituration, filtered through a 15 μm Nitex mesh (Tetko) to remove any remaining clumps of tissue, collected by centrifugation, and resuspended in Dulbecco's PBS (DPBS) containing 0.02% BSA and 125 U/ml DNase I and 1 μg/ml propidium iodide (PI) for fluorescence-activated cell sorting (FACS).
FACS isolation of astrocytes
Live astrocytes were isolated at room temperature by FACS at the Stanford Shared FACS Facility on the basis of their GFP expression on a BD Vantage running CellQuest software (for 1 d MCAO and sham samples) or a BD Aria II running BDFACSDiva software (for 1 d LPS and saline samples and 3 and 7 d MCAO samples). Cell suspensions were sorted twice sequentially using forward light scatter and SSC to gate single cells, followed by gating for GFP fluorescence in the absence of PI to select live astrocytes. The Flowjo software (Treestar) was used to analyze purity of the final astrocyte populations.
RNA purification, amplification, labeling, and hybridization
QIAGEN Qiashredder and microeasy spin columns were used to lyse purified astrocyte populations and to purify their total RNA. The integrity and concentration of the isolated total RNA was confirmed by analysis on a bioanalyzer (Agilent Technologies). Approximately 20 ng of total RNA from each sample was amplified and biotin labeled using the Two-cycle Target Labeling kit (Affymetrix) and hybridized to the GeneChip Mouse Genome 430 2.0 arrays (Affymetrix). Samples were processed by the Protein and Nucleic Acid Facility at the Stanford University School of Medicine. Samples were hybridized using a GeneChip Hybridization Oven 640 (Affymetrix), processed using a GeneChip Fluidics Station 450 (Affymetrix), and scanned using a GeneChip Scanner 3000 7G (Affymetrix). The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (Edgar et al., 2002) and are accessible through the GEO Series accession number GSE35338 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE35338).
Normalization and analysis of array signal intensities
The .cel files from the scanned GeneChip arrays were processed with Arraystar 4.0 software (DNAstar) using Robust Multichip Average (RMA) processing with quantile normalization. The .cel files from Cahoy et al. (2008) used in this study for comparison purposes were reprocessed along with the samples in this study to allow for direct comparison of signal intensities. The Significance Analysis of Microarrays (SAM) (Tusher et al., 2001) Excel add-in, two class unpaired response type was used on the normalized expression values in log2 to generate lists of significantly changed genes between quiescent and reactive astrocytes. Because of differences in variability between datasets, an false discovery rate (FDR) of <1% was used for the MCAO stroke comparison, an FDR of <3.5% was used for the LPS neuroinflammation comparison, and an FDR of <5.5% was used for the time course data using the signed area option. These cutoffs maximized the ratio of significantly changed probe sets to the calculated false positives. Significantly changed probe sets were reimported back into Arraystar 4.0 and consolidated to one probe set per gene for further analysis. Probe sets were filtered for a minimum average expression value in log2 of 8 in any astrocyte population to be included in the analysis. Significantly changed probe sets were analyzed for statistically enriched pathways using Ingenuity Pathway Analysis (IPA; Ingenuity Systems, www.ingenuity.com) and categorized for biological function using Amigo (The Gene Ontology Consortium, http://www.godatabase.org/cgi-bin/amigo/go.cgi.) accessed June-July, 2011.
Immunohistochemistry
Brain tissue for immunostaining was prepared as follows. Mice were deeply anesthetized with a ketamine/xylazine mixture and transcardially perfused with DPBS followed by 4% paraformaldehyde (PFA). Brains were removed and immersion fixed for 2–3 h on ice with 4% PFA. Fixed brains were cryoprotected by immersion in 30% sucrose in PBS overnight at 4°C. Cryoprotected brains were embedded in two parts O.C.T. compound (Tissue-Tek) to one part 30% sucrose in PBS. Embedded tissue was quick frozen and cut into 10 μm cryosections using a cryostat (Leica). For immunohistochemistry, the following antibodies were used: rabbit polyclonal to GFAP at 1:2000 (Dako) and rabbit polyclonal to nestin at 1:1000 (Abcam). For these antibodies, cryosections on microscope slides were fixed for an additional 10 min in 4% PFA. No additional fixation was used for chicken polyclonal against vimentin used at 1:2000 (Novus Biologicals) and rabbit polyclonal against Iba1 used at 1:500 (Wako). The GFP fluorescence was enhanced using chicken polyclonal against GFP used at 1:1000 (Millipore Corporation) where protocol allowed. Brain sections were rehydrated in PBS and blocked with 10% goat serum in PBS with 0.2% Triton X-100 for 1 h at room temperature. All primary antibody incubations were done overnight at 4°C. Immunostaining was visualized using Alexa secondary antibodies (Invitrogen). Quantification of the fluorescence area was done in NIH ImageJ 1.45s (W. S. Rasband, National Institutes of Health, Bethesda, MD; http://imagej.nih.gov/ij/, 1997–2011). Images from control and injured sections were thresholded within each experimental replicate. The percentage of area with signal above threshold was measured in representative areas of cortex. For MCAO sections, the area measured was immediately adjacent to the core as defined by loss of eGFP signal. The percentage area above threshold in each control and injured section was normalized to the average percentage area above threshold from all control sections within the experimental replicate to obtain fold increase in expression relative to control. The one-tailed t test was used on the resulting ratios to assess significance.
ISH histochemistry
For ISH, deeply anesthetized animals were perfused with DPBS. Brains were quick frozen in O.C.T., and cut into 10 μm cryosections using a cryostat (Leica). Colorimetric ISH was performed using digoxygenin (dig)-labeled RNA probes to target genes. Color development was done by anti-dig-AP Fab fragments from sheep (Roche) using NBT/BCIP (Roche). Fluorescent ISH was performed using FITC-labeled and Dig-labeled RNA probes. Fluorescence development was done by anti-FITC-POD and anti-dig-POD Fab fragments (Roche) using FITC-tyramide and Cy3-tyramide amplification (PerkinElmer Life and Analytical Sciences).
Results
MCAO and systemic LPS injection induce astrocyte reactive gliosis
We confirmed the induction of astrocyte reactive gliosis in two complementary brain injury models: focal ischemic stroke produced by transient MCAO and neuroinflammation induced by systemic LPS injection. One-hour MCAO leads to destruction in the ipsilateral hemisphere of parts of cortex, striatum, and hippocampus (Han et al., 2009; Xiong et al., 2011). The core of the lesion is marked by extensive cell, including astrocyte, death (Liu et al., 1999) surrounded by the relatively intact, but stressed, tissue of the lesion penumbra in which astrocytes become reactive and subsequently form the glial scar (Kindy et al., 1992; Yamashita et al., 1996). Neuroinflammation was induced by a single intraperitoneal injection of the bacterial endotoxin LPS. Although LPS itself mostly fails to cross the blood–brain barrier (Banks and Robinson, 2010), this treatment induces microglia activation in response to induction of inflammatory cytokines in the periphery (Buttini and Boddeke, 1995; Qin et al., 2007) that subsequently leads to astrocyte activation (Herx and Yong, 2001). Astrocytes, marked by eGFP expression driven by the Aldh1l1 promoter in GENSAT Bac Aldh1L1-eGFP mice (Anthony and Heintz, 2007; Cahoy et al., 2008), showed little GFAP immunoreactivity in sections from control, healthy cortices (Fig. 1 A,I). By 1 d after MCAO, increased GFAP immunoreactivity was seen in astrocytes in the MCAO penumbra of the ipsilateral cortex, which persisted for at least 7 d after MCAO (Fig. 1A–D). eGFP expression was reduced in astrocytes in the lesion core at both 1 d after MCAO (data not shown) and 7 d after MCAO (Fig. 1D, stars), consistent with rapid astrocyte death (Liu et al., 1999). By 1 d after injection, cortex from LPS-injected animals showed patches of increased GFAP immunoreactivity (Fig. 1J, arrowheads), which persisted for at least 1 week (Fig. 1I–L). Particularly strong activation was seen in astrocytes near the pial surface (Fig. 1L). The reactive astrocyte response fully resolved by 30 d after LPS injection (data not shown). There was no loss of eGFP expression in astrocytes from brains experiencing neuroinflammation in response to LPS, consistent with lack of brain cell death in this inflammation model (Deng et al., 2003). The fold increase in astrogliosis was quantified by the percentage of area with GFAP immunoreactivity above threshold after MCAO (Fig. 1Q) and LPS (Fig. 1S). Both MCAO and systemic LPS injection induced astrogliosis in the cortex, as indicated by increased GFAP expression.
Figure 1.
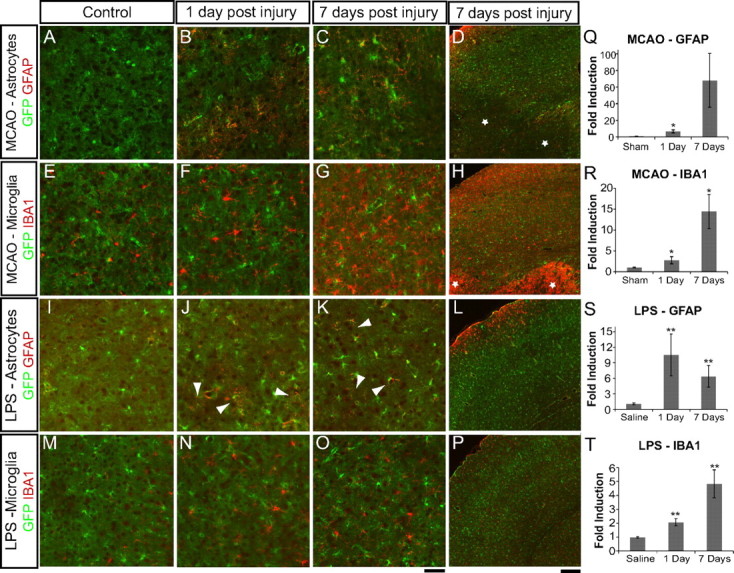
Middle cerebral artery occlusion and systemic LPS injection induce astrogliosis and microglia activation. Immunofluorescent detection of gliosis markers used 10 μm fixed cryosections from brains of Aldh1l1-eGFP mice that had undergone sham surgeries (A, E), MCAO (B–D, F–H), saline injection (I, M), or LPS injection (J–L, N–P). eGFP, in green, is expressed by and exclusively marks astrocytes. A–D, I–L, Reactive astrocytes, identified by GFAP immunostaining, in red, are present in the MCAO lesion penumbra (A–D) and cortex from LPS-treated mice (I–L) 1 d after injury and persist for over 1 week. J, K, Arrowheads indicate patches of reactive astrocytes in the cortex of LPS-treated mice. E–H, M–P, Activated microglia, identified by increased IBA1 immunoreactivity and process thickening, are evident over the same time course. eGFP expression in astrocytes is reduced in MCAO lesion core astrocytes (D, stars). Intense IBA1 expression in amoeboid cells in the MCAO lesion suggests peripheral macrophage infiltration into the core (H, stars). Scale bars: A–C, E–G, I–K, M–O, 50 μm; D, H, L, P, 200 μm. Q–T, Quantification of immunofluorescence signal was done using individual sections from control and injured brains. Q, Quantification of GFAP in brain sections (n = 4 for sham and 1 d, n = 5 for 7 d) shows the increase in reactive astrogliosis in the first week after MCAO-induced injury. R, Quantification of Iba1 (n = 4) in brain sections shows the increase in activated microglia in the first week after MCAO-induced injury. S, Quantification of GFAP in brain sections (n = 12 for saline, n = 13 for 1 d, and n = 4 for 7 d) shows the increase in reactive astrogliosis during LPS-induced neuroinflammation. T, Quantification of IBA1 in brain sections (n = 21 for saline, n = 22 for 1 d, and n = 9 for 7 d) shows the fold increase in activated microglia during LPS-induced neuroinflammation, with error bars representing SEM. *p < 0.05; **p < 0.01 for each injured time point relative to control. p = 0.055 for 7 d MCAO versus sham GFAP because of the high level of variance in response between animals.
Astrocyte activation was concomitant with activation of microglia, the endogenous brain macrophages. In healthy cortex, IBA1 lightly labels resting microglia cell bodies and their thin, highly ramified processes (Fig. 1E,M). At 1 d after MCAO in the lesion penumbra (Fig. 1F) and core (data not shown), microglia showed stronger immunoreactivity for IBA1, which increases after macrophage activation, and thickening of their processes, indicating a shift in state toward a more activated amoeboid morphology (Jonas et al., 2012). Since infiltration of peripheral macrophages into the brain is low for the first few days after MCAO (Schilling et al., 2003), IBA1-positive cells are likely to be mainly microglia. At 7 d after MCAO, strong activation in the penumbra was evident (Fig. 1G,H), and there were increased numbers of immune cells in the core of the lesion (Fig. 1H, stars), mostly reflecting immune cell infiltration from the periphery. Microglia activation was seen 1 d after LPS-injection in the cortex (Fig. 1, N vs M). Activation persisted for at least 7 d after LPS injection (Fig. 1P). The fold increase in microglia activation was quantified by percentage of area with IBA1 immunoreactivity above threshold after MCAO (Fig. 1R) and LPS (Fig. 1T). Increased IBA1 expression and morphology changes indicated that microglia became activated in both models.
FACS isolation of healthy and reactive pure astrocyte populations
FACS was used to acutely isolate pure populations of astrocytes from mice aged P30–P35 from control and injured Aldh1l1-eGFP brains on the basis of their astrocyte-restricted GFP expression using a simplified version of the protocol used to isolate astrocytes from S100β-eGFP mice (Cahoy et al., 2008). Astrocytes have reached their mature gene profiles by P30 (Cahoy et al., 2008), and the use of P30–P35 mice significantly enhanced the yield of viable astrocytes compared with mice of older ages. Two consecutive rounds of sorting enriched astrocytes from 17.5 ± 4.4% (SD) of the starting cell suspension (representative FACS plots in Fig. 2A,B) to 98.8 ± 1.3% (SD) of live cells in the final isolated population (representative FACS plots in Fig. 2C,D). We were routinely able to isolate 50,000–100,000 live GFP-positive astrocytes in the final cell population.
Figure 2.
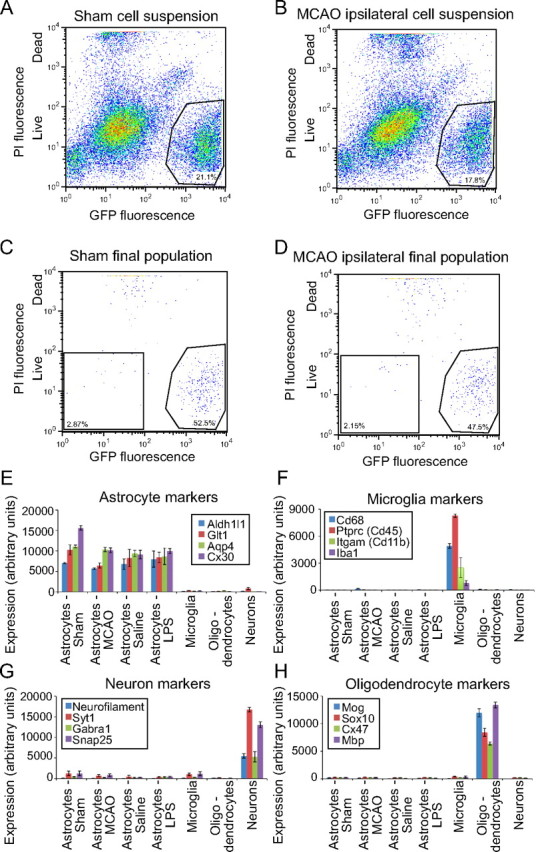
FACS isolation of GFP-positive cells from brain suspensions made from healthy and injured Aldh1l1-eGFP mice yields pure populations of astrocytes. A–D, FACS plots show the composition of starting single-cell suspensions (A, B) and final astrocyte populations (C, D). GFP fluorescence, which is astrocyte specific, is shown on the x-axis, and PI, which is taken up by dead cells, is shown on the y-axis. A, B, eGFP+ astrocytes are 15–25% of the starting cell population from healthy (A) and injured (B) brain tissues. C, D, Double sorting of cell suspensions for live eGFP+ cells yields 98.8 ± 1.1% pure final populations of astrocytes. Bar graphs comparing probe set expression levels between astrocytes and other brain cell types confirm low levels of contamination in the isolated astrocyte populations. E–H, Isolated cell populations express markers for astrocytes (E) but not markers for microglia (F), neurons (G), and oligodendrocytes (H). Neuron and oligodendrocyte .cel files from Cahoy et al. (2008) were renormalized with the astrocyte and microglia .cel files from this study to make the comparison.
Reactive astrocytes were no more or less amenable to isolation than resting astrocytes. Despite the lowered expression of GFP in the astrocytes of the MCAO core lesion (Fig. 1D), there was no significant difference between percentage of GFP-positive astrocytes present in starting cell suspensions made from MCAO-injured and sham control brains (p = 0.19, unpaired two-tailed t test). Likewise, there was no significant difference in percentage of GFP-positive astrocytes in cell suspensions made from brains from LPS-injected and saline-injected mice (p = 0.29, unpaired two-tailed t test). Astrocytes were 15.6 ± 5.4% (mean ± SD; n = 11) and 18.6 ± 3.3% (n = 10) in MCAO and sham suspensions, respectively, and 17.4 ± 1.9% (n = 5) and 20.2 ± 5.1% (n = 4) in LPS-injected and saline-injected brain suspensions, respectively. There were no significant differences between the final purity of MCAO astrocytes [98.5 ± 1.4% (S.D)] and sham control astrocytes [98.2 ± 1.4% (S.D)] (p = 0.56, unpaired two-tailed t test), or between LPS astrocytes [99.8 ± 0.3% (S.D)] and saline control astrocytes [99.9 ± 0.1% (S.D)] (p = 0.32, unpaired two-tailed t test). The healthy and injured brains yielded comparable populations of purified astrocytes.
We confirmed that we had isolated relatively pure populations of astrocytes, both quiescent and reactive, by semiquantitative RT-PCR for the astrocyte marker GFAP and additional cell-type-specific markers for oligodendrocyte lineage cells, neurons, endothelial cells, and microglia (data not shown) and then through analysis of the subsequent GeneChip expression levels for cell-type-specific markers. We normalized the quiescent and reactive astrocyte GeneChip expression files to the neuron and oligodendrocyte lineage cell expression profiles from previous work (Cahoy et al., 2008), as well as microglial profiles from the Aldh1l1-eGFP mice (J. L. Zamanian, B. A. Barres, and R. G. Giffard, unpublished observations). As expected for purified populations of astrocytes, GeneChip analysis of the isolated quiescent and reactive astrocyte populations showed high expression of the astrocyte markers Glt1, Aqp4, Connexin 30 (Cx30), and Aldh1l1, expression of which did not change between astrocytes isolated from healthy and injured brains (Fig. 2E). The purified astrocyte populations expressed only low levels of markers specific for neurons (neurofilament, Syt1, Gabra1, and Snap25; Fig. 2G), microglia (Cd68, Ptprc, Itgam, and Iba1; Fig. 2F), and oligodendrocytes [Mog, Sox10, connexin 47 (Cx47), and Mbp; Fig. 2H]. By GeneChip expression comparison, the astrocyte populations were contaminated by neurons to 4.0 ± 0.7% (SEM), by microglia to 1.6 ± 0.1% (SEM), and by oligodendrocytes by 1.8 ± 0.8% (SEM). Contamination levels were used to filter out confounding signals from the astrocyte dataset. We thus isolated highly pure populations of astrocytes by FACS from both healthy and injured mouse brains.
Astrocytes isolated from MCAO and LPS exposed brains are reactive
To confirm that we had isolated reactive astrocytes from injured brains, we next assessed changes in established markers of reactive astrocytes in the GeneChip expression profiles. Since we isolated astrocytes on the basis of their astrocyte-specific GFP expression and not on the basis of a reactive astrocyte marker, the astrocyte populations isolated from the injured brains will be a mix of quiescent and reactive astrocytes (the majority of astrocytes isolated, however, were reactive; see below). Classic reactive astrocyte markers GFAP and vimentin were strongly upregulated in both the MCAO and LPS reactive astrocyte populations (Fig. 3A). At 1 d, mRNAs for vimentin and GFAP were sevenfold increased in the MCAO reactive astrocyte population and fivefold increased in the LPS reactive astrocyte population, indicating a similar level of activation between the two stresses. GFAP and vimentin expression continued to rise for 3 d after MCAO injury and persisted for at least 1 week. Nestin, another intermediate filament protein that is upregulated in reactive astrocytes after stroke (Clarke et al., 1994; Duggal et al., 1997), was induced sevenfold at 1 d in the MCAO reactive astrocytes but was not induced in LPS reactive astrocytes. In contrast to GFAP and vimentin, induction of nestin expression does not persist and had returned to near baseline by 7 d after MCAO injury. A fourth marker, tenascin c, an extracellular matrix protein secreted by reactive astrocytes (Laywell et al., 1992), was induced only in reactive astrocytes from the MCAO model (data not shown). The identification of expression changes in well established reactivity markers in both MCAO and LPS astrocyte populations confirms that, by using these methods, we could successfully identify expression changes indicative of reactive astrocytes, and also provides a clear indication that astrogliosis differs depending on the nature of the inducing stimulus.
Figure 3.
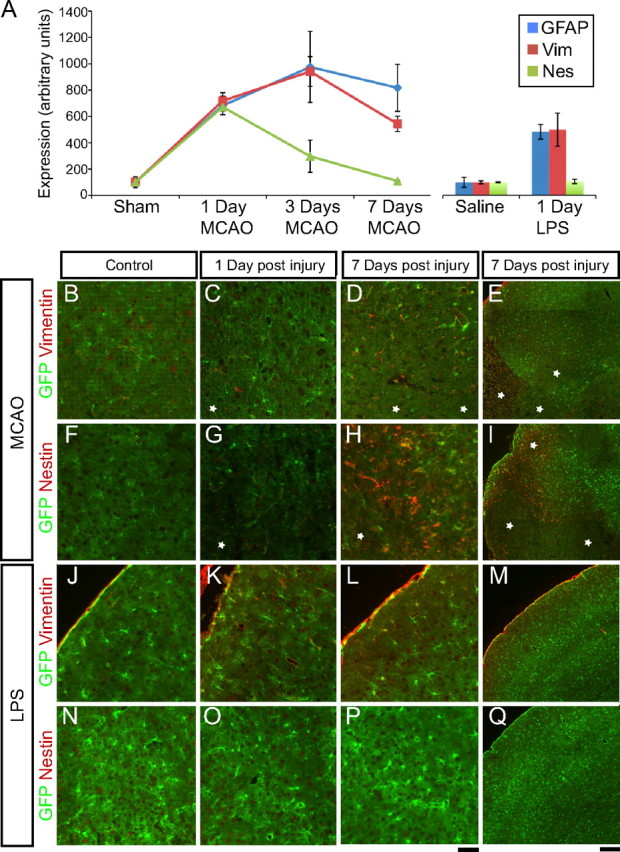
The isolated astrocytes express the classical markers of reactive astrocytes. A, Relative expression of reactive astrocyte genes in the GeneChip profiles from astrocytes isolated from healthy and injured brains is graphed over the course of 1 week. By 1 d after injury, GFAP and vimentin (Vim) are induced to similar degrees between MCAO and LPS reactive astrocytes. Nestin (Nes) is induced in MCAO reactive astrocytes but not LPS reactive astrocytes. GFAP and vimentin expression remains elevated over the course of 1 week after injury. In contrast, nestin expression is transient, decreasing to baseline levels by day 7 after injury. Error bars represent SEM. B–Q, Immunofluorescence for reactive astrocyte markers was performed on 10 μm fixed cryosections from the brains of Aldh1l1-eGFP mice that had undergone control surgeries (B, F), MCAO (C–E, G–I), saline injection (J, N), or LPS injection (K–M, O–Q). eGFP is expressed in and marks astrocytes. B, J, F, N, Reactive astrocyte markers are minimally expressed (vimentin; B, J) or not expressed (nestin; F, N) in healthy tissue. D, E, L, M, Vimentin expression is seen in astrocytes of the MCAO lesion penumbra strongly at 7 d (D, E) after injury and in astrocytes of the LPS-treated cortex after 7 d (L, M). H, I, MCAO penumbra astrocytes express nestin 7 d after injury. White stars indicate MCAO core regions in which astrocytes have lost GFP expression. O–Q, Nestin is not expressed in the LPS-treated cortex. Scale bars: B–D, F–H, J–L, N–P, 50 μm; E, I, M, Q, 200 μm.
We used immunohistochemistry to confirm these similar and divergent gene expression changes in reactive astrocytes identified by GeneChip expression profiling. Vimentin immunoreactivity is normally very low and restricted to the pial layer in control brain sections (Fig. 3J). One day after MCAO, vimentin immunoreactivity was modestly increased in penumbral astrocytes (Fig. 3C). By 7 d after MCAO, vimentin was strongly expressed in the astrocytes in the penumbra as seen by colocalization of vimentin with GFP-positive astrocytes (Fig. 3D,E). Induction of vimentin protein expression after LPS injection in astrocytes was seen most clearly in astrocytes near the pial layer at 7 d (Fig. 3K–M). No nestin protein was seen in healthy cortex (Fig. 3F,N). Despite induction of nestin transcription by 1 d after MCAO, little protein expression was seen by immunostaining at that time point (Fig. 3G). Strong nestin immunoreactivity in astrocytes in the penumbra was observed by 7 d after MCAO (Fig. 3H,I). Nestin-expressing reactive astrocytes were less widespread than GFAP-expressing astrocytes, restricted to those astrocytes closest to the lesion core (Fig. 3, asterisks) and absent in more distal regions. In contrast, GFAP expression at 7 d after MCAO was found in astrocytes more distal to the lesion (Fig. 1D). As predicted from the GeneChip expression profiling, no nestin immunoreactivity was seen in the cortex of LPS-injected animals (Fig. 3O–Q). The expression of established markers of reactive astrocytes is therefore heterogeneous in marker composition, localization, and the complement of markers expressed.
Reactive astrocyte transcriptomes show extensive induction of gene expression
Having established that we had isolated purified populations of reactive astrocytes and that we could use GeneChip expression profiling to identify gene expression differences between quiescent and reactive astrocyte populations, we conducted a comparison analysis between our healthy and stressed astrocyte populations to more thoroughly characterize the gene expression changes observed in reactive astrocytes. Expression in log2 of all 45,037 probe sets on the Affymetrix GeneChip Mouse Genome 430 2.0 arrays are represented on scatter plots comparing astrocytes from LPS-injected mice (LPS reactive astrocytes) to astrocytes from saline-injected animals (saline astrocytes; Fig. 4A) and astrocytes from mice that had undergone MCAO (MCAO reactive astrocytes) to astrocytes from mice that had undergone the sham surgery (sham astrocytes; Fig. 4B). Gene expression in LPS and MCAO reactive astrocytes differed, to a similar degree, when compared with their control populations. The R2 value for best fit to a straight line was 0.9686 for LPS versus saline astrocytes and 0.9566 for MCAO versus sham astrocytes. The scatter plots demonstrate that the vast majority of expression changes (fourfold cutoff), 206 of 220 genes for MCAO and 113 of 116 for LPS, involved induction of gene expression.
Figure 4.
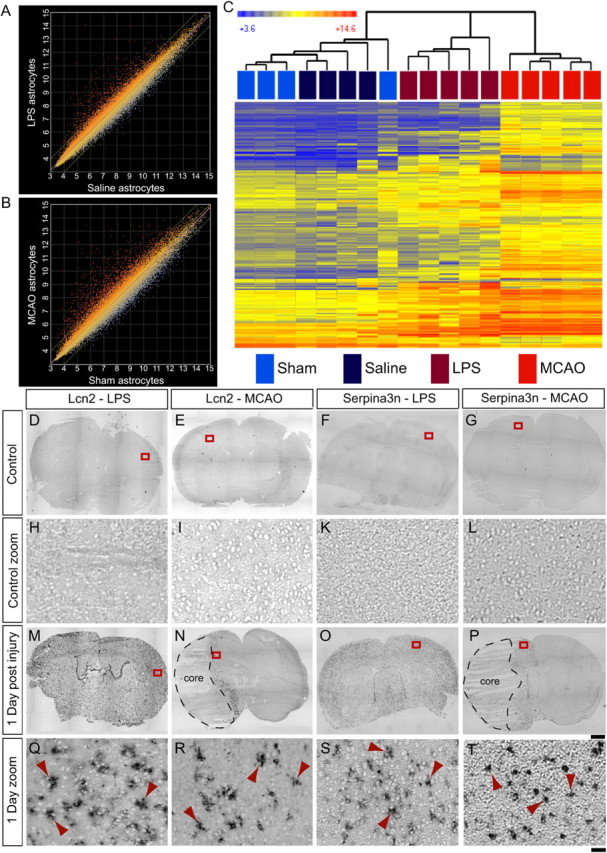
GeneChip analysis of reactive astrocyte populations suggests new markers of reactive astrocytes. A, B, The relative expression, on a log2 scale, of all probe sets from the Affymetrix GeneChip Mouse Genome 430 2.0 arrays is represented on scatter plots. Expression by astrocytes from LPS-injected animals was compared with that by astrocytes from saline-injected animals (A), and expression by astrocytes from animals that had undergone MCAO was compared with that by astrocytes from animals that had undergone sham surgeries (B). Reactive astrocyte populations on the y-axis are compared with quiescent astrocyte populations on the x-axis. Each dot represents a probe set. Probe sets that are induced in LPS and MCAO reactive astrocytes are represented by red dots; probe sets that are repressed in LPS and MCAO reactive astrocytes are represented by blue dots. C, A heat map was generated by hierarchical clustering using the 263 genes whose expression is significantly induced more than fourfold at 1 d after injury. The dendrogram of the quiescent and reactive astrocyte replicates is represented in Euclidean distance. The relative expression of each probe set is indicated by color intensity, where blue indicates lower expression and red indicates higher expression. D–T, In situ hybridization with probes for two identified reactive astrocyte markers was done on fresh frozen coronal brain sections from healthy (D–G, zoom in H–L) and injured (M–P, zoom in Q–T) mice. Control sections from saline-injected and sham-operated mice do not have expression of either Lcn2 (H and I are zoom of boxes in D and E) or Serpina3n (K and L are zoom of boxes in F and G). Lcn2 is expressed in astrocytes throughout the cortex 1 d after LPS injection (Q is zoom of box in M) and in the astrocytes of the lesion penumbra 1 d after MCAO (R is zoom of box in N). Serpina3n in expressed in astrocytes 1 d after LPS injection (S is the zoom of box in O) or MCAO (T is the zoom of box in P). Red arrowheads indicate cells with astrocyte stellate morphology (Q–T). Scale bars: D–G, M–P, 1000 μm; H–L, Q–T, 50 μm.
We identified 263 individual genes whose expression levels are significantly induced in astrocytes at least fourfold at 1 d after injury in reactive astrocytes: 206 by MCAO and 113 by LPS. The heat map generated by cluster analysis of the quiescent (n = 8) and reactive (n = 10) astrocyte population replicates using the identified reactive astrocyte genes revealed that injured astrocyte populations fall into distinct groups depending on how the astrocytes were made reactive (Fig. 4B). Clustering using the top 1057 genes significantly changed more than twofold in MCAO and/or LPS reactive astrocytes gave the same clustering result (data not shown). The corresponding quiescent astrocyte population replicates from the saline-injected and sham-operated animals were clustered away from both sets of reactive astrocyte replicates and interspersed with each other. Using the identified astrogliosis genes, cluster analysis demonstrates that different injuries produce different patterns of reactive astrocyte gene expression.
To validate the GeneChip expression profiling data, we chose two potential reactive astrocyte markers that were among the most highly expressed genes induced in both reactive astrocyte populations. Lcn2, a secreted lipophilic protein that is induced after infection and that limits bacterial growth by sequestering bacterial iron sidephores (Goetz et al., 2002; Flo et al., 2004), and which was recently implicated in astrocyte reactive gliosis (Lee et al., 2009; Chia et al., 2011), was induced 228-fold and 355-fold in MCAO and LPS reactive astrocytes, respectively, relative to their respective control astrocyte populations 1 d after treatment. Serpina3n, a secreted peptidase inhibitor whose expression is induced by inflammation and nerve injury (Takamiya et al., 2002; Gesase and Kiyama, 2007), was induced 9.1-fold in MCAO reactive astrocytes and 30-fold in LPS reactive astrocytes 1 d after injury. ISH confirmed that Lcn2 and Serpina3n were induced by injury in astrocytes based on the stellate morphology of many of the stained cells (Fig. 4, arrowheads) and colocalization with the astrocyte glutamate transporter Glast (Fig. 12). Lcn2 and Serpina3n could not be detected in healthy brain sections (Fig. 4D–G, zoom in H–L). Lcn2 (Fig. 4M, zoom in Q) and Serpina3n (Fig. 4O, zoom in S) were upregulated in astrocytes (Fig. 4Q,S, arrowheads) 1 d after LPS treatment in the cortex and throughout the brain (Fig. 4M,O). From the gene expression profiles, Serpina3n upregulation was specific to astrocytes after both LPS-induced neuroinflammation and MCAO (Zamanian, Barres, and Giffard, unpublished observations). Lcn2 was strongly induced after LPS injury, not only in astrocytes but also in endothelial cells (Zamanian, Barres, and Giffard, unpublished observations; Ip et al., 2011), strongly in choroid plexus (Fig. 4M and Marques et al., 2008) and, to a lesser extent, in microglia (Zamanian, Barres, and Giffard, unpublished observations; Ip et al., 2011). One day after MCAO, Lcn2 was induced in astrocytes (Fig. 4R, arrowheads) in the penumbra (Fig. 4N, zoom in R) and also in endothelial cells (Fig. 12H, white arrowhead). Induction of Serpina3n expression was more widespread, extending further than Lcn2 from the lesion (Fig. 4P, zoom in T). Thus, Lcn2 and Serpina3n gene induction are both markers of the early phase of astrocyte reactive gliosis in both models.
Figure 12.

Reactive astrocytes in the injured brain are heterogeneous. Double-fluorescent ISH with identified markers Lcn2 and Serpina3n was performed on 10 μm fresh frozen cryosections. A–H, Astrocytes are labeled for Glast in green. Reactive astrocyte markers, Lcn2 (A–D) or serpina3n (E–H), are labeled in red. Strongly reactive astrocytes (B, D, F, H, red arrows) can be seen adjacent to relatively quiescent astrocytes (B, D, F, H, red arrowheads). Expression of Lcn2 is also localized in endothelial cells (H, white arrowhead). I–L, The background signal from the FITC-tyramide and Cy3-tyramide amplifications can be seen. Scale bar, 50 μm.
Reactive astrocyte genes cluster into six distinct patterns of expression over time
To study how reactive astrocyte gene expression changes over time, we isolated astrocytes 3 d (n = 3) and 7 d (n = 3) after MCAO. Using SAM (Tusher et al., 2001), we identified the top 317 genes that changed significantly over time. We used cluster analysis to separate the 317 time courses of gene expression into six groups (Fig. 5A). Relative expression in log2 is shown for time points sham, 1 d, 3 d, and 7 d after MCAO. Group 1 contains 37 genes that were not induced at 1 d after MCAO but were induced at 3 d and which were moderating their expression by 7 d. Many genes suggestive of proliferation are present in this group, including late-phase cyclins b1 and b2 (Ccnb1 and Ccnb2), Cdk1, Top2a, and the proliferation marker Ki67 (Figs. 5A, 6). Group 2 contains 44 genes whose expression was induced at 1 d, increased further at 3 d, but was decreasing by 7 d after MCAO. This group contains the classic reactive gliosis marker vimentin (see also Fig. 3A), galectins Lgals3 and Lgals1, and osteopontin (Spp1). The largest cluster, group 3, contains 135 genes, including many of the most highly induced genes by fold induction. These genes were highly upregulated at 1 d, were decreasing by 3 d, and continued down, but remained elevated, by 7 d after MCAO. Lcn2, Serpina3n, tweak receptor (Tnfrsf12a), S1pr3, and all Ptx3 fall into this group (see also Fig. 5B). The 14 group 4 genes were up more modestly 1 d after MCAO, stayed elevated at 3 d, and decreased by 7 d. Group 5 contains 22 genes that were increased at 1 d, and remained elevated at 3 and 7 d after MCAO. The chemokines CXCL1, CXCL2, and CXCL10 (see also Fig. 5C), as well as universal reactive gliosis marker GFAP, fell into this group. Group 6 contains 65 genes that were induced at 1 d after MCAO but were rapidly decreased over 7 d to baseline or below. Bdnf, the oncostatin M receptor (Osmr), and transcription factor tumor suppressor klf6 fell into this category. The expression of genes with diverse functions was rapidly induced and moderated during reactive gliosis. Genes involved in adhesion, ECM modification, immune response and the neurotrophic cytokines all followed this trend (Fig. 5B). Chemokines were a major class of genes that were stably induced (Fig. 5C), retaining high expression even out to 30 d after MCAO (data not shown). Even within this class of genes, there was variation of expression course with the chemokine for monocytes, CCL2, following the rapidly moderating group 3 expression pattern whereas other cytokines, CXCL1, CXCL2, and CXCL10 (Cartier et al., 2005), remained elevated as part of a group 5 expression pattern. Overall, gene expression profiling of reactive astrocytes reveals a dramatic burst of induced expression that is rapidly moderated.
Figure 5.
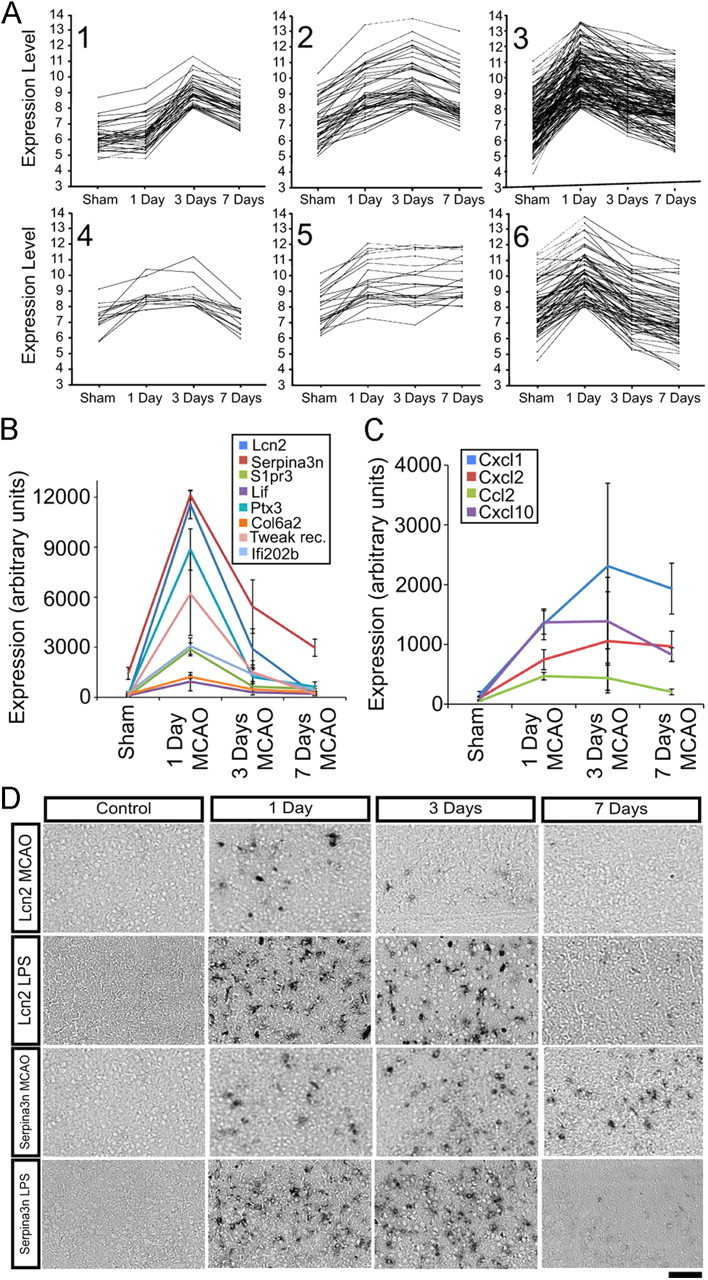
Expression of genes induced in reactive astrocytes over time. A, The top 317 genes significantly upregulated in MCAO reactive astrocytes over 7 d were clustered using the k-means method with the standard Pearson correlation coefficient. Each line represents the relative expression of one gene at time points sham, 1 d, 3 d, and 7 d after MCAO. Genes are grouped into six patterns of expression, containing 14–135 members, that are represented in graphs 1–6. B, Diverse genes in group 3 are strongly induced at 1 d and are rapidly downregulated to near-quiescent levels by 7 d. Lcn2 is downregulated to near-baseline levels. Serpina3n expression remains elevated at 7 d. C, Expression of a subset of chemokines (CXCL1, CXCL2, and CXCL10) remains elevated over the course of 1 week (group 5 expression pattern). In contrast, macrophage chemokine CCL2 follows a group 3 expression pattern in which expression is rapidly moderated. D, In situ hybridization on coronal brain sections from healthy and injured mice confirms the time course indicated by the GeneChip expression profiles. Rows 1 (MCAO) and 2 (LPS), Lcn2 is upregulated by 1 d after injury but is rapidly downregulated; rows 3 (MCAO) and 4 (LPS), Serpina3n is upregulated by 1 d after injury and is downregulated, but remains elevated, 7 d after injury. Scale bar, 100 μm.
Figure 6.

Reactive astrocytes show a delayed and transient upregulation of cell-cycle genes, suggesting modest proliferation after injury. The relative expression of cell-cycle genes in reactive astrocytes over time was represented on a line graph. The expression of early-phase cyclin D is upregulated at 1 d after MCAO. The expression of late-phase cell-cycle genes and proliferation marker ki67 is upregulated at 3 d. All cell-cycle gene expression is returning toward baseline by 7 d after MCAO.
We confirmed the rapid induction and reduction of expression by ISH on tissue sections from brain 1, 3, and 7 d after MCAO and LPS (Fig. 5D). Consistent with the GeneChip expression values, Lcn2 had the fastest time course for reduction in expression. Its expression was clearly reduced by 3 d after MCAO and was below detectable limits at 7 d (Fig. 5D, top row, B). Expression persisted for longer in the LPS tissue, present at 3 d, but was nearly absent by 7 d (Fig. 5D, second row). The rapid induction and decrease in gene expression of Lcn2 in astrocytes was similar to the time course of induction and repression in choroid plexus after LPS (Marques et al., 2008). Consistent with the expression profiling result (Fig. 5B), induction of Serpina3n expression persisted for longer, for at least 3 d after LPS (Fig. 5D, fourth row) and for at least 7 d after MCAO (Fig. 5D, third row, B), as seen by ISH in sections adjacent to those used for Lcn2. ISH on injured brain sections confirmed the time course of induction and moderation of expression of reactive astrogliosis genes seen in the gene expression profiles.
Gene expression changes suggest a delayed and brief burst of astrocyte proliferation after injury
Whether reactive astrocytes proliferate after injury or simply undergo hypertrophy has long been controversial (Sofroniew, 2009). We recently analyzed this question with BrdU labeling in these Aldh1l1-GFP mice and found significant numbers of astrocytes colabeling with BrdU on day 2 after MCAO, with only modest additional numbers of cells if labeling was extended through day 6 (Barreto et al., 2011). We analyzed the reactive astrocyte expression profiles for cell-cycle genes and markers of proliferation (Fig. 6). Early-phase cyclin D (Ccnd1) was induced fourfold to fivefold, and growth arrest gene Gas1 was repressed 50% by 1 d after injury in MCAO reactive astrocytes. Many cell-cycle genes, including the late-phase cyclin B (Ccnb1) and cyclin-dependent kinase Cdk1, were not induced at 1 d after MCAO but were elevated threefold to fourfold in MCAO reactive astrocytes 3 d later. By 7 d after MCAO, the cell-cycle genes were decreasing toward their baseline expression, consistent with our previous BrdU labeling study (Barreto et al., 2011). The expression of cell proliferation marker Ki67 was induced approximately fourfold at 3 d and was returning toward baseline by 7 d after MCAO. Similar changes in LPS reactive astrocyte gene expression occurred at 1 d after injection. Cyclin D1 expression was induced 3.4-fold, and Gas1 expression was decreased by 40%. These data support previous findings that reactive astrocytes divide with a brief delay after injury, but that this proliferation is limited.
Reactive astrocyte transcriptome depends on the nature of the inducing stimulus
Hierarchical clustering of the quiescent and reactive astrocyte populations by GeneChip expression revealed that reactive astrocytes are separated into groups depending on whether their activation was induced by MCAO or LPS (Fig. 4C). We also analyzed the similarities and differences between the two types of reactive astrocytes. Fifty-six of the more than fourfold induced reactive gliosis genes representing 50% of the genes induced by LPS and 25% of the genes induced by MCAO were shared between the two types of reactive astrocytes (Fig. 7A). We identified 57 genes whose expression was induced significantly at more than fourfold in LPS reactive astrocytes but not MCAO reactive astrocytes and 150 genes whose expression was induced significantly at more than fourfold in MCAO reactive astrocytes but not LPS reactive astrocytes. At a twofold cutoff for both reactive astrocyte gene sets, the Venn diagram was similar to that for fourfold. One hundred sixty-six genes were induced in both reactive astrocyte populations, representing 22% of genes (766) induced by MCAO and 57% of the genes (291) induced by LPS (data not shown). Some of the genes excluded from one reactive astrocyte gene set at a cutoff level were induced, to a lesser degree. Ninety of the 113 genes (80%) that were induced more than fourfold by LPS were induced by more than twofold by MCAO, and 82 of the 220 genes (37%) that were induced more than fourfold by MCAO were induced by more than twofold by LPS, indicating that reactive astrocyte gene induction by individual injuries varies in both gene representation and fold induction. The top 50 gene changes with fold induction are listed in Table 1, for MCAO reactive astrocytes, and Table 2, for LPS reactive astrocytes.
Figure 7.
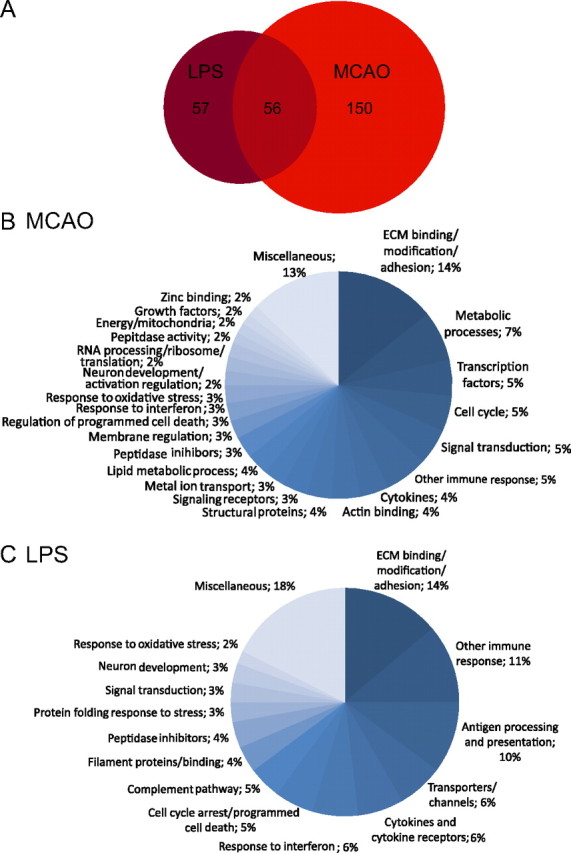
LPS and MCAO reactive astrocytes have overlapping but distinct sets of induced genes. A, The Venn diagram shows the distribution of the 263 genes significantly upregulated more than fourfold in the LPS and/or MCAO reactive astrocytes. There are 113 genes more than fourfold upregulated in LPS reactive astrocytes. There are 205 genes more than fourfold upregulated in MCAO reactive astrocytes. Fifty-six genes are common to both types of reactive astrocytes. B, C, Pie charts show the Gene Ontology categorization of the MCAO (B) and LPS (C) reactive gene sets. Extracellular matrix modification and immune response are prominent classes.
Table 1.
Top 50 changes in MCAO reactive astrocytes
| Probe set ID | Gene symbol | Sham astrocytes | 1 d MCAO astrocytes | Fold induction |
|---|---|---|---|---|
| 1427747_a_at | Lcn2 | 50 | 11,429 | 228.1 |
| 1457666_s_at | Ifi202b | 35 | 4757 | 134.5 |
| 1437270_a_at | Clcf1 | 15 | 1039 | 70.4 |
| 1460197_a_at | Steap4 | 41 | 2625 | 63.8 |
| 1451416_a_at | Tgm1 | 70 | 3878 | 55.4 |
| 1438658_a_at | S1pr3 | 263 | 12,023 | 45.6 |
| 1418666_at | Ptx3 | 192 | 8454 | 43.9 |
| 1456642_x_at | S100a10 | 67 | 2673 | 40.0 |
| 1451596_a_at | Sphk1 | 55 | 2209 | 40.0 |
| 1460227_at | Timp1 | 90 | 3422 | 38.2 |
| 1436346_at | Cd109 | 91 | 3230 | 35.4 |
| 1417262_at | Ptgs2 | 36 | 1246 | 34.4 |
| 1416529_at | Emp1 | 188 | 6352 | 33.7 |
| 1428776_at | Slc10a6 | 102 | 2630 | 25.7 |
| 1450958_at | Tm4sf1 | 538 | 11,522 | 21.4 |
| 1420994_at | B3gnt5 | 72 | 1527 | 21.1 |
| 1422943_a_at | Hspb1 | 350 | 6522 | 18.7 |
| 1417268_at | Cd14 | 129 | 2197 | 17.0 |
| 1429236_at | Galntl2 | 74 | 1226 | 16.6 |
| 1423062_at | Igfbp3 | 67 | 1034 | 15.4 |
| 1451021_a_at | Klf5 | 70 | 1057 | 15.0 |
| 1418930_at | Cxcl10 | 89 | 1323 | 14.8 |
| 1423760_at | Cd44 | 605 | 8779 | 14.5 |
| 1448239_at | Hmox1 | 89 | 1231 | 13.9 |
| 1426808_at | Lgals3 | 142 | 1926 | 13.5 |
| 1418674_at | Osmr | 838 | 10,764 | 12.9 |
| 1423537_at | Gap43 | 105 | 1325 | 12.6 |
| 1418949_at | Gdf15 | 82 | 1007 | 12.2 |
| 1447812_x_at | Flnc | 166 | 1994 | 12.0 |
| 1443721_x_at | Sbno2 | 107 | 1243 | 11.6 |
| 1419091_a_at | Anxa2 | 436 | 5050 | 11.6 |
| 1416431_at | Tubb6 | 161 | 1858 | 11.5 |
| 1426875_s_at | Srxn1 | 244 | 2672 | 10.9 |
| 1418571_at | Tnfrsf12a | 270 | 2937 | 10.9 |
| 1451095_at | Asns | 150 | 1624 | 10.9 |
| 1416953_at | Ctgf | 299 | 3241 | 10.8 |
| 1425503_at | Gcnt2 | 222 | 2391 | 10.8 |
| 1417494_a_at | Cp | 107 | 1080 | 10.1 |
| 1416184_s_at | Hmga1 | 670 | 6684 | 10.0 |
| 1449773_s_at | Gadd45b | 472 | 4637 | 9.8 |
| 1436094_at | Vgf | 274 | 2523 | 9.2 |
| 1460330_at | Anxa3 | 145 | 1337 | 9.2 |
| 1419100_at | Serpina3n | 1331 | 12,103 | 9.1 |
| 1435036_at | Aspg | 269 | 2441 | 9.1 |
| 1450641_at | Vim | 1258 | 10,917 | 8.7 |
| 1457273_at | Odz2 | 138 | 1163 | 8.4 |
| 1419706_a_at | Akap12 | 248 | 1956 | 7.9 |
| 1435084_at | C730049O14Rik | 260 | 2022 | 7.8 |
| 1426509_s_at | Gfap | 403 | 3056 | 7.6 |
| 1424067_at | Icam1 | 137 | 1033 | 7.5 |
Shown are the top 50 genes in order of fold induction with normalized linear expression in MCAO astrocytes >1000.
Table 2.
Top 50 changes in LPS reactive astrocytes
| Probe set ID | Gene symbol | Saline astrocytes | 1 d LPS astrocytes | Fold induction |
|---|---|---|---|---|
| 1427747_a_at | Lcn2 | 49 | 17,253 | 355.1 |
| 1460197_a_at | Steap4 | 29 | 2295 | 78.6 |
| 1449556_at | H2-T23 | 117 | 5270 | 45.0 |
| 1460227_at | Timp1 | 77 | 3461 | 44.9 |
| 1416625_at | Serping1 | 240 | 8146 | 34.0 |
| 1419100_at | Serpina3n | 526 | 16,269 | 31.0 |
| 1450170_x_at | H2-D1 | 125 | 3671 | 29.3 |
| 1418483_a_at | Ggta1 | 67 | 1279 | 19.0 |
| 1419043_a_at | Iigp1 | 104 | 1905 | 18.4 |
| 1435906_x_at | Gbp2 | 165 | 3022 | 18.3 |
| 1417494_a_at | Cp | 67 | 1175 | 17.6 |
| 1416164_at | Fbln5 | 120 | 2002 | 16.7 |
| 1426260_a_at | Ugt1a | 471 | 7686 | 16.3 |
| 1416125_at | Fkbp5 | 86 | 1397 | 16.2 |
| 1435036_at | Aspg | 257 | 3378 | 13.1 |
| 1422962_a_at | Psmb8 | 85 | 1099 | 12.9 |
| 1447927_at | Gbp10 | 249 | 3173 | 12.8 |
| 1417426_at | Srgn | 97 | 1096 | 11.3 |
| 1418930_at | Cxcl10 | 162 | 1829 | 11.3 |
| 1434601_at | Amigo2 | 284 | 3003 | 10.6 |
| 1422943_a_at | Hspb1 | 163 | 1576 | 9.7 |
| 1418674_at | Osmr | 373 | 2933 | 7.9 |
| 1426509_s_at | Gfap | 235 | 1758 | 7.5 |
| 1419647_a_at | Ier3 | 180 | 1308 | 7.3 |
| 1423754_at | Ifitm3 | 426 | 3050 | 7.2 |
| 1438658_a_at | S1pr3 | 185 | 1187 | 6.4 |
| 1447602_x_at | Sulf2 | 200 | 1207 | 6.0 |
| 1436332_at | Hspb6 | 293 | 1671 | 5.7 |
| 1449937_at | Endou | 245 | 1368 | 5.6 |
| 1427035_at | Slc39a14 | 716 | 3950 | 5.5 |
| 1421812_at | Tapbp | 1259 | 6881 | 5.5 |
| 1437056_x_at | Crispld2 | 409 | 2173 | 5.3 |
| 1421817_at | Gsr | 221 | 1123 | 5.1 |
| 1448276_at | Tspan4 | 303 | 1508 | 5.0 |
| 1457644_s_at | Cxcl1 | 433 | 2150 | 5.0 |
| 1418392_a_at | Gbp3 | 661 | 3279 | 5.0 |
| 1418825_at | Irgm1 | 417 | 2064 | 5.0 |
| 1425784_a_at | Olfm1 | 536 | 2625 | 4.9 |
| 1449875_s_at | H2-T10 | 327 | 1583 | 4.8 |
| 1451537_at | Chi3l1 | 1128 | 5317 | 4.7 |
| 1450641_at | Vim | 1067 | 4941 | 4.6 |
| 1418021_at | C4b | 1659 | 7652 | 4.6 |
| 1449289_a_at | B2m | 1443 | 6609 | 4.6 |
| 1434719_at | A2m | 531 | 2411 | 4.5 |
| 1417639_at | Slc22a4 | 261 | 1175 | 4.5 |
| 1423596_at | Nek6 | 601 | 2602 | 4.3 |
| 1425826_a_at | Sorbs1 | 706 | 2992 | 4.2 |
| 1431213_a_at | Gm3579 | 786 | 3302 | 4.2 |
| 1423760_at | Cd44 | 561 | 2347 | 4.2 |
| 1417141_at | Igtp | 823 | 3299 | 4.0 |
Shown are the top 50 genes in order of fold induction with normalized linear expression in LPS astrocytes >1000.
We analyzed the identified reactive astrogliosis genes using gene ontology (GO) classification (The Gene Ontology Consortium, http://www.godatabase.org/cgi-bin/amigo/go.cgi.). The categorization by class and/or biological process for genes more than fourfold induced is shown in pie charts for MCAO (Fig. 7B) and LPS (Fig. 7C). The gene constituents of each category induced more than fourfold in MCAO and LPS reactive astrocytes are listed in Table 3. Proteins involved in extracellular matrix modification and adhesion were the largest class for both types of reactive astrocytes. Constituents of this class (Fig. 8) included not only ECM proteins, such as collagen (Col12a1, Col6a1) and versican (Vcan), but proteins that interact with the ECM [such as thrombospondin (Thbs1) and fibulin 5 (Fbln5)], proteins involved in cell adhesion [such as Cd44 and neurofascin (Nfasc)], and enzymes that modify the carbohydrate side chains of extracellular molecules, such as Ggta1 and Galntl2. The dendrogram resulting from hierarchical clustering of extracellular matrix and adhesion proteins demonstrates that MCAO and LPS reactive astrocytes, while both showing gene induction strongly suggestive of modification of the extracellular space, differed greatly in the specifics of the changes (Fig. 8A). Whereas both types of reactive astrocytes exhibited a large array of induced genes, the degree to which any gene is induced depended on the stimulus (Fig. 8B). Prominently, collagen (Col6a1, Col12a1) and versican (Vcan) were more strongly induced by MCAO, as might be expected to seal off the dying tissue and form the glial scar. Conversely, other genes in the class, Fbln5 and Amigo2, were more strongly upregulated in LPS reactive astrocytes.
Table 3.
Groupings of induced genes based on gene ontology
| Gene Ontology grouping | Genes |
|---|---|
| Genes induced in astrocytes after MCAO | |
| ECM binding/modification/adhesion | CD24A, CD44, JUB, ChL4, COL6A1, IGFBP3, COL6A2, COL12A1, ICAM1, LGALS1, PVR, THBS1, THBS2, TGFBI, VCAN, CHI3L1, DPYSL3, ECM1, ADAM12, ADAMTS4, CELA1, ADAMTS5, GALNTL2, LGALS3, NAV2, FGL2, NETO2, OLFML3 |
| Basic metabolic processes | AKR1B8, ASPG, BCAT1, ODC1, ESD, PDE3B, ASNS, CYP1B1, B3GNT5, GCNT2, GGTA1, OASL2, CTPS, UCK2 |
| Transcription factors | BCL3, CDBPD, KLF5, KLF6, TGIF1, AHR, FOSL1, HMGA, FOSL2, SBNO2 |
| Cell cycle | NEK6, S100A6, ZWINT, CDT1, CCND1, CDK6, CDKN1A, GADD45A, HMGA2, EMP1 |
| Signal transduction | BDKRB2, GADD45B, SPHK1, SOCS3, CAMK2D, RHOJ, SPHKAP, ODZ2, SHISA6 |
| Unclassified immune response | CD14, C3, NUPR1, PROCR, THBD, TLR4, MPA2L, PLP2, PTX3, LCN2 |
| Cytokines | CLCF1, CCL2, CXCL1, CXCL10, CXCL2, IL6, LIF, SPP1 |
| Actin binding | ARPC1B, ACTN1, FSCN1, FLNC, FLNA, SYNPO, MSN, TAGLN2 |
| Metal ion transport | STEAP4, CP, STEAP1, SLC10A6, SLC39A14, SLC5A3 |
| Structural proteins | AKAP12, GFAP, LMNA, NES, TUBA1A, TUBB6, VIM |
| Lipid metabolic process | AGPAT9, CAV1, LASS6, PLA2G4A, CH25H, HPGD, PTGS2 |
| Peptidase inhibitors | CD109, A2M, SERPINA3N, SERPINE1, SERPING1, TIMP1 |
| Membrane regulation | ANXA1, ANXA2, ANXA7, ANXA3, S100A10, S100A11 |
| Response to oxidative stress | GPX1, HMOX1, SRXN1, TXNRD1 |
| Regulation of programmed cell death | GCH1, MCL1, LITAF, EDA2R, SULF1 |
| Response to interferon β | IFI202B, IFITM3, IFI203, IIGP1, GBP3 |
| Peptidase activity | PAPPA, PRSS23, USP18, LONRF1 |
| Neuron activity and development | GAP43, VGF, CACNG5, SYT4 |
| RNA processing and translation | NHP2, NOP58, EIF1A, PCBP3 |
| Signaling receptors | IL13RA1, IL6RA, OSMR, S1PR3, MET, TNFRSF12A |
| Growth factors | BDNF, CTGF, GDF15 |
| Zinc binding | NT5E, RNF125, RNF19B |
| Mitochondria | OCIAD2, MRPS6, MTHFD2 |
| Miscellaneous | CPNE8, EII2, FAM129B, GRB10, HSPB1, HSPB6, TMEM74, KLHDC8A, LRRC59, LRG1, TSPAN4, MEST, TM4SF1, LRRFIP1, STX11, TGM1, SPATA13, IER3, SLC44A3, SLC7A1, AHNAK, AHNAK2, H19, BTG3, CHAC1 |
| Genes induced in astrocytes after LPS treatment | |
| ECM binding/modification/adhesion | CD44, GALNTL2, SULF2, CRISPLD2, DCN, THBS2, LRG1, GGTA1, AMIGO2, FBL5, ICAM1, NFASC, SORBS1, CHI3L1 |
| Unclassified immune response | CD14, SRGN, S1PR3, TLR2, OASL2, MPA2L, SAA3.ZC3HAV1, TRIM30A, LCN2, TGTP1, IRGM1, IRGM1, TNFAIP2, PTX3 |
| Antigen processing and presentation | TAPBP, TAPBPL, B2M, H2-D1, H2-K1, H2-Q6, H2-T10, H2-T23, PMSB8, PSMB9, PROCR |
| Transporters/channels | STEAP4, CP, KCTD1, SLC22A4, SLC39A14, SLC1A5, SLC10A6 |
| Cytokines and cytokine receptors | CXCL1, CXCL10, CXCL2, SPP1, IL1R1, IL13RA, OSMR |
| Response to interferon beta | IFI202B, IFI44, IFITM3, IGTP, IIGP1, GBP2, GBP3 |
| Cell-cycle arrest/programmed cell death | BCL3, CDKN1A, PARP14, XAF1, NEK6 |
| Complement pathway | C1RB, C1RA, C3, SERPING1, C4B, C1S |
| Filament proteins/binding | GFAP, VIM, TAGLN2, SYNPO |
| Peptidase inhibitors | CD109, A2M, SERPINA3N, TIMP1 |
| Protein folding response to stress | FKBP5, HSP1, HSPB6 |
| Signal transduction | SPHKAP, MAP3K6, RHOJ |
| Neuron development | GAP43, OLFM1, SEMA4C |
| Response to oxidative stress | GPX3, GSR |
| Miscellaneous | ACSL5, HPGD, ASPG, CELA1, USP18, CPNE8, LY6E, UGT1A1, ENDOU, PLIN4, TSPO, TGM1, NT5E, ANGPT1, TGM2, S100A10, TSPAN4, SLC43A3, IER3 |
Reactive astrogliosis genes more than fourfold induced by MCAO and LPS and grouped by gene ontology. Bold is used for emphasis.
Figure 8.
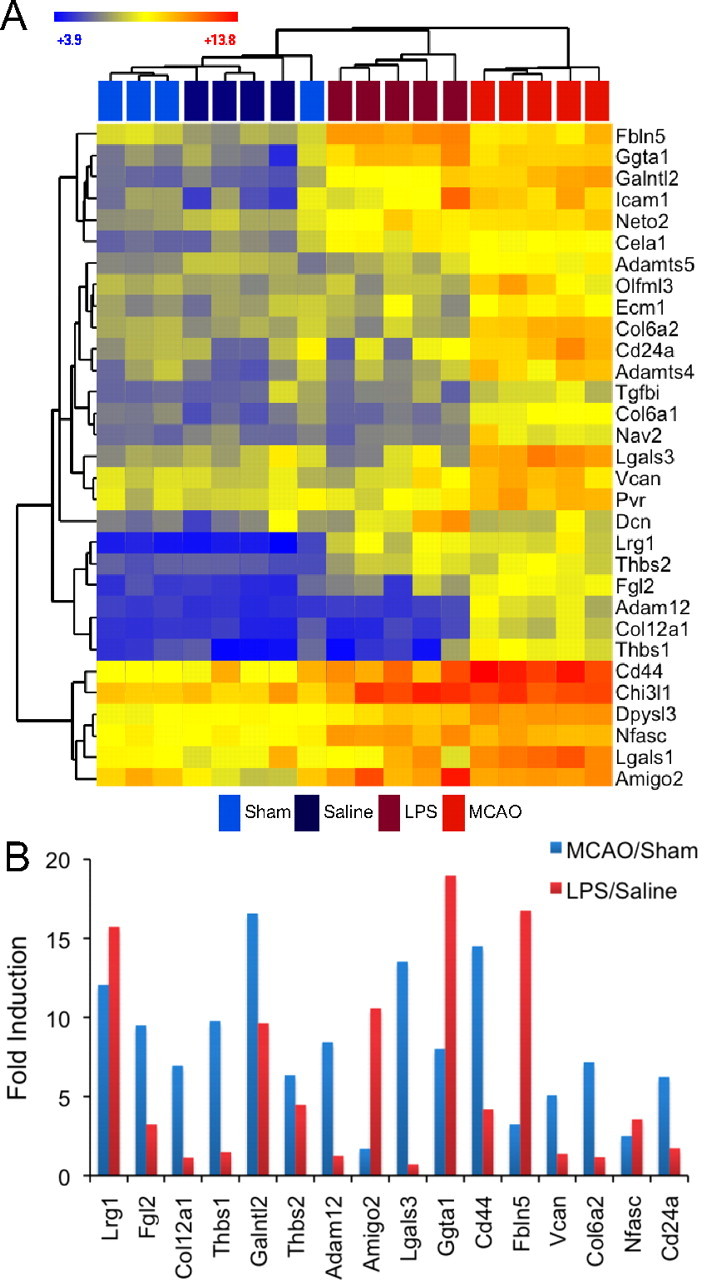
MCAO reactive astrocytes and LPS reactive astrocytes express differing levels of extracellular binding/adhesion/modification genes. A, A heat map was generated by hierarchical clustering using extracellular binding/adhesion/modification genes whose expression was significantly induced more than fourfold at 1 d after either injury. The dendrogram of the quiescent and reactive astrocyte replicates is represented in Euclidean distance. Relative expression is shown where blue indicates lower expression and red indicates higher expression. LPS reactive astrocytes and MCAO reactive astrocytes modify the extracellular space in distinct ways. B, The bar graph shows the fold induction of genes within the class in MCAO reactive astrocytes (blue) and LPS reactive astrocytes (red). All replicates within a class were averaged to obtain the fold induction. Although there are similarities between MCAO and LPS reactive astrocytes, there are extensive differences in gene expression induction patterns between them.
Proteins involved in transport, especially of metal ions and immune response, also figure prominently. In fact, a full 50% of all LPS and 25% of MCAO reactive astrocyte genes had a GO categorization that involved them in immune response. Cytokine signaling, in particular, was induced in both MCAO and LPS reactive astrocytes. Even within this gene class, differences in induction were clear (Fig. 9A). The C-X-C class of chemokines was induced, to a similar degree, by stroke and neuroinflammation. For instance, CXCL1, on average, was induced ∼5-fold in both types of astrocytes (CXCL2, ∼8-fold; CXCL10, 11- to 15-fold). Alternatively, CCL2, a macrophage chemokine, was more prominently upregulated by MCAO reactive astrocytes, eightfold versus twofold by LPS. The neurotrophic cytokines, LIF and CLCF1 (Bauer et al., 2007), were greatly induced in MCAO reactive astrocytes but only marginally induced in LPS reactive astrocytes. IL6, another cytokine known to be important in stroke, with both beneficial and deleterious effects depending on timing and context (Gadient and Otten, 1997; Monje et al., 2003; Suzuki et al., 2009; Voloboueva et al., 2010), also follows this pattern. The dendrogram made from hierarchical clustering (Fig. 9B) shows that part of this difference was attributable to variation in astrocyte response during LPS-induced neuroinflammation.
Figure 9.
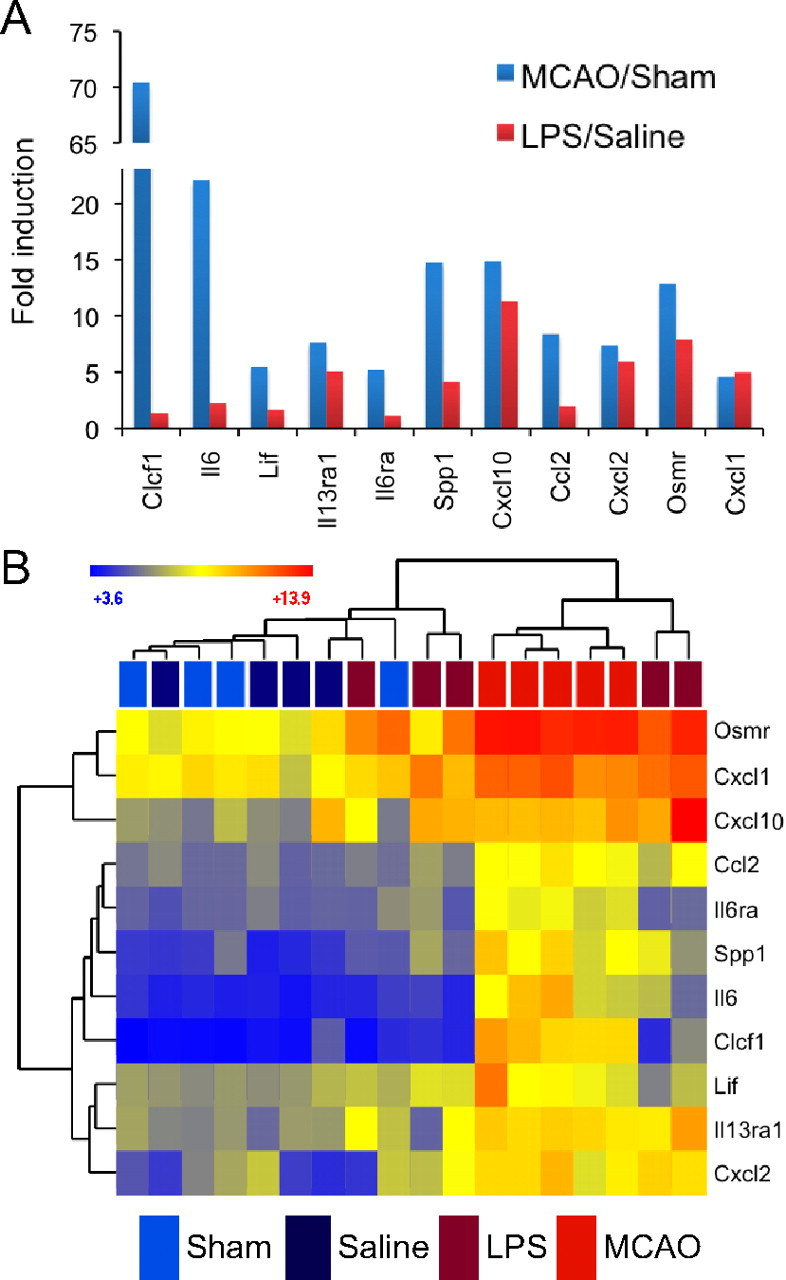
MCAO reactive astrocytes and LPS reactive astrocytes express differing levels of cytokine signaling genes. A, The bar graph shows fold induction of cytokine signaling genes in MCAO reactive astrocytes (blue) and LPS reactive astrocytes (red). All replicates within a class were averaged to obtain the fold induction. Pro-inflammatory cytokines CXCL1, CXCL2, and CXLC10 are similarly induced in both MCAO and LPS reactive astrocytes. The neurotrophic cytokines LIF, IL6, and CLCF1 and macrophage chemokine CCL2 are more strongly induced in MCAO reactive astrocytes. B, A heat map was generated by hierarchical clustering of resting and reactive astrocyte populations using cytokine signaling genes whose expression is significantly induced more than fourfold at 1 d after either injury. The dendrogram of astrocyte replicates is represented in Euclidean distance. The heat map indicates relative expression where blue indicates lower expression and red indicates higher expression. Variability in reactive astrocyte response to injury occurs even within an injury model. Two replicates of LPS reactive astrocytes cluster more closely with the MCAO reactive astrocytes, and one replicate has an expression pattern that places it within the quiescent astrocytes cluster.
Certain categories of genes were more prominently represented in one type of reactive astrocyte. Increased metabolic activity, cell-cycle genes, and transcription factors were prominent categories for MCAO reactive astrocytes (Fig. 7B) but not LPS reactive astrocytes (Fig. 7C). In contrast, the antigen presentation pathway, complement pathway, and response to interferon figured more prominently in the LPS reactive astrocytes (Figs. 7C, 10) than in the MCAO reactive astrocytes. After injury, genes within the antigen presentation pathway (York and Rock, 1996), including class I MHC molecules (H2-D1, H2-K1, H2-T10) and Tapbp and B2m, which participate in peptide processing and MHC association, were upregulated by 2- to 30-fold in LPS reactive astrocytes, but only by 10% to threefold in MCAO reactive astrocytes (Fig. 10A). Hierarchical clustering using probe sets for genes in the antigen presentation pathway showed that all but one replicate of MCAO reactive astrocytes cluster with quiescent astrocyte populations. One replicate showed strong induction of part of this pathway, again demonstrating variability in reactive gliosis even within an injury model (Fig. 10B). Interestingly, the genes of the initiating part of the complement cascade, C1r, C1s, C3, and C4B, as well as complement inhibitor Serping1 (Gasque, 2004), were all induced 4.5- to 34-fold (15-fold on average) in LPS reactive astrocytes, but only 2.5- to 7-fold (4-fold on average) in MCAO reactive astrocytes (Fig. 10C). Hierarchical clustering of complement pathway genes showed that four of our five LPS reactive astrocyte replicates cluster apart from all other astrocyte populations (Fig. 10D). Although MCAO and neuroinflammation both induced reactive gliosis based on classical markers, the characteristics of the activation greatly differed by inducing signal.
Figure 10.

LPS reactive astrocytes more highly induce the antigen presentation and complement pathways. A, The fold induction of antigen presentation pathway genes in MCAO reactive astrocytes (blue) and LPS reactive astrocytes (red) is shown. All replicates within a class were averaged to obtain the fold induction. Antigen presentation pathway genes are induced 2- to 30-fold in LPS reactive astrocytes but only 10% to threefold in MCAO reactive astrocytes. B, A heat map was generated by hierarchical clustering using antigen presentation pathway genes. The distance on the dendrogram between resting and reactive astrocyte population replicates is represented in Euclidean distance. The heat map indicates relative expression where blue indicates lower expression and red indicates higher expression. LPS reactive astrocytes cluster separate from other astrocyte populations. C, The fold induction of complement pathway genes in MCAO reactive astrocytes (blue) and LPS reactive astrocytes (red) is compared. Complement genes are induced 4.5- to 34-fold in LPS reactive astrocytes but only 2.4- to 6.8-fold in MCAO reactive astrocytes. D, Four of the five LPS reactive astrocyte replicate clusters separate from other astrocyte populations on the basis of their complement pathway gene expression. MCAO reactive astrocytes, one LPS replicate, and one quiescent replicate show an intermediate induction of complement pathway expression.
We also analyzed the reactive astrocyte genes using the canonical pathways analysis in IPA (Ingenuity Systems, www.ingenuity.com). To increase capture of induced pathways, we used the twofold cutoff gene set list from each reactive astrocyte subtype for analysis. The top 20 significant pathways, with significance value, are shown in Table 4. Constituent members of each pathway induced in the reactive astrocytes are also listed. IPA supports the finding that, although there are some pathways that are induced in both types of reactive astrocytes, astrogliosis is qualitatively different between the two inducing injuries. The acute-phase signaling and hepatic stellate cell activation, two pathways indicative of cellular activation, are induced in both reactive astrocyte populations. Prominently, the IL6 and IL10 signaling pathways, as well as aminosugar metabolism, which suggests increased metabolism, are enhanced in the MCAO reactive astrocytes. Conversely, the antigen presentation, complement, and response to interferon pathways are significantly induced in LPS reactive astrogliosis. IPA analysis supports the idea that astrogliosis differs depending on the inducing stimulus.
Table 4.
Ingenuity pathway analysis of pathways induced in reactive astrocytes
| Ingenuity canonical pathways | p value | Genes |
|---|---|---|
| Pathways induced in astrocytes after MCAO | ||
| Acute phase response signaling | 1.66E-08 | SERPING1, SAA1, SERPINF2, IL6R, C4B, STAT3, HMOX1, OSMR, C1R, NRAS, NFKBIE, KRAS, SERPINE1, IL6, MYD88, TNFRSF1A, A2M, IL1RAP, IL1R1, SERPINA3, CEBPB, C1S, SOCS5, RRAS2, SOCS3, CP, C3, HPX |
| IL-6 signaling | 1.45E-05 | TNFRSF1A, A2M, IL1RAP, IL6R, IL1R1, STAT3, CEBPB, TNFAIP6, HSPB1, CD14, RRAS2, NRAS, NFKBIE, MAP4K4, KRAS, IL6 |
| Hepatic fibrosis/hepatic stellate cell activation | 4.27E-05 | FAS, TNFRSF1A, A2M, ICAM1, IL1RAP, IL6R, IL1R1, CTGF, TLR4, CD14, TIMP1, MYL9, CXCL3, MYH9, CSF1, IGFBP4, IL6, IGFBP3, MET |
| Role of macrophages, fibroblasts, and endothelial cells in rheumatoid arthritis | 1.26E-04 | PLCE1, PLCD3, CEBPA, IL6R, CAMK2D, STAT3, C1R, MYC, NRAS, CSF1, NFKBIE, CEBPD, KRAS, IL6, TLR2, ROCK2, MYD88, TNFRSF1A, ICAM1, ATF4, IL1RAP, IL1R1, TLR4, CCND1, CEBPB, C1S, FZD3, RRAS2, ADAMTS4, SOCS3, NFATC2, CEBPG |
| Coagulation system | 2.40E-04 | BDKRB2, SERPINF2, A2M, PLAT, PROS1, PLAUR, SERPINE1, THBD |
| PI3K/AKT signaling | 2.51E-04 | PTGS2, GDF15, PPP2R2A, YWHAG, MCL1, CCND1, HSP90AA1, CDKN1A, RRAS2, EIF4EBP1, GAB2, NRAS, HLA-B, NFKBIE, KRAS, MAP3K8 |
| RAN signaling | 4.57E-04 | IPO5, RANBP1, XPO1, RAN, KPNB1 |
| Germ cell–Sertoli cell junction signaling | 4.79E-04 | ACTN1, RND3, SORBS1, TNFRSF1A, A2M, TUBA1A, IQGAP1, TUBB, TUBA1C, MAP3K6, RRAS2, NRAS, TUBB6, RHOJ, TUBA3E, KRAS, MAP3K8, ZYX |
| ERK5 signaling | 9.33E-04 | FOSL1, RRAS2, MYC, ATF4, YWHAG, NRAS, LIF, KRAS, MAP3K8, RPS6KA3 |
| HGF signaling | 1.23E-03 | PTGS2, CCND1, STAT3, CDKN1A, ELK3, ETS2, MAP3K6, RRAS2, NRAS, KRAS, IL6, MAP3K8, MET |
| Aminosugars metabolism | 1.38E-03 | PDE1B, PDE12, NPL, CYB5R3, PDE4B, HK1, PDE3B, GFPT2, CYB5R1, HK2, HKDC1 |
| 14-3-3-mediated signaling | 1.48E-03 | PLCE1, TNFRSF1A, YWHAG, PLCD3, TUBA1A, TUBB, TUBA1C, RRAS2, NRAS, TUBB6, TUBA3E, GFAP, VIM, KRAS |
| IL-10 signaling | 1.74E-03 | CD14, FCGR2B, SOCS3, IL1RAP, IL1R1, STAT3, MAP4K4, NFKBIE, HMOX1, IL6 |
| p53 signaling | 2.14E-03 | CCNG1, FAS, GADD45G, DRAM1, GNL3, GADD45A, RPRM, THBS1, CCND1, CDKN1A, GADD45B, PERP |
| PI3K signaling in B-lymphocytes | 2.34E-03 | PLCE1, PLCD3, ATF4, IRS2, TLR4, CAMK2D, FCGR2B, RRAS2, C3, NFATC2, NRAS, NFKBIE, ATF5, KRAS, ATF3 |
| Dendritic cell maturation | 3.39E-03 | TLR2, MYD88, TNFRSF1A, HLAC, ICAM1, ATF4, TLR4, COL18A1, FCGR2B, COL5A3, RELB, FSCN1, HLA-B, NFKBIE, IL6 |
| Thyroid cancer signaling | 3.72E-03 | BDNF, RRAS2, CXCL10, MYC, NRAS, CCND1, KRAS |
| Type I diabetes mellitus signaling | 5.13E-03 | SOCS5, FAS, MYD88, TNFRSF1A, SOCS3, HLAC, CASP8, PTPRN, IL1RAP, IL1R1, HLA-B, NFKBIE |
| ILK signaling | 5.37E-03 | ACTN1, RND3, PTGS2, TNFRSF1A, PPP2R2A, ATF4, PARVA, IRS2, CCND1, MYL9, MYC, FLNA, MYH9, RHOJ, VIM, FLNC |
| Oncostatin M signaling | 5.75E-03 | CHI3L1, OSMR, RRAS2, NRAS, STAT3, KRAS |
| Pathways induced in astrocytes after LPS treatment | ||
| Acute phase response signaling | 3.89E-08 | PIK3R1, SERPING1, SAA1, MYD88, A2M, RRAS, IL1R1, SERPINA3, C4B, C1S, OSMR, C1R, FN1, CP, C3, NFKBIE, LBP |
| Antigen presentation pathway | 3.98E-08 | B2M, HLA-C, HLA-E, PSMB9, HLA-B, PSMB8, TAP2, TAPBP |
| Interferon signaling | 1.74E-06 | IFITM1, IRF9, OAS1, IFI35, PSMB8, IFIT3, IFNAR2 |
| Hepatic fibrosis/hepatic stellate cell activation | 2.88E-06 | ICAM1, A2M, SMAD3, IL1R1, IFNAR2, TIMP1, CD14, TGFBR2, FN1, CXCL3, LBP, PGF, MET |
| Virus entry via endocytic pathways | 5.89E-05 | PRKCA, PIK3R1, B2M, HLAC, PRKCD, RRAS, FLNA, HLA-B, CAV1 |
| Glioma invasiveness signaling | 1.12E-04 | TIMP1, F2R, PIK3R1, RHOU, RRAS, RHOJ, CD44 |
| Role of pattern recognition receptors in recognition of bacteria and viruses | 1.20E-04 | IFIH1, PIK3R1, TLR2, MYD88, OAS1, C3, DDX58, PTX3 |
| Protein ubiquitination pathway | 1.26E-04 | USP18, UBE2V2, PSME1, B2M, HLA-C, PSMB9, HSPB8, PSMB10, TAP2, HSPB1, BIRC3, HLA-B, PSMB8, PSMB2, HSPB6 |
| Role of macrophages, fibroblasts, and endothelial cells in rheumatoid arthritis | 2.09E-04 | PIK3R1, TLR2, MYD88, ICAM1, PRKCD, RRAS, IL1R1, CAMK2D, CCND1, C1S, PRKCA, C1R, FN1, IL17RA, NFKBIE, CEBPD, PGF |
| Complement system | 2.19E-04 | C1S, SERPING1, C1R, C3, C4B |
| OX40 signaling pathway | 4.90E-04 | B2M, HLA-C, HLA-E, HLA-B, NFKBIE |
| Caveolar-mediated endocytosis signaling | 5.13E-04 | PRKCA, FLOT1, B2M, HLA-C, FLNA, HLA-B, CAV1 |
| LPS-stimulated MAPK signaling | 5.62E-04 | CD14, PRKCA, PIK3R1, PRKCD, RRAS, NFKBIE, LBP |
| HGF signaling | 7.76E-04 | PRKCA, PIK3R1, MAP3K6, PRKCD, RRAS, CCND1, CDKN1A, MET |
| Germ cell–Sertoli cell junction signaling | 8.13E-04 | PVRL2, PIK3R1, SORBS1, MAP3K6, TGFBR2, RHOU, A2M, RRAS, RHOJ, GSN |
| HER-2 signaling in breast cancer | 9.12E-04 | NRG1, PRKCA, PIK3R1, PRKCD, RRAS, CCND1, CDKN1A |
| fMLP signaling in neutrophils | 1.23E-03 | PRKCA, PIK3R1, PRKCD, RRAS, ARPC5, NFKBIE, ARPC1B, ITPR1 |
| Allograft rejection signaling | 1.35E-03 | B2M, HLA-C, HLA-E, HLA-B |
| Thrombin signaling | 1.35E-03 | F2R, ADCY9, PRKCA, PIK3R1, RHOU, PRKCD, RRAS, CAMK2D, MYLK, RHOJ, ITPR1 |
| Dendritic cell maturation | 1.58E-03 | PIK3R1, TLR2, RELB, MYD88, B2M, HLA-C, ICAM1, HLA-B, NFKBIE |
Shown are the top 20 canonical pathways identified using Ingenuity Pathway Analysis of genes more than twofold significantly enriched in MCAO and LPS reactive astrocytes. Bold is used for emphasis.
Validation of alternate forms of reactive astrocytes by ISH
Having discovered by GeneChip expression profiling that the character of reactive astrogliosis is different in response to different stimuli, we confirmed this using ISH on sections from injured brain tissue. We chose H2-D1, a class I MHC molecule, and Serping1, a C1q inhibitor, that is a critical regulator of complement activity (Cicardi et al., 2005), as representatives of the antigen presentation and complement pathways that expression profiling revealed were more strongly induced in astrocytes by LPS than by MCAO. H2-D1 is expressed at low levels in the healthy brain by all cell types in our expression profiling datasets. After injury, the most dynamically induced H2-D1 probe set was induced 30-fold by LPS, but only threefold by MCAO (Fig. 10A). ISH shows that H2-D1 was expressed in sparse cells in brain sections from healthy brain from saline-injected animals (Fig. 11A). After MCAO, H2-D1-expressing cells were present at higher density (Fig. 11B), but LPS increased the density to a still greater degree (Fig. 11C). Expression was observed not only in astrocytes, but based on morphology, other cell types. This is consistent with the recent findings that MCAO significantly induces H2-D1 and H2-K1 in neurons (Adelson et al., 2012). GeneChip expression profiling also identified the complement pathway as being induced, to a greater extent, in LPS reactive astrocytes. Serping1 is expressed at very low levels in the GeneChip expression profiles for all cell types. After injury, it was induced 6.5-fold in reactive astrocytes after MCAO and 34-fold in reactive astrocytes after LPS (Fig. 10C). ISH detected no expression in the cortex of healthy brain from a saline-injected animal (Fig. 11D). Very sparse cells expressed Serping1 after MCAO (Fig. 11E). After LPS, however, astrocytes throughout the cortex expressed Serping1 (Fig. 11F).
Figure 11.
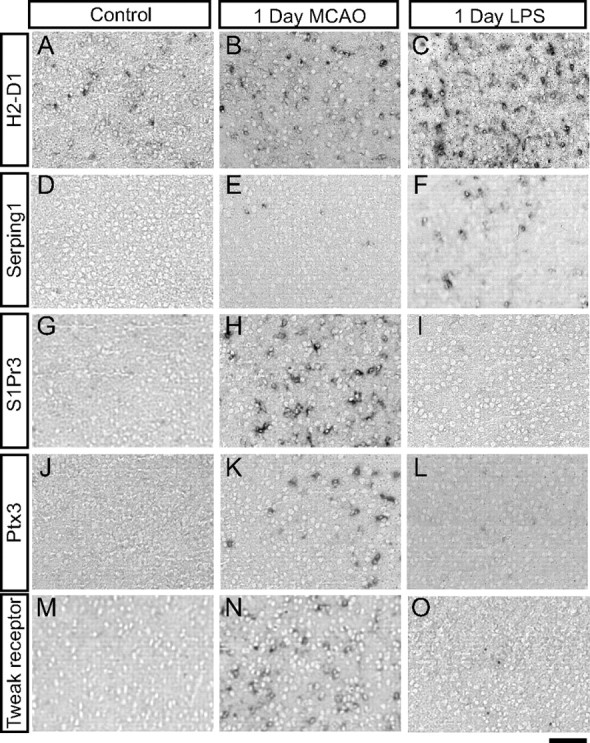
ISH confirms differences in expression of reactive astrocyte genes between injuries. In situ hybridization with potential markers of LPS and MCAO reactive astrocytes was performed on coronal brain sections from healthy (A, D, G, J, M) and injured (B, C, E, F, H, I, K, L, N, O) mice. A, D, H2-D1 (A) is expressed in sparse cells, and Serping1 (D) is not expressed in cortex of saline-injected mice. B, E, One day after MCAO, increased numbers of cells express H2-D1 (B) and very sparse cells express Serping1 (E). C, F, One day after LPS, a high density of cells, including some astrocytes, express H2-D1 (C) and Serping1 (F). G, J, M, S1Pr3 (G), Ptx3 (J), and tweak receptor (M) are not expressed in the healthy cortex from sham mice. H, I, K, L, N, O, S1PR3 (H), Ptx3 (K), and tweak receptor (N) are highly expressed in MCAO penumbral astrocytes 1 d after MCAO but are below detectable limits in the cortex of LPS-injected mice (I, L, A,). Scale bar, 100 μm.
Of the large number of other genes more highly expressed by astrocytes after MCAO than after LPS, many are involved in immune response, including the opsinin Ptx3 and signaling receptors for tweak and S1P. In our gene expression profiles, Ptx3 was induced 44-fold after MCAO but only 5.5-fold after LPS; tweak receptor (Tnfrsf12a) was induced by 14-fold after MCAO but only 3.3-fold by LPS; and S1P receptor 3 (S1Pr3) was induced 46-fold after MCAO but only 6.4-fold by LPS. ISH confirmed no detectable expression of Ptx3, tweak receptor, and S1Pr3 in the cortex of animals that had undergone a sham surgery (Fig. 11G,J,M) and demonstrated expression in the penumbra (Fig. 11H,K,N) 1 d after MCAO. Little to no expression of these markers is seen in the cortex of LPS-treated mice (Fig. I,L,O). The ISH studies thus confirmed that MCAO and LPS induced different subtypes of reactive astrocytes.
Individual astrocytes within the cortex respond differently to injury
Given the identified heterogeneity of reactive gliosis between stimuli, we also wondered whether there would be heterogeneity in astrocyte phenotype even within the response to a single inducing stimuli. To investigate this question, we used our newly identified markers of reactive astrogliosis to investigate the uniformity of the reactive astrocyte response. We used double fluorescent ISH to look at the extent and distribution of reactive astrocytes in the cortex after LPS injection and penumbra after MCAO. An ISH probe to the GLAST astrocytic glutamate transporter was used to mark the astrocytes in green in sections from healthy brain (Fig. 12A,C, E,G) and 1 d after injury (Fig. 12B,D,F,H). As expected based on the colorimetric ISH results, astrocytes throughout the cortex expressed Lcn2 (Fig. 12B) and Serpina3n (Fig. 12F), both shown in red, after LPS treatment but not in healthy cortex from saline-injected (Fig. 12A,E) and sham-operated (Fig. 12C,G) mice. Astrocytes in the MCAO lesion penumbra also express Lcn2 (Fig. 12D) and Serpina3n (Fig. 12H). Endothelial cells in this region also express Lcn2 (Fig. 12H white arrowhead). Reactive astrocytes, as defined by Lcn2 or Serpina3n expression (Fig. 12B,D,F,H, red arrows), were interspersed with adjacent quiescent and lightly reactive astrocytes (Fig. 12B,D,F, H, red arrowheads), demonstrating that neighboring astrocytes can differ in reactivity.
McCarthy-de Vellis astrocytes highly express many reactive astrocyte genes
A major finding from previous expression profiling of astrocytes is that cultured neonatal astrocytes produced by the McCarthy-de Vellis method (McCarthy and de Vellis, 1980) (MD-astrocytes) are highly dissimilar to mature astrocytes acutely purified from the healthy brain (astrocytes in vivo) (Cahoy et al., 2008; Foo et al., 2011). We were surprised to notice that many of our newly identified reactive astrocyte markers, including the markers Lcn2 and Serpina3n, common to both inflammation and MCAO injury-stimulated astrocytes, were also expressed at a much higher level in MD-astrocytes than in acutely purified postnatal and adult astrocytes in vivo isolated by FACS (Cahoy et al., 2008; Foo et al., 2011) and as determined by the Bac-Trap method (Doyle et al., 2008). In the expression profiling data from Cahoy et al. (2008), Lcn2 was expressed 593-fold higher and Serpina3n 11-fold higher in MD-astrocytes than in astrocytes in vivo. A full 60% (126 of 206) of MCAO reactive astrocyte genes were more than fourfold more highly expressed by MD-astrocytes than by astrocytes in vivo, whereas 47% (53 of 113) of LPS reactive astrocyte genes were more than fourfold more highly expressed by MD-astrocytes than by astrocytes in vivo (Fig. 13A). Hierarchical clustering using our most highly induced reactive astrocyte genes shows that MD-astrocyte replicates cluster with MCAO reactive astrocytes and away from both quiescent astrocytes and LPS reactive astrocytes (Fig. 13B). Using subsets of reactive astrocyte genes, MD-astrocytes cluster with MCAO reactive astrocytes and/or LPS reactive astrocytes depending on the specific pathways analyzed (Fig. 13C). When using ECM binding and adhesion genes, cytokine signaling molecules, and IL6 signaling pathway genes, MD-astrocytes clustered with MCAO reactive astrocytes. When using complement pathway genes, MD-astrocytes clustered with LPS reactive astrocytes. When using antigen presentation pathway genes, MD-astrocytes clustered with MCAO reactive astrocytes and quiescent astrocytes. When using peptidase inhibitors (closer to MCAO reactive astrocytes), transporters/channels (closer to LPS reactive astrocytes), acute phase signaling (closer to MCAO reactive astrocytes), and interferon response genes, MD-astrocytes clustered with both LPS and MCAO reactive astrocytes and away from quiescent astrocytes. These expression profiling analyses demonstrate the MD-astrocytes share many of the characteristics of reactive astrocytes.
Figure 13.

McCarthy-de Vellis astrocytes express elevated levels of reactive gliosis genes. A, The Venn diagram shows the distribution of genes significantly induced more than fourfold in LPS and MCAO reactive astrocytes and significantly expressed more than fourfold more in MD-astrocytes relative to astrocytes in vivo. More than 50% of astrocyte reactive gliosis genes are elevated in MD-astrocytes relative to astrocytes in vivo. B, The dendrogram represented in Euclidean distance was generated by hierarchical clustering of the quiescent, reactive, and MD-astrocyte expression profiles using the 263 more than fourfold induced reactive astrocyte genes. MD-astrocytes cluster most strongly with MCAO reactive astrocytes and away from all three groups of quiescent astrocytes. C, Hierarchical clustering represented in Euclidean distance of the quiescent, reactive, and MD-astrocyte expression profiles using class subsets of genes. Using the cytokine signaling genes, ECM binding/adhesion genes, and IL6 signaling pathway, MD-astrocytes cluster most closely with MCAO reactive astrocytes. Using the complement pathway and interferon response genes, MD-astrocytes cluster most closely with the LPS reactive astrocytes. Using transporter/channel genes, peptidase inhibitors, and acute phase signaling, MD-astrocytes cluster with both subtypes of reactive astrocytes. Using the antigen presentation pathway, MD-astrocytes cluster with quiescent astrocytes.
Discussion
Expression profiling identifies genes of diverse classes whose expression is induced in reactive astrocytes
The role of reactive astrocytes has long been mysterious. Expression profiling offers a powerful approach to understanding the molecular changes that characterize reactive astrocytes, which in turn provide a starting point for generating new hypotheses about their function. Our reactive astrocyte transcriptomes show that reactive astrocytes undergo extensive changes in gene expression relative to quiescent astrocytes. We identified over 1000 genes from across a broad spectrum of biological processes whose levels of expression were induced at least twofold. The extensive shift in gene expression supports the idea that reactive astrogliosis is a highly complex change in astrocyte cell state. The fold changes are dramatic, with 260 genes having more than fourfold induction of expression. Several of the most strongly expressed genes were induced over 100-fold, with several dozen genes over 10-fold induced. In brain tissue that had been injured by MCAO or activated by LPS exposure, most or all cell types in the brain undergo dramatic gene expression changes (Zamanian, Barres, and Giffard, unpublished observations). Separation of reactive astrocytes enables analysis of their relative contribution to the expression changes in CNS tissue. Although our protocol was not designed to separate reactive astrocytes from quiescent astrocytes, our in situ hybridization studies demonstrated that the majority of the astrocytes in the LPS-treated cortex and MCAO penumbra were in an activated state and contamination with quiescent astrocytes would only lead to underestimation of the observed changes in gene expression. The transcriptomes that we generated are therefore highly representative of the reactive astrocyte state and offer new insight into their phenotypes and possible functions.
One well studied role for reactive astrocytes is the modification of the extracellular milieu. Reactive astrocyte modification of the extracellular matrix can inhibit axon regeneration but also limit spread of damage after injury by formation of the glial scar. In accordance with this known activity, extracellular matrix genes were among the most highly represented in our induced gene expression profiles. In addition, many of the induced peptidases and peptidase inhibitors are secreted and will also influence the extracellular space. Upregulation of intermediate filament proteins such as GFAP, vimentin, and nestin, and changes in actin cytoskeleton binding proteins, are also a well represented class in the reactive astrocyte transcriptome, as would be expected based on the well described hypertrophy and morphological alterations of these cells. Many cytokines that are known to powerfully control the immune system were also represented in our gene profiles, suggesting that reactive astrocytes may play particularly important roles in controlling and interacting with immune cells. Interestingly, many of the reactive astrocyte genes are well described genes that are induced after injury and in wound healing in peripheral tissues, suggesting important commonalities between the way the brain and other tissue systems respond to injury. For instance, AHNAK, Anxa2, and s100a10 were highly upregulated and recently have been described to form a complex that promotes the sealing of damaged membranes (Lennon et al., 2003; Rezvanpour et al., 2011).
The wide array of genes induced demonstrates the complexity of reactive astrocyte influence on these biological processes and suggests new targets for manipulating reactive astrocytes after injury. Transporters, especially those that regulate metal ion homeostasis, and channels whose activities regulate intercellular ionic homeostasis are also well represented. One day after injury, there were, surprisingly, few changes in transcription factors. No transcription factor was more than fourfold induced in LPS reactive astrocytes. One of the few transcription factors identified as strongly induced in the MCAO reactive astrocytes was Klf6, which has recently been identified as being induced in astrocytes in an epilepsy injury model (Jeong et al., 2011). Signal transduction changes, which are essentially posttranslational modification, are primarily not picked up by gene expression profiling, but one central player in reactive gliosis, Stat3 (Sriram et al., 2004; Okada et al., 2006; Herrmann et al., 2008), is increased twofold in both types of reactive astrocytes and exhibits a sustained 50% elevation over the course of 1 week. Of great interest, many signaling receptors were very strongly upregulated in reactive astrocytes, including the oncostatin receptor Osmr, S1Pr3, Tnfrsf12a, and c-Met. These signaling pathways are thus new candidates to investigate for their roles in inducing or maintaining reactive astrogliosis.
Our findings also demonstrate that neonatal astrocyte cultures (MD-astrocytes) prepared by the method of McCarthy and de Vellis (1980) share many aspects of the reactive astrocyte phenotype, as shown by comparison of transcriptomes. They express over 100 genes not normally expressed by mature astrocytes in vivo (Cahoy et al., 2008; Foo et al., 2011) but do express 56% of the genes that are strongly induced in reactive astrocytes at elevated levels. These MD-astrocytes are also very highly proliferative compared with prospectively isolated mature brain astrocytes and may thus represent glial progenitor cells in a reactive state.
Most gene expression changes associated with reactive gliosis are transient
Although prolonged GFAP expression is widely reported after injury, and particularly in scar-forming astrocytes (Sofroniew, 2009), it has not been clear to what extent the reactive astrocyte state is stable, transient, or represents multiple phenotypes. To get at this question, we examined reactive astrocyte gene expression over 1 week. We found that during 1 week after MCAO or LPS exposure, broad classes of genes with roles in many different processes all demonstrated a strong burst of increased gene expression that was subsequently rapidly moderated. Neurotrophic cytokines and growth factors (Bauer et al., 2007) are among those genes rapidly downregulated. Conversely, pro-inflammatory cytokines are among the most persistently elevated genes. The powerful macrophage chemoattractant chemokine, CCL2, is rapidly induced and then downregulated. Recruitment of neutrophils and macrophages may be beneficial to clear bacteria and debris from dying cells. An interesting question raised by this observation is whether drugs that alter the time course of these reactive changes could be beneficial.
Identification of new markers for reactive astrocytes
Reactive astrocytes are frequently identified by their strong expression of the intermediate filament protein GFAP, considered a universal marker for reactive astrocytes. However, GFAP is also normally expressed by many astrocytes, and it would be useful for analysis of human brain injuries and diseases to have reactive astrocyte-specific markers that do not identify quiescent astrocytes. Our gene expression profiling shows that there is a core group of genes, including GFAP, that is induced in both of our populations of reactive astrocytes. We identified the acute phase protein, Lcn2, and proteinase inhibitor, Serpina3n, as markers common to reactive astrocytes induced by MCAO and LPS exposure. Their expression has been shown previously to be induced in several other injuries, including inflammation and excitotoxicity for Lcn2 (Chia et al., 2011) and after nerve injury (Gesase and Kiyama, 2007) and amyotrophic lateral sclerosis (Fukada et al., 2007) for Serpina3n. Lcn2 may be a marker of the earliest part of the reactive astrogliosis response as its expression is rapidly downregulated after initial induction. Serpina3n may be a useful marker of the more persistent reactive gliosis response because its expression remains relatively elevated at least over the course of 1 week. Given their expression after diverse injuries, they are possible universal markers for reactive astrocytes. Ptx3, Tgm1, and Cd109 are additional potential markers of reactive astrocytes. Like Serpina3n, the expression of these genes in our expression profile dataset is specific to astrocytes. Although they have low induced expression in LPS reactive astrocytes, they are induced, to a high degree, in MCAO reactive astrocytes and may be markers of this reactive astrocyte subtype.
Reactive astroglial phenotype depends on the nature of the inducing stimulus
It has been unclear whether there is one primary type of reactive astrocyte or whether there are multiple different reactive phenotypes depending on response to different acute and chronic diseases and injuries. Despite a core set of gene expression changes, we found that reactive astrocyte gene expression differed dramatically depending on whether the inducing stimulus was MCAO or LPS. Of a total of 263 reactive glial genes that we identified, 150 of these were preferentially expressed by MCAO reactive astrocytes, 57 were preferentially expressed by LPS reactive astrocytes, and only 56 were shared. For instance, although MCAO reactive astrocytes and LPS reactive astrocytes each had extensive induction of genes involved in modifying the extracellular space and interaction with the immune system, the particular genes within each of these classes differed. This is likely caused by differences in the inducing signals for each of these types of reactive astrocytes. Whereas LPS activates TLR4 receptors on microglia and macrophages (Buttini and Boddeke, 1995; Poltorak et al., 1998; Olson and Miller, 2004), which in turn secrete signals that induce reactive astrogliosis, MCAO exposes all brain cells within its territory to ischemia and causes direct cell death and breakdown of the blood–brain barrier, allowing serum influx. In MCAO, reactive astrogliosis is likely triggered by a combination of signals, such as signals from dying cells, hypoxia, acidosis, serum factors, direct trauma, and immune system signals. MD-astrocytes, which are grown in serum, express many of the MCAO reactive astrocyte genes (Cahoy et al., 2008; Foo et al., 2011). Different inducing signals thus induce different phenotypes of reactive astrocytes.
Our findings clearly demonstrate that there are distinct subtypes of reactive astrocytes and raise important new questions. How many types of reactive astrocytes are there, and how are their functions distinguished? With immune cells, the concept of functionally different subclasses of cell types, such as helper and suppressor lymphocytes or M1 and multiple M2 macrophages, is well established. Analogously, it will be important in future work to begin to understand the roles of the different types of reactive astrocytes. We identified many reactive astrocyte upregulated genes that were unique to the MCAO subtype or the LPS subtype of reactive astrocytes. Ptx3, S1Pr3 and tweak receptor are markers for the MCAO subtype of reactive astrocytes and H2-D1 and Serping1 are markers for the LPS subtype of reactive astrocytes. These genes will be useful as new markers of distinct reactive astrocyte subtypes so that these subtypes can now be identified and explored in different brain injuries and diseases.
Are different reactive astrocyte subtypes either relatively protective or relatively destructive?
A longstanding question concerning reactive astrocytes is whether they are beneficial or detrimental. The pioneering studies of Sofroniew (2009) and colleagues has established that scar-forming astrocytes induced by ischemia play a critical role in repairing the blood–brain barrier, limiting immune cell influx, and minimizing neuron death (Sofroniew, 2009). In analogy with the immune system, the gene profiles we have elucidated raise the hypothesis that reactive astrocytes can be polarized in a relatively protective orientation or, instead, in a relatively destructive orientation. Consistent with the work of Sofroniew and colleagues, our gene profiles strongly suggest that MCAO reactive astrocytes are protective. They express high levels of neurotrophic factors and cytokines, including CLCF1, LIF, and IL6, and thrombospondins that may help repair and rebuild lost synapses (Christopherson et al., 2005; Eroglu et al., 2009). In contrast, LPS reactive astrocytes more strongly upregulate the initial part of the classical complement cascade (C1r, C1s, C3, and C4) that is involved in synapse pruning during development (Stevens et al., 2007) and is hypothesized to drive the synapse loss, which drives neuron loss in neurodegenerative disease (Stevens et al., 2007; Stephan et al., 2012). LPS injection itself is sufficient to causes degeneration of the dopaminergic neurons that mimics Parkinson's disease (Herrera et al., 2000; Qin et al., 2007). Similarly, inflammation caused by bacterial infection, which can be experimentally mimicked by LPS injection, is known to be a priming factor for neurodegenerative disease (Perry, 2004). Because LPS upregulates the classical complement cascade components, which together with microglial-derived C1q provide a fully active complement system, it is possible that LPS is sufficient to activate complement-mediated destruction of synapses. Could activation of astrocytes in this detrimental orientation thereby sensitize the CNS to neurodegeneration? Using the markers of these potentially protective and detrimental reactive astrocyte subtypes identified in this work, this hypothesis can begin to be tested. If so, a future therapeutic avenue may be to develop new drugs that control the polarization of astrocytes, inhibiting the detrimental orientation and enhancing the protective orientation.
Footnotes
The authors declare no competing financial interests.
This work was supported by NIH Grants R01 NS059893 (B.A.B.) and RO1 GM49831 (R.G.G.), the Myelin Repair Foundation (B.A.B.), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (B.A.B.), the Christopher & Dana Reeve Foundation (B.A.B.), and an A-Star Fellowship (L.C.F.).
References
- Adelson JD, Barreto GE, Xu L, Kim T, Brott BK, Ouyang YB, Naserke T, Djurisic M, Xiong X, Shatz CJ, Giffard RG. Neuroprotection from stroke in the absence of MHCI or PirB. Neuron. 2012;73:1100–1107. doi: 10.1016/j.neuron.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alilain WJ, Horn KP, Hu H, Dick TE, Silver J. Functional regeneration of respiratory pathways after spinal cord injury. Nature. 2011;475:196–200. doi: 10.1038/nature10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony TE, Heintz N. The folate metabolic enzyme ALDH1L1 is restricted to the midline of the early CNS, suggesting a role in human neural tube defects. J Comp Neurol. 2007;500:368–383. doi: 10.1002/cne.21179. [DOI] [PubMed] [Google Scholar]
- Banks WA, Robinson SM. Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav Immun. 2010;24:102–109. doi: 10.1016/j.bbi.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto GE, Sun X, Xu L, Giffard RG. Astrocyte proliferation following stroke in the mouse depends on distance from the infarct. PLoS One. 2011;6:e27881. doi: 10.1371/journal.pone.0027881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S, Kerr BJ, Patterson PH. The neuropoietic cytokine family in development, plasticity, disease and injury. Nat Rev Neurosci. 2007;8:221–232. doi: 10.1038/nrn2054. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Hurtado A, Persaud T, Esham K, Pearse DD, Oudega M, Bethea JR. Transgenic inhibition of astroglial NF-kappa B leads to increased axonal sparing and sprouting following spinal cord injury. J Neurochem. 2009;110:765–778. doi: 10.1111/j.1471-4159.2009.06190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Buttini M, Boddeke H. Peripheral lipopolysaccharide stimulation induces interleukin-1 beta messenger RNA in rat brain microglial cells. Neuroscience. 1995;65:523–530. doi: 10.1016/0306-4522(94)00525-a. [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. 2005;48:16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Chia WJ, Dawe GS, Ong WY. Expression and localization of the iron-siderophore binding protein lipocalin 2 in the normal rat brain and after kainate-induced excitotoxicity. Neurochem Int. 2011;59:591–599. doi: 10.1016/j.neuint.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Cicardi M, Zingale L, Zanichelli A, Pappalardo E, Cicardi B. C1 inhibitor: molecular and clinical aspects. Springer Semin Immunopathol. 2005;27:286–298. doi: 10.1007/s00281-005-0001-4. [DOI] [PubMed] [Google Scholar]
- Clarke SR, Shetty AK, Bradley JL, Turner DA. Reactive astrocytes express the embryonic intermediate neurofilament nestin. Neuroreport. 1994;5:1885–1888. doi: 10.1097/00001756-199410000-00011. [DOI] [PubMed] [Google Scholar]
- Deng H, Han HS, Cheng D, Sun GH, Yenari MA. Mild hypothermia inhibits inflammation after experimental stroke and brain inflammation. Stroke. 2003;34:2495–2501. doi: 10.1161/01.STR.0000091269.67384.E7. [DOI] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggal N, Schmidt-Kastner R, Hakim AM. Nestin expression in reactive astrocytes following focal cerebral ischemia in rats. Brain Res. 1997;768:1–9. doi: 10.1016/s0006-8993(97)00588-x. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch MT, Silver J. CNS injury, glial scars, and inflammation: inhibitory extracellular matrices and regeneration failure. Exp Neurol. 2008;209:294–301. doi: 10.1016/j.expneurol.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- Foo LC, Allen NJ, Bushong EA, Ventura PB, Chung WS, Zhou L, Cahoy JD, Daneman R, Zong H, Ellisman MH, Barres BA. Development of a method for the purification and culture of rodent astrocytes. Neuron. 2011;71:799–811. doi: 10.1016/j.neuron.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada Y, Yasui K, Kitayama M, Doi K, Nakano T, Watanabe Y, Nakashima K. Gene expression analysis of the murine model of amyotrophic lateral sclerosis: studies of the Leu126delTT mutation in SOD1. Brain Res. 2007;1160:1–10. doi: 10.1016/j.brainres.2007.05.044. [DOI] [PubMed] [Google Scholar]
- Gadient RA, Otten UH. Interleukin-6 (IL-6)–a molecule with both beneficial and destructive potentials. Prog Neurobiol. 1997;52:379–390. doi: 10.1016/s0301-0082(97)00021-x. [DOI] [PubMed] [Google Scholar]
- Gasque P. Complement: a unique innate immune sensor for danger signals. Mol Immunol. 2004;41:1089–1098. doi: 10.1016/j.molimm.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Gesase AP, Kiyama H. Peripheral nerve injury induced expression of mRNA for serine protease inhibitor 3 in the rat facial and hypoglossal nuclei but not in the spinal cord. Ital J Anat Embryol. 2007;112:157–168. [PubMed] [Google Scholar]
- Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10:1033–1043. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- Han RQ, Ouyang YB, Xu L, Agrawal R, Patterson AJ, Giffard RG. Postischemic brain injury is attenuated in mice lacking the beta2-adrenergic receptor. Anesth Analg. 2009;108:280–287. doi: 10.1213/ane.0b013e318187ba6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, Greengard P, Heintz N. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz N. Gene expression nervous system atlas (GENSAT) Nat Neurosci. 2004;7:483. doi: 10.1038/nn0504-483. [DOI] [PubMed] [Google Scholar]
- Herrera AJ, Castaño A, Venero JL, Cano J, Machado A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis. 2000;7:429–447. doi: 10.1006/nbdi.2000.0289. [DOI] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herx LM, Yong VW. Interleukin-1 beta is required for the early evolution of reactive astrogliosis following CNS lesion. J Neuropathol Exp Neurol. 2001;60:961–971. doi: 10.1093/jnen/60.10.961. [DOI] [PubMed] [Google Scholar]
- Ip JP, Noçon Al, Hofer MJ, Lim SL, Müller M, Campbell IL. Lipocalin 2 in the central nervous system host response to systemic lipopolysaccharide administration. J Neuroinflammation. 2011;8:124. doi: 10.1186/1742-2094-8-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong KH, Lee KE, Kim SY, Cho KO. Upregulation of Kruppel-like factor 6 in the mouse hippocampus after pilocarpine-induced status epilepticus. Neuroscience. 2011;186:170–178. doi: 10.1016/j.neuroscience.2011.02.046. [DOI] [PubMed] [Google Scholar]
- Jonas RA, Yuan TF, Liang YX, Jonas JB, Tay DK, Ellis-Behnke RG. The spider effect: morphological and orienting classification of microglia in response to stimuli in vivo. PLoS One. 2012;7:e30763. doi: 10.1371/journal.pone.0030763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindy MS, Bhat AN, Bhat NR. Transient ischemia stimulates glial fibrillary acid protein and vimentin gene expression in the gerbil neocortex, striatum and hippocampus. Brain Res Mol Brain Res. 1992;13:199–206. doi: 10.1016/0169-328x(92)90027-9. [DOI] [PubMed] [Google Scholar]
- Laywell ED, Dorries U, Bartsch U, Faissner A, Schachner M, Steindler DA. Enhanced expression of the developmentally regulated extracellular matrix molecule tenascin following adult brain injury. Proc Natl Acad Sci U S A. 1992;89:2634–2638. doi: 10.1073/pnas.89.7.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Park JY, Lee WH, Kim H, Park HC, Mori K, Suk K. Lipocalin-2 is an autocrine mediator of reactive astrocytosis. J Neurosci. 2009;29:234–249. doi: 10.1523/JNEUROSCI.5273-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon NJ, Kho A, Bacskai BJ, Perlmutter SL, Hyman BT, Brown RH., Jr Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J Biol Chem. 2003;278:50466–50473. doi: 10.1074/jbc.M307247200. [DOI] [PubMed] [Google Scholar]
- Liu D, Smith CL, Barone FC, Ellison JA, Lysko PG, Li K, Simpson IA. Astrocytic demise precedes delayed neuronal death in focal ischemic rat brain. Brain Res Mol Brain Res. 1999;68:29–41. doi: 10.1016/s0169-328x(99)00063-7. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat Neurosci. 2006;9:443–452. doi: 10.1038/nn1654. [DOI] [PubMed] [Google Scholar]
- Marques F, Rodrigues AJ, Sousa JC, Coppola G, Geschwind DH, Sousa N, Correia-Neves M, Palha JA. Lipocalin 2 is a choroid plexus acute-phase protein. J Cereb Blood Flow Metab. 2008;28:450–455. doi: 10.1038/sj.jcbfm.9600557. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon RJ, Schreiber RC, Rudge JS, Silver J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. 1991;11:3398–3411. doi: 10.1523/JNEUROSCI.11-11-03398.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun. 2004;18:407–413. doi: 10.1016/j.bbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvanpour A, Santamaria-Kisiel L, Shaw GS. The S100A10-annexin A2 complex provides a novel asymmetric platform for membrane repair. J Biol Chem. 2011;286:40174–40183. doi: 10.1074/jbc.M111.244038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol. 2003;183:25–33. doi: 10.1016/s0014-4886(03)00082-7. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K, Benkovic SA, Hebert MA, Miller DB, O'Callaghan JP. Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: key signaling pathway for astrogliosis in vivo? J Biol Chem. 2004;279:19936–19947. doi: 10.1074/jbc.M309304200. [DOI] [PubMed] [Google Scholar]
- Stephan A, Stevens B, Barres BA. The complement system: an unexpected role in synapse regression. Ann Rev Neurosci. 2012;35:369–389. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Tanaka K, Suzuki N. Ambivalent aspects of interleukin-6 in cerebral ischemia: inflammatory versus neurotrophic aspects. J Cereb Blood Flow Metab. 2009;29:464–479. doi: 10.1038/jcbfm.2008.141. [DOI] [PubMed] [Google Scholar]
- Takamiya A, Takeda M, Yoshida A, Kiyama H. Inflammation induces serine protease inhibitor 3 expression in the rat pineal gland. Neuroscience. 2002;113:387–394. doi: 10.1016/s0306-4522(02)00198-7. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloboueva LA, Lee SW, Emery JF, Palmer TD, Giffard RG. Mitochondrial protection attenuates inflammation-induced impairment of neurogenesis in vitro and in vivo. J Neurosci. 2010;30:12242–12251. doi: 10.1523/JNEUROSCI.1752-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, Sofroniew MV. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci. 2009;29:11511–11522. doi: 10.1523/JNEUROSCI.1514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsson U, Bushong EA, Price DL, Smarr BL, Phung V, Terada M, Ellisman MH, Pekny M. Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc Natl Acad Sci U S A. 2006;103:17513–17518. doi: 10.1073/pnas.0602841103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Barreto GE, Xu L, Ouyang YB, Xie X, Giffard RG. Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke. 2011;42:2026–2032. doi: 10.1161/STROKEAHA.110.593772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita K, Vogel P, Fritze K, Back T, Hossmann KA, Wiessner C. Monitoring the temporal and spatial activation pattern of astrocytes in focal cerebral ischemia using in situ hybridization to GFAP mRNA: comparison with sgp-2 and hsp70 mRNA and the effect of glutamate receptor antagonists. Brain Res. 1996;735:285–297. doi: 10.1016/0006-8993(96)00578-1. [DOI] [PubMed] [Google Scholar]
- York IA, Rock KL. Antigen processing and presentation by the class I major histocompatibility complex. Annu Rev Immunol. 1996;14:369–396. doi: 10.1146/annurev.immunol.14.1.369. [DOI] [PubMed] [Google Scholar]