

Abstract
Abstract
The direct arylation of N-(2-pyridyl) substituted anilines is described. Arylation takes place in ortho position to the amine functionality and is directed by the pyridine N-substituent. Remarkably, N-arylation was never observed as a competing process even though conditions also suitable for Buchwald–Hartwig reactions were applied. The scope of the reaction was investigated in terms of aryl donors as well as the electronic nature of the substrate. Good yields were obtained for most examples through an operationally simple procedure, which did not require inert conditions or even glove box techniques. Pd(OAc)2 was applied as a cheap catalyst and boronic acids as readily available aryl donors. To obtain full conversion, 1,4-benzoquinone and a silver salt (e.g., Ag2O) were required as additives and reacted at relatively mild temperatures (e.g., 80 °C). Additionally, the pyridine-directing group was cleaved after the reaction to give ortho-arylated aniline derivatives.
Keywords: amines, C—H activation, heterocycles, homogeneous catalysis, palladium
Introduction
The formation of C—C bonds is a process of utmost importance in organic synthesis. Catalytic methods can realize such reactions. Prominent reactions in this regard are metal-catalyzed cross-coupling reactions of organo (pseudo) halides and various metal organyls.1 The great success of these reactions led to a Nobel Prize for Akira Suzuki, Richard Heck, and Ei-ichi Negishi in 2010. In recent years, a new trend has emerged in organic synthesis that attempts to further facilitate and simplify the transition metal catalyzed bond-forming process in synthetic strategy. Metal-catalyzed direct functionalization of C—H bonds (generally called C—H activation) strives to avoid at least one of the prefunctionalized building blocks typically used in cross-coupling reactions.2 This represents a significant advantage, as C—H bonds are ubiquitous in organic molecules and synthetic steps to obtain a required (pseudo) halide or organo-metal species can be avoided. Synthetic sequences become shorter and more time, resource, energy, and atom efficient, which falls perfectly in line with the principles of green chemistry.3 Such C—H activation reactions have been reported mainly for the functionalization of sp3 and sp2 C—H bonds (the Sonogashira reaction can be considered as a C—H activation reaction of an sp C—H bond). Methods to create C—C,4 C—O,5 C—N,6 C—S,7 and C—X8 bonds have been reported in recent years regarding the direct functionalization of sp2 hybridized C—H bonds. Since C—H bonds are omnipresent in organic molecules, this opens great opportunities for C—H activation chemistry, but also raises problems regarding the selectivity between this manifold of C—H bonds. Methods have been found to target one C—H bond of many to obtain a specific product from a direct functionalization reaction. The presence of a suitable directing group in the molecule is one way to achieve this: the metal catalyst is directed to a specific position, in which it activates one C—H bond. Regioselective functionalizations of C—H bonds have been reported on arenes by using directing groups, such as pyridine,[9] pyridine N-oxide,4e oxazoline,10 isoxazole,11 carboxylic acids,12 anilides,13 aldehydes and ketones (or their imine derivatives),14 and amides,15 to name the most prominent ones. Although this provides a large arsenal of possible directing groups, most of these groups cannot be readily cleaved, which presents a major limitation. Palladium, ruthenium, and rhodium have been most frequently applied as catalysts. In an ongoing project to use pyridine as a directing group for the arylation of C—H bonds, we have demonstrated the feasibility of sp3 arylation under Ru-catalysis.16 Here, we report on the direct ortho-arylation of aniline derivatives directed by a 2-pyridyl N-substituent, which can also be cleaved after direct arylation.
Results and Discussion
In our investigations towards regioselective and orthogonal cross-coupling methodologies on pyridine systems,17 an interesting and unexpected byproduct was observed, which clearly originated from a direct arylation after C—H activation (Scheme 1). Although the expected product of a Suzuki–Miyaura cross-coupling reaction in position 3 of pyridine was isolated as a major compound, ortho-arylation of the N-phenyl ring was also observed with a concomitant dechlorination. Dehalogenations in the presence of palladium are not uncommon, indeed they are exploited synthetically,18 however, the observed arylation represented an unusual and interesting result that indicated a directing effect by pyridine.
Scheme 1.
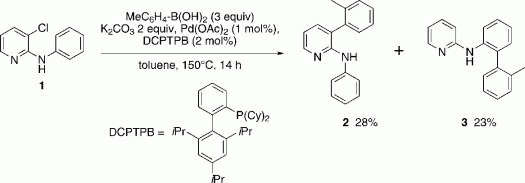
Initial finding of a pyridine directed direct arylation.
Remarkably, this transformation tolerated the presence of a free amino-group, as N-arylation would have also been possible under the applied reaction conditions. After scanning the literature for similar reactions, we found that ortho-arylation of 2-phenoxypyridines19 and 2-phenoxypyrimidines20 had been reported recently. In the case of 2-phenoxypyridines, potassium trifluoroborates were used as aryl donors, together with a complex mixture of solvents and additives (Scheme 2, 6 to 7). Owing to the high reaction temperature of 130–140 °C, the reactions had to be performed in an autoclave and lasted typically for 48 h. In the reaction with 2-phenoxypyrimidines, the more convenient boronic acids could be used, as well as toluene as the sole solvent at 120 °C, which did not require the use of an autoclave (Scheme 2, 4→5). The catalyst Pd(OAc)2 and the additives Ag2O and Cu(OTf)2 were used. Very recently, Wu and co-workers also reported a method with N-aryl-pyridine-2-amines as substrate (Scheme 2, 8→9+10).21 Under their optimized conditions, arylation of the N-aryl substituent took place in the ortho position and the resultant intermediate reacted further to give the corresponding N-2-pyridyl-carbazoles 10 to form the major product. This showed the CH activation reaction under Pd catalysis in the presence of a free amino functionality to be challenging because C—N bond formation could also take place under these conditions. Daugulis and co-workers reported direct arylation of 15 in ortho-position to the carbamate group. After deprotection, ortho-arylated anilines 16 were obtained.13d Very recently, an RuII-catalyzed protocol was also reported.22
Scheme 2.
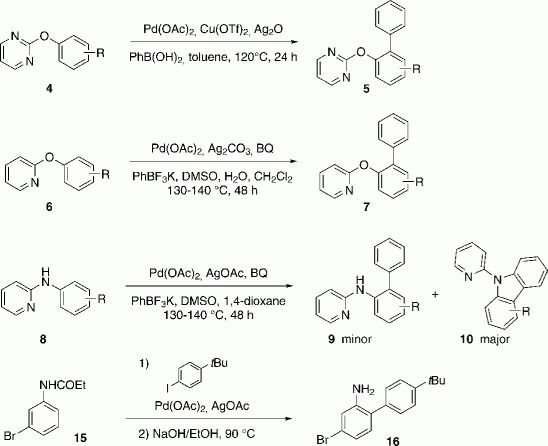
Literature procedures.
Based on these reported procedures, we started an optimization effort to further exploit the secondary process observed in our initial experiments. It was quickly found that the condition successful in the arylation of 2-phenoxypyridines was not suitable for arylation of N-phenylpyridine-2-amines. The use of Ph—BF3K as aryl donor did not lead to the complete conversion of the starting material and biphenyl formation was the most prominent process (Table 1, entries 1 and 2). This side reaction was also the reason for the use of 2.5 equiv. of aryl donor in the literature.19 To disfavor homocoupling, the presence of oxygen was excluded by performing the reaction under an argon atmosphere, however no improvement was detected in this or other experiments that compared reactions in air to those in an inert atmosphere. On using a solvent that did not require the use of an autoclave, the conversion could only be increased to 61 % if toluene was used (entry 2). Use of 1,2-dichlorobenzene as solvent (DCB, T=140 and 180 °C) led to complete failure of the reaction; use of 1,2-dichloroethane (DCE) also proved inefficient (data not shown).
Table 1.
Optimization of the pyridine directed direct arylation of 11 a.
| Entry | Solvent | T [°C] | Additives [equiv.] | Conversion[a,b] | |
|---|---|---|---|---|---|
| 1[c] | DCM | 135 | BQ (1.0) DMSO (4.0) | Ag2CO3 (2.0) H2O (8.0) | 50 |
| 2[c] | toluene | 120 | BQ (1.0) DMSO (4.0) | Ag2CO3 (2.0) H2O (8.0) | 61 |
| 3[d] | DCM | 135 | BQ (1.0) DMSO (4.0) | Ag2CO3 (2.0) H2O (8.0) | 60 |
| 4[d] | DCB | 140 | BQ (1.0) DMSO (4.0) | Ag2CO3 (2.0) H2O (8.0) | 42 (40) |
| 5[d] | DCE | 120 | BQ (1.0) DMSO (4.0) | Ag2CO3 (2.0) H2O (8.0) | 100 (62) |
| 6[d] | toluene | 120 | BQ (1.0) DMSO (4.0) | Ag2CO3 (2.0) H2O (8.0) | 100 (67) |
| 7[d] | toluene | 120 | BQ (1.0) | Ag2CO3 (2.0) | 78 (24) |
| 8[d] | toluene | 120 | Cu(OTf)2 (1.0) | Ag2O (1.0) | 44 |
| 9[d] | toluene | 120 | Cu(OTf)2 (1.0) | Ag2O (1.0) | 58 |
| 10[d] | toluene | 120 | BQ (0.5) | Ag2O (1.0) | 47 (10) |
| 11[d] | THF | 60 | BQ (0.5) | Ag2O (1.0) | 54 |
| 12[d] | THF | 60 | BQ (0.5) | Ag2O (1.0) | 82 |
| 13[d] | THF | 80 | BQ (0.5) | Ag2O (1.0) | 100 (69) |
| 14[d] | THF | 80 | Ag2O (1.0) | n.c.[e] | |
| 15[d] | THF | 80 | BQ (0.5) | 30 | |
| 16[d] | THF | 80 | BQ (1.0) | 59 |
[a] Determined by GC–MS with dodecane as the internal standard. [b] Isolated yield in parentheses. [c] 2.5 equiv. Ph—BF3K as aryl donor. [d] 3.0 equiv. Ph—B(OH)2 as aryl donor. [e] n.c.=No conversion.
This prompted us to change the aryl donor to phenylboronic acid, which led to significantly better results. If DCM was used as the solvent at 135 °C (autoclave), an increased conversion of 60 % was detected (entry 3); DCB also produced the desired product if the boronic acid was used as an aryl donor and 40 % of product was isolated (entry 4). However, biphenyl was a major byproduct and, hence, an excess of phenylboronic acid had to be used. On switching to dichloroethane, a full conversion was obtained for the first time (entry 5), and the product was isolated in 62 % yield. Full conversion was also obtained with toluene and the product was isolated in 67 % yield (entry 6). In this experiment, traces of bisarylated product were also detected according to GC–MS, but could not be isolated. If no water or DMSO were added, the conversion and isolated yields dropped significantly (entry 7).
Subsequently, we also investigated the conditions reported for the arylation of 2-phenoxypyrimidines. Using the same conditions, we found that it led to only 44 % conversion (entry 8), which improved if the 3 equivalents of boronic acid were added in several portions (58 %, entry 9). Substituting Cu(OTf)2 for the cheaper BQ, had no dramatic effect (entry 10). One remaining drawback of the conditions investigated thus far was that, according to TLC, the reactions were not very clean and several spots were always present, which complicated isolation by column chromatography. Based on the literature, THF was reported as a suitable solvent for pyridine-directed alkylation reactions at sp2 centers.[23a] Indeed, this led to a reaction that was much cleaner. The initial experiment at 60 °C gave 54 % conversion (entry 11), which could be improved to 82 % if the boronic acid was added in portions (entry 12). By increasing the temperature to 80 °C, full conversion was obtained and the product was isolated in 69 % yield (entry 13). Finally, we tested whether a combination of Ag2O and BQ was required to achieve full conversion. Using solely Ag2O gave no product formation at all (entry 14). However, 30 % conversion to the desired product was observed in the absence of Ag2O with 0.5 equiv. of BQ (entry 15). If the amount of BQ was increased (1.0 equiv.) 59 % conversion were detected. In both cases, significant biphenyl formation was observed, which was a typical result (entry 16). A further increase in BQ led to the predominant formation of the bisarylated product (not shown).
Based on the above summarized optimization results, we extended the following conditions for substrate scope investigations: 3 equiv. of boronic acid, 10 mol % Pd(OAc)2, 0.5 equiv. 1,4-benzoquinone (BQ), and 1 equiv. Ag2O in dry THF as solvent. As biphenyl formation could not be suppressed under an argon atmosphere, reactions were conducted in air for greater operational simplicity. Adding the arylboronic acid in serial portions suppressed the biphenyl formation to some extent and, hence, a periodic administration (1 equivalent every 6 h) was employed.
Initially, N-phenylpyridin-2-amine was arylated by using different boronic acids (Table 2). It was found that boronic acids carrying electron-donating (entries 2 and 3) and electron-withdrawing substituents (entries 4 and 5) were well-tolerated, giving yields between 62 % and 74 %. Actually, the most electron-deficient boronic acid (3-nitrophenylboronic acid) gave the highest yield (entry 5). Then, we investigated the influence of the electronic nature of the substrate. If the aryl ring for arylation was electron-rich, such as in N-(4-methoxyphenyl)pyridin-2-amine, improved yields were generally found (entries 6–10). This could be attributed to a facilitated oxidative addition, owing to the higher electron density of the phenyl ring. Introduction of the phenyl group gave 83 % yield (entry 6, cf. entry 1: 69 %), p-tolyl 76 % (entry 7, cf. entry 2: 63 %), and 3-nitrophenyl 88 % (entry 8, cf. entry 5: 74 %). 4-acetylphenylboronic acid and 4-fluorophenylboronic acid also produced good yields (entries 9 and 10).
Table 2.
Substrate scope of the pyridine directed direct arylation of compounds 11.
| Entry[a] | Substrate | Boronic acid | Product | Yield [%] |
|---|---|---|---|---|
| 1 |  |
 |
 |
69 |
| 2 | |
 |
 |
63 |
| 3 | |
 |
 |
65 |
| 4 | |
 |
 |
62 |
| 5 | |
 |
 |
74 |
| 6 | 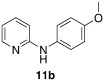 |
|
 |
83 |
| 7 | |
|
 |
76 |
| 8 | |
|
 |
88 |
| 9 | |
 |
 |
57 |
| 10 | |
 |
 |
77 |
| 11 | 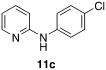 |
|
 |
63 |
| 12 | |
|
 |
57 |
| 13 | 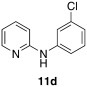 |
|
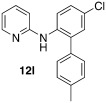 |
45 |
| 14 | 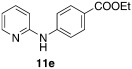 |
|
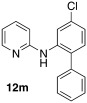 |
43 |
| 15 | 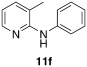 |
|
 |
50[a] |
[a] Bisarylated product 13 formed as a byproduct.
Switching to starting materials with electron-withdrawing substituents gave different results. In the case of N-(4-chlorophenyl)pyridine-2-amine, yields were slightly lower compared to the unsubstituted substrate (entries 11 and 12). This effect was even more pronounced if the chlorine was located at position 3 of the phenyl ring of the starting material (entry 13). In this case, two different products could be formed, owing to the presence of two different ortho-positions. However, only arylation of the sterically less demanding position was observed. Also carboxylic ester functionality in the starting material led to a decreased yield of 43 % (entry 14), which was the lowest in the whole series, but in our opinion still synthetically useful. In these substrates the decreased electron density of the phenyl ring was, of course, detrimental for oxidative addition and, hence, lower reactivity and lower yields were observed. Notably, the reaction was ineffective if sterically demanding o-tolylboronic acid was used and had a negligible conversion. This was true for substrate 11 a and the usually more reactive substrate 11 b.
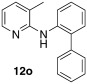
Using starting material 11 f, significant amounts of bisarylated product 13 were observed, which was not the case in any of the previous examples (Scheme 3). The formation of the bisarylated product can be explained by sterics. After monoarylation, the methyl group of the pyridine-directing group and the newly introduced phenyl ring incline to arrange away from each other, exposing the second ortho position to the directing group facilitating a second arylation reaction. By using 6 equiv. of boronic acid (in 1 equiv. portions over 24 h), mono- and bisarylated products were formed in a ratio of approximately 1:1. The isolated yield of the monoarylated product was 45 %, whereas 46 % of the bisarylated product was obtained.
Scheme 3.
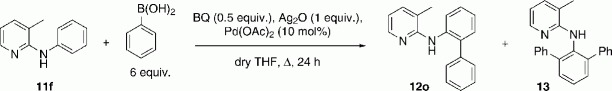
Bisarylation of substrate 11 f.
Mechanistic proposal
The mechanism proposed in Scheme 4 is supported by recent literature findings.21 The pyridine nitrogen initially coordinates the metal catalyst and facilitates insertion into the aryl C—H bond in the ortho position of the amino group, likely giving rise to an intermediate PdII complex such as A. The boronic acid then undergoes transmetalation with this complex to form AcO—B(OH)2 and B, still a PdII complex. Finally, reductive elimination from B delivers the product and a Pd0 species which is reoxidized by BQ or a combination of BQ and Ag2O to PdII and then re-enters the catalytic cycle.
Scheme 4.
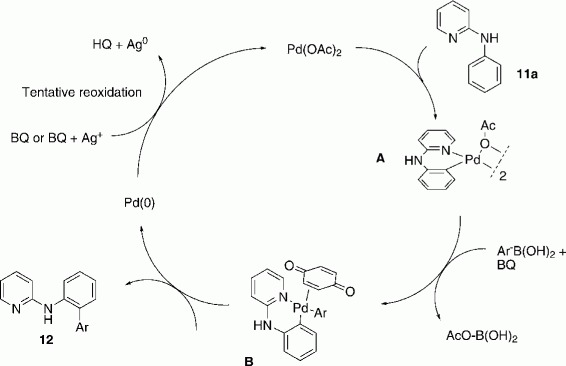
Proposed mechanism for the direct arylation of substrates 11.
It was stated in the literature that BQ is required for the C—H activation and reductive elimination step23 and for reoxidation of Pd0 to PdII.10b If BQ alone was needed for the reoxidation of Pd0 to PdII, at least some conversion (maximum 10 % for one full turnover of catalyst) should have been observed in its absence. However, our findings did not confirm this (Table 1, entry 14). Hence, BQ might also have played an important role as a ligand.21, 9a Several literature reports state that BQ promotes reoxidation of Pd0 to PdII in combination with a silver salt.9f, 24 Experiments in the absence of Ag2O suggested that the silver salt is actually not mandatory, although it facilitates this oxidation step (Table 1, entries 15 and 16). We found that 0.5 and 1.0 equiv. of BQ led to 30 and 59 % conversion, respectively. The oxidation potential of BQ was high enough to reoxidize Pd0, also in the absence of silver, but a combination of these two reagents worked significantly better. Additionally, it was reported that silver salts promoted the transmetalation step, which might also be the case in our transformation.9f Interestingly, we found that only parts of BQ were reduced to hydroquinone, as BQ could still be detected by GC–MS after full conversion.
Finally, we showed that the pyridine-directing group could be cleaved according to a literature-known procedure in two steps (Scheme 5).25 Pd-catalyzed hydrogenation and subsequent treatment with NH2NH2 and HCl furnished 2-aminobiphenyl 14 in good yield (78 %). This demonstrated that pyridine could be considered as the removable directing group and 2-aminobiphenyls were formed from this reaction sequence. These are important ligands for metal catalysts26 and structural motifs in organic electroluminescent devices.27
Scheme 5.
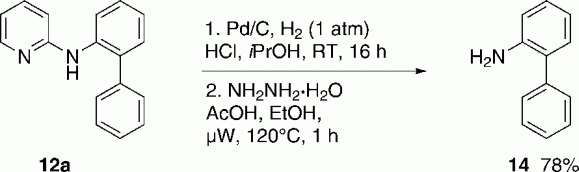
Cleavage of the pyridyl directing group.
Conclusions
An efficient method has been developed for arylation in the ortho position of anilines directed by pyridine. The reaction is robust regarding electronic effects because the electron rich substrates are more reactive. Regarding the aryl donors, electron poor boronic acids give higher yields compared to their electron rich counterparts. Sterically demanding boronic acids were not tolerated. Introducing steric bulk in the pyridine-directing group of the substrate leads to the bisarylation product. The reaction does not require inert conditions and is operationally simple. A mild temperature of 80 °C can be used and boronic acids act as readily available aryl donors. Cleavage of the pyridine directing group is also demonstrated and gives a good yield of 78 % over two steps. A plausible mechanism is proposed based on our observations.
After completion of our optimization experiments and substrate scope investigations, Wu and co-workers published a procedure in which N-2-pyridyl-carbazoles were formed from the type of starting materials used here.21 Also in this transformation Pd(OAc)2, a silver salt (AgOAc) and BQ were used together with Ar—BF3K salts as the aryl donor. The silver salt in a threefold excess and a full equivalent of BQ were required. 1,4-Dioxane and 4 equiv. DMSO were identified as the most effective solvents. Eventually, by increasing the amounts of oxidants and by using our boronic acid method disclosed in this contribution, it could be possible to form carbazoles as well. We intend to investigate this in the near future.
Experimental Section
Unless otherwise noted, chemicals were purchased from commercial suppliers and used without further purification. Microwave reactions were performed on a Biotage Initiator Sixty microwave unit. Flash column chromatography was performed on silica gel 60 from Merck (40–63 μm), whereas most separations were performed by using a Büchi SepacoreTM MPLC system with a 45 g column. For TLC aluminum-backed silica gel was used. Melting points were determined by using a Kofler-type Leica Galen III micro hot stage microscope and are uncorrected. HR-MS for compounds unknown in the literature were performed by E. Rosenberg at Vienna University of Technology, Institute for Chemical Technologies and Analytics; all samples were analyzed by LC-IT-TOF-MS in only positive ion detection mode with the recording of MS and MS/MS spectra. NMR-spectra were recorded in CDCl3 with TMS as internal standard on a Bruker AC 200 (200 MHz) spectrometer and chemical shifts are reported in ppm. For assignment of 13C multiplicities standard 13C and DEPT spectra were recorded. GC–MS runs were performed on a Thermo Finnigan Focus GC/DSQ II with a standard capillary column BGB 5 (ID=30 m×0.32 mm).
General method A: Preparation of starting materials
2-Bromopyridine (1 equiv.), amine (1.2 equiv.), NaOtBu (2.0 equiv.) or K2CO3 (10 equiv., in case of 1 d), Pd(OAc)2 (2 mol %) and (+/−)-BINAP (2 mol %) were taken in a closed vial and the reaction vessel was flushed with argon. Dry toluene was added to it through the septum and then it was placed in a heating block at 120 °C overnight. Purification by column chromatography was performed by using light petroleum (LP)/EtOAc to obtain the desired starting material 11 a,28 11 b,28 11 c,29 11 d,29 11 e,28 or 11 f,30 in excellent yield.
General method B: Direct arylation
N-aryl-2-aminophenyl pyridine (1 equiv.), aryl boronic acid (total 3 equiv. in 1. equiv. portions), BQ (0.5 equiv.), Ag2O (1 equiv.), and Pd(OAc)2 (10 mol %) were placed in a screw-cap vial with dry THF (4 mL) and the reaction mixture was stirred on a heating block at 80 °C for 24 h. A portion of aryl boronic acid (1 equiv.), was added at the start, followed by a portion (1 equiv.) after both 6 and 12 h. Reactions were monitor by TLC and GC—MS. Purification by column chromatography was performed by using LP/EtOAc. The following compounds were prepared using this method.
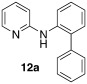
N-([1,1′-biphenyl]-2-yl)pyridin-2-amine (12 a): Substrate 11 a (50 mg, 0.29 mmol), phenylboronic acid (35 mg, 0.29 mmol), Ag2O (67 mg, 0.29 mmol), BQ (16 mg, 0.15 mmol), Pd(OAc)2 (6.5 mg, 0.029 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 69 % (50 mg, 0.20 mmol) yellow oil. Rf=0.26 (LP/EtOAc=10:1). GC–MS: 246 (69, M+), 245 (100), 169 (64), 230 (29), 167 (13). δ=6.46 (s, 1 H), 6.64–6.74 (m, 1 H), 6.79 (d, J=8.4 Hz, 1 H), 7.07–7.17 (m, 1 H), 7.23–7.51 (m, 8 H), 7.76 ppm (d, J=8.2 Hz, 1 H). 13C NMR (CDCl3, 50 MHz): δ=108.6 (d), 115.1 (d), 120.6 (d), 122.9 (d), 127.6 (d), 128.2 (d), 128.8 (d), 129.3 (d), 130.8 (d), 133.3 (s), 137.4 (s), 137.6 (d), 138.8 (s), 148.4 (d), 155.9 ppm (s). HR-MS: Predicted [MH]+=247.1230; Measured [MH]+=247.1220 (diff. in ppm=−4.05).
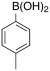
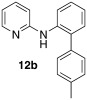
N-(4′-methyl-[1,1′-biphenyl]-2-yl)pyridin-2-amine (12 b): Substrate 11 a (50 mg, 0.29 mmol), 4-methylphenylboronic acid (39 mg, 0.29 mmol), Ag2O (67 mg, 0.29 mmol), BQ (16 mg, 0.15 mmol), Pd(OAc)2 (6.5 mg, 0.029 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 63 % (48 mg, 0.18 mmol) yellow oil. Rf=0.25 (LP/EtOAc=10:1). GC–MS: 260 (78, M+), 259 (100), 244 (40), 169 (54), 129 (20). 1H NMR (CDCl3, 200 MHz): δ=2.39 (s, 3 H), 6.45 (s, 1 H), 6.65–6.74 (m, 1 H), 7.05–7.15 (m, 1 H), 7.17–7.34 (m, 6 H), 7.35–7.57 ppm (m, 1 H). 13C NMR (CDCl3, 50 MHz): δ=21.2 (q), 108.6 (d), 115.0 (d), 120.6 (d), 122.0 (d), 129.7 (d), 129.6 (d), 130.8 (d), 133.4 (s), 135.8 (s), 137.3 (s), 137.5 (s), 137.7 (d), 148.3 (d), 156.0 ppm (s). HR-MS: Predicted [MH]+=261.1386; Measured [MH]+=261.1374 (diff. in ppm=−4.60).
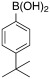
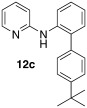
N-(4′-(tert-butyl)-[1,1′-biphenyl]-2-yl)pyridin-2-amine (12 c): Substrate 11 a (50 mg, 0.29 mmol), 4-tert-butylphenylboronic acid (52 mg, 0.29 mmol), Ag2O (67 mg, 0.29 mmol), BQ (16 mg, 0.15 mmol), Pd(OAc)2 (6.5 mg, 0.029 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 65 % (63 mg, 0.22 mmol) yellow oil. Rf=0.47 (LP/EtOAc=10:1). GC–MS: 302 (60, M+), 301 (51), 286 (40), 169 (100), 129 (51). 1H NMR (CDCl3, 200 MHz): δ=1.10 (s, 9 H), 6.20 (s, 1 H), 6.46 (dd, J1=7.0 Hz, J2=1.6 Hz, 1 H), 6.62 (d, J=8.4 Hz, 1 H), 6.79–6.90 (m, 1 H), 7.01–7.12 (m, 1 H), 7.14–7.27 (m, 3 H), 7.52 (d, J=8.0 Hz, 1 H), 7.92 ppm (dd, J1=4.9 Hz, J2=1.4 Hz, 1 H). 13C NMR (CDCl3, 50 MHz): δ=31.3 (q), 34.6 (s), 108.7 (d), 115.0 (d), 120.4 (d), 125.8 (d), 129.0 (d), 130.9 (d), 133.2 (s), 135.7 (s), 137.5 (s), 137.7 (s), 148.4 (d), 150.5 (s), 155.9 ppm (s). HR-MS: Predicted [MH]+=303.1862; Measured [MH]+=303.1856 (diff. in ppm=1.98).
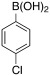
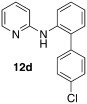
N-(4′-chloro-[1,1′-biphenyl]-2-yl)pyridin-2-amine (12 d): Substrate 11 a (50 mg, 0.29 mmol), 4-chlorophenylboronic acid (45 mg, 0.29 mmol), Ag2O (67 mg, 0.29 mmol), BQ (16 mg, 0.15 mmol), Pd(OAc)2 (6.5 mg, 0.029 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 62 % (51 mg, 0.18 mmol) yellow oil. Rf=0.49 (LP/EtOAc=10:1). GC–MS: 281 (32, M+), 280 (36), 264 (25), 143 (21), 169 (100). 1H NMR (CDCl3, 200 MHz): δ=6.33 (s, 1 H), 6.66 −6.81 ( m, 2 H), 7.07–7.18 (m, 7 H), 7.74 (d, J=7.8 Hz, 1 H), 8.10–8.19 ppm (m, 1 H). 13C NMR (CDCl3, 50 MHz): δ=108.4 (d), 115.2 (d), 121.3 (d), 123.4 (d), 128.6 (d), 129.0 (d), 130.6 (d), 132.5 (s), 133.6 (s), 137.3 (s), 137.4 (s), 137.7 (d), 148.4 (d), 155.8 ppm (s). HR-MS: Predicted [MH]+=281.0840; Measured [MH]+=281.0843 (diff. in ppm=1.07).
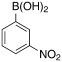
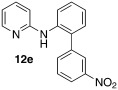
N-(3′-nitro-[1,1′-biphenyl]-2-yl)pyridin-2-amine (12 e): Substrate 11 a (50 mg, 0.29 mmol), 3-nitrophenylboronic acid (48 mg, 0.29 mmol), Ag2O (67 mg, 0.29 mmol), BQ (16 mg, 0.15 mmol), Pd(OAc)2 (6.5 mg, 0.029 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 74 % (63 mg, 0.22 mmol) yellow oil. Rf=0.16 (LP/EtOAc=10:1). GC–MS: 291 (34, M+), 290 (34,), 244 (32), 243 (44), 169 (100). 1H NMR (CDCl3, 200 MHz): δ=6.36 (s, 1 H), 7.06–7.19 (m, 1 H), 7.20–7.49 (m, 4 H), 7.55–7.72 (m, 2 H), 7.95–8.11 (m, 2 H), 8.15–8.21 ppm (m, 1 H). 13C NMR (CDCl3, 50 MHz): δ=108.3 (d), 115.3 (d), 122.4 (d), 122.7 (d), 124.2 (d), 129.5 (d), 129.6 (d), 130.8 9d), 132.2 (s), 135.4 (s), 137.5 (s), 137.8 (d), 140.8 (s), 148.4 (d), 148.5 (s), 155.9 ppm (s). HR-MS: Predicted [MH]+=292.1081; Measured [MH]+=292.1081 (diff. in ppm=0).
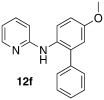
N-(5-methoxy-[1,1′-biphenyl]-2-yl)pyridin-2-amine (12 f): Substrate 11 b (50 mg, 0.25 mmol), phenylboronic acid (30.5 mg, 0.25 mmol), Ag2O (57 mg, 0.25 mmol), BQ (14 mg, 0.13 mmol), Pd(OAc)2 (5.6 mg, 0.025 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 83 % (57 mg, 0.21 mmol) yellow oil. Rf=0.32 (LP/EtOAc=5:1). GC–MS: 276 (100, M+), 275 (51), 261 (42), 199 (52), 116 (25). 1H NMR (CDCl3, 200 MHz): δ=3.84 (s, 3 H), 6.21 (s, 1 H), 6.57–6.70 (m, 2 H), 6.86–6.98 (m, 2 H), 7.27–7.48 (m, 6 H), 7.50–7.61 (m, 1 H), 8.05–8.15 ppm (m, 1 H). 13C NMR (CDCl3, 50 MHz): δ=55.6 (q), 107.5 (d), 113.8 (d), 114.2 (d), 115.8 (d), 125.1 (d), 127.6 (d), 128.6 (d), 129.9 (d), 130.2 (d), 136.8 (s), 137.6 (d), 138.8 (s), 148.2 (d), 156.3 (s), 157.1 ppm (s). HR-MS: Predicted [MH]+=277.1335; Measured [MH]+=277.1331 (diff. in ppm=−1.44).
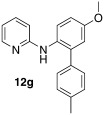
N-(5-methoxy-4′-methyl-[1,1′-biphenyl]-2-yl)pyridin-2-amine (12 g): Substrate 11 b (50 mg, 0.25 mmol), 4-methyl boronic acid (34 mg, 0.25 mmol), Ag2O (57 mg, 0.25 mmol), BQ (14 mg, 0.13 mmol), Pd(OAc)2 (5.6 mg, 0.025 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 76 % (55 mg, 0.19 mmol) yellow oil. Rf=0.18 (LP/EtOAc=10:1). GC–MS: 290 (100, M+), 289 (51), 275 (39), 199 (42), 78 (43). 1H NMR (CDCl3, 200 MHz): δ=2.40 (s, 3 H), 3.82 (s, 3 H), 6.17 (s, 1 H), 6.58 −6.69 (m, 2 H), 6.84–6.96 (m, 2 H), 7.15–7.30 (m, 4 H), 7.35–7.47 (m, 1 H), 7.50–7.60 (m, 1 H), 8.05–8.14 ppm (m, 1 H). 13C NMR (CDCl3, 50 MHz): δ=21.1 (q), 55.6 (q), 107.4 (q), 113.6 (d), 114.2 (d), 115.8 (d), 124.9 (d), 128.9 (d), 129.4 (d), 130.3 (s), 135.8 (s), 136.8 (s), 137.4 (d), 148.2 (d), 157.2 (s), 156.2 ppm (s). HR-MS: Predicted [MH]+=291.1492; Measured [MH]+=291.1491 (diff. in ppm=−0.34).
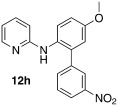
N-(5-methoxy-3′-nitro-[1,1′-biphenyl]-2-yl)pyridin-2-amine (12 h): Substrate 11 b (50 mg, 0.25 mmol), 3-nitrophenylboronic acid (42 mg, 0.25 mmol), Ag2O (57 mg, 0.25 mmol), BQ (14 mg, 0.13 mmol), Pd(OAc)2 (5.6 mg, 0.025 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 88 % (71 mg, 0.22 mmol) yellow oil. Rf=0.18 (LP/EtOAc=10:1). GC–MS: 321 (100, M+), 320 (36), 306 (29), 207 (28), 199 (72). 1H NMR (CD3OD, 200 MHz): δ=3.84 (s, 3 H), 6.34–6.47 (m, 1 H), 6.49–6.66 (m, 3 H), 6.91–7.10 (m, 2 H), 7.27–7.41 (m, 2 H), 7.43–7.56 (m, 1 H), 7.73–7.86 (m, 2 H), 8.01–8.12 (m, 1 H), 8.21–8.32 ppm (m, 1 H). 13C NMR (CD3OD, 50 MHz): δ=56.1 (q), 109.7 (d), 114.7 (d), 116.8 (d), 122.9 (d), 124.8 (d), 130.4 (d), 131.3 (s), 136.4 (d), 138.8 (s), 139.2 (d), 142.8 (s), 151.3 (s), 159.4 ppm (s). HR-MS: Predicted [MH]+=322.1186; Measured [MH]+=322.1194 (diff. in ppm=2.84).
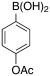
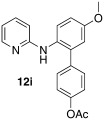
5′-Methoxy-2′-(pyridin-2-ylamino)-[1,1′-biphenyl]-4-yl acetate (12 i): Substrate 11 b (50 mg, 0.25 mmol), 4-acetylphenylboronic acid (41 mg, 0.25 mmol), Ag2O (57 mg, 0.25 mmol), BQ (14 mg, 0.13 mmol), Pd(OAc)2 (5.6 mg, 0.025 mmol). Column chromatography 5:1 LP/EtOAc, followed by a preparative TLC by using LP/EtOAc=3:1 and 1 % NEt3. Yield: 57 % (45 mg, 0.18 mmol) yellow oil. Rf=0.15 (LP/EtOAc=5:1). GC–MS: 318 (100), 317 (50), 303 (38), 199 (66), 78 (42). 1H NMR (CDCl3, 200 MHz): δ=2.60 (s, 3 H), 3.84 (s, 3 H), 6.10 (s, 1 H), 6.53–6.69 (m, 2 H), 6.87–7.01 (m, 2 H), 7.35–7.60 (m, 4 H), 7.95 (d, J=8.2 Hz, 2 H), 8.05 ppm (d, J=4.9 Hz, 1 H). 13C NMR (CDCl3, 50 MHz): δ=26.6 (q), 55.6 (q), 107.2 (d), 114.4 (d), 114.5 (d), 115.7 (d), 126.1 (d), 128.6 (d), 129.3 (d), 130.1 (s), 136.0 (s), 136.2 (s), 137.7 (d), 143.9 (s), 148.4 (d), 156.6 (s), 157.2 (s), 197.7 ppm (s). HR-MS: Predicted [MH]+=319.1441; Measured [MH]+=319.1450 (diff. in ppm=2.82).
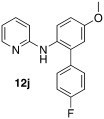
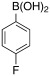
N-(4′-fluoro-5-methoxy-[1,1′-biphenyl]-2-yl)pyridin-2-amine (12 j): Substrate 11 b (50 mg, 0.25 mmol), 4-fluorophenylboronic acid (35 mg, 0.25 mmol), Ag2O (57 mg, 0.25 mmol), BQ (14 mg, 0.13 mmol), Pd(OAc)2 (5.6 mg, 0.025 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 77 % (36.7 mg, 0.19 mmol) yellow oil. Rf=0.16 (LP/EtOAc=10:1). GC–MS: 294 (100, M+), 293 (55), 279 (42), 199 (51), 78 (61). 1H NMR (CDCl3, 200 MHz): δ=3.84 (s, 3 H), 6.04 (s, 1 H), 6.54–6.71 (m, 2 H), 6.84–6.98 (m, 2 H), 6.98 −7.46 (m, 3 H), 7.52 (d, J=8.6 Hz, 1 H), 8.10 ppm (d, J=3.5 Hz, 1 H). 13C NMR (CDCl3, 50 MHz): δ=55.6 (t), 107.2 (d), 113.9 (d), 114.3 (d), 115.6 (d, JCF=21.4 Hz), 115.8 (d), 125.6 (d), 130.2 (s), 130.7 (d, JCF=8.0 Hz), 134.8 (s, JCF=3.4 Hz), 136.2 (s), 137.6 (d), 148.4 (d), 156.4 (s), 157.2 (s), 162.3 ppm (d, JCF=247 Hz). HR-MS: Predicted [MH]+=295.1241; Measured [MH]+=295.1241 (diff. in ppm=0.0).
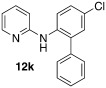
N-(5-chloro-[1,1′-biphenyl]-2-yl)pyridin-2-amine (12 k): Substrate 11 c (50 mg, 0.24 mmol), phenylboronic acid (29 mg, 0.24 mmol), Ag2O (55 mg, 0.24 mmol), BQ (13 mg, 0.12 mmol), Pd(OAc)2 (5.4 mg, 0.024 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 63 % (43 mg, 0.15 mmol) yellow oil. Rf=0.26 (LP/EtOAc=10:1). GC–MS: 281 (30, M+), 280 (51), 264 (23), 143 (19), 169 (100). 1H NMR (CDCl3, 200 MHz): δ=6.25–6.41 (s, 1 H), 6.63–6.80 (m, 2 H), 7.22–7.33 (m, 2 H), 7.34–7.53 (m, 6 H), 7.86 (d, J=8.2 Hz, 1 H), 8.12–8.22 ppm (m, 1 H). 13C NMR (CDCl3, 50 MHz): δ=109.1 (d), 115.4 (d), 121.6 (d), 127.4 (s), 128.0 (d), 128.1 (d), 129.0 (d), 129.2 (d), 130.3 (d), 134.4 (s), 136.2 (s), 137.5 (s), 137.7 (d), 148.3 (d), 155.5 ppm (s). HR-MS: Predicted [MH]+=281.0840; Measured [MH]+=281.0844 (diff. in ppm=1.42).
N-(5-chloro-4′-methyl-[1,1′-biphenyl]-2-yl)pyridin-2-amine (12 l): Substrate 11 c (50 mg, 0.24 mmol), 4-methylphenylboronic acid (34 mg, 0.24 mmol), Ag2O (55 mg, 0.24 mmol), BQ (13 mg, 0.12 mmol), Pd(OAc)2 (5.4 mg, 0.024 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 57 % (41 mg, 0.14 mmol) yellow oil. Rf=0.43 (LP/EtOAc=10:1). GC–MS: 296 (33), 295 (44, M+), 294 (100), 193 (89), 278 (51). 1H NMR (CDCl3, 200 MHz): δ=2.39 (s, 3 H), 6.37 (s, 1 H), 6.66–6.78 (m, 2 H), 7.19–7.32 (m, 6 H), 7.41–7.52 (m, 1 H), 7.80–7.89 (m, 1 H), 8.13–8.21 ppm (m, 1 H). 13C NMR (CDCl3, 50 MHz): δ=109.1 (d), 115.3 (d), 121.5 (d), 127.4 (s), 129.0 (d), 129.7 (d), 130.3 (d), 134.5 (s), 136.2 (s), 137.7 (d), 137.9 (s), 148.2 (d), 155.5 ppm (s). HR-MS: Predicted [MH]+=295.0097; Measured [MH]+=295.1003 (diff. in ppm=2.03).
N-(4-chloro-[1,1′-biphenyl]-2-yl)pyridin-2-amine (12 m): Substrate 11 d (50 mg, 0.24 mmol), phenylboronic acid (29 mg, 0.24 mmol), Ag2O (55 mg, 0.24 mmol), BQ (13 mg, 0.12 mmol), Pd(OAc)2 (5.4 mg, 0.024 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 45 % (31 mg, 0.11 mmol) yellow oil. Rf=0.37 (LP/EtOAc=10:1). GC–MS: 281 (30, M+), 280 (60), 279 (69), 203 (100), 121 (34). 1H NMR (CDCl3, 200 MHz): δ=6.47 (s, 1 H), 6.69–6.83 (m, 2 H), 7.00–7.10 (m, 1 H), 7.15–7.29 (m, 2 H), 7.33–7.58 (m, 6 H), 8.03 (d, J=2.5 Hz, 1 H), 8.16–8.26 ppm (m, 1 H). 13C NMR (CDCl3, 50 MHz): δ=109.6 (d), 115.7 (d), 119.3 (d), 122.3 (d), 127.9 (d), 129.0 (d), 129.3 (d), 130.6 (s), 131.5 (d), 133.8 (s), 137.7 (s), 137.8 (d), 138.7 (s), 148.1 (d), 154.9 ppm (s). HR-MS: Predicted [MH]+=281.0840; Measured [MH]+=281.0845 (diff. in ppm=1.78).
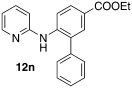
Ethyl 6-(pyridin-2-ylamino)-[1,1′-biphenyl]-3-carboxylate (12 n): Substrate 11 e (50 mg, 0.21 mmol), phenylboronic acid (25.6 mg, 0.21 mmol), Ag2O (48 mg, 0.21 mmol), BQ (12 mg, 0.11 mmol), Pd(OAc)2 (4.7 mg, 0.021 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 43 % (28 mg, 0.09 mmol) yellow oil. Rf=0.17 (LP/EtOAc=10:1). GC–MS: 318 (100, M+), 317 (81), 302 (24), 289 (39), 241 (51). 1H NMR (CDCl3, 200 MHz): δ=1.38 (t, J=7.2 Hz, 3 H), 4.36 (q, J=7.2 Hz, 2 H), 6.72 (s, 1 H), 6.78–6.88 (m, 2 H), 7.35–7.59 (m, 6 H), 7.91–8.15 (m, 3 H), 8.24 ppm (d, J=4.5 Hz, 1 H). 13C NMR (CDCl3, 50 MHz): δ=14.4 (q), 60.7 (t), 110.6 (q), 116.4 (d), 117.1 (d), 123.3 (s), 128.1 (d), 129.2 (d), 129.4 (d), 130.1 (d), 130.8 (d), 132.1 (d), 137.8 (d), 142.0 (s), 148.2 (d), 154.4 (s), 166.4 ppm (s). HR-MS: Predicted [MH]+=319.1441; Measured [MH]+=319.1453 (diff. in ppm=3.76).
N-([1,1′-biphenyl]-2-yl)-3-methylpyridin-2-amine (12 o): Substrate 11 f (50 mg, 0.27 mmol), 2-methylphenylboronic acid (33 mg, 0.27 mmol), Ag2O (62 mg, 0.27 mmol), BQ (16 mg, 0.15 mmol), Pd(OAc)2 (6.0 mg, 0.027 mmol). Column chromatography 10:1 LP/EtOAc. Yield: 50 % (35 mg, 0.13 mmol) yellow oil. Rf=0.48 (LP/EtOAc=10:1). GC–MS: 260 (58, M+), 259 (100), 183 (90), 152 (14), 129 (15). 1H NMR (CDCl3, 200 MHz): δ=1.85 (s, 3 H), 6.36 (s, 1 H), 6.68 (dd, J1=7.2 Hz, J2=5.1 Hz, 1 H), 7.01–7.11 (m, 1 H), 7.21–7.31 (m, 2 H), 7.32–7.49 (m, 6 H), 8.06–8.15 (m, 1 H), 8.40 ppm (d, J=8.02 Hz, 1 H). 13C NMR (CDCl3, 50 MHz): δ=16.9 (q), 115.1 (d), 118.5 (s), 120.0 (d), 121.7 (d), 127.7 (d), 128.3 (d), 129.4 (d), 129.9 (d), 131.6 (s), 137.8 (d), 139.0 (s), 145.1 (d), 153.9 ppm (s). HR-MS: Predicted [MH]+=261.1386; Measured [MH]+=261.1374 (diff. in ppm=−4.60).
N-([1,1′:3′,1′′-terphenyl]-2′-yl)-3-methylpyridin-2-amine (13): 1 b (50 mg, 0.27 mmol), 2-methylphenylboronic acid (33 mg, 0.27 mmol), Ag2O (62 mg, 0.27 mmol), BQ (16 mg, 0.15 mmol), Pd(OAc)2 (6.0 mg, 0.027 mmol). Prepared according to general procedure B, except with 6 equiv. of boronic acid. Column chromatography 10:1 LP/EtOAc, followed by preparative HPLC (n-heptane/iPrOH=90:10). Yield: 46 % (25 mg, 0.07 mmol) yellow oil. Rf=0.40 (LP/EtOAc=10:1). GC–MS: 336 (13, M+), 260 (23), 259 (100), 258 (5), 257 (5). 1H NMR (CDCl3, 200 MHz): δ=1.82 (s, 3 H), 5.51 (s, 1 H), 6.37 (dd, J1=7.2 Hz” J2=5.1 Hz, 1 H), 6.97–7.07 (m, 1 H), 7.12–7.30 (m, 6 H), 7.31–7.44 (m, 7 H), 7.70–7.77 ppm (m, 1 H). 13C NMR (CDCl3, 50 MHz): δ=17.0 (t), 114.3 (d), 117.8 (s), 125.5 (d), 126.8 (d), 127.9 (d), 128.8 (d), 130.1 (d), 135.2 (s), 136.3 (d), 138.5 (s), 140.3 (s), 145.4 (d), 154.8 ppm (s). HR-MS: Predicted [MH]+=261.1386; Measured [MH]+=261.1374 (diff. in ppm=−4.60).
2-phenylaniline 14: A 3-neck flask was charged with Pd/C (80 mg; 10 % Pd-basis) and iPrOH (5 mL) and the mixture was stirred for 5 min under N2. Afterwards, 2 a (246 mg, 1 mmol), which was dissolved in iPrOH (10 mL), and 2 n HCl (3 mL, 6 mmol, 6 equiv.) were added to the solution. The resulting mixture was flushed with H2 three times and then stirred under H2 (1 atm, 101 kPa) at 50 °C overnight. Then, the solids were removed by filtration through Celite, and the solution was evaporated to dryness. Afterwards, 1 M NaOH solution (8 mL) was added, and the reaction mixture was extracted with DCM (3×10 mL). The combined organic layers were dried over Na2SO4, filtered and evaporated to dryness. The crude product (231 mg) was dissolved in 5 mL NH2NH2⋅H2O/AcOH (2.5:0.7 M in EtOH) in a microwave vial and flushed with argon. The vial was heated up to 120 °C for 1 h in microwave. The reaction mixture was allowed to cool to ambient temperature and the volatiles were removed under reduced pressure. After addition of 1 n NaOH solution (5 mL), the mixture was extracted with Et2O (3×10 mL). The combined organic layers were dried over Na2SO4, filtered, and evaporated to dryness. The product was dried under high vacuum to give the pure amine 14 (131 mg) in 78 % yield as a pale yellow solid. M.p.=48–49 °C; The spectral data is in agreement with literature values.31 1H NMR (CDCl3, 200 MHz): δ=3.69 (br s, 2 H), 6.73–6.85 (m, 2 H), 7.11–7.18 (m, 2 H), 7.33–7.45 ppm (m, 5 H). 13C NMR (CDCl3, 50 MHz): δ=115.7 (d), 118.7 (d), 127.3 (d), 127.7 (s), 128.6 (d), 128.9 (d), 129.2 (d), 130.6 (d), 139.6 (s), 143.6 ppm (s).
Acknowledgments
We acknowledge the Austrian Science Foundation (FWF, project P21202-N17) for financial support of this work.
Supplementary material
References
- 1a.Negishi E, de Meijere A, editors. Handbook of Organopalladium Chemistry for Organic Synthesis; Vol. 1–2. Hoboken, NJ: Wiley; 2002. For selected reviews see: [Google Scholar]; Suzuki A. Angew. Chem. 2011;123:6854–6869. [Google Scholar]; Angew. Chem. Int. Ed. 2011;50:6722–6737. doi: 10.1002/anie.201101379. [DOI] [PubMed] [Google Scholar]; de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions, 2nd ed., Vol. 1–2. Weinheim, Germany: Wiley-VCH; 2004. [Google Scholar]; Schnürch M, Flasik R, Khan AF, Spina M, Mihovilovic MD, Stanetty P. Eur. J. Org. Chem. 2006:3283–3307. [Google Scholar]; Kambe N, Iwasaki T, Terao J. Chem. Soc. Rev. 2011;40:4937–4947. doi: 10.1039/c1cs15129k. [DOI] [PubMed] [Google Scholar]; Schröter S, Stock C, Bach T. Tetrahedron. 2005;61:2245–2267. [Google Scholar]; Negishi E-i. Angew. Chem. 2011;123:6870–6897. [Google Scholar]; Angew. Chem. Int. Ed. 2011;50:6738–6764. doi: 10.1002/anie.201101380. [DOI] [PubMed] [Google Scholar]
- 2a.Ritleng V, Sirlin C, Pfeffer M. Chem. Rev. 2002;102:1731–1769. doi: 10.1021/cr0104330. [DOI] [PubMed] [Google Scholar]; Shilov AE, Shul′pin GB. Chem. Rev. 1997;97:2879–2932. doi: 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]; Murai S, editor. Topics in Organometal Chemistry, Vol. 3: Activation of unreactive bonds and organic synthesis. Berlin: Springer; 1999. [Google Scholar]; Miura M, Nomura M. In: Topics in Current Chemistry. Miyaura N, editor. Vol. 219. Heidelberg: Springer; 2002. pp. 211–241. [Google Scholar]; Labinger JA, Bercaw JE. Nature. 2002;417:507–514. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]; Godula K, Sames D. Science. 2006;312:67–72. doi: 10.1126/science.1114731. [DOI] [PubMed] [Google Scholar]; Kakiuchi F, Chatani N. Adv. Synth. Catal. 2003;345:1077–1101. [Google Scholar]; Dyker G, editor. Handbook of C—H Transformations: Applications in Organic Synthesis Vol. 1–2. Wiley-VCH Weinheim; 2005. [Google Scholar]; Su Y-X, Sun L-P. Mini-Rev. Org. Chem. 2012;9:87–117. [Google Scholar]; Schnürch M, Dastbaravardeh N, Ghobrial M, Mrozek B. Curr. Org. Chem. 2011;15:2694–2730. [Google Scholar]; McGlacken GP, Bateman LM. Chem. Soc. Rev. 2009;38:2447–2464. doi: 10.1039/b805701j. [DOI] [PubMed] [Google Scholar]; Ackermann L, Vicente R, Kapdi AR. Angew. Chem. 2009;121:9976–10011. doi: 10.1002/anie.200902996. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:9792–9826. doi: 10.1002/anie.200902996. [DOI] [PubMed] [Google Scholar]; Colby DA, Bergman RG, Ellman JA. Chem. Rev. 2010;110:624–655. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yu J-Q, Shi Z, editors. C—H Activation, Topics in Current Chemistry 292. Heidelberg: Springer; 2010. [PubMed] [Google Scholar]
- 3a.Anastas P, Eghbali N. Chem. Soc. Rev. 2010;39:301–312. doi: 10.1039/b918763b. [DOI] [PubMed] [Google Scholar]; Sheldon RA, Arends I, Hanefeld U, editors. Green Chemistry and Catalysis. Weinheim: Wiley-VCH; 2007. [Google Scholar]; Crabtree RH, Anastas PT, editors. Handbook of Green Chemistry, Vol. 1–6. Weinheim: Wiley-VCH; 2009. [Google Scholar]
- 4a.Dipannita K, Deprez NR, Deprez LV, Sanford MS. J. Am. Chem. Soc. 2005;127:7330–7331. doi: 10.1021/ja051402f. For selected references, see: [DOI] [PubMed] [Google Scholar]; Hull KL, Sanford MS. J. Am. Chem. Soc. 2007;129:11904–11905. doi: 10.1021/ja074395z. [DOI] [PubMed] [Google Scholar]; Deng GJ, Zhao L, Li C-J. Angew. Chem. 2008;120:6374–6378. [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:6278–6282. doi: 10.1002/anie.200801544. [DOI] [PubMed] [Google Scholar]
- e.Zhao XD, Yu ZK. J. Am. Chem. Soc. 2008;130:8136–8137. doi: 10.1021/ja803154h. [DOI] [PubMed] [Google Scholar]; Campeau L-C, Schipper DJ, Fagnou KC. J. Am. Chem. Soc. 2008;130:3266–3267. doi: 10.1021/ja710451s. [DOI] [PubMed] [Google Scholar]; Alberico D, Scott ME, Lautens M. Chem. Rev. 2007;107:174–238. doi: 10.1021/cr0509760. [DOI] [PubMed] [Google Scholar]; Hachiya H, Hirano K, Satoh T, Miura M. Org. Lett. 2009;11:1737–1740. doi: 10.1021/ol900159a. [DOI] [PubMed] [Google Scholar]; Canivet J, Yamaguchi J, Ban I, Itami K. Org. Lett. 2009;11:1733–1736. doi: 10.1021/ol9001587. [DOI] [PubMed] [Google Scholar]; Ackermann L, Mulzer M. Org. Lett. 2008;10:5043–5045. doi: 10.1021/ol802252m. [DOI] [PubMed] [Google Scholar]
- 5a.Dick AR, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]; Wang G-W, Yuan T-T, Wu X-L. J. Org. Chem. 2008;73:4717–4720. doi: 10.1021/jo8003088. [DOI] [PubMed] [Google Scholar]; Kalyani D, Sanford MS. Org. Lett. 2005;7:4149–4152. doi: 10.1021/ol051486x. [DOI] [PubMed] [Google Scholar]
- 6.Thu H-Y, Yu W-Y, Che C-M. J. Am. Chem. Soc. 2006;128:9048–9049. doi: 10.1021/ja062856v. [DOI] [PubMed] [Google Scholar]
- 7.Zhao XD, Dimitrijevic E, Dong VM. J. Am. Chem. Soc. 2009;131:3466–3467. doi: 10.1021/ja900200g. [DOI] [PubMed] [Google Scholar]
- 8a.Kalyani D, Dick AR, Anani WQ, Sanford MS. Org. Lett. 2006;8:2523–2526. doi: 10.1021/ol060747f. [DOI] [PubMed] [Google Scholar]; Wan X, Ma Z, Li B, Zhang K, Cao S, Zhang S, Shi Z. J. Am. Chem. Soc. 2006;128:7416–7417. doi: 10.1021/ja060232j. [DOI] [PubMed] [Google Scholar]; Mei T-S, Giri R, Maugel N, Yu J-Q. Angew. Chem. 2008;120:5293–5297. doi: 10.1002/anie.200705613. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:5215–5219. doi: 10.1002/anie.200705613. [DOI] [PubMed] [Google Scholar]
- 9a.Chu J-H, Tsai S-L, Wu M-J. Synthesis. 2009:3757–3764. [Google Scholar]; Jia X, Zhang S, Wang W, Luo F, Cheng J. Org. Lett. 2009;11:3120–3123. doi: 10.1021/ol900934g. [DOI] [PubMed] [Google Scholar]; Gou F-R, Wang X-C, Huo P-F, Bi H-P, Guan Z-H, Liang Y-M. Org. Lett. 2009;11:5726–5729. doi: 10.1021/ol902497k. [DOI] [PubMed] [Google Scholar]; Ackermann L, Born R, Alvarez-Bercedo P. Angew. Chem. 2007;119:6482–6485. doi: 10.1002/anie.200701727. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:6364–6367. doi: 10.1002/anie.200701727. [DOI] [PubMed] [Google Scholar]
- 9f.Chen X, Hao X-S, Goodhue CE, Yu J-Q. J. Am. Chem. Soc. 2006;128:6790–6791. doi: 10.1021/ja061715q. [DOI] [PubMed] [Google Scholar]; Chen X, Goodhue C, Yu J-Q. J. Am. Chem. Soc. 2006;128:12634–12635. doi: 10.1021/ja0646747. [DOI] [PubMed] [Google Scholar]; Oi S, Fukita S, Hirata N, Watanuki N, Miyano S, Inoue Y. Org. Lett. 2001;3:2579–2581. doi: 10.1021/ol016257z. [DOI] [PubMed] [Google Scholar]; Oi S, Fukita S, Inoue Y. Chem. Commun. 1998:2439–2440. [Google Scholar]
- 10a.Ackermann L, Novak P. Org. Lett. 2009;11:4966–4969. doi: 10.1021/ol902115f. [DOI] [PubMed] [Google Scholar]
- b.Xiao C, Li J, Goodhue CE, Charles E, Yu J-Q. J. Am. Chem. Soc. 2006;128:78–79. doi: 10.1021/ja0570943. [DOI] [PubMed] [Google Scholar]; Ackermann L, Althammer A, Born R. Angew. Chem. 2006;118:2681–2685. doi: 10.1002/anie.200504450. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:2619–2622. doi: 10.1002/anie.200504450. [DOI] [PubMed] [Google Scholar]
- 11.Chu J-H, Chen C-C, Wu MJ. Organometallics. 2008;27:5173–5176. [Google Scholar]
- 12a.Giri R, Maugel N, Li J-J, Wang D-H, Breazzano SP, Saunders LB, Yu J-Q. J. Am. Chem. Soc. 2007;129:3510–3511. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]; Chiong HA, Pham Q-N, Daugulis O. J. Am. Chem. Soc. 2007;129:9879–9884. doi: 10.1021/ja071845e. [DOI] [PubMed] [Google Scholar]; Miura M, Tsuda T, Satoh T, Pivsa-Art S, Nomura M. J. Org. Chem. 1998;63:5211–5215. [Google Scholar]; Giri R, Yu J-Q. J. Am. Chem. Soc. 2008;130:14082–14083. doi: 10.1021/ja8063827. [DOI] [PubMed] [Google Scholar]
- 13a.Shi Z, Li B, Wan X, Cheng J, Fang Z, Cao B, Qin C, Wang Y. Angew. Chem. 2007;119:5650–5654. doi: 10.1002/anie.200700590. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:5554–5558. doi: 10.1002/anie.200700590. [DOI] [PubMed] [Google Scholar]; Li B, Tian S, Fang Z, Shi Z. Angew. Chem. 2008;120:1131–1134. [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:1115–1118. doi: 10.1002/anie.200704092. [DOI] [PubMed] [Google Scholar]; Yang S, Li B, Wan X, Shi Z. J. Am. Chem. Soc. 2007;129:6066–6067. doi: 10.1021/ja070767s. [DOI] [PubMed] [Google Scholar]
- 13d.Shabashov D, Daugulis O. J. Org. Chem. 2007;72:7720–7725. doi: 10.1021/jo701387m. [DOI] [PubMed] [Google Scholar]; Zaitsev VG, Daugulis O. J. Am. Chem. Soc. 2005;127:4156–4157. doi: 10.1021/ja050366h. [DOI] [PubMed] [Google Scholar]; Tremont SJ, Rahman HU. J. Am. Chem. Soc. 1984;106:5759–5760. [Google Scholar]
- 14a.Thirunavukkarasu VS, Parthasarathy K, Cheng C-H. Angew. Chem. 2008;120:9604–9607. doi: 10.1002/anie.200804153. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:9462–9465. doi: 10.1002/anie.200804153. [DOI] [PubMed] [Google Scholar]; Desai LV, Malik HA, Sanford MS. Org. Lett. 2006;8:1141–1144. doi: 10.1021/ol0530272. [DOI] [PubMed] [Google Scholar]; Desai LV, Hull LK, Sanford MS. J. Am. Chem. Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]; Satoh T, Kametani Y, Terao Y, Miura M, Nomura M. Tetrahedron Lett. 1999;40:5345–5348. [Google Scholar]; Terao Y, Kametani Y, Wakui H, Satoh T, Miura M, Nomura M. Tetrahedron. 2001;57:5967–5974. [Google Scholar]; Ackermann L. Org. Lett. 2005;7:3123–3125. doi: 10.1021/ol051216e. [DOI] [PubMed] [Google Scholar]; Yoshikai N, Matsumoto A, Norinder J, Nakamura E. Angew. Chem. 2009;121:2969–2972. doi: 10.1002/anie.200900454. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:2925–2928. doi: 10.1002/anie.200900454. [DOI] [PubMed] [Google Scholar]
- 15.Li B-J, Yang S-D, Shi Z-J. Synlett. 2008:949–957. [Google Scholar]
- 16.Dastbaravardeh N, Schnürch M, Mihovilovic MD. Org. Lett. in print, DOI:10.1021/ol300627p. [DOI] [PubMed]
- 17a.Koley M, Wimmer L, Schnürch M, Mihovilovic MD. Eur. J. Org. Chem. 2011:1972–1979. [Google Scholar]; Koley M, Schnürch M, Mihovilovic MD. Synlett. 2010:1505–1510. [Google Scholar]; Stanetty P, Schnürch M, Mihovilovic MD. Synlett. 2003:1862–1864. [Google Scholar]; Stanetty P, Hattinger G, Schnürch M, Mihovilovic MD. J. Org. Chem. 2005;70:5215–5220. doi: 10.1021/jo0505223. [DOI] [PubMed] [Google Scholar]
- 18.Alonso F, Beletskaya IP, Yus M. Chem. Rev. 2002;102:4009–4091. doi: 10.1021/cr0102967. For a review, see: [DOI] [PubMed] [Google Scholar]
- 19.Chu J-H, Lin Pi-S, Wu M-;J. Organometallics. 2010;29:4058–4065. [Google Scholar]
- 20.Gu S, Chen C, Chen W. J. Org. Chem. 2009;74:7203–7206. doi: 10.1021/jo901316b. [DOI] [PubMed] [Google Scholar]
- 21.Chu J-H, Lin P-S, Lee Y-M, Shen W-T, Wu M-J. Chem. Eur. J. 2011;17:13613–13620. doi: 10.1002/chem.201101528. [DOI] [PubMed] [Google Scholar]
- 22.Ackermann L, Diers E, Manvar A. Org. Lett. 2012;14:1154–1157. doi: 10.1021/ol3000876. [DOI] [PubMed] [Google Scholar]
- 23a.Shi BF, Maugel N, Zhang Y-H, Yu J-Q. Angew. Chem. 2008;120:4960–4964. doi: 10.1002/anie.200801030. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:4882–4886. doi: 10.1002/anie.200801030. [DOI] [PubMed] [Google Scholar]; Chen MS, Prabagaran N, Labenz NA, White MC. J. Am. Chem. Soc. 2005;127:6970–6971. doi: 10.1021/ja0500198. [DOI] [PubMed] [Google Scholar]; Albéniz AC, Espinet P, Martin-Ruiz B. Chem. Eur. J. 2001;7:2481–2489. doi: 10.1002/1521-3765(20010601)7:11<2481::aid-chem24810>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 24.Boele MDK, van Strijdonck GPF, de Vries AHM, Kamer PCJ, de Vries JG, van Leeuwen PWNM. J. Am. Chem. Soc. 2002;124:1586–1587. doi: 10.1021/ja0176907. [DOI] [PubMed] [Google Scholar]
- 25.Prokopcová H, Bergman SD, Aelvoet K, Smout V, Herrebout W, Van der Veken B, Meerpoel L, Maes BUW. Chem. Eur. J. 2010;16:13063–13067. doi: 10.1002/chem.201001887. [DOI] [PubMed] [Google Scholar]
- 26.Gorelsky SI, Lapointe D, Fagnou K. J. Org. Chem. 2012;77:658–668. doi: 10.1021/jo202342q. [DOI] [PubMed] [Google Scholar]
- 27.Hatakeyama T, Hashimoto S, Seki S, Nakamura M. J. Am. Chem. Soc. 2011;133:18614–18617. doi: 10.1021/ja208950c. [DOI] [PubMed] [Google Scholar]
- 28.Masters KS, Rauws TRM, Yadav AK, Herrebout WA, Van der Veken B, Maes BUW. Chem. Eur. J. 2011;17:6315–6320. doi: 10.1002/chem.201100574. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Wang Y, Peng C, Zhang J, Zhu Q. J. Am. Chem. Soc. 2010;132:13217–13219. doi: 10.1021/ja1067993. [DOI] [PubMed] [Google Scholar]
- 30.Chen J, Pang Q, Sun Y, Li X. J. Org. Chem. 2011;76:3523–3526. doi: 10.1021/jo1025546. [DOI] [PubMed] [Google Scholar]
- 31.Ishikawa S, Manabe K. Angew. Chem. 2010;122:784–787. doi: 10.1002/anie.200905544. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2010;49:772–775. doi: 10.1002/anie.200905544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.