

Abstract
Although it is well known that there are mutation hot spots in mtDNA, whether there are damage hot spots remain elusive. In this study, the regional DNA damage of mitochondrial genome after ionizing radiation was determined by real-time quantitative PCR. The mtDNA damage level was found to be dose-dependent and regional unequal. The control region was the most susceptible region to oxidative damage. GGG, as an typical hole trap during charge transport, was found to be disproportionally enriched in the control region. A total of 107 vertebrate mitochondrial genomes were then analyzed to testify whether the GGG enrichment in control region was evolutionary conserved. Surprisingly, the triple G enrichment can be observed in most of the homeothermal animals, while the majority of heterothermic animals showed no triple G enrichment. These results indicated that the triple G enrichment in control region was related to the mitochondrial metabolism during evolution.
The human mitochondrial genome is a small (16569-bp) closed-circular, duplex molecule that is present at a multiple number per cell. MtDNA contains 37 genes including 13 structural genes for the components of the respiratory-chain enzyme complexes, 2 ribosomal RNAs, and a complete set of 22 tRNAs required for the translation of mtDNA-encoded mRNAs1. MtDNA is believed to be more susceptible to DNA damage and consequently acquires mutations at a higher rate than does nuclear DNA. The differences in its susceptibility could be caused by lack of protective histones, limited DNA repair capacity, and high level of reactive oxygen species produced during oxidative phosphorylation2,3,4. It is estimated that the mutational rate of mtDNA is at least 10 times higher that of nuclear DNA. High polymorphism is characteristic of the mitochondrial genome. Transitions C-T and G-A occur at the rate of 2*10−7 per site per year in mammals5. On the other hand, GC-rich mtDNA sequences and especially palindromic regions are highly sensitive to mutagenesis6,7. Especially, there are positions and short sequences with an extremely high mutation rate8,9. One of the direct causes of mutations is the specific DNA damage at certain positions. As the mutation “hot spots” are widely indentified in mitochondrial genome, it is of interest to figure out whether there are mtDNA damage “hot spots” correspondingly.
MtDNA damage can be measured by Southern Blotting, 8-oxoG damage, or a comprehensive scanning of the mitochondrial genome by RFLP or TTGE analyses. However, these approaches are labor intensive and require large amounts of DNA. To overcome the difficulties in mtDNA damage evaluation, Santos et.al. established a long-run quantitative PCR method that could permitted the amplification of the whole mtDNA genome10. The rationale is that any lesion on DNA strands will interfere the PCR amplification. On that basis, real-time QPCR was developed for better quantification of mtDNA damage11. The semi-long run real-time PCR approach allowing analysis of differential DNA damage in the mitochondrial genome12. Although region-specific mtDNA damage has been investigated by Rothfuss et.al.12, only four regions in mtDNA genome have been analyzed and the whole picture of regional mtDNA damage is still lacking. In addition to the reports of Rothfuss et.al., Merino et.al. found that damage and mutations are preferentially funneled to the conserved sequence block II within the control region of the mitochondrial genome through DNA charge transport13,14,15. Charge migration in DNA has been a long lasting interest of the radiation community due to its relevance for the mechanisms of DNA oxidative damage16. The relative simplicity of mtDNA due to its prokaryotic origin provided favorable conditions to investigate the mechanism involved in the regional mtDNA damage.
Here, we developed a semi-long run real-time PCR method for the accurate quantification of mtDNA damage induced by ionizing radiation (IR). For the first time the regional mtDNA damage covered the whole mitochondrial genome was investigated. The mtDNA damage was found to be dose-dependent and regional unequal. The control region was found to be the most susceptible region to oxidative damage. A specific nucleotide motif, GGG was disproportionally enriched in the control region. A total of 107 mitochondria genome were then analyzed to testify whether the GGG enrichment in control region was genetic conserved.
Taken together, the first mitochondrial genomic mapping of DNA damage induced by IR was reported. Experimental data and bioinformatics analysis provided data supporting that charge transport play an important role in the radiation-induced region-specific mitochondria DNA damage. The triple G enrichment in control region of homeothermal animals provided evidence of mtDNA evolution in vertebrates.
Results
The D-loop region is most vulnerable to radiation induced mtDNA damage
To investigate the regional mtDNA damage induced by ionizing radiation, 11 pairs of primers were used to amplify the whole mitochondrial genome. Each of the amplicons was about 1.8 kb, with one exception that was 1077 bp (region 13902-14979). The results of mtDNA damage by SLR rt-PCR analysis were shown in Figure 1. Dose-dependent increase of mtDNA damage can be observed in all the regions analyzed. The SLR rt-PCR analysis revealed a similar lesion rate in all the encoding regions from position 1196 to 14979. Initial mtDNA damage in MCF-7 cells treated with 2 Gy X-ray irradiation ranged from 0.87 lesions per 10kb DNA in the region 4487-6286 to 1.51 lesions per 10 kb DNA in the region 7266-9077. A steady increase of mtDNA lesions was observed with the increase dosage of X-ray irradiation from 2–8 Gy. Strikingly, there were approximately 3.8-fold and 2.5-fold increase in region 14898-151 and 16488-1677, respectively with 2 Gy irradiation. The significant elevation of mtDNA lesions in these two regions can also be observed in MCF-7 cells with 4 Gy and 8 Gy irradiation, although the fold increase was alleviated. Interestingly, both region 14898-151 and 16488-1677 overlapped with non-coding region of mtDNA (16024-576), while all of the less-damaged areas (1196-14979) were within the coding region of mtDNA.
Figure 1. Radiation-induced dose-dependent and regional specific mtDNA damage.

(A) Quantification of mtDNA damage per 10 kb DNA by SLR rt-PCR amplification of total DNA isolated from MCF-7 cells exposed to 2–8 Gy X-ray irradiation showing an steadily increasing mtDNA damage with increased dosage in all tested mtDNA regions. Error bars designate standard deviation (at least three independent experiments). Region 14898-151 and 16488-1677 exhibited significant increased mitochondrial damage, overlapping the control region of human mitochondrial genome. (B) Quantitative measurement of mitochondrial DNA damage using Long-run quantitative PCR. Representation of the DNA lesions of the 8.9 kb and 9.4 kb mitochondrial fragments. Lesion frequencies of treated samples were calculated per amplicon size and expressed per 10 kb of mitochondrial genome. Error bars represent the SD, each done in at least triplicate. *Statistical significant at p < 0.05; **Statistical significant at p < 0.01.
To testify whether the regional mtDNA damage was cell line dependent, HeLa and HepG2 cells were employed. Similar results of initial mtDNA damage in each region was observed in HeLa and HepG2 cells after 4 Gy irradiation (data not shown), indicating that the regional mtDNA damage was not cell line specific.
To testify whether there was regional mtDNA damage after irradiation, as shown by SLR rt-PCR, a classic mtDNA damage assay, Long-run quantitative PCR was employed. As shown in Fig. 1. B, the mtDNA lesions of the 9.4 kb fragment harboring the D-loop region were much higher than the 8.4 kb amplicon that containing no D-loop region after 4 Gy irradiation. These results confirmed that there were preferentially damaged regions in mitochondrial genome after ionizing radiation.
The triple G was enriched in D-loop region
The complete map of mtDNA damage after oxidative insult provided evidence supporting that D-loop played a role of sacrifice during oxidative stress. So why most of the oxidative burden was loaded on the D-loop? It has been long recognized that radiation-induced DNA damage are not randomized17,18. Due to the different oxidation potential of DNA bases, guanine was thought to be the most easily oxidized base among all four bases of DNA19. It was therefore reasonable to speculate that region enriched with GC content could be more vulnerable to oxidative damage. However, both the severely damaged regions (14898-151 and 16488-1677) showed no overall biased GC enrichment (48% and 45.4%, respectively). Another explanation was that the non-randomness for DNA damage induction could be dependent on the primary structure of the adjacent DNA sequences20. If so, specific nucleotide motifs, rather than individual nucleotide positions, are targets of oxidative insult.
To investigate whether there were specific nucleotide motifs selectively accumulated in the most damaged region of mtDNA, computational scanning of the motif combinations in mtDNA was carried out. Initially, the complete 16569 bp mtDNA was divided by 1841 bp×9. The scanning began at the position 14898 according to the SLR rt-PCR results. The outputs showed that while most of the three code combination of nucleotides were distributed randomly along the mtDNA circle (Supplement materials), the GGG motif was unevenly accumulated in region 14898-169 (Fig. 2A). The enrichment of G-stretch in this region can also be detected by the 4–6 codons scanning (data not shown). Since D-loop has been proposed to be the most vulnerable region to oxidative damage in mtDNA, scanning began at position 16024, which coincided with the beginning of D-loop, was carried out. As shown in Fig. 2B, significant increased number of triple G in D-loop region was detected.
Figure 2. Computational scanning of the triple G across the mitochondrial genome.

The 16569 bp mtDNA was divided into 9 sections, each contained an equal number of 1841 bp fragment. The scanning began at position 14898 according to the SLR rt-PCR data (A) or began at position 16024 of the beginning of control region (B). The scanning program was design by C language.
Triple G enrichment in control region was restricted in homeothermal animals
If charge transport does play an important role in mitochondrial biogenesis, it would not be a special case for human. To further confirm the biological importance of charge transport in mtDNA, phylogenetic analyses combined with the screen of triple G enrichment in control region were carried out. 107 vertebrate species were analyzed and the result was illustrated in figure 3. Strikingly, most homeothermal animals (30/31) tested here showed triple G enrichment in control region, which were ranging from 96.41%–473.09%. The average abundance ratio for homeothermal animals was 211.95%. while most of the heterothermic animals (71/76) showed lower triple G distribution in control region (less than 100%), as measured by triple G abundance ratio (Fig. 3). The average abundance ratio for heterothermic animals was 56.30%.
Figure 3. Computational score of the abundance ratio of triple G in D-loop region of 107 vertebrates.
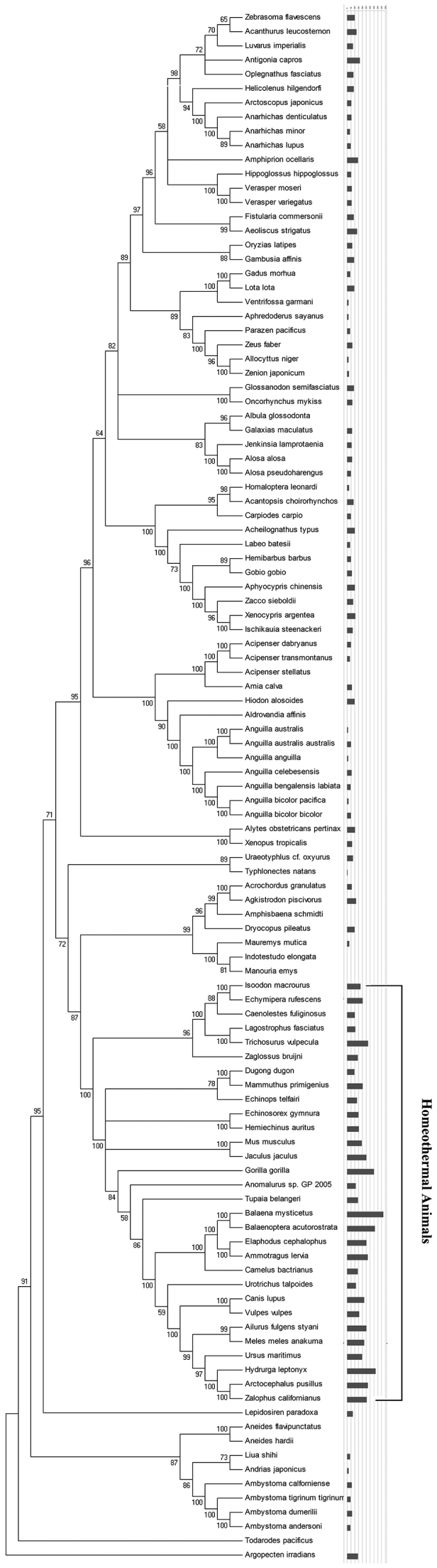
The scanning program was design by C language. The phylogenetic tree of vertebrates were constructed based on 107 complete vertebrate mitochondrial genome available in GenBank.
Discussion
The mitochondrial genome is known to be more fragile than nuclear DNA under ROS attack. Moreover, there has been speculation about a variable vulnerability to oxidative stress along the mitochondrial genome. Rothfuss et.al. first reported that D-loop was more prone to ROS-derived damage than other three region along mtDNA12. In line with their study, our current results showed that control is the one and only hot spot along mtDNA, as the complete mapping of regional mtDNA damage was provided. The control region is the most polymorphic region of mtDNA genome, it is therefore not surprise that initial damage inflicted by IR was most extensive in this area. Since mtDNA control region play a key regulatory role of mtDNA transcription and replication, the preferential funneling of damage to control region could served as a passive checkpoint for mtDNA dynamic control.
In radiation society, it has been a decades-long mystery that radiation-induced DNA fragmentation was not randomized. Nuclear DNA was too complex to investigate, but mtDNA may provide a more simpler model to investigate the mechanism involved. In our study we found that GGG was selectively enriched in control region. Triple G acted as the hole trap during charge transport (CT)21,22. Accordingly, oxidative damage could be funneled to the triple G site via charge transport. Our current findings underscored the importance of charge transport on mtDNA damage and mutations, as proposed by Merino et.al.14,15,23. The preferential funneling of damage to the control region via CT may provide a checkpoint for mtDNA replication and transcription, since D-loop regulated the replication and transcription of mtDNA. In addition, with the positive charge has a high probability of migrating towards the non-encoding area of mtDNA, the gene essentially for the electron transport chain was thus protected against the mutations.
The role of selection in mtDNA evolution attracted little attention until recently24,25,26. Current results pointed to the importance of charge transport in mtDNA during evolution progress. Homeothermal animals need to sustain high metabolic rate for the maintenance of body temperature (36–38°C for most mammals and 39–42°C for most birds). Mitochondrial oxidative phosphorylation (OXPHOS) generates not ATP but also heat, which is essentially important for the maintenance of body temperature in homeothermal animals. Shen et.al. suggested that strongly locomotive birds would have experienced stronger evolutionary constraints to eliminate deleterious mutations in mtDNA to maintain efficient energy metabolism, as compared to weak locomotive birds27. If so, homeothermal animals, which require more energy metabolism to sustain body temperature than heterothermic animals, should have also subject to stronger evolutionary constraints to minimize the deleterious mutations. The funneling of mtDNA damage to the non-encoding region via charge transport, thus provide additional weapon to defend the oxidative stress during energy metabolism in homeothermal animals. It is interesting to note that while triple G enrichment in the control region of mitochondrial genome was discovered in current study, triple G enrichment was obviously enriched at the end of linear chromosomes, called telomeres. Telomere repeat, TTAGGG, which was evolutionary conserved in vertebrates28, was also susceptible to oxidative stress29. Further investigation is needed for better understanding of the effect of charge transport on genome evolution.
Methods
Cell culture and irradiation treatment
The human breast cancer cell line, MCF-7, liver hepatocellular cells, HepG2 and cervix carcinoma cells, HeLa were purchased from the American Type Culture Collection. Cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM; Gibco) supplemented with 10% fetal bovine serum. Cells were cultured in 5% CO2 in humidified air at 37°C.
X-ray were generated with an X-ray machine (FAXITRON RX650, USA) operated at 130 keV. An exposure-rate meter (AE-1321 M, Applied Engineering Inc, Japan) was used for the dosimetry. The dose rates were 1.3 Gy/min. Cells in exponential growth were irradiated at room temperature, with non-irradiated culture cells (control) which were handled in parallel with the irradiated samples.
DNA isolation and purification
Samples after irradiation were immediately subjected to DNA isolation. Total DNA was purified using DNA Blood and Tissue Kit (Tiangen, China) from cells with/without ionizing radiation, respectively, and DNA quantity and purity was determined by spectrometric analysis. The isolated DNA showed a high purity (A260/A280 > 1.8) and was stored at 4°C according to standard procedures.
Semi-long run rt-PCR
The SLR rt-PCR amplifications were conducted in the FTC-3000 qPCR system (FUNGLYN, Canada), and the amplification was monitored and analyzed by measuring the intercalation of the fluorescent dye to double-stranded DNA supplied by the MightyAMP ver.2 kit (Takara, Japan). The mtDNA regions, primers and their PCR efficiency for rt-PCR are displayed in Table 1.
Table 1.
| MtDNA region | Primer sequences | PCR efficiency (%) | |
|---|---|---|---|
| 5464–7287 | F: TTACCACGCTACTCCTACCTAT | 1823 | 83.2 |
| R: GAGAAATGAATGAGCCTACAGAT | |||
| 4487–6286 | F: ACCCGTCATCTACTCTACCATC | 1799 | 79.4 |
| R: ACTGTTCAACCTGTTCCTGCT | |||
| 7266–9077 | F: TCTGTAGGCTCATTCATTTCTC | 1811 | 88 |
| R: ATGGTTGATATTGCTAGGGTG | |||
| 8910–10648 | F : CTTACCACAAGGCACACCTACA | 1738 | 85.1 |
| R:ATGGACCACAAGGCACACCTACA | |||
| 10294–12107 | F:TACAAACAACTAACCTGCCACT | 1815 | 83.6 |
| R: GTTGAGGGATAGGAGGAGAAT | |||
| 11977–13830 | F:CTCCCTCTACATATTTACCACAAC | 1853 | 87.8 |
| R:AAGTCCTAGGAAAGTGACAGCGA | |||
| 13902–14979 | F: CTCCAACATACTCGGATTCTAC | 1047 | 92 |
| R: ATTCAGCCATAATTTACGTCTC | |||
| 14898–151 | F: TAGCCATGCACTACTCACCAGA | 1849 | 84.8 |
| R:GGATGAGGCAGGAATCAAAGAC | |||
| 16488–1677 | F: CTGTATCCGACATCTGGTTCCT | 1785 | 81.1 |
| R : GTTTAGCTCAGAGCGGTCAAGT | |||
| 1196–2998 | F: AGAGGAGCCTGTTCTGTAATCG | 1802 | 84.7 |
| R: ATCCAACATCGAGGTCGTAAA | |||
| 2716–4658 | F: GACGAGAAGACCCTATGGAGC | 1942 | 78.7 |
| R: TTGCGTGAGGAAATACTTGATGG | |||
| 14620–14841[10] | F:CCCCACAAACCCCAT TACTAAACCCA | 222 | 99.5 |
| R:TTTCATCATGCGGAGATGTTGGATGG | |||
| 401–490 | F: CACAGACATCATAACAAAAAATTTCC | 90 | 99.9 |
| R: GGTGTTAGGGTTCTTTGTTTTTGG | |||
| 3459–3562 | F: ACGCCATAAAACTCTTCACCAAAG | 104 | 98.9 |
| R: TAGTAGAAGAGCGATGGTGAGAGCTA | |||
| 8456–8557 | F:ACAAACTACCACCTACCTCCC | 102 | 99.9 |
| R:CAATGAATGAAGCGAACAGAT | |||
| 15015–6140 | F:ATTCTTTATCTGCCTCTTCCTAC | 9417 | n/a |
| R: TCGTTGACCTCGTCTGTTATG | |||
| 5999–1484110 | F:TCTAAGCCTCCTTATTCGAGCCGA | 8843 | n/a |
| R:TTTCATCATGCGGAGATGTTGGATGG |
The PCR conditions for the different fragments were optimized to achieve similar amplification efficiencies required to compare different amplicons. The product specificity was monitored by melting curve analysis and sequenced by Sangon, Shanghai for identification (data not shown). The reaction mix (total volume V = 20 µl) consists of 1 × MightyAMP real time mix (Takara, Japan), 500 nM each forward and reverse primer and the equivalent quantities of template DNA (10 ng of total DNA). The cycling conditions include a pre-incubation phase of 10 min at 95°C followed by 40 cycles of 10 s 95°C, 10 s 55°C, and 60 s 72°C. Each sample was assayed in quadruplicate, fluorescence was continuously monitored versus cycle numbers and threshold cycle (Ct) values were calculated automatically.
Data analysis is based on the measurement of the Ct. Isolated total DNA from untreated sample was taken as reference. For each of the 11 mtDNA regions the difference in the Ct ΔCt (IR/non-IR) was used as a measure of the relative mtDNA damage with the 2−ΔCt method in correlation to the amplification size of the long fragment19. The Ct of IR-damaged mtDNA versus the Ct of non-irradiated mtDNA was directly employed to represent the level of mtDNA damage in each region. The short segment of each region was not employed for reference due to the fact that even the amplification of short amplicon could be affected by IR (Figure 4). The DNA damage was calculated as lesion per 10 kb DNA of each mtDNA region by including the size of the respective long fragment and displayed as average of at least three independent experiments.
Figure 4. Reduced amplification of different amplicons after ionizing radiation.

Analysis was performed immediately after exposure to radiation. IR/non-IR ratios were calculated by dividing the IR-damaged mtDNA signal for amplicon by the untreated control signal and expressing the ratio as a percentage of the untreated control set at 100%. Error bars represent the SD, each done in at least triplicate. *Statistical significant at p < 0.05.
Long-run quantitative PCR for mtDNA damage assay
long-run quantitative PCR method was conducted to validate the results generated by the SLR re-PCR method. Two amplicons were employed, the 8.9 kb amplicons were from Santos and colleagues10, and additional 9260 bp amplicon was used for confirmation of region specific mtDNA damage. The primer information was listed in table 1. The experiment procedures were carried out according to the protocol of Santos et.al.10.
Computational analysis of motif distribution in human mtDNA
A computer program was designed for the scanning of sequence motifs distribution on mtDNA. The input parameter included the length of motifs and the fragment size to divide the whole mtDNA. The output will be counting number of each motif in each fragment.
Computational scanning of triple G abundance ratio in D-loop region of vertebrates
The GGG abundance ratio was determined by the ratio of the number of triple G of D-loop to the total number of triple G in the mitochondrial genome. The scanning program was design by C language. A total of 107 vertebrate complete mtDNA sequences were analyzed.
Phylogenetic analyses
A total of 107 vertebrate mtDNA sequences were randomly chosen from NCBI nucleotide database, which fitted for both triple-G abundance ratio scoring and phylogenetic analysis. Sequences were aligned first using the program Clustal_X30 and refined manually. Phylogenetic analysis was done using the MEGA4.031. Distance matrices were constructed using the Kimura two-parameter model and trees generated by the neighbor-joining (NJ) distance method. The bootstrap values were estimated from 1,000 replications.
Statistical analysis
Statistical analysis was performed on the means of the data obtained from at least three independent experiments. Data are presented as means±SD. Student’s t-test program in Microsoft Excel was used to detect statistical significance. p<0.05 was considered to be statistically significant.
Author Contributions
Conceived and designed the experiments: HZ and XZ. Performed the experiments: XZ, XZ and YH. Software development: XL. Data analysis: RZ and XZ. Reviewed the paper: QL and ZW. Wrote the paper: XZ. HZ takes responsibility for the integrity of the data analysis.
Supplementary Material
Software package information
Acknowledgments
This work was supported by HIMAC project B364 which is supported by National Institute of Radiological Sciences, grants from the National Basic Research Program of China (973 Program) (2010CB834202), National Natural Science Foundation of China (10835011, 10675151, 10805064), the Scientific Technology Research Projects of Gansu Province (0702NKDA045, 0806RJYA020) and Western Talent Program of Chinese Academy of Sciences (0760160XBO, 0860260XBO).
References
- Shadel G. S. & Clayton D. A. Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem 66, 409–435, doi:10.1146/annurev.biochem.66.1.409 (1997). [DOI] [PubMed]
- Croteau D. L. & Bohr V. A. Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J Biol Chem 272, 25409–25412 (1997). [DOI] [PubMed] [Google Scholar]
- Beal M. F. Mitochondria, free radicals, and neurodegeneration. Curr Opin Neurobiol 6, 661–666 (1996). [DOI] [PubMed] [Google Scholar]
- Lightowlers R. N., Chinnery P. F., Turnbull D. M. & Howell N. Mammalian mitochondrial genetics: heredity, heteroplasmy and disease. Trends Genet 13, 450–455 (1997). [DOI] [PubMed] [Google Scholar]
- Lightowlers R. N., Howitt S. M., Hatch L., Gibson F. & Cox G. B. The proton pore in the Escherichia coli F0F1-ATPase: a requirement for arginine at position 210 of the a-subunit. Biochim Biophys Acta 894, 399–406 (1987). [DOI] [PubMed] [Google Scholar]
- Malyarchuk S. & Harrison L. DNA repair of clustered uracils in HeLa cells. J Mol Biol 345, 731–743, 10.1016/j.jmb.2004.10.079 (2005). [DOI] [PubMed] [Google Scholar]
- Cann R. L., Brown W. M. & Wilson A. C. Polymorphic sites and the mechanism of evolution in human mitochondrial DNA. Genetics 106, 479–499 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz S., Weisser H. J., Heizmann J. & Pollak S. Location and frequency of polymorphic positions in the mtDNA control region of individuals from Germany. Int J Legal Med 111, 67–77 (1998). [DOI] [PubMed] [Google Scholar]
- Myers K. A., Saffhill R. & O'Connor P. J. Repair of alkylated purines in the hepatic DNA of mitochondria and nuclei in the rat. Carcinogenesis 9, 285–292 (1988). [DOI] [PubMed] [Google Scholar]
- Santos J. H., Meyer J. N., Mandavilli B. S. & Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol 314, 183–199, doi:10.1385/1-59259-973-7:183 (2006). [DOI] [PubMed] [Google Scholar]
- Edwards J. G. Quantification of mitochondrial DNA (mtDNA) damage and error rates by real-time QPCR. Mitochondrion 9, 31–35, 10.1016/j.mito.2008.11.004 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothfuss O., Gasser T. & Patenge N. Analysis of differential DNA damage in the mitochondrial genome employing a semi-long run real-time PCR approach. Nucleic Acids Res 38, e24, doi:10.1093/nar/gkp1082 (2010). [DOI] [PMC free article] [PubMed]
- Merino E. J., Davis M. L. & Barton J. K. Common Mitochondrial DNA Mutations Generated through DNA-Mediated Charge Transport. Biochemistry 48, 660–666, doi:Doi 10.1021/Bi801570j (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino E. J. & Barton J. K. DNA oxidation by charge transport in mitochondria. Biochemistry 47, 1511–1517, doi:Doi 10.1021/Bi701775s (2008). [DOI] [PubMed] [Google Scholar]
- Merino E. J. & Barton J. K. Oxidation by DNA charge transport damages conserved sequence block II, a regulatory element in mitochondrial DNA. Biochemistry 46, 2805–2811, doi:Doi 10.1021/Bi062024+ (2007). [DOI] [PubMed] [Google Scholar]
- Chatterjee A. & Holley W. R. Computer simulation of initial events in the biochemical mechanisms of DNA damage. Advances in radiation biology 17, 181–226 (1993). [DOI] [PubMed] [Google Scholar]
- Rydberg B. Clusters of DNA damage induced by ionizing radiation: formation of short DNA fragments. II. Experimental detection. Radiat Res 145, 200–209 (1996). [PubMed] [Google Scholar]
- Lobrich M., Cooper P. K. & Rydberg B. Non-random distribution of DNA double-strand breaks induced by particle irradiation. Int J Radiat Biol 70, 493–503 (1996). [DOI] [PubMed] [Google Scholar]
- Steenken S. Electron transfer in DNA? Competition by ultra-fast proton transfer? Biol Chem 378, 1293–1297 (1997). [PubMed] [Google Scholar]
- Prise K. M., Pinto M., Newman H. C. & Michael B. D. A review of studies of ionizing radiation-induced double-strand break clustering. Radiat Res 156, 572–576 (2001). [DOI] [PubMed] [Google Scholar]
- Bixon M. et al. Long-range charge hopping in DNA. Proc Natl Acad Sci U S A 96, 11713–11716 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese B. et al. Memory of Chirality in Photochemistry. Angew Chem Int Ed Engl 38, 2586–2587 (1999). [PubMed] [Google Scholar]
- Merino E. J., Boal A. K. & Barton J. K. Biological contexts for DNA charge transport chemistry. Curr Opin Chem Biol 12, 229–237, DOI 10.1016/j.cbpa.2008.01.046 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoe T. A., Jiang Z. J., Gu W., Wang Z. O. & Pollock D. D. Adaptive evolution and functional redesign of core metabolic proteins in snakes. PLoS One 3, e2201, 10.1371/journal.pone.0002201 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Fonseca R. R., Johnson W. E., O'Brien S. J., Ramos M. J. & Antunes A. The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics 9, 119, 10.1186/1471-2164-9-119 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Pesini E., Mishmar D., Brandon M., Procaccio V. & Wallace D. C. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 303, 223–226, 10.1126/science.1088434 (2004). [DOI] [PubMed] [Google Scholar]
- Shen Y. Y., Shi P., Sun Y. B. & Zhang Y. P. Relaxation of selective constraints on avian mitochondrial DNA following the degeneration of flight ability. Genome Research 19, 1760–1765, doi:DOI 10.1101/gr.093138.109 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyne J., Ratliff R. L. & Moyzis R. K. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc Natl Acad Sci U S A 86, 7049–7053 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A., Rossi M. L., Aulds J., Croteau D. & Bohr V. A. Telomeric D-loops containing 8-oxo-2'-deoxyguanosine are preferred substrates for Werner and Bloom syndrome helicases and are bound by POT1. J Biol Chem 284, 31074–31084, doi:10.1074/jbc.M109.027532 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F. & Higgins D. G. The CLUSTAL_X Windows Interface: Flexible Strategies for Multiple Sequence Alignment Aided by Quality Analysis Tools. Nucleic Acids Research 25, 4876-4882, doi:10.1093/nar/25.24.4876 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Dudley J., Nei M. & Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24, 1596–1599, doi:10.1093/molbev/msm092 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Software package information