

SUMMARY
Glycogen storage disease type IIIa (GSD IIIa) is an autosomal recessive disease caused by deficiency of glycogen debranching enzyme (GDE) in liver and muscle. The disorder is clinically heterogeneous and progressive, and there is no effective treatment. Previously, a naturally occurring dog model for this condition was identified in curly-coated retrievers (CCR). The affected dogs carry a frame-shift mutation in the GDE gene and have no detectable GDE activity in liver and muscle. We characterized in detail the disease expression and progression in eight dogs from age 2 to 16 months. Monthly blood biochemistry revealed elevated and gradually increasing serum alanine transaminase (ALT), aspartate transaminase (AST) and alkaline phosphatase (ALP) activities; serum creatine phosphokinase (CPK) activity exceeded normal range after 12 months. Analysis of tissue biopsy specimens at 4, 12 and 16 months revealed abnormally high glycogen contents in liver and muscle of all dogs. Fasting liver glycogen content increased from 4 months to 12 months, but dropped at 16 months possibly caused by extended fibrosis; muscle glycogen content continually increased with age. Light microscopy revealed significant glycogen accumulation in hepatocytes at all ages. Liver histology showed progressive, age-related fibrosis. In muscle, scattered cytoplasmic glycogen deposits were present in most cells at 4 months, but large, lake-like accumulation developed by 12 and 16 months. Disruption of the contractile apparatus and fraying of myofibrils was observed in muscle at 12 and 16 months by electron microscopy. In conclusion, the CCR dogs are an accurate model of GSD IIIa that will improve our understanding of the disease progression and allow opportunities to investigate treatment interventions.
INTRODUCTION
Mutations in glycogen debranching enzyme (GDE) gene cause glycogen storage disease type III (GSD III), resulting in accumulation of cytoplasmic glycogen in liver and muscle, the two major tissues for glycogen metabolism (Illingworth and Cori, 1952; Illingworth et al., 1956). GDE is a bifunctional protein having two distinct enzymatic activities: 1,4-α-D-glucan:1,4 α-D-glucan 4-α-D-glycosyltransferase (EC 2.4.1.25) and amylo-1,6-glucosidase (EC 3.2.1.33) (Taylor et al., 1975; Nakayama et al., 2001). Together with glycogen phosphorylase, GDE is responsible for complete degradation of cytoplasmic glycogen. More than 80% of GSD III patients have debranching enzyme deficiencies in both liver and muscle (type IIIa), and most of the rest manifest only liver involvement (type IIIb) (Van Hoof and Hers, 1967; Kishnani et al., 2010).
General clinical manifestations of GSD IIIa include hepatomegaly, fasting hypoglycemia, hyperlipidemia, growth retardation, and variable myopathy and cardiomyopathy. However, disease phenotypes vary widely in patients, most probably caused by different GDE mutations specific to individual families on different genetic and environmental backgrounds (Hobson-Webb et al., 2010; Kishnani et al., 2010). Liver symptoms often appear in childhood and typically improve after puberty, but liver cirrhosis and hepatic adenoma or hepatocellular carcinoma have been reported in some cases (Haagsma et al., 1997; Labrune et al., 1997; Siciliano et al., 2000; Cosme et al., 2005; Demo et al., 2007). Progressive myopathy is the major cause of morbidity in GSD IIIa patients. Muscle weakness is usually not a prominent feature during childhood but can progress with age, rendering some patients wheelchair bound in their third or fourth decade of lives (Momoi et al., 1992; Lucchiari et al., 2007; Kishnani et al., 2010). Periodic acid-Schiff stain (PAS)-positive glycogen storage can be observed in adult patients along with distorted myofiber structures (Kim et al., 2008; Schoser et al., 2008). Glycogen deposition in cardiac muscle has been recognized since 1968 and ventricular hypertrophy is common in GSD IIIa patients (Pearson, 1968; Moses et al., 1989; Lee et al., 1997). Patients with cardiac involvement are at risk of heart failure and life-threatening arrhythmias, yet the actual incidence is relatively low (Miller et al., 1972; Moses et al., 1989; LaBarbera et al., 2010). Consistent with liver and muscle damage, laboratory tests also show elevated serum alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP) and creatine phosphokinase (CPK) activities (Coleman et al., 1992; Lee et al., 1995; Lucchiari et al., 2007; Karwowski et al., 2011).
Because the search for an effective treatment for GSD IIIa is ongoing and the pathophysiology of the disease and mechanisms of clinical variability are not well understood, an appropriate animal model that mimics human disease is needed. Recently, GSD IIIa was identified in curly-coated retrievers (CCR) (Gregory et al., 2007). The affected dogs carry a frame-shift mutation predicting deletion of the C-terminal 126 amino acids of GDE and resulting in a GSD IIIa phenotype. Investigation on two affected dogs showed no detectable GDE enzyme activity in liver and muscle. Analysis of liver biopsies revealed severe glycogen accumulation but there was no evidence of inflammation or fibrosis in the liver. PAS-positive glycogen deposits were observed in a skeletal muscle biopsy taken from one of them at age 14 months (Gregory et al., 2007). However, a thorough characterization of this model and of disease characteristics has not been performed. We have established a breeding colony of the CCR dogs to better understand the phenotype, and to allow for better understanding of disease progression. In this article, we describe this canine GSD IIIa model in extensive biochemical and histological details.
TRANSLATIONAL IMPACT.
Clinical issue
Liver and muscle are the major affected tissues in humans with glycogen storage disease type IIIa (GSD IIIa), a hereditary disease caused by mutations in the gene encoding glycogen debranching enzyme (GDE). Liver symptoms often appear in childhood and typically improve after puberty in most individuals, but liver cirrhosis and hepatic adenoma or hepatocellular carcinoma have been reported in some cases. Progressive myopathy and cardiomyopathy are a major cause of morbidity in adults. The natural history of GSD IIIa disease progression has not been well established in human patients and there is no effective treatment. The absence of an adequate animal model is a major obstacle in progressing our understanding of the pathological mechanisms of the disease and evaluating new therapies. Recently, GSD IIIa was identified in curly-coated retrievers (CCRs) that carry a frame-shift mutation predicted to delete the C-terminal 126 amino acids of GDE. The dogs showed no detectable GDE enzyme activity in liver and muscle, but a thorough characterization of these dogs has not been reported.
Results
In this study, the authors characterized in detail the expression and progression of GSD IIIa in affected CCRs. Abnormally high glycogen deposition was found in liver and muscle, and, consistent with liver and muscle damage, high and gradually increasing activity of enzymes including AST, ALT, ALP and CPK were found in serum. In muscle, increased glycogen deposition was accompanied by disruption of the contractile apparatus and fraying of myofibrils. Progressive, age-related liver fibrosis and muscle damage caused by glycogen accumulation were the major features of GSD IIIa in affected dogs.
Implications and future directions
Canine models are emerging as powerful tools to study hereditary diseases and the development of new therapeutic approaches. This work shows that CCRs with GSD IIIa closely resemble human patients, and will be a valuable model for future studies of disease progression, biomarker discovery and treatment interventions. The authors are aiming to evaluate potential treatments such as high-protein diet management, gene therapy, enzyme replacement therapy and blocking glycogen synthesis with existing drugs in this animal model.
RESULTS
Serum biochemistry
Monthly routine serum biochemistry panels of eight affected dogs revealed gradually increasing liver enzyme activities in these dogs. ALT (normal range 12–118 U/l) increased from 144±36 U/l at 2 months to a peak of more than 700 U/l at 15 months (Fig. 1A). AST (normal range 15–66 U/l) was slightly higher than normal before 6 months (70–80 U/l), then gradually increased to 283±67 U/l at 13 months, followed by a jump to nearly 600 U/l at 14 months (Fig. 1B). The average value of ALP (normal range 5–131 U/l) fluctuated between 200 and 400 U/l, with a trend of increasing with age (Fig. 1C). The CPK level (normal range 59–895 U/l) in the tested dogs was in the normal range before age 10 months, became slightly above normal from 10 to 12 months, and then continually increased to above 2000 U/l at 16 months (Fig. 1D). Triglycerides (Fig. 1E) and cholesterol (Fig. 1F) concentrations were normal in all dogs throughout the study. All other parameters including glucose, bilirubin, albumin, urea nitrogen, and creatinine were within normal ranges. There was no difference in the growth curves of affected dogs and their normal littermates
Fig. 1.
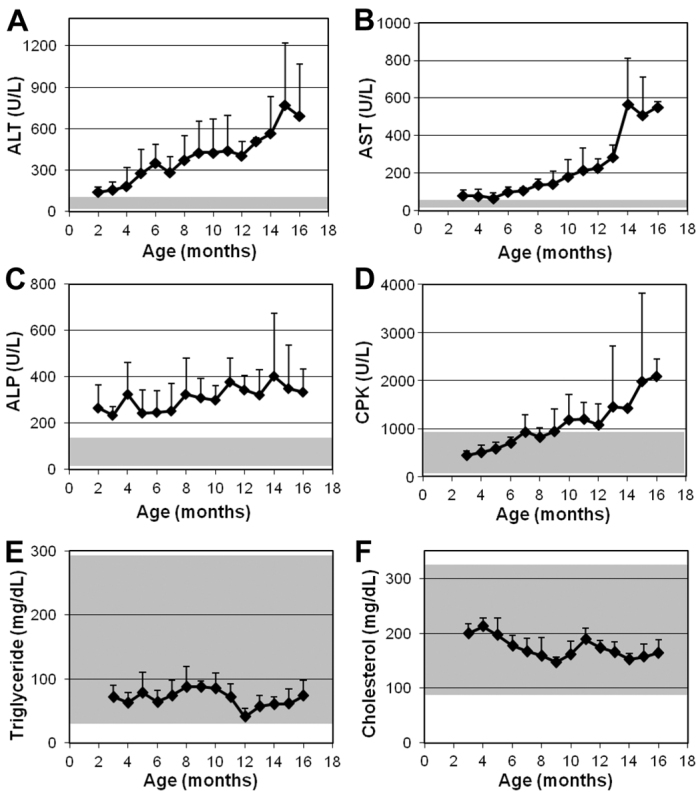
Serum enzyme activities and lipid levels in GSD IIIa-affected CCRs. (A–F) Blood was collected every month and routine serum biochemistry determinations were performed as indicated. Values show mean + s.d. of 3–6 dogs at each time point. Shaded areas indicate normal ranges.
Liver and muscle glycogen contents
Liver and muscle biopsies were performed on GSD IIIa dogs after overnight fasting at ages of 4, 12 and 16 months. Liver glycogen content (Fig. 2A) was 209±47 μmol glucose/g tissue at 4 months of age, more than fourfold of that found in a normal dog (∼47 μmol glucose/g). The value increased to 269±44 μmol glucose/g at 12 months and dropped to 180±59 μmol glucose/g at 16 months. The decrease in liver glycogen content from age 12 to 16 months was probably due to large-scale replacement of hepatocytes by fibrous tissue, as described later, and correlates well with the observed accelerated increase of serum ALT and AST activities in the same time frame (Fig. 1A,B). Gradually increasing muscle glycogen content was observed in all dogs. As shown in Fig. 2B, muscle glycogen contents were 81±11, 104±12 and 168±49 μmol glucose/g tissue at ages 4, 12 and 16 months, respectively, compared with ∼23 μmol glucose/g tissue in a normal dog. The damaging effect of high glycogen content on muscle is evidenced by the sharply increased serum CPK and AST activities after 12 months (Fig. 1) and by histopathological analyses described later.
Fig. 2.
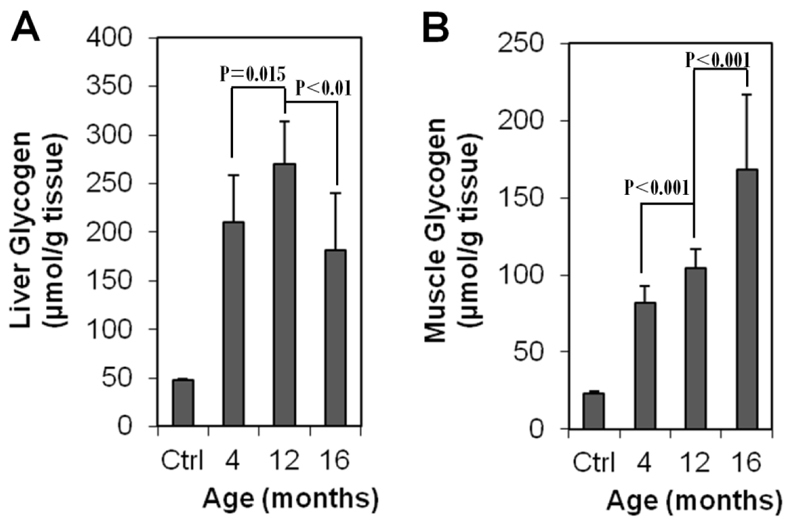
Fasting glycogen contents in liver and skeletal muscle of GSD IIIa-affected CCRs. (A) Liver glycogen and (B) muscle glycogen at each time point were measured in four dogs at 4 and 12 months, and three dogs at 16 months, with two pieces of the same tissue for each dog. Means + s.d. are shown. Ctrl, average of triplicate specimens from a 6-month-old normal dog.
Gross and histological appearance of liver
Liver and muscle are two major tissues affected by glycogen storage. High glycogen turnover rates in liver (Magnusson et al., 1994) predispose this organ to glycogen deposition even at young ages in GSD IIIa. At age 4 months when the first liver biopsy was performed, the livers were enlarged and fragile with relatively smooth surfaces in all dogs (n=4). At 12 months, the livers were severely enlarged and firmer, with small nodules scattered on the surface (n=4). At 16 months, the severely and diffusely enlarged livers were partially or fully involved with large nodules and cirrhosis (n=3). Histological analysis of liver specimens revealed marked glycogen accumulation in hepatocytes at all ages and a gradual disturbance of hepatocellular organization with age. As shown in Fig. 3, hematoxylin and eosin (H&E)-stained paraffin sections (Fig. 3A–C) exhibited the typical vacuolated appearance of glycogen-filled hepatocytes; in high-resolution light microscopy (HRLM) sections stained with PAS-Richardson’s stain (Fig. 3D–F), pools of glycogen appear light purple and are well-preserved within hepatocytes. The hepatic architecture appeared normal at 4 months (Fig. 3A,D), mildly altered in local areas at 12 months (Fig. 3B,E) and very distorted at age 16 months, with increased fibrous areas (Fig. 3C,F). A low power view of trichrome-stained paraffin sections illustrates the progressive hepatic fibrosis in canine GSD IIIa (Fig. 4A–C): from periportal fibrosis at 4 months, bridging fibrosis at 12 months and cirrhosis at 16 months. The evolution of these pathological processes was highlighted at higher magnification (Fig. 4D–F).
Fig. 3.

Marked glycogen accumulation is present in hepatocytes at 4, 12 and 16 months of age. (A–C) Paraffin-embedded, H&E-stained liver sections illustrate the typical vacuolated appearance of glycogen-filled hepatocytes at 4, 12 and 16 months. (D–F) In HRLM sections stained with PAS-Richardson’s stain, the glycogen is well preserved and appears light purple. Dense fibrosis is evident in F; fibroblasts stain light blue. Scale bars: 50 μm (A,B,F), 100 μm (C) and 30 μm (D,E).
Fig. 4.

Progressive hepatic fibrosis is a feature of canine GSD3. (A–C) Progression from periportal fibrosis, to bridging fibrosis to cirrhosis at 4, 12 and 16 months, respectively (paraffin sections, trichrome stain). (D–F) Evolution of these pathological processes at higher magnification (paraffin sections, trichrome stain). Scale bars: 300 μm (A–C) and 100 μm (D–F).
Progressive muscle damage caused by gradually increased glycogen accumulation
Progressive glycogen accumulation and tissue damage with age was detected in skeletal muscle of GSD IIIa dogs (Fig. 5). At age 4 months, only low levels of glycogen accumulated within the cytoplasm of myocytes (Fig. 5A), which is consistent with the observation that symptomatic myopathy is not commonly seen in young patients (Kishnani et al., 2010). All muscle cells were similarly affected. Under electron microscopy, the glycogen granules dispersed among the myofibrils, and small blebs of sarcolemma containing glycogen deposits were readily seen beneath the cytoplasmic membrane (Fig. 5D). At 12 months, the cytoplasmic glycogen began to pool around the periphery of the myotubes. Most cells were involved in glycogen accumulation, with a range from mild to severe involvement (Fig. 5B). The large glycogen pools disrupted the contractile apparatus and caused fraying of myofibrils (Fig. 5E). At 16 months, regions of the sarcoplasm were entirely occupied by cytoplasmic glycogen, displacing the contractile elements (Fig. 5F). The histological findings matched the pattern of gradually increasing glycogen content measured in the muscle tissues (Fig. 2B), and were also in concert with the trend of serum CPK activity (Fig. 1D).
Fig. 5.

Progressive cytoplasmic glycogen accumulation occurs in skeletal of GSD3 dogs. (A–C) Progressive accumulation of glycogen in skeletal muscle over time. MetaMorph measurements were 6.5±3.1%, 20.3±7.6% and 17.3±4.7% tissue area occupied by glycogen at 4, 12 and 16 months, respectively (HRLM, PAS-Richardson’s stain). (D–F) Ultrastructural changes that occur over time. At 4 months, glycogen accumulates in the cytoplasm and dissects in between myofibrils and just beneath the cytoplasmic membrane, forming small blebs (D). At 12 months, the cytoplasmic glycogen begins to pool and disrupts the contractile apparatus, causing fraying of myofibrils (E). At 16 months, entire regions of cells are filled with glycogen, displacing all contractile elements, leaving only mitochondria to float within the pools of glycogen (F). Arrows indicate glycogen pools. Scale bars: 50 μm (A–C), 2 μm (D), 5 μm (E) and 6 μm (F).
Glycogen deposition in adipocytes in muscle tissues
Fig. 6 demonstrates the glycogen accumulation in adipocytes present in a muscle biopsy from one of the GSD IIIa dogs at 16 months of age. The appearance of PAS-positive substances in adipocytes was an isolated event in our study, but it drew our attention to a potential disturbance of glycogen metabolism in adipocytes in GSD IIIa.
Fig. 6.

Accumulation of cytoplasmic glycogen within adipocytes is apparent in 16 month biopsies. (A) Glycogen stains purple at the periphery of fat globules within adipocytes present in skeletal muscle biopsies (HRLM, PAS-Richardson’s stain). (B) Electron microscopy demonstrates the finely granular ultrastructure of the glycogen surrounding fat globules in adipocytes. Arrows indicate glycogen pools. Scale bars: 30 μm (A) and 5 μm (B).
DISCUSSION
GSD III is one of the most common glycogen storage diseases. Currently, disease progression and pathology are not well characterized. Other than symptomatic management, no therapy is available for this condition (Kishnani et al., 2010). There is an urgent need for an animal model to study disease progression and to develop effective therapies that are definitive or targeted and relevant to human treatment modalities. In the past decade, canine models have emerged as a powerful tool for studying hereditary diseases and for the development of new therapeutic approaches. For example, a canine model of GSD I has been established and successfully used for studying disease pathophysiology, long-term complications, and development of gene therapy (Kishnani et al., 2001; Koeberl et al., 2008). The naturally occurring GDE frame-shift mutation in CCR was first identified in 2007 (Gregory et al., 2007). The initial study of two affected dogs confirmed glycogen accumulation in liver and muscle and both dogs showed similar clinical signs to those of the human disease (Gregory et al., 2007). A breeding colony was established to obtain a larger cohort of affected dogs with the aim of understanding pathophysiological disease progression and developing novel therapies. The current study was designed to investigate in detail the natural history of the disease in this canine model.
Hypoglycemia and hyperlipidemia are dominant features in patients with GSD III in infancy and childhood (Hershkovitz et al., 1999; Geberhiwot et al., 2007; Bernier et al., 2008; Kishnani et al., 2010). Hyperlipidemia in human patients is possibly caused by increased lipid flux from adipose tissue to the liver as an alternative source of fuel in the setting of hypoglycemia (Bernier et al., 2008). However, none of the eight affected dogs ever displayed signs of hypoglycemia during a 12-hour fasting, which could explain the normal concentrations of triglycerides and cholesterol throughout the study. In addition, there is a great difference in lipoprotein profiles and lipids metabolism between human and dog (Xenoulis and Steiner, 2010).
Liver and muscle damages are common features in GSD IIIa patients, and serum enzyme activities related to these organs are often elevated in the patients. Though abnormalities in serum enzyme activities have been repeatedly reported in previous clinical studies on GSD IIIa (Lucchiari et al., 2007; Karwowski et al., 2011), detailed correlation between enzyme levels and the states of disease progression has not been established. In this study of eight affected dogs, we showed that measurements of both ALT and AST activity were elevated at a young age and continually increased throughout the experiment, indicating progressive liver damage. Though elevations of the two transaminases both indicate liver impairment, ALT is a more direct indicator because it is predominantly found in liver, whereas AST also broadly exists in other tissues, especially muscle (Goessling and Friedman, 2005). The steeper increase of the two enzymes after 12 months of age coincided with the increased liver fibrosis and cirrhosis, as confirmed by histology, though increased AST could have also come from damaged muscle where this enzyme exists in significant amounts (Weibrecht et al., 2010). High levels of ALP were noted in the dogs at different ages. Human ALP exists in several isoforms and various conditions can lead to elevated serum ALP activity, but very high ALP activities are often of liver origin and caused by severe intrahepatic cholestasis or bile duct obstruction (Sapey et al., 2000). Consistently elevated serum ALP activity in the GSD IIIa dogs appears primarily a result of hepatocyte swelling due to cytoplasmic glycogen accumulation, and had little correlation with the extent of fibrosis. CPK catalyzes the conversion of creatine to phosphocreatine, an energy reservoir for the rapid regeneration of ATP through the reverse reaction, in muscle and some neuronal tissues (Wallimann et al., 1992). High serum CPK activity is usually caused by injury or stress to the muscle or heart tissue (Arts et al., 2007). The accelerated increase of CPK after 12 months, correlating with increased muscle glycogen content and damage, implies the disruption of myocyte integrity that mimics the onset of phenotypic myopathy in adult patients. These parameters are useful in clinical diagnosis and evaluation of disease status, but whether they can be used as disease biomarkers needs to be further evaluated in this animal model.
High levels of glycogen were detected in liver at 4 months of age in all dogs. Liver glycogen content increased at 12 months, followed by a significant decrease at 16 months. Progressive liver fibrosis was observed in this dog model. Liver fibrosis was minor at 4 months of age but extensive micronodular and macronodular cirrhosis was found in all three dogs analyzed at 16 months. The decline in glycogen content in liver from age 12 to 16 months was probably due to large-scale replacement of hepatocytes by fibrous tissue, and correlated well with the observed accelerated increase of serum ALT and AST activities. Muscle glycogen content gradually increased with age. Disruption of the contractile apparatus and fraying of myofibrils were directly observed at 12 and 16 months as a result of large cytoplasmic glycogen deposition and correlated with sharply increased serum CPK and AST activities.
In addition to skeletal muscle, varied cardiac muscle involvement has been reported in patients with GSD IIIa. Ventricular hypertrophy is a frequent finding, but overt cardiac dysfunction or symptomatic cardiomyopathy is rare (Moses et al., 1989; Labrune et al., 1991; Hobson-Webb et al., 2010). A recent study demonstrated that a high-protein diet dramatically decreased the left ventricular mass index and serum creatine kinase levels and reversed cardiomyopathy in a patient with GSD IIIa (Dagli et al., 2009), indicating that this treatment is a beneficial therapeutic choice for GSD IIIa patients with cardiomyopathy. Because it is not practical to perform frequent myocardial biopsies on the dogs, cardiac muscle involvement was not a focus in this study. However, we did perform a less invasive electrophysiology study to test the electrical conduction system of the heart in four affected dogs at age 7–8 months, using a single catheter situated within the heart through femoral vein. Of the four dogs studied, two had atrial fibrillation upon electrical stimulation but all other conduction system characteristics were normal. The other two dogs were both within normal limits. Thus, at this time, there is no inclusive conclusion of cardiac involvement in this model.
It is interesting to find significant glycogen accumulation in adipocytes in an affected dog. Adipose tissue is a primary site for lipid storage. Glycogen is also found in adipocytes at a much lower concentrations than in liver and muscle, though its exact role remains unclear (Jurczak et al., 2007; Markan et al., 2010). Early studies suggested that glycogen is converted into fat in the adipose tissue and the dynamic regulation of adipose glycogen may serve as an energy sensor in coordinating glucose and lipid metabolism during the fasted to fed transition (Markan et al., 2010). In normal and nutritionally balanced states, glycogen is histologically invisible in adipose tissue in rats (Fawcett, 1948). However, when rats are fed on a carbohydrate-rich diet after prolonged starvation, adipose glycogen is markedly increased (Tuerkischer and Wertheimer, 1942). In addition, administration of insulin in rats or dogs can result in a transient glycogen accumulation in the adipocytes (Fawcett, 1948). The ability to accumulate glycogen suggests that the synthesis of glycogen in adipocytes is also a dynamic process in GSD IIIa dogs. The clearance of adipocyte glycogen seems to be impaired by the lack of GDE activity in the affected dogs, suggesting that glycogen catabolism takes place through a mechanism similar to that in muscle and liver. To our knowledge, glycogen accumulation in adipose tissues has not been reported in patients with GSD IIIa and other GSDs, though one of us (P.S.K.) has palpated a lipoma-like structure in a GSD III individual. The existence of significant glycogen accumulation in adipocytes is unusual; whether the involvement of adipose tissue in GSD IIIa dogs is a species-specific event or a common feature needs to be further studied.
In this study, we have demonstrated that the naturally occurring GSD IIIa dog model in CCR has a phenotype that closely resembles the human disease, with glycogen accumulation in liver and skeletal muscle that leads to progressive hepatic fibrosis and myopathy. This disease model will help us better understand GSD IIIa disease progression, identify new biomarkers for the disease and develop effective therapies such as enzyme replacement therapy, gene therapy and substrate reduction therapy.
METHODS
Animals
The CCR breeding colony was maintained at Michigan State University. Housing. Mating, rearing of offspring and transport of dogs were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at MSU. Diagnosis of GSD IIIa was confirmed by mutation analysis (Gregory et al., 2007). A total of eight affected dogs of ages ranging from 2 to 6 months were transported to Duke University Medical Center for this study. The animals were reared on a normal canine diet at the Division of Laboratory Animal Resources (DLAR) of Duke University. All animal experiments were approved by the Institutional Animal Care and Use Committee at Duke University and were in accordance with the National Institutes of Health guidelines. Throughout the study, all dogs were on a regular diet that contained approximately 25–30% protein and 45% carbohydrate. For the first year of life, the dogs were fed with Science Diet puppy large breed dry food (Hill’s Pet Nutrition, Topeka, KS) by ad-libitum feeding for a few months and then got 2.25 cups twice a day (4.5 cups/day) along with one can of puppy canned food (Science Diet Puppy Gourmet Chicken Entrée) per day. After 1 year, all dogs were switched to normal adult food (Purina Lab Canine Diet 5006; Purina Mills, St Louis, MO). Each dog was given 3 cups of the dry food twice a day (6 cups/day) and also one can of Purina ProPlan Chicken and Rice Entrée (Nestlé Purina PetCare Company, St Louis, MO) wet food per day.
Routine laboratory testing and tissue biopsy
Blood (5 ml) was collected from saphenous or jugular veins for each dog every 4 weeks. Samples were sent to a commercial laboratory for a panel of routine biochemical tests, which included ALT, AST, ALP, CPK, glucose, triglycerides, cholesterol, bilirubin, albumin, urea nitrogen, γ-glutamyl transpeptidase and creatinine. Liver biopsies by laparotomy and skeletal muscle (quadriceps) biopsies were performed on each dog at specified ages under general anesthesia. Fresh tissue specimens were immediately frozen on dry ice and stored at −80°C until used for biochemical analysis, or placed in 3% glutaraldhyde or 10% neutral buffered formalin (NBF) for histology. All dogs were fasted, but offered water for 12 hours prior to the surgical procedures.
Tissue glycogen analysis
Tissue glycogen content was assayed enzymatically using a protocol modified from Kikuchi et al. (Kikuchi et al., 1998). Frozen liver or muscle tissues (50–100 mg) were homogenized in ice-cold de-ionized water (20 ml water/g tissue) and sonicated three times for 20 seconds with 30-second intervals between pulses, using a Misonix XL2020 ultrasonicator. Homogenates were clarified by centrifugation at 12,000 g for 20 minutes at 4°C. Supernatant (20 μl) was mixed with 55 μl of water, boiled for 3 minutes and cooled to room temperature. Amyloglucosidase (#A1602; Sigma) solution (25 μl diluted 1:50 into 0.1 M potassium acetate buffer, pH 5.5) was added and the reaction incubated at 37°C for 90 minutes. Samples were boiled for 3 minutes to stop the reaction and centrifuged at top speed for 3 minutes in a bench-top microcentrifuge. Supernatant (30 μl) was mixed with 1 ml of Infinity Glucose reagent (Thermo Scientific) and left at room temperature for at least 10 minutes. Absorbance at 340 nm was measured using a Shimadzu UV-1700 spectrophotometer. A reaction without amyloglucosidase was used for background correction for each sample. A standard curve was generated using standard glucose solutions in the reaction with Infinity Glucose reagent (0-120 μM final glucose concentration in the reaction).
Histopathology
Fresh tissues were cut into 1-mm cubes and immersion-fixed overnight in 3% glutaraldehyde in 0.2 M sodium cacodylate buffer, pH 7.4 (Electron Microscopy Sciences, Hatfield, PA). The fixed tissues were then processed into Epon resin for HRLM (Lynch et al., 2005). Tissue sections were cut at 1 μm and stained with PAS-Richardson’s stain for glycogen observation. Additional ultrathin sections (70 nm) were cut, stained and examined by electron microscopy. In addition, small pieces of fresh tissue were fixed in 10% NBF and processed into paraffin blocks, cut into 5 μm sections and stained with H&E or trichrome stains.
Statistical analysis of glycogen content
The significance of differences between two different time points was assessed using two-tailed, unequal variance Student’s t-test; P<0.05 was considered to be statistically significant.
Acknowledgments
We wish to acknowledge inspiration and support from the Workman family of Lowell, Indiana. We deeply appreciate assistance from the Duke DLAR staff in animal care and procedures. We wish to acknowledge excellent technical support from Elizabeth Drake, Keri Fredrickson and Stuti Das, and expert advice from Yuan-Tsong Chen.
Footnotes
FUNDING
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
H.Y. and B.S. performed the major experiments and wrote the manuscript; B.L.T. performed histology of tissue biopsies; S.C. performed surgical procedures; J.F. maintained and genotyped the CCR colony; P.S.K., D.D.K. and S.A. provided clinical advice and technical support. B.S. and P.S.K. designed the experiments and supervised the study.
REFERENCES
- Arts M. P., Nieborg A., Brand R., Peul W. C. (2007). Serum creatine phosphokinase as an indicator of muscle injury after various spinal and nonspinal surgical procedures. J. Neurosurg. 7, 282–286 [DOI] [PubMed] [Google Scholar]
- Bernier A. V., Sentner C. P., Correia C. E., Theriaque D. W., Shuster J. J., Smit G. P., Weinstein D. A. (2008). Hyperlipidemia in glycogen storage disease type III: effect of age and metabolic control. J. Inherit. Metab. Dis. 31, 729–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman R. A., Winter H. S., Wolf B., Chen Y. T. (1992). Glycogen debranching enzyme deficiency: long-term study of serum enzyme activities and clinical features. J. Inherir. Metab. Dis. 15, 869–881 [DOI] [PubMed] [Google Scholar]
- Cosme A., Montalvo I., Sanchez J., Ojeda E., Torrado J., Zapata E., Bujanda L., Gutierrez A., Arenas I. (2005). [Type III glycogen storage disease associated with hepatocellular carcinoma]. Gastroenterol. Hepatol. 28, 622–625 [DOI] [PubMed] [Google Scholar]
- Dagli A. I., Zori R. T., McCune H., Ivsic T., Maisenbacher M. K., Weinstein D. A. (2009). Reversal of glycogen storage disease type IIIa-related cardiomyopathy with modification of diet. J. Inherit. Metab. Dis. doi: 10.1007/s10545-009-1088-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demo E., Frush D., Gottfried M., Koepke J., Boney A., Bali D., Chen Y. T., Kishnani P. S. (2007). Glycogen storage disease type III-hepatocellular carcinoma a long-term complication? J. Hepatol. 46, 492–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett D. W. (1948). Histological observations on the relation of insulin to the deposition of glycogen in adipose tissue. Endocrinology 42, 454–467 [DOI] [PubMed] [Google Scholar]
- Geberhiwot T., Alger S., McKiernan P., Packard C., Caslake M., Elias E., Cramb R. (2007). Serum lipid and lipoprotein profile of patients with glycogen storage disease types I, III and IX. J. Inherit. Metab. Dis. 30, 406. [DOI] [PubMed] [Google Scholar]
- Goessling W., Friedman L. S. (2005). Increased liver chemistry in an asymptomatic patient. Clin. Gastroenterol. Hepatol. 3, 852–858 [DOI] [PubMed] [Google Scholar]
- Gregory B. L., Shelton G. D., Bali D. S., Chen Y. T., Fyfe J. C. (2007). Glycogen storage disease type IIIa in curly-coated retrievers. J. Vet. Intern. Med. 21, 40–46 [DOI] [PubMed] [Google Scholar]
- Haagsma E. B., Smit G. P., Niezen-Koning K. E., Gouw A. S., Meerman L., Slooff M. J., The Liver Transplant Group (1997). Type IIIb glycogen storage disease associated with end-stage cirrhosis and hepatocellular carcinoma. Hepatology 25, 537–540 [DOI] [PubMed] [Google Scholar]
- Hershkovitz E., Donald A., Mullen M., Lee P. J., Leonard J. V. (1999). Blood lipids and endothelial function in glycogen storage disease type III. J. Inherit. Metab. Dis. 22, 891–898 [DOI] [PubMed] [Google Scholar]
- Hobson-Webb L. D., Austin S. L., Bali D. S., Kishnani P. S. (2010). The electrodiagnostic characteristics of Glycogen Storage Disease Type III. Genet Med. 12, 440–445 [DOI] [PubMed] [Google Scholar]
- Illingworth B., Cori G. T. (1952). Structure of glycogens and amylopectins. III. Normal and abnormal human glycogen. J. Biol. Chem. 199, 653–660 [PubMed] [Google Scholar]
- Illingworth B., Cori G. T., Cori C. F. (1956). Amylo-1,6-glucosidase in muscle tissue in generalized glycogen storage disease. J. Biol. Chem. 218, 123–129 [PubMed] [Google Scholar]
- Jurczak M. J., Danos A. M., Rehrmann V. R., Allison M. B., Greenberg C. C., Brady M. J. (2007). Transgenic overexpression of protein targeting to glycogen markedly increases adipocytic glycogen storage in mice. Am. J. Physiol. Endocrinol. Metab. 292, E952–E963 [DOI] [PubMed] [Google Scholar]
- Karwowski C., Galambos C., Finegold D., Shneider B. L. (2011). Markedly elevated serum transaminases in glycogen storage disease type III. J. Pediatr. Gastroenterol. Nutr. 52, 621–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T., Yang H. W., Pennybacker M., Ichihara N., Mizutani M., Van Hove J. L., Chen Y. T. (1998). Clinical and metabolic correction of pompe disease by enzyme therapy in acid maltase-deficient quail. J. Clin. Invest. 101, 827–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. O., Lee H. J., Choi J. W., Eun J. R., Choi J. H. (2008). An adult case of glycogen storage disease type IIIa. Korean J. Hepatol. 14, 219–225 [DOI] [PubMed] [Google Scholar]
- Kishnani P. S., Faulkner E., VanCamp S., Jackson M., Brown T., Boney A., Koeberl D., Chen Y. T. (2001). Canine model and genomic structural organization of glycogen storage disease type Ia (GSD Ia). Vet. Pathol. 38, 83–91 [DOI] [PubMed] [Google Scholar]
- Kishnani P. S., Austin S. L., Arn P., Bali D. S., Boney A., Case L. E., Chung W. K., Desai D. M., El-Gharbawy A., Haller R., et al. (2010). Glycogen storage disease type III diagnosis and management guidelines. Genet. Med. 12, 446–463 [DOI] [PubMed] [Google Scholar]
- Koeberl D. D., Pinto C., Sun B., Li S., Kozink D. M., Benjamin D. K., Jr, Demaster A. K., Kruse M. A., Vaughn V., Hillman S., et al. (2008). AAV vector-mediated reversal of hypoglycemia in canine and murine glycogen storage disease type Ia. Mol. Ther. 16, 665–672 [DOI] [PubMed] [Google Scholar]
- LaBarbera M., Milechman G., Dulbecco F. (2010). Premature coronary artery disease in a patient with glycogen storage disease III. J. Invasive Cardiol. 22, E156–E158 [PubMed] [Google Scholar]
- Labrune P., Huguet P., Odievre M. (1991). Cardiomyopathy in glycogen-storage disease type III: clinical and echographic study of 18 patients. Pediatr. Cardiol. 12, 161–163 [DOI] [PubMed] [Google Scholar]
- Labrune P., Trioche P., Duvaltier I., Chevalier P., Odievre M. (1997). Hepatocellular adenomas in glycogen storage disease type I and III: a series of 43 patients and review of the literature. J. Pediatric Gastroenterol. Nutr. 24, 276–279 [DOI] [PubMed] [Google Scholar]
- Lee P., Burch M., Leonard J. V. (1995). Plasma creatine kinase and cardiomyopathy in glycogen storage disease type III. J. Inherit. Metab. Dis. 18, 751–752 [DOI] [PubMed] [Google Scholar]
- Lee P. J., Deanfield J. E., Burch M., Baig K., McKenna W. J., Leonard J. V. (1997). Comparison of the functional significance of left ventricular hypertrophy in hypertrophic cardiomyopathy and glycogenosis type III. Am. J. Cardiol. 79, 834–838 [DOI] [PubMed] [Google Scholar]
- Lucchiari S., Santoro D., Pagliarani S., Comi G. P. (2007). Clinical, biochemical and genetic features of glycogen debranching enzyme deficiency. Acta Myol. 26, 72–74 [PMC free article] [PubMed] [Google Scholar]
- Lynch C. M., Johnson J., Vaccaro C., Thurberg B. L. (2005). High-resolution light microscopy (HRLM) and digital analysis of Pompe disease pathology. J Histochem. Cytochem. 53, 63–73 [DOI] [PubMed] [Google Scholar]
- Magnusson I., Rothman D. L., Jucker B., Cline G. W., Shulman R. G., Shulman G. I. (1994). Liver glycogen turnover in fed and fasted humans. Am. J. Physiol. 266, E796–E803 [DOI] [PubMed] [Google Scholar]
- Markan K. R., Jurczak M. J., Brady M. J. (2010). Stranger in a strange land: roles of glycogen turnover in adipose tissue metabolism. Mol. Cell. Endocrinol. 318, 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. G., Alleyne G. A., Brooks S. E. (1972). Gross cardiac involvement in glycogen storage disease type 3. Br. Heart J. 34, 862–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momoi T., Sano H., Yamanaka C., Sasaki H., Mikawa H. (1992). Glycogen storage disease type III with muscle involvement: reappraisal of phenotypic variability and prognosis. Am. J. Med. Genet. 42, 696–699 [DOI] [PubMed] [Google Scholar]
- Moses S. W., Wanderman K. L., Myroz A., Frydman M. (1989). Cardiac involvement in glycogen storage disease type III. Eur. J. Pediatr. 148, 764–766 [DOI] [PubMed] [Google Scholar]
- Nakayama A., Yamamoto K., Tabata S. (2001). Identification of the catalytic residues of bifunctional glycogen debranching enzyme. J. Biol. Chem. 276, 28824–28828 [DOI] [PubMed] [Google Scholar]
- Pearson C. M. (1968). Glycogen metabolism and storage diseases of types III, IV and V. Am. J. Clin. Pathol. 50, 29–43 [DOI] [PubMed] [Google Scholar]
- Sapey T., Mendler M. H., Guyader D., Morio O., Corbinais S., Deugnier Y., Brissot P. (2000). Respective value of alkaline phosphatase, gamma-glutamyl transpeptidase and 5′ nucleotidase serum activity in the diagnosis of cholestasis: a prospective study of 80 patients. J. Clin. Gastroenterol. 30, 259–263 [DOI] [PubMed] [Google Scholar]
- Schoser B., Glaser D., Muller-Hocker J. (2008). Clinicopathological analysis of the homozygous p.W1327X AGL mutation in glycogen storage disease type 3. Am. J. Med. Genet. 146A, 2911–2915 [DOI] [PubMed] [Google Scholar]
- Siciliano M., De Candia E., Ballarin S., Vecchio F. M., Servidei S., Annese R., Landolfi R., Rossi L. (2000). Hepatocellular carcinoma complicating liver cirrhosis in type IIIa glycogen storage disease. J. Clin. Gastroenterol. 31, 80–82 [DOI] [PubMed] [Google Scholar]
- Taylor C., Cox A. J., Kernohan J. C., Cohen P. (1975). Debranching enzyme from rabbit skeletal muscle. Eur. J. Biochem. 51, 105–115 [DOI] [PubMed] [Google Scholar]
- Tuerkischer E., Wertheimer E. (1942). Glycogen and adipose tissue. J. Physiol. 100, 385–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoof F., Hers H. G. (1967). The subgroups of type 3 glycogenosis. Eur. J. Biochem. 2, 265–270 [DOI] [PubMed] [Google Scholar]
- Wallimann T., Wyss M., Brdiczka D., Nicolay K., Eppenberger H. M. (1992). Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem. J. 281, 21–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibrecht K., Dayno M., Darling C., Bird S. B. (2010). Liver aminotransferases are elevated with rhabdomyolysis in the absence of significant liver injury. J. Med. Toxicol. 6, 294–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xenoulis P. G., Steiner J. M. (2010). Lipid metabolism and hyperlipidemia in dogs. Vet. J. 183, 12–21 [DOI] [PubMed] [Google Scholar]