

Abstract
The soluble epoxide hydrolase (sEH) enzyme was discovered while investigating the metabolism of xenobiotic compounds in the Casida laboratory. However an endogenous role of sEH is to regulate the levels of a group of potent bioactive lipids, epoxygenated fatty acids (EFAs) that have pleiotropic biological activities. The EFAs, in particular the arachidonic acid derived EETs, are established autocrine and paracrine messengers. The most recently discovered outcome of inhibition of sEH and increased EFAs are their effects on the sensory system and in particular their ability to reduce pain. The inhibitors of sEH block both inflammatory and neuropathic pain. Elevation of EFAs, both in the central and peripheral nervous systems, blocks pain. Several laboratories have now published a number of potential mechanisms of action for the pain reducing effects of EFAs. Here we provide a brief history of the discovery of the sEH enzyme and argue that inhibitors of sEH through several independent mechanisms display pain reducing effects.
As illustrated by this collection of manuscripts, the laboratory of John Casida is renowned for the number of new fields which have emerged from the basement of Wellman Hall at U.C. Berkeley. One could accuse the laboratory of being a nebula for the birthplace of new ideas in toxicology. There are probably many reasons for this phenomenon. Certainly one reason is John Casida's approach of scouring the literature for interesting problems, and then presenting an interesting question to a new student or postdoctoral fellow. We see this approach repeated many times with fields including the ryanodine story, GABA agonists, neonicotinoids, pyrethroids, absinthe mechanism, insecticide resistance, cytochrome P450 and other areas. A second reason is John's willingness to give scientists in his laboratory a free reign to approach a problem in their own way. Certainly a major reason is John's insatiable curiosity. He certainly does not jump from one field to another, rather he moves from one scientific strength to another strength where an established approach is applied to new problems and then these problems stimulate the development of new approaches and techniques. Many scientists are lacking either the courage or skill to move on to new things, but John Casida has a career brimming with courage, skill and creativity. Of course his students and colleagues wonder why he deserts such lucrative fields after a period of years to move on to more exciting areas. Is he giving his protégées a new field of endeavor or is John just so very curious that he must try something new? As he commonly says the most interesting project he has ever worked on is “the next project.”
Sarjeet Gill and I (BDH) arrived in John's laboratory as graduate students on the same day, and neither of us had bothered to warn him that we were coming to work for him. Sarjeet may have gotten more encouragement. However with me, John was exceptionally nice, then he explained that “graduate students were not cost effective, space effective or time effective” and went on to say that he was “limited by resources, space and time.” Of course neither of us left John's laboratory, and we shared the low ceiling room behind Irene next to the house fly room as both office and laboratory. To complicate John's life further we both had read Carroll Williams' Scientific American paper on third generation pesticides (1) and were determined not only to work with John but also to work on insect juvenile hormone. After several months John noticed that we both filled out paper work that he was our major professor, and accepted us with some reluctance. After both of us failed at projects including isolating juvenile hormone active compounds from plants, isolating mitochondria from mites, pyrethroid metabolism and a few other projects, he then suggested that we work on an experimental insecticide from Stauffer Chemical Company that mimicked insect juvenile hormone and was termed R-20458.
Shortly before Christmas of 1969 the compound was radiolabeled in two positions with tritium (2). Sarjeet was assigned to do mammalian metabolism and environmental degradation while Bruce was assigned to synthesize possible metabolites and carry out insect metabolism work. These research boundaries were poorly respected resulting in collaboration and entertainment that has lasted 40 years.
Sarjeet Gill found that R-20458 produced a vast array of metabolites both in mammalian liver homogenates and in vivo. As in insects, Sarjeet found that a major mammalian metabolite of R-20458 was from the hydrolytic opening of the 6,7-epoxide to the corresponding diol (3). The surprise was that in contrast to the excellent work pioneered in John Daley's laboratory at NIH and Gerry Brooks in England (4), the enzyme responsible was largely in the 100,000 g soluble fraction of tissue homogenates. Over several years multiple attempts were made by Gill and Hammock to disprove the diol structure, disprove the enzyme's subcellular localization and disprove that an epoxide hydrolase was involved in its production. When these efforts failed, no objection was raised when a soluble epoxide hydrolase was suggested to be involved in the production of one of a very large number of metabolites of R-20458 (5). Gill and Hammock for several years after leaving the Casida laboratory worked on the hypothesis that the soluble epoxide hydrolase had been overlooked because the enzyme favored trisubstituted lipophilic substrates including terpene oxides such as farnesol 10,11 or squalene 2,3 epoxides. However, Susanne Mumby prepared a series of epoxides similar to R-20458 to find that the enzyme was very active on both mono substituted epoxides and on cis- and trans-1,2-disubstitued epoxides. The resulting manuscript may have set a record for the number of journals that rejected it before Elvins Spencer stated that young faculty `deserve the freedom to make fools of themselves' and accepted the paper with three negative reviews (6). This surprising substrate selectivity quickly led to a study explaining why other laboratories had missed the enzyme (7) and a report by Gill and Hammock (8) that the enzyme rapidly turned over epoxy fatty acids. The properties of the enzyme as well as its activity on fatty acids quickly led to the hypothesis that epoxides of arachidonic acid could be important chemical mediators and that the soluble epoxide hydrolase was degrading them just as insect juvenile hormone esterase degraded the insect chemical mediator juvenile hormone. During this period Hammock carried vials of epoxides of arachidonate (epoxyeicosanoids, EETs) and their mimics from U.C. Riverside around the world begging people to test them. The general failure to find biological activity in retrospect may have been due to the rapid metabolism of the EETs by the soluble epoxide hydrolase (compounded by lack of enthusiasm from collaborators). In spite of the hypothesis that the epoxide hydrolase and EETs were involved in chemical mediation, funding for work on the enzyme was almost exclusively for its role in xenobiotic metabolism for many years.
Through the years the enzyme was purified, characterized, cloned and expressed. Also during this period there were a number of attempts to design effective inhibitors, but none of these compounds was sufficiently stable in vivo to provide convincing evidence of a role of the soluble epoxide hydrolase in chemical mediation. When the mechanism of catalysis of the enzyme was determined to be a two step process like all known α/ß-hydrolase fold enzymes (9), Morisseau et al., were able to design ureas, carbamates and amides that were potent transition state mimics of the enzyme (10). These inhibitors of the sEH or sEHIs were both potent and stable enough that they could be used to demonstrate unequivocally a role for the soluble epoxide hydrolase in vivo in the hydrolysis of lipid epoxide chemical mediators. Several of the sEHIs commonly used in animal models are shown in Figure 1. In parallel over this period, evidence was building that arachidonic acid epoxides and other fatty acid epoxides could have endogenous roles in chemical mediation. Thus, samples of epoxide hydrolase inhibitors joined samples of EETs being delivered to laboratories around the world. An early collaboration with Deanna Kroetz resulted in a demonstration of blood pressure lowering in spontaneously hypertensive rats (11) but it was the recruitment of John Imig (12) and later Rudi Busse (13) as well respected cardiovascular scientists in the U.S. and Europe when the effectiveness of soluble epoxide hydrolase inhibitors and thus EETs became more widely recognized in cardiovascular biology (14–16).
Figure 1.
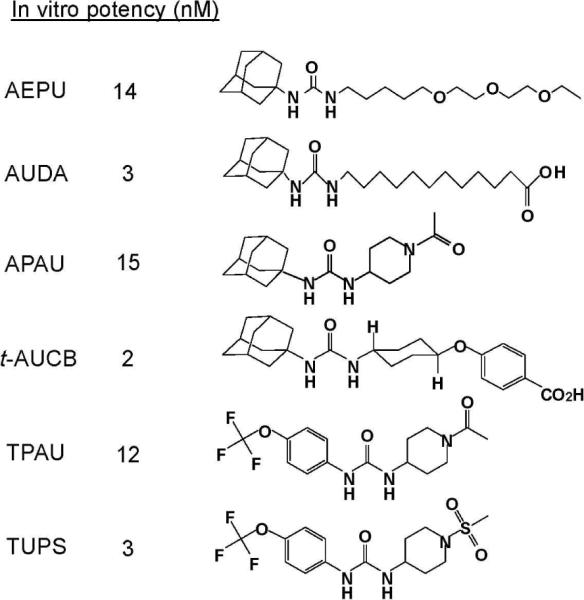
sEHI commonly used in animal models. Accepted abbreviation and potency are indicated with structures. In vitro potency (IC50) was determined using the fluorescent substrate CMNPC, an α-cyanocarbonate, on baculovirus expressed and affinity purified recombinant human sEH.
Producing sEH inhibitors that were of increased potency as sEH inhibitors proved easier than making compounds that were easy to administer to animals and gave good pharmacokinetics. Much of the inspiration in this effort was drawn from agricultural chemistry (4). However, there have been a number of studies through the years to develop such compounds (17–18). Figure 1 shows a sequence of some of the structures from this laboratory that has yielded tools for generating `chemical knock outs' of the sEH catalytic activity. These inhibitors coupled with the wider acceptance of EETs and epoxides of other fatty acids (EFAs) and the wider availability of reagents led more scientists to try these reagents to demonstrate biological activity of EFA's. These biological activities included protection against end organ damage (19), pulmonary inflammation (20), acute inflammation (21), vascular inflammation (22), stroke (23), heart attack (24), and other biologies. More recently there seems to be involvement of sEH inhibition in moderating the effects of diabetes (25) and atherosclerosis (26–27). Several reviews have covered aspects of the physiological events influenced with sEH inhibitors (9, 12, 14, 17).
Since sEH inhibitors were found to reduce inflammation in multiple systems it seemed quite logical to try sEH inhibitors and EETs in inflammatory pain models. It was no surprise when the pain was reduced in multiple systems (28–29). Since it was not anticipated that sEH inhibitors would influence neuropathic pain this biological model was tried as an anticipated negative control. Surprisingly the compounds were very powerful in reducing even neuropathic pain, and this exciting biological observation is the focus of the novel science reported here.
Soluble epoxide hydrolase and epoxy-fatty acids
Many classes of lipids are bioactive molecules (30). Likewise epoxy fatty acids (EFAs) have pleiotropic biological roles (14, 19, 21, 31). Over the past few decades incremental advances in detecting and quantifying these molecules has culminated in a better view of their functionality and their importance in cellular signaling. More importantly the realization that the soluble epoxide hydrolase (sEH, EPHX2) is the major enzyme metabolizing these endogenous EFAs was pivotal to our understanding of their biological roles (32). It was not until at least 15 years later that selective inhibitors of the sEH enabled the ability to modulate their levels in vivo (10). Although the recently described beneficial neurobiological effects are the primary focus of this paper, studies dating back to the early 1980s indicate fundamental roles for EFAs in the central nervous system (33). Among the multiple actions of EFAs, the pain reducing effects are most unexpected. EFAs do not fulfill the criteria of neurotransmitters, i.e. presynaptic presence, release due to calcium dependent depolarization, and receptors on post synaptic cells. Furthermore they are not known ligands of nervous system modulating ion channels. Despite the lack of these properties, they modulate a number of physiological and pathophysiological processes in the nervous system.
Three major classes of enzymes, cyclooxygenase (COX), lipoxygenases (LOX), and cytochrome P450 monoepoxygenases (CYP450) use arachidonic acid (AA) as a substrate, all of which have a number of isoforms producing a plethora of bioactive lipids. Overall a significant portion of current pharmaceuticals act on the AA cascade (34) making this cascade a traditional target for mitigating pain and inflammation. The sEH is a downstream enzyme in the less well studied CYP450 branch of the AA cascade. In contrast to selective COX inhibitors (coxibs), NSAIDS and LOX inhibitors that block the production of pro-inflammatory lipid mediators, inhibition of sEH stabilizes and elevates anti-inflammatory and analgesic EFAs. Although this third branch of the AA cascade was first discovered in the 1970s its importance is not broadly recognized. Currently there are no approved drugs in the market that specifically modulate the activities of the CYP450 epoxygenases, sEH or their products.
The AA derived epoxy eicosatrienoic acids (EETs) have so far been the subject of most investigations concerning EFAs. Nevertheless epoxygenation of free fatty acids by epoxygenases will yield epoxy fatty acids from linoleic, linolenic, arachidonic, eicosapentaenoic, docosahexaenoic acids each with multiple regioisomers (Figure 2). The number of monoepoxy regioisomers formed in these cases depends on the number of olefin bonds in the parent fatty acid. From each regioisomer two possible enantiomers may be produced, resulting in the formation of a multitude of bioactive molecules (35–36).
Figure 2.
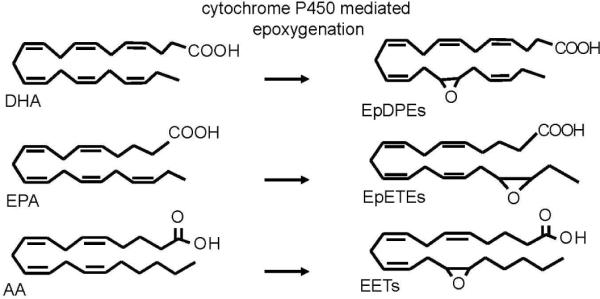
Selected free fatty acids and their corresponding epoxygenated fatty acid (EFA) metabolites generated by CYP450 monoepoxygenases. Each olefin bond can be epoxidized to yield 6 regioisomers from DHA, 5 from EPA and 4 from AA.
The distribution, quantities and biological roles of EFAs vary depending on their location and the biological context (14). Importantly, these lipid epoxides are atypical compared to xenobiotic epoxides because they are neither reactive nor mutagenic (37).
The EETs and other EFAs are stable in the absence of enzymes such as the sEH which degrades them (38–39). The sEH enzyme transforms the EETs to their corresponding vicinal diols, the dihydroxyeicosatrienoic acids (DHETs), metabolites deemed mostly inactive (40). In ex vivo preparations of endothelial tissue radiolabeled free AA is converted to EETs and further to DHETs (41). In vitro, recombinant sEHs from multiple species convert EETs to DHETs (10). In vivo, inhibitors of sEH prevent the degradation of EETs and increase the plasma EET to DHET ratio (21). The substrates of sEH are of course not limited to the EETs but extend to other EFAs. Though most studies cite EETs as the bioactive metabolites stabilized by sEHI, this is an oversimplification. Therefore we will refer to EFAs (which include EETs) for the remainder of this paper.
The sEH enzyme is widely distributed in human tissues (42). It is well expressed in the central and peripheral nervous systems, including in astrocytes, and neurons (43–44). The epoxygenases, EETs, sEH and DHETs are all present in the nervous system (45–46). The endogenous levels of the EETs and other EFAs in the CNS are in the low nanomolar range (46) and they have exceedingly short half-lives on the order of several seconds (38). Interestingly, the spatial distribution of epoxygenases does not entirely overlap with the presence of sEH. More interestingly, sEH seems to be expressed in a brain region selective manner (43, 47). Overall these observations highlight the EFAs' potential role as signaling molecules. It is yet to be determined how these expression patterns relate to functional outcomes.
EFAs and sEH inhibitors block inflammatory and neuropathic pain
Throughout the past few decades the neurobiological effects of EETs have been described albeit sporadically (33, 48–49). Here we expand on the novel hypothesis that a major role of EETs and other EFAs is the attenuation of pain. Thus inhibitors of the sEH by stabilizing EFA's reduce pain through several mechanisms that are not well explored. The fact that few mechanisms are described signals an exciting future for this field.
The possible pain reducing effects of EFAs were first conceived as a result of targeted metabolomic analysis while we were investigating the anti-inflammatory activity of sEH inhibitors (21). These in vivo studies demonstrated the sEH inhibitors cause a dramatic reduction in morbidity and mortality in mice treated with lipopolysaccharide (LPS). An increase in epoxide to diol ratios of EFAs supported the mechanism of action of the inhibitors and showed target engagement (28). However we were surprised that the reduction in the inflammatory eicosanoid prostaglandin E2 (PGE2) was far more dramatic than the effects on EFAs. Since PGE2 causes both pain and inflammation it was logical to try sEH inhibitors as therapeutics for inflammatory pain (28–29). The initial indication that EETs and possibly other EFAs are powerful anti-inflammatory molecules came from observations made by Nakashima et al., who described the anti-pyretic properties of endogenous epoxygenase metabolites in the brain (48). This was further supported by Node et al. who demonstrated a mechanism of action for EETs in vitro involving the NFkB pathway (50). We then tested the effects of EFAs in vivo by chemically inhibiting the sEH (21). These in vivo studies demonstrated that sEH inhibitors reduce the formation of metabolites of the COX-2 enzyme in liver and plasma, specifically the pro-inflammatory and pain producing molecule PGE2.
Our initial work in testing this hypothesis focused on intraplantar injection of a small amount of LPS (lipopolysaccharide, 10 μg) into one hind paw of rats (29). This pain model was an extension of the mouse sepsis model induced by a large dose of LPS (10 mg/kg), in which sEH inhibitors were highly efficacious in reducing PGE2 and improving survival of mice (21). Intraplantar LPS (Figure 3 and 4) instigates an inflammatory response that elicits a disproportionate sensitivity to normally innocuous stimuli (allodynia) and increased sensitivity to moderately painful stimuli (hyperalgesia) (51). These two types of sensory information are relayed by different types of neurons that are specialized to receiving and transmitting pain related information (nociceptors) (52). Many pain reducing (antihyperalgesic) compounds including NSAIDs and narcotic analgesics variably affect these two sensory parameters. It is interesting to observe that all sEH inhibitors tested were highly efficacious in the LPS elicited inflammatory pain model (Figure 4). Importantly, despite being efficacious in pain models, none of the sEHIs tested at doses highly effective in inflamed animals changed acute pain thresholds or motor responses of healthy animals (53). This indicates that sEHIs do not act like narcotics.
Figure 3.
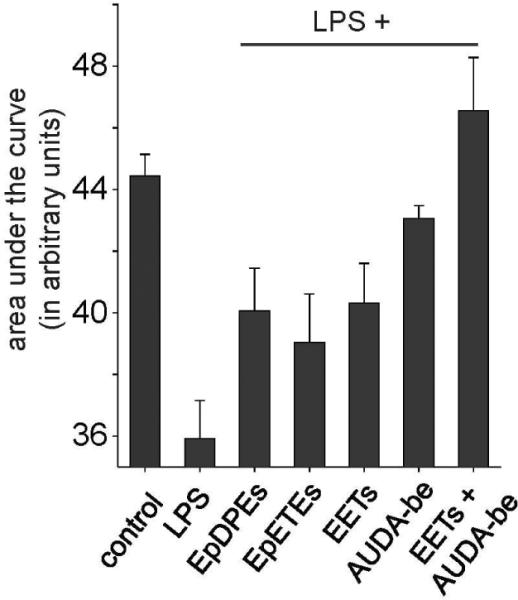
EFAs block inflammatory pain. Area under the curve (efficacy vs. time) for key EFAs with and without the sEHI AUDA-be (butyl ester) using the Hargreaves thermal hyperalgesia test (n=4 per group) is shown. Control animals did not receive LPS, and thus are not inflamed. LPS group received a single intraplantar injection of LPS (10 μg) in one hind paw. All EFAs and sEHI were formulated in Vanicream® and administered topically at a dose of (50 mg/kg) as described (29). LPS injection dramatically reduces the threshold for pain decreasing the latency of thermal withdrawal responses, thus this group has a lower AUC. EFAs of DHA, EPA and AA were equally effective in reducing pain. The sEHI AUDA-butyl ester was also highly efficacious in eliminating inflammatory pain. Co administration of EETs with sEHI prolonged the efficacy of the EETs and produced an additive effect in reducing pain.
Figure 4.
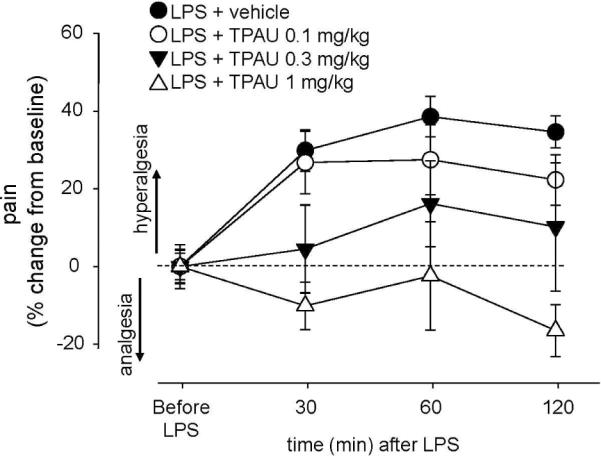
The sEHIs block inflammatory pain. Line graph showing the elimination of inflammatory pain by TPAU using the Hargreaves thermal hyperalgesia test (n=4 per group). LPS + vehicle group received a single intraplantar injection of LPS (10 μg) in one hind paw following baseline measurements. Immediately thereafter TPAU formulated in PEG400 was administered subcutaneously at the doses indicated. LPS injection over time dramatically increased pain from baseline thermal withdrawal latency responses. TPAU was effective in a dose dependent manner. TPAU at 1mg/kg of dose was sufficient to completely eliminate inflammatory pain with the dotted line showing the control thermal withdrawal latency.
Inhibition of sEH is both anti-hyperalgesic and anti-allodynic in tests measuring thermal and mechanical stimuli both separately in the inflamed paw. The time course of the anti-hyperalgesic effect of sEH inhibition in these experiments demonstrate that pain reduction is both rapid onset and of long duration. Interestingly, therapeutic and prophylactic administrations of sEHIs are equally effective in reducing pain implying the involvement of multiple mechanisms of action in mediating their anti-hyperalgesic effects.
The design, synthesis and characterization of chemically different, potent and bioavailable sEH inhibitors over the past several years allowed us to test and support the hypothesis that sEHI induced pain reduction is not a compound related observation but is due to inhibition of the sEH. Consistent with this hypothesis, not only EETs but also other EFAs of DHA and EPA origin are highly efficacious (Figure 3) in reducing the hyperalgesia elicited by LPS (29). Furthermore co-administration of EETs with a sEHI results in an additive increase in antihyperalgesia. This result supports the argument that sEHI stabilize EETs and other EFAs and the observed biological effects are produced by these bioactive lipid metabolites.
Our original hypothesis was that sEHIs reduce inflammatory pain by reducing painful mediators of inflammation. An unexpected observation came about when we used a model of diabetes induced neuropathic pain (DNP) as a negative control while testing sEHIs. A number of sEH inhibitors displayed impressive pain reducing effects in this model, one of which is reported (53). Until this observation, there was no indication that the pain reducing effects of EFAs or sEHI would extend beyond inflammatory pain. In this model a single injection of the microbial toxin streptozocin primarily kills pancreatic β-islet cells to generate a hyperglycemic state and measurable allodynia as soon as two days after toxin administration (54). In the DNP model NSAIDs or selective coxibs are not efficacious, though debate about the inflammatory nature of this model is ongoing. A key part of the etiology of the DNP is hyperglycemia (55–56). Thus it will be important to test if inhibition of sEH reduces this neuropathy by way of regulating hyperglycemia or insulin signaling, in particular given a recent report suggesting that sEH may be involved in regulating insulin secretion (25). Overall, inhibition of sEH seems to have broader efficacy profile than that of coxibs and NSAIDs. Notably co-administration of sEHI with either coxibs or NSAIDS results in a synergistic reduction in plasma PGE2 during LPS elicited inflammation (28). Thus sEHI are not only efficacious as standalone analgesics but also offer a number of advantages in making the coxibs and NSAIDs more safe and efficacious, such as restoring the plasma prostacyclin to thromboxane ratio of rofecoxib treated mice (28).
Peripheral and central nervous systems are both affected by sEHI
The powerful pain reducing effects of EFAs and sEHI in inflammatory and neuropathic pain models naturally brings up a fundamental question in regard to their site of action. The peripheral nervous system (PNS) activity of sEH inhibitors is clearly apparent via local administration of very low amounts of sEHI attenuating both thermal and mechanical pain. To test if inhibition of sEH in the CNS would also attenuate pain we administered the sEHI, AEPU into the spinal cord in a carrageenan elicited rat pain model (53). Intraplantar carrageenan much like LPS leads to an intense and sustained pain state in which animals display both hyperalgesia and allodynia though these effects are local and are largely limited to the carrageenan administered paw. The response thresholds for mildly painful and innocuous stimuli are drastically reduced (about 5 fold) in the carrageenan administered paw. The sEHIs effectively attenuate carrageenan elicited pain in a dose dependent manner whether administered by dermal or intraspinal routes (53). Interestingly, thermal hyperalgesia was more favorably reduced than mechanical allodynia in these experiments implying selective effects of inhibition of sEH in the CNS.
LPS or carrageenan mediated inflammation in the hind paw of the rat generates locally mediated hyperalgesia and allodynia. However, this changes after a period of continuous transmission of pain signals. A barrage of nociceptor cell firings from the paw (periphery area) to the spinal cord and the brain (central area) leads to “central sensitization” where neurons in the spinal cord and brain (CNS) become more responsive to signals that are smaller in magnitude. The etiology of this phenomenon is beyond the scope of this paper though one component of central sensitization, the transcriptional plasticity of genes that produce proinflammatory molecules, is worth mention. In the LPS elicited inflammatory pain model LPS injection to the hind paw leads to upregulation of the cyclooxygenase-2 message and protein both locally, and in the spinal cord and brain, which results in increased production of pro-inflammatory prostanoids, particularly PGE2 (57). The spinally produced PGE2 generates further pain signaling by directly acting on cells expressing E-prostanoid receptors and independently makes cells more responsive to incoming signals from the periphery (58). This process amplifies the pain signals and results in increased perceived pain. Some selective inhibitors of COX-2 penetrate through the blood brain barrier (59) and therefore reduce the production of PGE2 both in the periphery and the spinal cord (60). Interestingly, both EFAs and sEHI act to suppress the induction of COX-2 message and protein. The first in vivo indication of this characteristic action was the observation that sEHIs had effects consistent with anti-inflammatory activity in the mouse sepsis model (21). In this model sEHI reduced the plasma levels of PGE2 which was through suppression of the induction of COX-2 protein in the liver. When we tested if this held true in the spinal cord, we found that sEHIs were highly effective in suppressing LPS elicited upregulation of COX-2 (27 fold) in the spinal cord (53). Thus peripherally administered sEHIs directly modulated CNS transcriptional plasticity. Given that Node et al. demonstrated in vitro that EETs prevent the nuclear translocation of NFkB, it is likely that sEHIs suppress spinal COX-2 through this mechanism (50).
Overall the systemic versus intraspinal efficacies of sEHI seem to be at least 100-fold different in favor of intraspinal sEHI. Furthermore, all sEHI tested so far penetrate through the blood brain barrier. For several compounds brain concentrations were about 10% of blood concentrations. These sEHI were detected in quantities in the brain well above their in vitro potency (IC50) on the affinity purified recombinant rat sEH enzyme. These observations strongly suggest that sEHI are both peripherally and centrally acting pain reducing agents. Further support towards the central mechanism of action of EETs and thus sEHI come from two recent studies to be discussed later (61–62).
Multiple mechanisms are in play for sEHI mediated analgesia
Suppression of spinal COX-2
The suppression of COX-2 gene expression in the CNS by sEHI is certainly an interesting part of the mechanism of action of sEH inhibitors. The time course of spinal COX-2 suppression described earlier was surprisingly not correlated with the observed decreases in pain related behavior in inflamed rats. This inconsistency led us to hypothesize that suppression of spinal or peripheral COX-2 induction may not be the sole mechanism of action for sEHIs. Consistent with this idea, in the DNP model sEHIs show considerable bioefficacy while even selective coxibs that penetrate through the blood brain barrier do not reduce DNP. Therefore it is unlikely that sEHIs reduce DNP solely by suppressing spinal COX-2 upregulation. The extent of the spinal upregulation of COX-2 in DNP is in fact very minor compared to intraplantar LPS elicited upregulation of spinal COX-2 (3 vs. 27 fold) (53, 63).
Release of endogenous analgesic peptides
Recently, in the rat Terashvili et al. demonstrated that direct administration of EETs into the ventrolateral periaquaductal gray (vlPAG) of the brain leads to decreased acute pain responses (61). This analgesic effect seems to be mediated through the enhanced release of beta endorphin and met enkephalin, two important endogenous opioid peptides. These are highly valuable observations given that the authors monitored acute pain responses in the absence of an underlying painful state. In our experiments, using very potent sEHI (IC50 in the low nM range) we have not encountered effects on acute pain thresholds in the absence of induced pain even when sEHI were administered at doses at least 10 times higher than that was required to reduce existing pain. These doses result in plasma sEHI concentrations that are many times (100–1000) above their in vitro IC50 and they are found in the brain tissue at levels at least 10 times above their in vitro IC50. It seems that the different thermal and mechanical threshold pain assays utilized by Terashvili et al. and our laboratory is the key to the differences between observations. In future studies it will be important to establish the relationship between the tissue inhibitor level, the tissue EFA level and bioefficacy. Nevertheless the findings of Terashvili et al. support the hypothesis that EETs are analgesic molecules that act on the CNS (61). This exciting mechanism of action was more recently supported by the work of Conroy et al. who demonstrated that genetic deletion of brain CYP450 reductase or pharmacological inhibition CYP450 epoxygenases in mice and rats abolished morphine analgesia (62). These authors suggest that epoxygenases in the vlPAG region are required for the analgesic actions of morphine and endogenous opioid peptides that stimulate and activate the descending analgesic pathway which is known to inhibit spinal nociception (64). Given that EETs are one of the important products of the CYP450 epoxygenases it is conceivable that yet another mechanism of action responsible for our observations is through activation of the descending analgesic pathway.
Over 25 years ago EET regioisomers were found to increase the release of somatostatin peptide in vitro (33). EETs are reported secretagogues for modulatory peptides including somatostatin in the hypothalamus as well as having possible action on insulin and pituitary adrenocorticotropic hormone (ACTH) secretions (65–66). Somatostatin in the brain and the spinal cord (67) has direct pain reducing effects (68). Peptide neurotransmitters in the CNS are usually found in the same secretory vesicles (69–70). Thus release of somatostatin would simultaneously occur with the release of endogenous opioid peptides. EETs may induce the release of opioid peptides in the CNS as argued by Terashvili et al. which would be consistent with somatostatin release and points to another possible mechanism of action of the EETs (61). While we were assessing the spinal COX-2 transcriptional response to sEH inhibition we used the somatostatin mRNA as a biomarker of inflammation driven increase in cAMP levels (53). Somatostatin expression is highly responsive to intracellular cAMP levels and is increased by cAMP or inflammation (71). However inhibition of sEH mediated a drastic further increase in the transcription of somatostatin mRNA over that produced by inflammation (Figure 5). All together the observations from Capdevila et al. (19) demonstrating the direct release of somatostatin peptide and from our laboratory demonstrating a drastic increase in the mRNA levels indicates that somatostatin secretion is potentially another mechanism responsible for the analgesic effects of EETs and sEHI.
Figure 5.
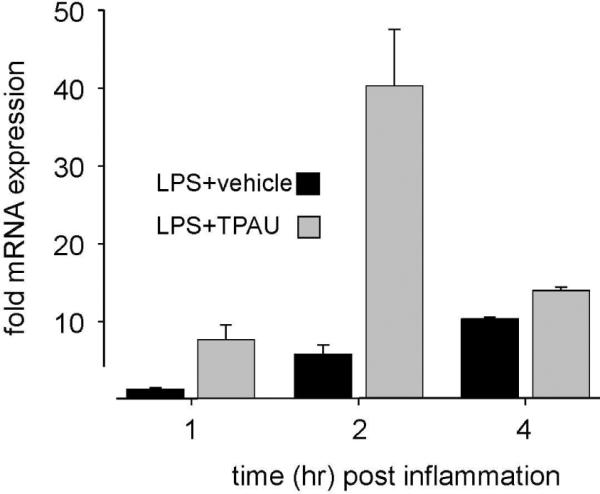
Inhibition of sEH dramatically elevates spinal somatostatin mRNA expression. Male rats received a single intraplantar injection of 10 μg of LPS (lipopolysaccharide) in one hind paw. The sEHI group received TPAU (10 mg/kg) subcutaneously, dissolved in PEG400 while the LPS group received PEG400 only. Another group of rats received intraplantar saline and PEG400 but not LPS or TPAU. This third group was used as calibrators for the quantitative RT-PCR. Spinal cords were extracted at indicated times, mRNAs were isolated and subjected to qRT-PCR using GAPDH as the endogenous control as described (53). Data are expressed as mean± sem, n=4 per group for each time point.
Modulation of CNS neurohormone synthesis
In an effort to understand the mechanism(s) of action of EETs we used a routine screening approach aimed at identifying potential receptors for EETs. This screening yielded a number of interesting and previously not known potential target receptors (72). Among these receptors EETs had a striking concentration dependent and regioisomer selective effect on the peripheral benzodiazepine receptor renamed TSPO (73). This protein is an intra-mitochondrial transporter for cholesterol that translocates cholesterol molecules from the outer to the inner mitochondrial membrane (74). This translocation is a required and rate limiting step for all steroid biosynthesis (75). The EETs displaced a high affinity radioligand, [3H] PK11195, from the TSPO. Based on the known function of TSPO in steroid synthesis we tested if that blocking general steroid synthesis at a downstream step from TSPO would affect the efficacy of sEHI. It was surprising to demonstrate that inhibition of steroid synthesis would produce full antagonism to the analgesic effects of sEHI. These findings strongly suggest the involvement of steroid synthesis, in particular CNS steroid synthesis in the mechanism of action of EETs and sEHI. In parallel to our results, Wang et al. proposed that EETs enhance the expression of another key protein in the steroid synthesis pathway, the steroidogenic acute regulatory protein (StARD1) that shuttles cholesterol to TPSO (76). We tested if administration of sEHI modulated spinal levels of the StARD1 mRNA. Expectedly spinal StARD1 was significantly induced in the presence of stabilized EETs or sEHI in rats treated with intraplantar LPS. Given the positive interaction between StARD1 and intracellular cAMP we tested if spinal administration of a cAMP analogue together with a EETs or sEHI would change StARD1 expression. Administration of inhibitors of sEH and EETs were ineffective in increasing StARD1 expression by themselves but did so when co-administered with 8-bromo cAMP both in the brain and the spinal cord. Future studies will address the possibility of synergism of combined elevated cAMP and sEHI treatment. Regardless these observations indicate the presence of another unique mechanism independent from those already reported.
The case for pain reducing effects of EFAs and sEHIs now is well supported through observations from a number of laboratories. These effects seem sufficiently different from all known pain reducing agents that the sEHIs as well as EFAs and their mimics can be placed in a class of their own. Specifically, sEHIs reduce the levels of pro-inflammatory lipid mediators during inflammation but do not inhibit either of the COX isozymes. The sEHIs suppress the induction of COX-2 transcription but do not share other properties with steroidal anti-inflammatory drugs. Finally, sEHIs are effective in models of pain where NSAIDs, coxibs and steroidal anti-inflammatory drugs fail. It is also clear that the pain reducing effects are governed by several independent mechanisms. So far work in this area has been descriptive because the effects are novel and unexpected. Future work is bound to yield even more interesting and unexpected results. Of particular interest will be the use of EFAs, EFA mimics and antagonists as well as sEHIs to probe the fundamental mechanisms of pain detection and suppression. Regardless, the mechanisms of action of these effects must be investigated to learn how, when and where EFAs and sEHIs modulate physiological events. On the other hand it is possible that this discovery can be relatively rapidly moved towards clinical testing. Neuropathic pain in particular, but also inflammatory pain associated with co-morbidities both remain unmet therapeutic needs. Despite the importance of pain, it is interesting that the rate of discovery for new molecular entities and drugs to treat pain is not increasing (77). Inhibition of sEH appears to be a viable approach for treating inflammatory and neuropathic pain. Thus, work initiated in the Casida laboratory 40 years ago on xenobiotic metabolism and insect metamorphosis may by a circuitous path lead to treatment of such maladies as diabetic neuropathic pain.
Acknowledgments
Support for this work was provided by NIEHS Grant R01 ES002710, NIEHS Superfund Basic Research Program P42 ES004699 and NIH/NHLBI R01 HL059699 to BDH and NIEHS T32ES007059 to KMW. BDH is a George and Judy Marcus Senior Fellow of the American Asthma Foundation.
References
- 1.Williams CM. Third-generation pesticides. Sci Am. 1967;217(1):13–7. doi: 10.1038/scientificamerican0767-13. [DOI] [PubMed] [Google Scholar]
- 2.Kamimura H, Hammock BD, Yamamoto I, Casida JE. Potent juvenile hormone mimic, 1-(4'-ethylphenoxy)-6,7-epoxy-3,7-dimethyl-2-octene, labeled with tritium in either the ethylphenyl- or geranyl-derived moiety. Journal of Agricultural and Food Chemistry. 1972;20(2):439–442. [Google Scholar]
- 3.Gill SS, Hammock BD, Yamamoto I, Casida JE. Preliminary chromatographic studies on the metabolites and photodecomposition products of the juvenoid 1-(4'-ethylphenoxy-6,7-epoxy-3,7- dimethyl-2-octene. Academic Press; New York: 1972. [Google Scholar]
- 4.Morisseau C, Hammock BD. Gerry Brooks and epoxide hydrolases: four decades to a pharmaceutical. Pest Manag Sci. 2008 doi: 10.1002/ps.1583. [DOI] [PubMed] [Google Scholar]
- 5.Gill SS, Hammock BD, Casida JE. Mammalian metabolism and environmental degradation of the juvenoid 1-(4'-ethylphenoxy)-3,7-dimethyl-6,7-epoxy-trans-2-octene and related compounds. J Agric Food Chem. 1974;22(3):386–95. doi: 10.1021/jf60193a058. [DOI] [PubMed] [Google Scholar]
- 6.Mumby SM, Hammock BD. Substrate selectivity and stereochemistry of enzymatic epoxide hydration in the soluble fraction of mouse liver. Pesticide Biochemistry and Physiology. 1979;11(1–3):275–284. [Google Scholar]
- 7.Ota K, Hammock BD. Cytosolic and microsomal epoxide hydrolases: differential properties in mammalian liver. Science. 1980;207(4438):1479–81. doi: 10.1126/science.7361100. [DOI] [PubMed] [Google Scholar]
- 8.Gill SS, Hammock BD. Hydration of cis- and trans-epoxymethyl stearates by the cytosolic epoxide hydrase of mouse liver. Biochem Biophys Res Commun. 1979;89(3):965–71. doi: 10.1016/0006-291x(79)91872-2. [DOI] [PubMed] [Google Scholar]
- 9.Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol. 2005;45:311–33. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- 10.Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc Natl Acad Sci U S A. 1999;96(16):8849–54. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87(11):992–8. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 12.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39(2 Pt 2):690–4. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 13.Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension. 2005;45(4):759–65. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- 14.Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8(10):794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension. 2005;46(4):975–81. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol. 2005;289(3):F496–503. doi: 10.1152/ajprenal.00350.2004. [DOI] [PubMed] [Google Scholar]
- 17.Shen HC. Soluble epoxide hydrolase inhibitors: a patent review. Expert Opin Ther Pat. 2010;20(7):941–56. doi: 10.1517/13543776.2010.484804. [DOI] [PubMed] [Google Scholar]
- 18.Tsai HJ, Hwang SH, Morisseau C, Yang J, Jones PD, Kasagami T, Kim IH, Hammock BD. Pharmacokinetic screening of soluble epoxide hydrolase inhibitors in dogs. Eur J Pharm Sci. 2010;40(3):222–38. doi: 10.1016/j.ejps.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol. 2004;15(5):1244–53. [PubMed] [Google Scholar]
- 20.Smith KR, Pinkerton KE, Watanabe T, Pedersen TL, Ma SJ, Hammock BD. Attenuation of tobacco smoke-induced lung inflammation by treatment with a soluble epoxide hydrolase inhibitor. Proc Natl Acad Sci U S A. 2005;102(6):2186–91. doi: 10.1073/pnas.0409591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci U S A. 2005;102(28):9772–7. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JY. The antiinflammatory effect of laminar flow: the role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci U S A. 2005;102(46):16747–52. doi: 10.1073/pnas.0508081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorrance AM, Rupp N, Pollock DM, Newman JW, Hammock BD, Imig JD. An epoxide hydrolase inhibitor, 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA), reduces ischemic cerebral infarct size in stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2005;46(6):842–8. doi: 10.1097/01.fjc.0000189600.74157.6d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seubert JM, Sinal CJ, Graves J, DeGraff LM, Bradbury JA, Lee CR, Goralski K, Carey MA, Luria A, Newman JW, Hammock BD, Falck JR, Roberts H, Rockman HA, Murphy E, Zeldin DC. Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ Res. 2006;99(4):442–50. doi: 10.1161/01.RES.0000237390.92932.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo P, Chang HH, Zhou Y, Zhang S, Hwang SH, Morisseau C, Wang CY, Inscho EW, Hammock BD, Wang MH. Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J Pharmacol Exp Ther. 2010 doi: 10.1124/jpet.110.167544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ulu A, Davis BB, Tsai HJ, Kim IH, Morisseau C, Inceoglu B, Fiehn O, Hammock BD, Weiss RH. Soluble epoxide hydrolase inhibitors reduce the development of atherosclerosis in apolipoprotein e-knockout mouse model. J Cardiovasc Pharmacol. 2008;52(4):314–23. doi: 10.1097/FJC.0b013e318185fa3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang YX, Ulu A, Zhang LN, Hammock B. Soluble epoxide hydrolase in atherosclerosis. Curr Atheroscler Rep. 2010;12(3):174–83. doi: 10.1007/s11883-010-0108-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmelzer KR, Inceoglu B, Kubala L, Kim IH, Jinks SL, Eiserich JP, Hammock BD. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103(37):13646–51. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inceoglu B, Jinks SL, Schmelzer KR, Waite T, Kim IH, Hammock BD. Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci. 2006;79(24):2311–9. doi: 10.1016/j.lfs.2006.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hla T. Genomic insights into mediator lipidomics. Prostaglandins & Other Lipid Mediators. 2005;77(1–4):197–209. doi: 10.1016/j.prostaglandins.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Smith AD, Dorrance AM. Arachidonic acid induces augmented vasoconstriction via cyclooxygenase 1 in the aorta from rats fed a high-fat diet. Prostaglandins Leukot Essent Fatty Acids. 2006;75(1):43–9. doi: 10.1016/j.plefa.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Chacos N, Capdevila J, Falck JR, Manna S, Martin-Wixtrom C, Gill SS, Hammock BD, Estabrook RW. The reaction of arachidonic acid epoxides (epoxyeicosatrienoic acids) with a cytosolic epoxide hydrolase. Arch Biochem Biophys. 1983;223(2):639–48. doi: 10.1016/0003-9861(83)90628-8. [DOI] [PubMed] [Google Scholar]
- 33.Capdevila J, Chacos N, Falck JR, Manna S, Negro-Vilar A, Ojeda SR. Novel hypothalamic arachidonate products stimulate somatostatin release from the median eminence. Endocrinology. 1983;113(1):421–3. doi: 10.1210/endo-113-1-421. [DOI] [PubMed] [Google Scholar]
- 34.Lednicer D. New drug discovery and development. Hoboken, N.J.; Wiley-Interscience: 2007. p. xii.p. 190. [Google Scholar]
- 35.Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation. Molecular and functional properties of the arachidonate monooxygenase. J Lipid Res. 2000;41(2):163–81. [PubMed] [Google Scholar]
- 36.Spector AA. Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res. 2009;50(Suppl):S52–6. doi: 10.1194/jlr.R800038-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiamvimonvat N, Ho CM, Tsai HJ, Hammock BD. The soluble epoxide hydrolase as a pharmaceutical target for hypertension. J Cardiovasc Pharmacol. 2007;50(3):225–37. doi: 10.1097/FJC.0b013e3181506445. [DOI] [PubMed] [Google Scholar]
- 38.Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292(3):C996–1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- 39.Fang X, Kaduce TL, Weintraub NL, Harmon S, Teesch LM, Morisseau C, Thompson DA, Hammock BD, Spector AA. Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells. Implications for the vascular effects of soluble epoxide hydrolase inhibition. J Biol Chem. 2001;276(18):14867–74. doi: 10.1074/jbc.M011761200. [DOI] [PubMed] [Google Scholar]
- 40.Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276(39):36059–62. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
- 41.Fang X, Kaduce TL, Weintraub NL, Spector AA. Cytochrome P450 metabolites of arachidonic acid: rapid incorporation and hydration of 14,15-epoxyeicosatrienoic acid in arterial smooth muscle cells. Prostaglandins, Leukotrienes and Essential Fatty Acids. 1997;57(4–5):367–371. doi: 10.1016/s0952-3278(97)90412-9. [DOI] [PubMed] [Google Scholar]
- 42.Enayetallah AE, French RA, Thibodeau MS, Grant DF. Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2J2 in human tissues. J Histochem Cytochem. 2004;52(4):447–54. doi: 10.1177/002215540405200403. [DOI] [PubMed] [Google Scholar]
- 43.Bianco RA, Agassandian K, Cassell MD, Spector AA, Sigmund CD. Characterization of transgenic mice with neuron-specific expression of soluble epoxide hydrolase. Brain Res. 2009;1291:60–72. doi: 10.1016/j.brainres.2009.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marowsky A, Burgener J, Falck JR, Fritschy JM, Arand M. Distribution of soluble and microsomal epoxide hydrolase in the mouse brain and its contribution to cerebral epoxyeicosatrienoic acid metabolism. Neuroscience. 2009;163(2):646–61. doi: 10.1016/j.neuroscience.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 45.Amruthesh SC, Falck JR, Ellis EF. Brain synthesis and cerebrovascular action of epoxygenase metabolites of arachidonic acid. J Neurochem. 1992;58(2):503–10. doi: 10.1111/j.1471-4159.1992.tb09749.x. [DOI] [PubMed] [Google Scholar]
- 46.Buczynski MW, Svensson CI, Dumlao DS, Fitzsimmons BL, Shim JH, Scherbart TJ, Jacobsen FE, Hua XY, Yaksh TL, Dennis EA. Inflammatory hyperalgesia induces essential bioactive lipid production in the spinal cord. J Neurochem. 2010 doi: 10.1111/j.1471-4159.2010.06815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang W, Koerner IP, Noppens R, Grafe M, Tsai HJ, Morisseau C, Luria A, Hammock BD, Falck JR, Alkayed NJ. Soluble epoxide hydrolase: a novel therapeutic target in stroke. J Cereb Blood Flow Metab. 2007;27(12):1931–40. doi: 10.1038/sj.jcbfm.9600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakashima T, Harada Y, Miyata S, Kiyohara T. Inhibitors of cytochrome P-450 augment fever induced by interleukin-1 beta. Am J Physiol. 1996;271(5 Pt 2):R1274–9. doi: 10.1152/ajpregu.1996.271.5.R1274. [DOI] [PubMed] [Google Scholar]
- 49.Kozak W, Kluger MJ, Tesfaigzi J, Kozak A, Mayfield KP, Wachulec M, Dokladny K. Molecular mechanisms of fever and endogenous antipyresis. Ann N Y Acad Sci. 2000;917:121–34. doi: 10.1111/j.1749-6632.2000.tb05376.x. [DOI] [PubMed] [Google Scholar]
- 50.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285(5431):1276–9. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanaan SA, Saade NE, Haddad JJ, Abdelnoor AM, Atweh SF, Jabbur SJ, Safieh-Garabedian B. Endotoxin-induced local inflammation and hyperalgesia in rats and mice: a new model for inflammatory pain. Pain. 1996;66(2–3):373–9. doi: 10.1016/0304-3959(96)03068-0. [DOI] [PubMed] [Google Scholar]
- 52.Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413(6852):203–10. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- 53.Inceoglu B, Jinks SL, Ulu A, Hegedus CM, Georgi K, Schmelzer KR, Wagner K, Jones PD, Morisseau C, Hammock BD. Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc Natl Acad Sci U S A. 2008;105(48):18901–6. doi: 10.1073/pnas.0809765105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahlgren SC, Levine JD. Mechanical hyperalgesia in streptozotocin-diabetic rats. Neuroscience. 1993;52(4):1049–55. doi: 10.1016/0306-4522(93)90551-p. [DOI] [PubMed] [Google Scholar]
- 55.Calcutt NA, David T. International Review of Neurobiology. Volume 50. Academic Press; 2002. Potential mechanisms of neuropathic pain in diabetes; pp. 205–228. [DOI] [PubMed] [Google Scholar]
- 56.Edwards JL, Vincent AM, Cheng HT, Feldman EL. Diabetic neuropathy: mechanisms to management. Pharmacol Ther. 2008;120(1):1–34. doi: 10.1016/j.pharmthera.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bingham S, Beswick PJ, Blum DE, Gray NM, Chessell IP. The role of the cylooxygenase pathway in nociception and pain. Seminars in Cell & Developmental Biology. 2006;17(5):544–554. doi: 10.1016/j.semcdb.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 58.Zeilhofer HU. Prostanoids in nociception and pain. Biochem Pharmacol. 2007;73(2):165–74. doi: 10.1016/j.bcp.2006.07.037. [DOI] [PubMed] [Google Scholar]
- 59.Dembo G, Park SB, Kharasch ED. Central nervous system concentrations of cyclooxygenase-2 inhibitors in humans. Anesthesiology. 2005;102(2):409–15. doi: 10.1097/00000542-200502000-00026. [DOI] [PubMed] [Google Scholar]
- 60.Zeilhofer HU, Brune K. Analgesic strategies beyond the inhibition of cyclooxygenases. Trends Pharmacol Sci. 2006;27(9):467–74. doi: 10.1016/j.tips.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 61.Terashvili M, Tseng LF, Wu HE, Narayanan J, Hart LM, Falck JR, Pratt PF, Harder DR. Antinociception produced by 14,15-epoxyeicosatrienoic acid is mediated by the activation of beta-endorphin and met-enkephalin in the rat ventrolateral periaqueductal gray. J Pharmacol Exp Ther. 2008;326(2):614–22. doi: 10.1124/jpet.108.136739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Conroy JL, Fang C, Gu J, Zeitlin SO, Yang W, Yang J, VanAlstine MA, Nalwalk JW, Albrecht PJ, Mazurkiewicz JE, Snyder-Keller A, Shan Z, Zhang S-Z, Wentland MP, Behr M, Knapp BI, Bidlack JM, Zuiderveld OP, Leurs R, Ding X, Hough LB. Opioids activate brain analgesic circuits through cytochrome P450/epoxygenase signaling. Nat Neurosci. 2010;13(3):284–286. doi: 10.1038/nn.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Freshwater JD, Svensson CI, Malmberg AB, Calcutt NA. Elevated spinal cyclooxygenase and prostaglandin release during hyperalgesia in diabetic rats. Diabetes. 2002;51(7):2249–55. doi: 10.2337/diabetes.51.7.2249. [DOI] [PubMed] [Google Scholar]
- 64.Basbaum AI, Clanton CH, Fields HL. Opiate and stimulus-produced analgesia: functional anatomy of a medullospinal pathway. Proc Natl Acad Sci U S A. 1976;73(12):4685–8. doi: 10.1073/pnas.73.12.4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Junier MP, Dray F, Blair I, Capdevila J, Dishman E, Falck JR, Ojeda SR. Epoxygenase products of arachidonic acid are endogenous constituents of the hypothalamus involved in D2 receptor-mediated, dopamine-induced release of somatostatin. Endocrinology. 1990;126(3):1534–40. doi: 10.1210/endo-126-3-1534. [DOI] [PubMed] [Google Scholar]
- 66.Capdevila JH, Falck JR. Biochemical and molecular characteristics of the cytochrome P450 arachidonic acid monooxygenase. Prostaglandins Other Lipid Mediat. 2000;62(3):271–92. doi: 10.1016/s0090-6980(00)00085-x. [DOI] [PubMed] [Google Scholar]
- 67.Chapman V, Dickenson AH. The effects of sandostatin and somatostatin on nociceptive transmission in the dorsal horn of the rat spinal cord. Neuropeptides. 1992;23(3):147–52. doi: 10.1016/0143-4179(92)90115-d. [DOI] [PubMed] [Google Scholar]
- 68.Carlton SM, Du J, Davidson E, Zhou S, Coggeshall RE. Somatostatin receptors on peripheral primary afferent terminals: inhibition of sensitized nociceptors. Pain. 2001;90(3):233–44. doi: 10.1016/S0304-3959(00)00407-3. [DOI] [PubMed] [Google Scholar]
- 69.Mains RE, Eipper BA. The tissue-specific processing of Pro-ACTH/Endorphin recent advances and unsolved problems. Trends Endocrinol Metab. 1990;1(8):388–94. doi: 10.1016/1043-2760(90)90097-m. [DOI] [PubMed] [Google Scholar]
- 70.Rouille Y, Martin S, Steiner DF. Differential processing of proglucagon by the subtilisin-like prohormone convertases PC2 and PC3 to generate either glucagon or glucagon-like peptide. J Biol Chem. 1995;270(44):26488–96. doi: 10.1074/jbc.270.44.26488. [DOI] [PubMed] [Google Scholar]
- 71.Montminy MR, Sevarino KA, Wagner JA. Identification of a cyclic-AMP-responsive element within the rat somatostatin gene. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(18):6682–6686. doi: 10.1073/pnas.83.18.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Inceoglu B, Schmelzer KR, Morisseau C, Jinks SL, Hammock BD. Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (EETs) Prostaglandins Other Lipid Mediat. 2007;82(1–4):42–9. doi: 10.1016/j.prostaglandins.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapere JJ, Lindemann P, Norenberg MD, Nutt D, Weizman A, Zhang MR, Gavish M. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27(8):402–9. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 74.Papadopoulos V, Lecanu L, Brown RC, Han Z, Yao ZX. Peripheral-type benzodiazepine receptor in neurosteroid biosynthesis, neuropathology and neurological disorders. Neuroscience. 2006;138(3):749–56. doi: 10.1016/j.neuroscience.2005.05.063. [DOI] [PubMed] [Google Scholar]
- 75.Stocco DM. StAR protein and the regulation of steroid hormone biosynthesis. Annu Rev Physiol. 2001;63:193–213. doi: 10.1146/annurev.physiol.63.1.193. [DOI] [PubMed] [Google Scholar]
- 76.Wang X, Shen CL, Dyson MT, Yin X, Schiffer RB, Grammas P, Stocco DM. The involvement of epoxygenase metabolites of arachidonic acid in cAMP-stimulated steroidogenesis and steroidogenic acute regulatory protein gene expression. J Endocrinol. 2006;190(3):871–8. doi: 10.1677/joe.1.06933. [DOI] [PubMed] [Google Scholar]
- 77.Munos B. Lessons from 60 years of pharmaceutical innovation. Nat Rev Drug Discov. 2009;8(12):959–68. doi: 10.1038/nrd2961. [DOI] [PubMed] [Google Scholar]