

Abstract
The amyloid protein aggregation associated with diseases such as Alzheimer’s, Parkinson’s, and type II diabetes (among many others), features a bewildering variety of β-sheet-rich structures in transition from native proteins to ordered oligomers and fibers. The variation in the amino acid sequences of the β-structures presents a challenge to developing a model system of β-sheets for the study of various amyloid aggregates. Here we introduce a family of robust β-sheet macrocycles that can serve as a platform to display a variety of heptapeptide sequences from different amyloid proteins. We have tailored these amyloid β-sheet mimics (ABSMs) to antagonize aggregation of various amyloid proteins, thereby reducing the toxicity of amyloid aggregates. We describe the structures and inhibitory properties of ABSMs containing amyloidogenic peptides from Aβ associated with Alzheimer’s disease, β2-microglobulin associated with dialysis-related amyloidosis, α-synuclein associated with Parkinson’s disease, islet amyloid polypeptide associated with type II diabetes, human and yeast prion proteins, and Tau, which forms neurofibrillary tangles.
Introduction
Amyloid aggregation is associated with many intractable protein aggregation diseases, notably including Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, type II diabetes, and prion diseases1–3. Amyloid fibrils with characteristic highly ordered cross-β structures are the ultimate products of amyloid aggregation. More than 30 proteins have been linked to amyloidogenesis and they exhibit enormous variations in sequences and polymorphic fibril structures1,4–6. Fibril formation of a given polypeptide, however, greatly depends on its specific residue order7,8. Crystallographic structures of amyloidlike fibrils formed by amyloidogenic peptide fragments suggest that the formation of highly-ordered parallel or antiparallel β-sheets and a steric-zipper interface between β-sheets are two essential elements for amyloid fibril formation9,10.
While amyloid fibrils are the most visible evidence of pathology, soluble oligomers are proving to be more important in amyloid toxicity11,12. Although increasing evidence shows that these transient, unstable structures are rich in β-sheets, their dynamic and polymorphic properties make amyloid oligomers difficult to study at the atomic level13–15. Additional tools are needed to study amyloid oligomers and aggregation and to shed light on controlling these processes.
β-Sheet mimics that can display amyloid β-strands provide a means to study amyloid oligomers and aggregation. We previously introduced 42-membered ring macrocyclic β-sheets containing pentapeptide fragments from amyloid-β peptide (Aβ) and tau protein (Tau) to mimic amyloidlike β-sheets and shed a light on the structures of transient amyloid oligomers16,17. We have also used these macrocyclic β-sheets to inhibit aggregation of the peptide Ac-VQIVYK-NH2 (AcPHF6) derived from Tau to provide insights into the aggregation process18.
The development of a robust chemical model of β-sheets that can tolerate a variety of amino acid sequences has been challenging because amyloidogenic sequences vary enormously and because folding of β-sheet mimics largely depends on the amino acid sequence1,19. In this paper, we introduce a new class of β-sheet macrocycles that can tolerate a wide range of amino acid sequences from amyloid proteins and still fold into β-sheet structures. We call these macrocycles amyloid β-sheet mimics (ABSMs).
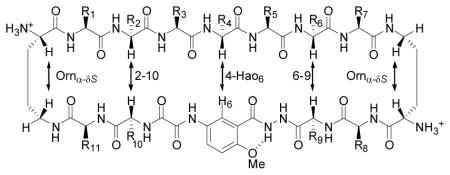
ABSM 1 is a 54-membered ring and comprises a heptapeptide β-strand (the upper strand), one Hao unit flanked by two dipeptides (the lower strand), and two δ-linked ornithine (δOrn) turns (Figure 1a). The “upper” β-strand of ABSM 1 incorporates different heptapeptide fragments from Aβ, Tau, yeast Sup35 prion protein (Sup35), human prion protein (hPrP), human β2-microglobulin (hβ2M), human α-synuclein (hαSyn), and human islet amyloid polypeptide (hIAPP). Hao is a tripeptide β-strand mimic that not only serves as a template for intramolecular hydrogen bonding but also minimizes the exposed hydrogen-bonding functionality of the “lower” strand20. This structural design of Hao helps prevent ABSMs 1 from aggregating in solution to form an infinite network of in-register β-sheets; instead, ABSMs 1 dimerize and then further self-assemble into oligomers. The “upper” and “lower” strands of ABSM 1 are connected by two δOrn β-turn mimics21.
Figure 1.

Design of amyloid β-sheet mimic 1. (a) Representation of ABSM 1 illustrating the upper β-strand (recognition β-strand), the δ-linked ornithine (δOrn) turn unit, and the Hao amino acid blocker unit. (b) Representation of ABSM 1 recognizing and blocking amyloid aggregation through β-sheet interactions.
We envisioned that ABSM 1 would fold well because it is conformationally constrained by cyclicity and has a Hao template to promote intramolecular hydrogen bonding and two δOrn β-turn mimics to promote turn formation. We also envisioned that four pairs of side chains (R1–R11, R2–R10, R6–R9, and R7–R8) would provide stabilizing transannular interactions. We anticipated that the flexibility of the dipeptides flanking Hao in the “lower” strand would better accommodate the flatness of the Hao template and thus minimize the kinks in the β-strands that we had previously observed in the 42-membered ring macrocycles17.
We designed ABSMs 1 to display exposed heptapeptide β-strands so that these β-strands can recognize and bind their parent amyloid proteins (Figure 1b). We envisioned recognition between ABSMs 1 and their parent amyloid proteins to take place through the β-sheet interactions observed in amyloid aggregation.
Here, we present structural studies of these ABSMs 1 and describe their effect upon amyloid aggregation and toxicity.
Results
1. Design of Amyloid β-Sheet Mimics 1
To test the folding of ABSMs 1, we selected 16 amyloidogenic heptapeptide β-strands from seven β-sheet-rich amyloid proteins for positions 1–7 in the “upper” strands (Table 1). ABSMs 1a–g contain heptapeptide sequences from two important hydrophobic and fibril-forming regions of Aβ associated with Alzheimer’s disease, residues 16–23 and 29–405, 22. ABSMs 1a–d and f contain native heptapeptide sequences, while ABSMs 1e and 1g are G33F and G37F mutants, in which the aromatic residue across from Hao promotes better folding16. ABSM 1h contains residues 7–13 from Sup35, which has been widely used as a model to study amyloid formation9. ABSM 1i contains residues 116–122 from hPrP, which is the infectious agent of prion diseases23. ABSM 1j contains residues 305–311 from Tau, which forms neurofibrillary tangles24. ABSM 1k–m contain residues 62–68 and 63–69 from hβ2M associated with dialysis-related amyloidosis25. ABSMs 1n and 1o contain residues 69–75 and 75–81 from hαSyn associated with Parkinson’s disease26. ABSMs 1p and 1q contain residues 11–17 and 26–32 from hIAPP associated with type II diabetes27. We chose polar and hydrophobic residues at positions 8–11 in the “lower” strands of ABSMs 1 to promote solubility in water and to increase hydrophobic residues that favor β-sheet formation.
Table 1.
 Amino Acid Sequences and Key NOEs of ABSMs 1a–q. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sequence | R1–R7 | R8–R11 | Ornα-δS | 2–10 | 4-Hao6 | 6–9 | Ornα–δS | Folding | ||
| 1a | Aβ16–22 | KLVFFAE | KLIE | Sa | —b | S | S | S | good | |
| 1b | Aβ17–23 | LVFFAED | KLIE | S | S | S | S | S | good | |
| 1c | Aβ29–35 | GAIIGLM | KFYK | S | S | S | S | S | good | |
| 1d | Aβ30–36 | AIIGLMV | KFYK | S | S | S | S | S | good | |
| 1e | Aβ30–36 G33F | AIIFLMV | KFYK | S | S | S | S | S | good | |
| 1f | Aβ34–40 | LMVGGVV | KFYK | S | S | Wa | —c | S | moderate | |
| 1g | Aβ34–40 G37F | LMVFGVV | KFYK | S | S | S | S | S | good | |
| 1h | Sup357–13 | GQQNNQY | KFYK | W | —c | —c | —c | W | poor | |
| 1i | hPrP116–122 | AAAGAVV | KFYK | W | W | —c | —c | W | poor | |
| 1j | Tau305–311 | SVQIVYK | EFYK | S | S | S | S | S | good | |
| 1k | hβ2M62–68 | FYLLYYT | KNSA | S | S | S | —b | S | good | |
| 1l | hβ2M63–69 | YLLYYTE | FKVS | W | —c | —c | —c | W | poor | |
| 1m | hβ2M63–69 | YLLYYTE | KVVK | S | —d | S | —d | S | good | |
| 1n | hαSyn69–75 | AVVTGVT | KFYV | S | S | S | —d | S | good | |
| 1o | hαSyn75–81 | TAVANKT | VFYK | S | S | S | —d | S | good | |
| 1p | hIAPP11–17 | RLANFLV | KFYK | S | S | S | S | S | good | |
| 1q | hIAPP26–32 | ILSSTNV | KFYK | S | S | S | S | S | good | |
| 1r | Aβ30–36 | AIIGLMV | KFFBr | K | ||||||
S: strong NOE; W: weak NOE.
NOE not observed due to overlap of proton resonances.
NOE not observed.
NOE not able to be observed due to overlap with HOD.
2. Synthesis of Amyloid β-Sheet Mimics 1
ABSMs 1 were prepared by synthesizing the corresponding protected linear peptides, followed by solution-phase cyclization and deprotection28. The protected linear peptide precursors were synthesized on 2-chlorotrityl chloride resin by conventional Fmoc-based solid-phase peptide synthesis. Macrocyclization was typically performed using HCTU and N,N-diisopropylethylamine in DMF at ca. 0.5 mM concentration. The ABSMs 1 were isolated in ca. 20–30% overall yield after HPLC purification and lyophilization. Each synthesis produces tens of milligrams of ABSMs 1 as fluffy white solids. (For details, see the Supplementary Information Text.)
3. X-Ray Crystallographic Studies of Amyloid β-Sheet Mimic 1r
X-ray crystallography of ABSM 1r validates the design of ABSMs 1 (Figure 2). ABSM 1r is a homologue of ABSM 1d in which the Tyr residue in the “lower” strand is replaced with 4-bromophenylalanine for crystallographic phase determination. ABSM 1r adopts a β-sheet structure in which the “upper” and “lower” strands are intramolecularly hydrogen-bonded to form eight hydrogen bonds (Figure 2a). The two δOrn residues of ABSM 1r fold into β-turn like conformations, Hao mimics a tripeptide β-strand, and the “upper” strand displays an exposed heptapeptide β-sheet edge.
Figure 2.

X-ray crystallographic structure of ABSM 1r, which contains the heptapeptide sequence AIIGLMV (Aβ30–36). (a) The monomer. (b) The dimer: top view. (c) The dimer: side view. (d) Stacked layers of dimer in the crystal lattice. Note that the view in b is perpendicular to the β-sheet (top view), whereas the view in c and d is 90° away, parallel to the β-sheet (side view) and shows the hydrophobic contacts. Some side chains in c and d have been omitted for clarity.
ABSM 1r forms a dimer in the crystal lattice in which the two recognition β-strands come together in an antiparallel β-sheet fashion (Figure 2b). The β-strands of the dimerization interface are shifted out of register, forming only six hydrogen bonds instead of the eight that would form through in-register contact.
The dimers stack in the crystal lattice, with hydrophobic contacts between the layers of the stack. The Ile, Leu, and Val, at positions 3, 5, and 7 on the “top” face of the dimer pack together in one set of hydrophobic contacts “above” the dimer, while the Met and Phe at positions 6 and 9 on the “bottom” face of the dimer pack together in another set of hydrophobic contacts “below” the dimer (Figures 2c and 2d). The hydrophobic contacts between the dimer layers appear to be important in the crystallization and supramolecular assembly of ABSM 1r and may explain the formation of the out-of-register interface within the dimer.
4. 1H NMR Studies of Amyloid β-Sheet Mimics 1
1H NMR studies of ABSMs 1a–q in D2O solution further validate the design of ABSMs 1 and establish that ABSMs 1 generally adopt folded β-sheet structures in solution. The 1H NMR spectra of ABSMs 1 show sharp, disperse resonances at submillimolar and low millimolar concentrations in D2O solution, suggesting ABSMs 1 to be non-aggregating in water. Antiparallel β-sheets have close contacts between the α-protons of the non-hydrogen-bonded pairs of amino acids, which generally give strong interstrand NOE cross-peaks (NOEs). In ABSMs 1, these close contacts should involve the α-protons of residues 2 and 10 (2–10) and residues 6 and 9 (6–9). There should also be homologous contacts involving the α-proton of residue 4 and H6 of Hao (4-Hao6) and the α- and pro-S δ-protons of the δOrn turns (Orn α-δS). Table 1 illustrates these contacts.
All ABSMs, except 1h, 1i, and 1l, exhibit most of these key NOEs (Table 1). ABSMs 1a–e, 1g, 1j, 1k, and 1m–q show strong 2–10, 6–9, 4-Hao6, and Ornα-δS NOEs and thus exhibit good folding. ABSM 1f shows strong Ornα-δS and 2–10 NOEs and a weak 4-Hao6 NOE and thus exhibits moderate folding. ABSMs 1h, 1i, and 1l show only Ornα-δS NOEs and thus exhibit weak folding. Although the lack of the interstrand NOEs indicates poor folding of ABSMs 1h, 1i, and 1l, the Ornα-δS NOEs suggest that their δOrn residues fold at least partially into a β-turn like conformation. Table 1 summarizes the observed key NOEs and the folding of ABSMs 1.
5. Inhibition of Amyloid Aggregation by Amyloid β-Sheet Mimics 1
Thioflavin T (ThT) fluorescence assays and transmission electron microscopy (TEM) studies show that the ABSMs containing amyloidogenic sequences can inhibit aggregation of amyloid proteins. We studied inhibition of Aβ40 and Aβ42 aggregation by ABSM 1a, inhibition of hβ2M aggregation by ABSM 1m, and inhibition of truncated human α-synuclein (hαSyn1–100) aggregation by ABSM 1o.
ThT fluorescence assays show that ABSMs 1a, 1m, and 1o effectively delay aggregation of their parent proteins at sub-stoichiometric concentrations in a dose-dependent manner (Figures 3a–d). ABSM 1a delays Aβ40 and Aβ42 aggregation by 280% and 350% respectively at 0.2 equivalents and by 430% and 600% at 0.5 equivalents (Figures 3a and 3b). Although ThT fluorescence assays show that Aβ aggregation exhibits comparable lag times at 0.5 and 1.0 equivalents of ABSM 1a, the growth phases of the aggregation are much slower at 1.0 equivalent than at 0.5 equivalents. (* in Figures 3a and 3b. For details, see Figures S1 and S2.) ABSM 1m delays hβ2M aggregation by 160% at 0.2 and 0.5 equivalents and by 340% at 1.0 equivalent (Figure 3c). ABSM 1o delays hαSyn1–100 aggregation by 150% at 0.2 equivalents (Figure 3d). Although hαSyn1–100 aggregation exhibits longer lag times with 0.5 and 1.0 equivalents of ABSM 1o than with 0.2 equivalents, some runs showed complete suppression of aggregation, while other runs showed typical sigmoidal curves. Because of this scatter in the data, the precise lag times are not reported. (* in Figure 3d. For details, see Figure S4.) TEM studies of samples taken directly from the ThT assays show that Aβ, hβ2M, and hαSyn1–100 form fibrils without ABSMs and do not form fibrils with ABSMs (1.0 equivalent) during the delayed lag time (Figures 3eh).
Figure 3.

Effect of ABSMs on inhibition of Aβ40, Aβ42, hβ2M, and hαSyn1–100 aggregation monitored by thioflavin T fluorescence assays and transmission electron microscopy. (a) Lag time of Aβ40 (20 μM) aggregation in the absence and presence of ABSM 1a. (b) Lag time of Aβ42 (20 μM) aggregation in the absence and presence of ABSM 1a. (c) Lag time of hβ2M (30μM) aggregation in the absence and presence of ABSM 1m. (d) Lag time of hαSyn1–100 (50μM) aggregation in the absence and presence of ABSM 1o. (e) TEM of Aβ40 (20 μM) after incubation for 6 h without ABSM 1a (top) and incubation for 6 h with 1.0 equivalent of ABSM 1a (bottom). (f) TEM of Aβ42 (20 μM) after incubation for 7 h without ABSM 1a (top) and incubation for 7 h with 1.0 equivalent of ABSM 1a (bottom). (g) TEM of hβ2M (30 μM) after incubation for 2 h without ABSM 1m (top) and incubation for 2 h with 1.0 equivalent of ABSM 1m (bottom). (h) TEM of hαSyn1–100 (50 μM) after incubation for 72 h without ABSM 1o (top) and incubation for 72 h with 1.0 equivalent of ABSM 1o (bottom). * For explanation, see the text and Figures S1, S2, and S4. Error bars correspond to standard deviation of four or greater sets of experiments. For experimental details, see the Supplementary Information Text.
Aβ has been shown to cross-interact with different amyloidogenic proteins containing similar primary sequences29–31. To investigate cross-interaction of Aβ with ABSMs, we compared the interaction of Aβ with ABSM 1a to that with ABSM 1m, which has a closely homologous sequence, and to that with ABSM 1o, which does not (Figure S5). ThT fluorescence assays show that ABSM 1m inhibits Aβ aggregation, like ABSM 1a, while ABSM 1o has little or no inhibitory effect (Figure S5). This result suggests that structurally homologous ABSMs can not only interact with their parent amyloid proteins but can also cross interact with different amyloid proteins.
To further investigate the effect of sequence on inhibition, we compared the interaction of ABSM 1a with Aβ40 to that of ABSMs 1b, 1c, 1d, and 1f with Aβ40. ThT fluorescence assays show that ABSM 1b is effective against Aβ40 aggregation, while ABSMs 1c, 1d, and 1f cause little or no inhibition (Figure S6). The inhibition of Aβ40 aggregation by both ABSMs 1a and 1b indicates that the central hydrophobic sequence Aβ17–21 is critical to the activity of ABSMs against Aβ40 aggregation. This result supports the role of Aβ17–21 in Aβ aggregation and suggests that strong interaction of this sequence in these ABSMs with that of the Aβ oligomers delays Aβ aggregation22, 32.
6. Detoxification of Aβ by Amyloid β-Sheet Mimic 1a
Cell viability (MTT) assays establish that ABSM 1a reduces the toxicity of Aβ40 and Aβ42 in PC-12 cells (Figure 4) and that ABSMs 1a, 1m, and 1o exhibit little or no toxicity (Figure S9). We examined the effect of ABSM 1a on the toxicity of Aβ40 and Aβ42, because ABSM 1a exhibits the best inhibitory activity among those studied. We first incubated Aβ monomers (5μM) without ABSM 1a to allow Aβ oligomers and fibrils to form. The resulting Aβ mixtures were directly used in cell viability assays. These assays show that the Aβ40 and Aβ42 preincubated without ABSM 1a kills 42% and 46% of the PC-12 cells respectively, relative to controls in which the cells are incubated in only PBS buffer solutions (Figure 4).
Figure 4.

Effect of ABSM 1a on Aβ40 and Aβ42 toxicity toward PC-12 cells. Addition of Aβ decreases cell survival when PC-12 cells are cultured for 24 h with preincubated Aβ. Cell survival increases when cells are cultured for 24 h with a preincubated mixture of ABSM 1a and Aβ in 0.2, 1.0, and 5 molar ratios. Cell survival is given as a percentage relative to controls in which only phosphate-buffered saline (PBS) is added. The cell survival of the PBS controls is taken to be 100%. Error bars correspond to standard deviation of four sets of experiments. For experimental details, see the Supplementary Information Text.
Cell viability assays further establish that preincubation of Aβ with ABSM 1a rescues the cells in a dose-dependent manner. Preincubation of Aβ40 and Aβ42 with 0.2 equivalents of ABSM 1a reduces the death of PC-12 to 29% and 38% respectively, while preincubation with 1.0 equivalent reduces cell death to 27% and 30% and preincubation with 5 equivalents reduces cell death to 14% and 6%. The rescue of these neuron like cells by ABSM 1a suggests that ABSMs may reduce the production of toxic amyloid oligomers or bind the oligomers and reduce their toxicity.
Discussion
Amyloid β-sheet mimics 1 provide a unique tool with which to elucidate the process of amyloid aggregation. Although many of the details of amyloid aggregation still remain unclear, nucleation-dependent polymerization, in which seeding to form a β-structured nucleus is the rate-determining step, is widely accepted1, 22. Based on the nucleation-dependent polymerization, we propose a model for the potent inhibition of Aβ aggregation by ABSM 1a. In this model, ABSM 1a binds early β-structured oligomers and blocks Aβ nucleation (Figure 5a). Without ABSM 1a, the unstructured monomer forms β-structured oligomers which, in the rate-determining step, go on to form a β-structured nucleus that ultimately assembles to form cross-β fibrils. The solid line in Figure 5a illustrates this pathway. ABSM 1a creates a new aggregation pathway for the early β-structured oligomers. In this pathway, ABSM 1a binds the β-structured oligomers to form Aβ-oligomer-ABSM-1a complexes and blocks the Aβ oligomer-to-nucleus transition. The dashed line in Figure 5a illustrates this pathway.
Figure 5.

β-Sheet interactions of Aβ peptides and ABSM 1a. (a) Proposed model of inhibition of Aβ aggregation by ABSM 1a. The solid curve corresponds to a pathway in which Aβ aggregates without ABSM 1a. The dashed curve corresponds to an alternative pathway in which ABSM 1a inhibits Aβ aggregation by binding Aβ oligomers. (b) Crystal structure of a macrocyclic peptide containing pentapeptide sequence LVFFA17. (PDB ID: 3Q9H) The magenta and green structures correspond to parallel and antiparallel β-sheet dimers formed by the macrocyclic peptide. The side view shows hydrophobic contacts formed between the parallel and antiparallel β-sheet dimers. (c) Crystal structure of the linear peptide KLVFFA35. (PDB ID: 3OW9) The orange and purple structures correspond to different layers within the crystal structure. The side view shows hydrophobic contacts between the layers.
It is significant that ABSM 1a substantially delays the aggregation of Aβ at sub-stoichiometric concentrations (as low as 1 μM), e.g. 0.05 equivalents of ABSM 1a per equivalent of Aβ (Figure S2), while simple linear peptide fragments derived from Aβ generally show substantial inhibitory effects at stoichiometric or greater concentrations33,34. This observation suggests that ABSM 1a binds a larger oligomer, not the monomer or a smaller oligomer, such as a dimer, trimer, or tetramer. ABSM 1a binds the early β-structured oligomers more strongly than the unstructured monomers bind oligomers, because the recognition β-strand of ABSM 1a is preorganized. This preorganization thus promotes formation of Aβ-oligomer-ABSM-1a complexes. The complexation may occur through edge-to-edge interactions between the hydrogen-bonding edge of ABSM 1a and exposed hydrogen-bonding groups of the Aβ oligomers and through face-to-face hydrophobic interactions between ABSM 1a and the hydrophobic surfaces of the Aβ oligomers. These types of interactions should take place between the hydrophobic sequence Aβ17–21 of ABSM 1a and that of the Aβ oligomers, as observed in the amyloid-related oligomers containing the pentapeptide sequence LVFFA shown in Figure 5b and the amyloidlike fibrils from the hexapeptide KLVFFA shown in Figure 5c. Similar interactions should also occur in the interactions of other ABSMs with their parent amyloidogenic peptides and proteins. The stabilization of these complexes creates a higher energy barrier to formation of the β-structured nucleus and thus delays or halts fibril formation. Because ABSM 1a cannot sequester all of the equilibrating Aβ oligomers, the Aβ monomers and oligomers eventually succumb to thermodynamics and form Aβ fibrils.
The X-ray crystallographic structure of ABSM 1r may provide insights not only into the stabilization of the dimerization and higher-order supramolecular assembly of ABSMs, but also into the stabilization and structure of intermediates formed during amyloid aggregation. The hydrophobic contacts formed by the Ile, Leu, and Val, at positions 3, 5, and 7 of ABSM 1r are akin to the steric zipper of amyloidlike fibrils formed by fragments Aβ16–21, Aβ30–35, Aβ35–40, and Aβ37–4210, 35. Both the layered crystal structure of ABSM 1r and the amyloidlike fibrils are stabilized by hydrophobic contacts. These observations suggest that maximization of both hydrophobic contact and hydrogen bonding is key to stabilizing not only amyloid fibrils but also transient amyloid oligomers36.
Conclusion
The amyloid β-sheet mimics 1 described herein provide a single platform with which to display a variety of amyloidogenic heptapeptide β-strands and provide a rational design for inhibitors to control amyloid aggregation. X-ray crystallographic and 1H NMR studies validate that the design of ABSMs 1, including the cyclicity, Hao template, two δOrn β-turn mimics, and paired side chains, promotes the formation of β-sheets in which the folding is largely independent of the amino acid sequence.
ABSMs 1 can be tailored to inhibit aggregation of different amyloid proteins. The inhibition of Aβ, hβ2M, and hαSyn1–100 aggregation by ABSMs 1 indicates that ABSMs containing one hydrogen-bonding edge and one blocking edge are an effective design for inhibitors of amyloid aggregation. The ability of ABSMs 1a, 1m, and 1o to inhibit amyloid aggregation and to detoxify amyloid aggregates suggests the potential for therapeutic applications in amyloid-related diseases.
Materials and Methods
Synthetic Aβ40 was purchased from GL Biochem (Shanghai) Ltd. Aβ42, hβ2M, and hαSyn1–100 were expressed in E. coli. (For details, see the Supplementary Information Text.) ABSMs 1 were synthesized as described above. (For details, see the Supplementary Information Text.) 1H NMR, 2D TOCSY, and 2D ROESY experiments of ABSMs 1 were performed in D2O with DSA as an internal standard at 500 MHz at 298 K. (For details, see the Supplementary Information Text.) Crystallization, data collection, and structure determination of ABSM 1r are described in the Supplementary Information Text. ThT fluorescence assays and TEM studies of Aβ, hβ2M, and hαSyn1–100 aggregation with ABSMs 1a, 1m, and 1o are described in the Supplementary Information Text. Cell viability assays to establish the toxicity of ABSMs 1a, 1m, and 1o toward HeLa, HEK-293, and PC-12 cells are described in the Supplementary Information Text.
Supplementary Material
Acknowledgments
We thank the NIH (5R01 GM049076, 1R01 GM097562, and 1R01 AG029430), NSF (CHE-1112188, CHE-0750523, and MCB-0445429), and HHMI for support, Arnie Berk and Dawei Gou for help with tissue culture experiments, and Suzanne Blum for suggestions to Figure 5a.
Footnotes
Author contributions
Author contribution: P.-N.C., C.L., D.E., and J.S.N. designed research; P.-N.C., C.L., and M.Z. performed research; P.-N.C., C.L., M.Z., D.E., and J.S.N. analyzed data; and P.-N.C., C.L., M.Z., D.E., and J.S.N. wrote the paper.
The authors declare no conflict.
References
- 1.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9:237–248. doi: 10.1038/nrd3050. [DOI] [PubMed] [Google Scholar]
- 3.Bartolini M, Andrisano V. Strategies for the inhibition of protein aggregation in human diseases. ChemBioChem. 2010;11:1018–1035. doi: 10.1002/cbic.200900666. [DOI] [PubMed] [Google Scholar]
- 4.Greenwald J, Riek R. Biology of amyloid: structure, function, and regulation. Structure. 2010;18:1244–1260. doi: 10.1016/j.str.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 5.Tycko R. Solid-state NMR studies of amyloid fibril structure. Annu Rev Phys Chem. 2011;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eichner T, Radford SE. A diversity of assembly mechanisms of a generic amyloid fold. Mol Cell. 2011;43:8–18. doi: 10.1016/j.molcel.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 7.Lopez de la Paz M, Serrano L. Sequence determinants of amyloid fibril formation. Proc Natl Acad Sci USA. 2004;101:87–92. doi: 10.1073/pnas.2634884100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci USA. 2010;107:3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson R, et al. Structure of the cross-β spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sawaya MR, et al. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 11.Conway KA, et al. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc Natl Acad Sci USA. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature. 2002;418:291–291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 13.Chimon S, et al. Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer’s β-amyloid. Nat Struct Mol Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- 14.Bernstein SL, et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ono K, Condron MM, Teplow DB. Structure–neurotoxicity relationships of amyloid β-protein oligomers. Proc Natl Acad Sci USA. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woods RJ, et al. Cyclic modular β-sheets. J Am Chem Soc. 2007;129:2548–2558. doi: 10.1021/ja0667965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu C, et al. Characteristics of amyloid-related oligomers revealed by crystal structures of macrocyclic β-sheet mimics. J Am Chem Soc. 2011;133:6736–6744. doi: 10.1021/ja200222n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng J, et al. Macrocyclic β-sheet peptides that inhibit the aggregation of a tau-protein-derived hexapeptide. J Am Chem Soc. 2011;133:3144–3157. doi: 10.1021/ja110545h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gellman SH. Minimal model systems for β-sheet secondary structure in proteins. Curr Opin Chem Biol. 1998;2:717–725. doi: 10.1016/s1367-5931(98)80109-9. [DOI] [PubMed] [Google Scholar]
- 20.Nowick JS, et al. An unnatural amino acid that mimics a tripeptide β-strand and forms β-sheet like hydrogen-bonded dimers. J Am Chem Soc. 2000;122:7654–7661. [Google Scholar]
- 21.Nowick JS, Brower JO. A new turn structure for the formation of β-hairpins in peptides. J Am Chem Soc. 2003;125:876–877. doi: 10.1021/ja028938a. [DOI] [PubMed] [Google Scholar]
- 22.Finder VH, Glockshuber R. Amyloid-β aggregation. Neurodegener Dis. 2007;4:13–27. doi: 10.1159/000100355. [DOI] [PubMed] [Google Scholar]
- 23.Walsh P, Simonetti K, Sharpe S. Core structure of amyloid fibrils formed by residues 106 126 of the human prion protein. Structure. 2009;17:417–426. doi: 10.1016/j.str.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 24.Friedhoff P, von Bergen M, Mandelkow EM, Davies P, Mandelkow E. A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci USA. 1998;95:15712–15717. doi: 10.1073/pnas.95.26.15712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Platt GW, Routledge KE, Homans SW, Radford SE. Fibril growth kinetics reveal a region of β2-microglobulin important for nucleation and elongation of aggregation. J Mol Biol. 2008;378:251–263. doi: 10.1016/j.jmb.2008.01.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vilar M, et al. The fold of α-synuclein fibrils. Proc Natl Acad Sci USA. 2008;105:8637–8642. doi: 10.1073/pnas.0712179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luca S, Yau WM, Leapman R, Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: Constraints from Solid-State NMR. Biochem. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng PN, Nowick JS. Giant macrolactams based on β-sheet peptides. J Org Chem. 2011;76:3166–3173. doi: 10.1021/jo102598n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan LM, Velkova A, Tatarek-Nossol M, Andreetto E, Kapurniotu A. IAPP mimic blocks Aβ cytotoxic self-assembly: cross-suppression of amyloid toxicity of Aβ and IAPP suggests a molecular link between Alzheimer’s disease and Type II diabetes. Angew Chem, Int Ed. 2007;46:1246–1252. doi: 10.1002/anie.200604056. [DOI] [PubMed] [Google Scholar]
- 30.Seeliger J, et al. Cross-amyloid interaction of Aβ and IAPP at lipid membranes. Angew Chem, Int Ed. 2012;51:679–683. doi: 10.1002/anie.201105877. [DOI] [PubMed] [Google Scholar]
- 31.Ma B, Nussinov R. Selective molecular recognition in amyloid growth and transmission and cross-species barriers. J Mol Biol. doi: 10.1016/j.jmb.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller Y, Ma B, Nussinov R. Polymorphism in Alzheimer Aβ amyloid organization reflects conformational selection in a rugged energy landscape. Chem Rev. 2010;110:4820–4838. doi: 10.1021/cr900377t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stains CI, Mondal K, Ghosh I. Molecules that Target beta-Amyloid. ChemMedChem. 2007;2:1674–1692. doi: 10.1002/cmdc.200700140. [DOI] [PubMed] [Google Scholar]
- 34.Sciarretta KL, Gordon DJ, Meredith SC. Peptide-based inhibitors of amyloid assembly. Methods Enzymol. 2006;413:273–312. doi: 10.1016/S0076-6879(06)13015-3. [DOI] [PubMed] [Google Scholar]
- 35.Colletier JP, et al. Molecular basis for amyloid-β polymorphism. Proc Natl Acad Sci USA. 2011;108:16938–16943. doi: 10.1073/pnas.1112600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laganowsky A, et al. Atomic view of a toxic amyloid small oligomer. Science. 2012;335:1228–1231. doi: 10.1126/science.1213151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.