

Abstract
We have positionally cloned and characterized a new calcium channel auxiliary subunit, α2δ-2 (CACNA2D2), which shares 56% amino acid identity with the known α2δ-1 subunit. The gene maps to the critical human tumor suppressor gene region in chromosome 3p21.3, showing very frequent allele loss and occasional homozygous deletions in lung, breast, and other cancers. The tissue distribution of α2δ-2 expression is different from α2δ-1, and α2δ-2 mRNA is most abundantly expressed in lung and testis and well expressed in brain, heart, and pancreas. In contrast, α2δ-1 is expressed predominantly in brain, heart, and skeletal muscle. When co-expressed (via cRNA injections) with α1B and β3 subunits in Xenopus oocytes, α2δ-2 increased peak size of the N-type Ca2+ currents 9-fold, and when co-expressed with α1C or α1G subunits in Xenopus oocytes increased peak size of L-type channels 2-fold and T-type channels 1.8-fold, respectively. Anti-peptide antibodies detect the expression of a 129-kDa α2δ-2 polypeptide in some but not all lung tumor cells. We conclude that the α2δ-2 gene encodes a functional auxiliary subunit of voltage-gated Ca2+ channels. Because of its chromosomal location and expression patterns, CACNA2D2 needs to be explored as a potential tumor suppressor gene linking Ca2+ signaling and lung, breast, and other cancer pathogenesis. The homologous location on mouse chromosome 9 is also the site of the mouse neurologic mutant ducky (du), and thus, CACNA2D2 is also a candidate gene for this inherited idiopathic generalized epilepsy syndrome.
Electrophysiological and molecular cloning studies have revealed an incredible diversity of voltage-gated calcium channels. They are formed by heteromultimeric complexes of α1, α2δ, β, and γ subunits. The α1 subunits contain the channel pore, voltage sensors, and the receptors for various classes of drugs and toxins (1). There are three families of α1 subunits: the L-type, Cav1, family, composed of α1S, α1C (Cav1.2), α1D, and α1F; the non-L-type high voltage-activated, or Cav2, family, which contains the P/Q-types encoded by α1A, the N-type encoded by α1B (Cav2.2), and R-types encoded by α1E; and the T-type family, or Cav3, encoded by α1G (Cav3.1), α1H, and α1I (2). The β subunit family is less diverse, with only four genes cloned so far (3). Co-expression studies have established two physiological roles of β subunits in high voltage-activated Ca2+ channels: they dramatically increase α1 expression at the plasma membrane, and they alter the biophysical properties of the channel currents. In general, β subunits have little effect on the expression of low voltage-activated currents (4). Although only one γ and α2δ subunit have been characterized biochemically, recent evidence suggests that there may be additional members of these gene families (5–7). The γ1 subunit was shown to be part of the skeletal muscle L-type channel (8); coexpression studies have indicated that it aids in the formation of L-type channels, as assayed by dihydropyridine binding (9), and may play a role in channel inactivation (10).
The α2δ subunit (α2δ-1) was first identified in biochemical studies of skeletal muscle L-type Ca2+ channels (reviewed in Ref. 1). Using antibodies, it has also been shown to be part of the cardiac L-type and neuronal N-type channels (11, 12). α2δ-1 cDNA has been cloned from skeletal muscle and brain cDNA libraries (13–15). The 175-kDa protein product is post-translationally cleaved to form disulfide-linked α2 and δ peptides, both of which are heavily glycosylated. Biochemical and mutation analysis supports a single transmembrane domain in the δ subunit that anchors the α2δ protein to the membrane (16). Coexpression of α2δ-1 with both high voltage-activated and low voltage-activated α1 subunits facilitates the assembly of channels in the plasma membrane (4, 9, 17). Coexpression studies also indicate that α2δ-1 can alter the pharmacological properties of L-type channels (18). In contrast to the β subunits that have a dramatic effect on gating of all high voltage-activated channel in many expression systems, the effects of α2δ-1 are more controversial, perhaps depending on the α1 subunit used or the expression system. For example, α2δ-1 has little or no effect on either L-type (18, 19) or N-type currents expressed in Xenopus oocytes (17) but appears to affect inactivation of L-type channels expressed in mammalian cells (20, 21). The opposite result occurred in studies on α1E-mediated currents, where no effect was observed in mammalian cells (22) and effects on channel inactivation were observed in Xenopus oocytes (23). The α2δ-1 subunit has a high affinity binding site for the anti-epileptic drug gabapentin (16). Gabapentin has been shown to modestly inhibit (~30%) neuronal Ca2+ currents, although it is unclear if this is its mechanism of action (24).
We have been attempting to identify a new human tumor suppressor gene in chromosome region 3p21.3, where frequent allele loss and occasional homozygous deletions have been found in lung, breast, and other human tumors (25). Several genes in the region have been identified using positional cloning strategies. The sequence of one of the genes and its mRNA splicing variants in the region (α2δ-2; GenBank™ numbers AF040709, AF042792, and AF042793; CACNA2D2) showed extensive homology with the known calcium channel α2δ-1 subunit. We have studied the tissue distribution of expression of this new α2δ-2 gene and tested the function of the gene product in Xenopus oocytes by coexpressing α2δ-2 cRNAs along with a representative member of the three families of calcium subunit α1 subunits. We find a pattern of expression different from the other α2δ subunit, whereas the α2δ-2 enhances the activity of the calcium subunit α1 subunits.
EXPERIMENTAL PROCEDURES
Positional Cloning of α2δ-2 cDNA
A contig1 of 22 cosmids covering 600 kb localized to the 3p21.3 small cell lung cancer homozygous deletions isolated from a human placental cosmid library have been described previously (25). The entire contig was sequenced by the joint effort of the Sanger Center (UK) and the Washington University Genome Sequence Center. The obtained sequence information was analyzed by BLAST and GENSCAN Informatics as well as the integrated informatic software package developed by the Garner lab at UT Southwestern, PANORAMA, and we found that cosmid LUCA#11 harbored an EST clone N53512 (Genome Systems) containing a portion of the 3′ end as well as putative exons of what would be α2δ-2. Further Southern blot analysis showed that various exons of the α2δ-2 gene are located on cosmid LUCA06 (GenBank™ number Z84493), LUCA07 (GenBank™ number Z84494), LUCA08 (GenBank™ number Z84495), LUCA09 (GenBank™ number Z75743) LUCA10 (GenBank™ number Z75742), and LUCA11 (GenBank™ number Z84492). Based on GENSCAN predictions, a primer set of LUCA11pr5, 5′-CTGAGAGTGAGGATGTGGAA-3′(sense primer), and LUCA11pr18, 5′-GTGCATCCTCATACACGTTG-3′ (antisense primer), was used for reverse transcriptase-polymerase chain reaction amplification for normal lung cDNA template, and a 960-base pair product was successfully amplified. The 1.5-kb NotI/HindIII fragment of the EST clone N53512 and the 960-base pair product were used as probes on human multiple tissue Northern blots (CLONTECH). The screening of a million clones from a lung cDNA library (CLONTECH) with the 960-base pair reverse transcriptase-polymerase chain reaction product yielded 120 positive clones, which were also screened by probing clone N53512 to obtain the clones with long inserts. Five clones randomly selected as single positives for the 960-base pair probe and 5 clones selected as double positive for both probes were subcloned and sequenced. All of the 10 clones had the sequence of α2δ-2, suggesting that all of the 120 clones were α2δ-2. Two clones (pY720c21 and pY724c95) that covered the longest sequence were assembled and further inserted into a plasmid expression vector pcDNA3.1 (Invitrogen) by standard methods.
Northern Blot Analysis
Human multiple tissue Northern blots (CLONTECH) were hybridized and washed according to the manufacturer’s recommendation. The three short probes were generated using polymerase chain reaction. All probes were labeled with a random-oligonucleotide priming kit (Rad Prime DNA labeling System, Life Technologies, Inc.).
In Vitro Translation and Transient Transfection Studies
α2δ-2 cDNA inserted into plasmid pcDNA3.1 (Invitrogen) was used for in vitro translation using [35S]methionine in an in vitro transcription/translation system (TNT Coupled Reticulocyte Systems, Promega). For transfection experiments, non-small cell lung cancer NCI-H1299 cells (3 × 105) were seeded in 3.5-cm culture dishes for 24 h in RPMI 1640 containing 5% fetal bovine serum, and then 1 μg of cloned DNA was introduced into the cells using the LipofectAMINE reagent (Life Technologies, Inc.). For protein expression after transfection, cells were harvested 48 h later and were lysed in 80 μl of sample buffer (50 mM Tris, pH 6.8, 1% SDS, 10% glycerol, and 0.3M of β-mercaptoethanol). NCI-H1299 cells were used for these studies because they do not express endogenous α2δ-2 mRNA or protein (see “Results”) and are homozygous for multiple polymorphic markers in the 600-kb homozygous deletion region (26) and, thus, have undergone loss of heterozygosity for this region.
Antibodies and Western Blots
Peptide A (YYDAKADAELDDPESEDVERG), corresponding to amino acids 161–181 of α2δ-2 (GenBank™ number AF040709), was synthesized, and rabbit polyclonal antibodies were raised using a commercial source (Alpha Diagnostic, San Antonio, TX). Antibodies were affinity-purified using this peptide conjugated to agarose beads (amino link immobilization kit; Pierce). Horseradish peroxidase-labeled anti-rabbit antibody and chemiluminescent substrates were used to detect the positive signal.
Electrophysiologic Studies
For the study of coexpression with α1B and β3 subunits, complementary RNA (cRNA) encoding human brain α1B (27), rabbit skeletal muscle α2δ-1(28), rabbit β3 (29), and α2δ-2 subunits was synthesized in vitro using T7 RNA polymerase, resuspended in water at a final concentration of ~1 mg/ml, and stored at −80 °C until injection. Xenopus oocytes harvested by standard methods (30) were injected with a mixtures of the following transcripts: α1B+β3, α1B+β3+α2δ-1, α1B+β3+α2δ-2, or α2δ-2 alone (approximately 50 ng of total cRNA/oocyte). Two days later oocytes were analyzed using standard two-electrode voltage-clamp technique with 5 mM Ba2+ as a charge carrier (31). The holding potential was −120 mV. Currents were recorded in response to test potentials ranging from −110 to +100 mV, filtered at 200 Hz, then analyzed using pClamp 6.04 (Axon Instruments) and Origin (Microcal) software. Leak and capacitance currents were subtracted on-line with a P/4 protocol.
For the studies of coexpression with α1C and α1G subunits, cRNA of either α1C, α1G, or α2δ-2 cDNA was synthesized using Ambion Megascript kit according to the supplier’s protocol (Ambion, Austin, TX). Due to low expression of wild-type α1C, we used the modified cDNA ΔN60, which is truncated by 60 amino acids at the N-terminal end of the rabbit cardiac α1 subunit (32). The rat brain α1G cDNA (33) was contained in the vector pGEM-HE (34). Fifty nl of cRNA (5 ng for α1C, 5 ng for α1G, and 2.5 ng for α2δ-2) of either α1C alone, α1C plus α2δ-2, α1G alone, or α1G plus α2δ-2 were injected into each oocyte using a Drummond Nanoject pipette injector (Parkway, PA). Expression of injected cRNA was measured from the 4th day after injection for α1C alone or α1C plus α2δ-2 and the 6 –7th day after injection for α1G alone or α1G plus α2δ-2 using the two-electrode voltage clamp method. Currents were measured in either 40 mM Ba2+ solution (40 mM Ba(OH)2, 50 mM NaOH, 1 mM KOH, and 5 mM HEPES, adjusted to pH 7.4 with methanesulfonic acid) for L-type currents or 10 mM Ba2+ solution (10 mM Ba(OH)2, 80 mM NaOH, 1 mM KOH, and 5 mM HEPES, adjusted to pH 7.4 with methanesulfonic acid) for T-type currents. Data were sampled at either 2 kHz for L-type currents or 5 kHz for T-type currents using the pClamp 6 system via a Digidata 1200 A/D converter (Axon Instrument, Foster city, CA). Leak currents were subtracted using a P/+4 for L-type currents or a P/−6 for T-type currents.
RESULTS
Characteristics of α2δ-2 cDNA and Its Predicted Amino Acid Sequence
Human chromosome 3p21.3 is deleted in many small cell lung cancers. While searching for a putative tumor suppressor gene in this region, we identified a gene (GenBank™ number AF040709, AF042792, and AF042793) that appeared to encode a homolog of theα2δ subunit of Ca2+ channels. An open reading frame of 3,435 nucleotides encoding 1,145 amino acids was identified. The molecular mass of the deduced amino acid sequence is 129,343 Da. BLAST searches and homology alignment revealed that the predicted protein shares 56% amino acid sequence identity with the human auxiliary α2δ-1 subunit (GenBank™ number M76559) of voltage-gated Ca2+ channels (13). Therefore we refer to the gene product as α2δ-2 and the gene as CACNA2D2. Notably, 17 out of 22 cysteines in α2δ-2 are conserved with α2δ-1, suggesting that the two proteins share similar overall secondary structure. Similar to the α2δ-1 subunit, the α2δ-2 sequence contains multiple putative N′-glycosylation sites and is likely to be glycosylated.
Tissue Specificity of α2δ-2 Expression
Tissue distribution of α2δ-2 expression was examined by Northern blot hybridization of the human multiple tissue blots (CLONTECH) using the entire coding region (Fig. 1, A and B) as well as three different short probes (nucleotides 510 – 653 (Fig. 1C) and nucleotides 993–1152 (Fig. 1D) and 2729 –3293 (Fig. 1E). An approximately 5.5-kb α2δ-2 mRNA was found and appeared most abundant in lung and testis, abundant in brain, heart, and pancreas, and detected at low amounts in prostate and skeletal muscle in all of the four Northern blot analysis. The significance of the results will be discussed in the discussion section.
Fig. 1. Expression of α2δ-2 in normal human tissues.

Human multiple tissue blots (CLONTECH) were hybridized with 32P-labeled cDNA synthesized from the entire coding sequence of α2δ-2 (A and B). Sizes of the RNA markers are indicated on the left. PBL, peripheral blood lymphocyte. Probes for C, D, and E are indicated at the bottom of each figure.
Biochemical Properties of the α2δ-2 Protein
To characterize the biochemical properties of α2δ-2 protein, we translated the α2δ-2 cDNA in vitro and in vivo. The product of in vitro translation is a single band with the molecular mass of ~130 kDa (Fig. 2A), which is consistent with the calculated molecular mass of 129,268 Daltons. For in vivo expression, the α2δ-2 coding sequence was inserted into the mammalian expression vector pcDNA3.1 (Invitrogen) and transfected into non-small cell lung cancer cell line NCI-H1299, which does not express α2δ-2 mRNA or its protein (Fig. 2C). An affinity-purified anti-α2δ-2 peptide antibody detected an ~150-kDa protein in the lysate from α2δ-2-transfected cells but not in cells transfected with the vector control (Fig. 2B). Most likely, the increase in the apparent molecular mass (129 to 150 kDa) compared with the conceptually translated protein is the result of N′-glycosylation, in agreement with multiple putative N-glycosylation sites in the α2δ-2 sequence, which represent known properties of the α2δ-1 protein (35). Endogenous α2δ-2 protein of 150 kDa was also detected in some lung tumor cell lines using the same antibody (Fig. 2C), which further confirmed our conceptual translation and anti-peptide antibody preparation were correct.
Fig. 2.
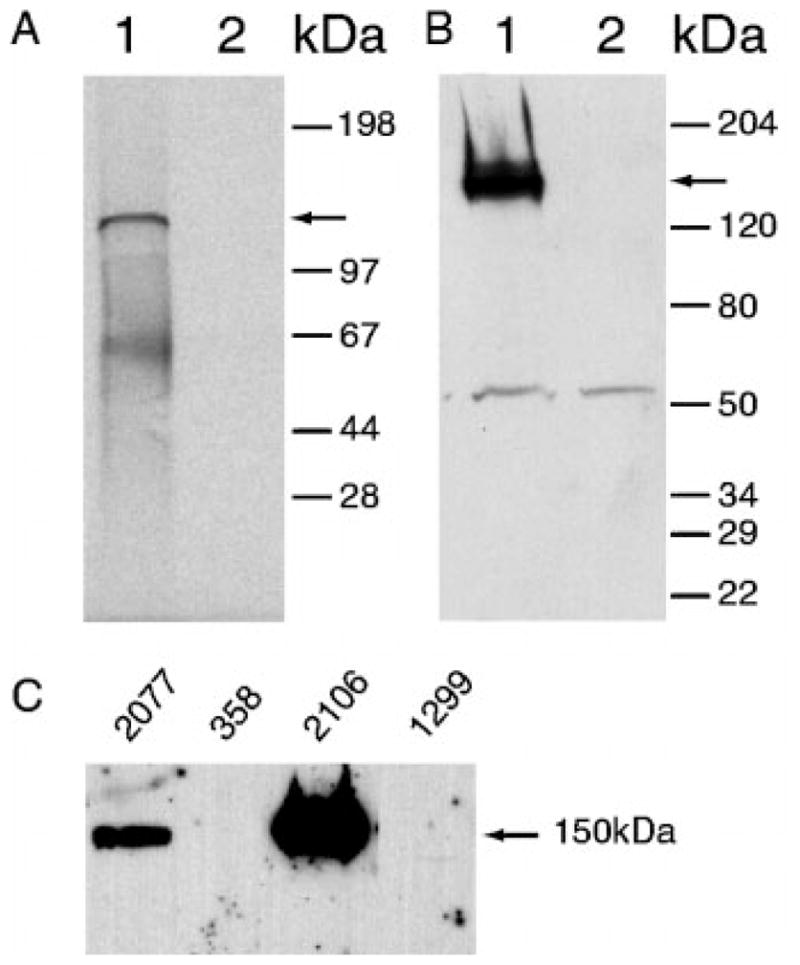
A, in vitro transcription and translation of α2δ-2. Lane 1, in vitro transcription and translation of α2δ-2 in expression vector pcDNA3.1. Lane 2, no DNA was added in the same reaction. The arrow indicates the expected 130 kDa product. B, Western blot analysis of transfection of NCI-H1299 cells with α2δ-2. Lane 1, transfection of NCI-H1299 cells with α2δ-2 in expression vector pcDNA3.1. Lane 2, transfection of NCI-H1299 cells with pcDNA3.1 vector alone. Affinity-purified anti-α2δ-2 peptide antibody was used to detect the protein product. The arrow indicates the expected protein product. Sizes of the prestained protein molecular weight markers are indicated on the right. C, 40 μg of protein from tumor cell lysates were loaded in each lane. Lane 1, NCI-H2O77 (adenocarcinoma); lane 2, NCI-H358 (adenocarcinoma); lane 3, NCI-H2106 (large cell neuroendocrine carcinoma); lane 4, NCI-H1299 (large cell carcinoma) cells.
Functional Properties of α2δ-2
To test for functional expression of the α2δ-2 subunit, we performed a series of two-electrode voltage clamp experiments using the Xenopus oocyte heterologous expression system. Injection of oocytes with cRNA encoding the pore-forming human α1B subunit together with an auxiliary β3 subunit resulted in expression of functional N-type calcium channels in oocyte plasma membranes with a peak current of 1.0 ± 0.1 μA (n = 4) (Fig. 3A). Channel activity was indicated as representative inward barium currents observed in response to 0 mV and +20 mV test potentials. The magnitude of N-type currents was increased 9-fold to 9.1 ± 1.4 μA (n = 10) when α1B and β3 were coexpressed with the rabbit skeletal muscle α2δ-1 subunit (Fig. 3B). When co-expressed with α1B and β3 subunits, the α2δ-2 subunit exerted a similar effect on N-type channel expression, increasing peak current size to 7.6 ± 0.6 μA (n = 10) (Fig. 3C). No channel activity was observed after injection of α2δ-2 cRNA alone (data not shown). By varying the test potential in the range from −100 mV to +100 mV we established that the shape and position of current-voltage relationships was similar for all three subunit combinations, with the maximum current at 0 mV test potential and reversal potential at + 50 mV (Fig. 3D).
Fig. 3. Representative records of barium currents evoked by step depolarization from −120 to 0 mV and +20 mV.

Oocytes were injected with cRNA encoding: A, α1B+β3; B, α1B+β3+α2δ-1; C, α1B+β3+α2δ-2. Residual capacitance transients at the end of test pulses were removed. D, mean current-voltage curves from two independent injections (mean ± S.E.) with α1B+β3 (open circles), α1B+β3+α2δ-1 (filled triangles), and α1B+β3+α2δ-2 (filled circles) cRNA combinations. E, current-voltage relationships of α1C alone (filled circles) and α1C/α2δ-2 (circles) induced currents. Currents were evoked by a series of test pulses of −50 mV to +70 mV from a holding potential of −70 mV in 40 mM Ba2+ solution. Average α1C currents were collected from 33 oocytes; α1C/α2δ-2 currents were from 31 oocytes isolated from three different frogs. Data represent the mean ± S.E. F, current-voltage relationships of α1G (filled squares)- and α1G/α2δ-2 (squares)-induced currents. Currents were elicited by test pulses of −70 mV to +50 mV from a holding potential of minus;90 mV in 10 mM Ba2+ solution. Average α1G currents were collected from 33 oocytes; α1G/α2δ-2 currents were from 32 oocytes isolated from four frogs. G, average stimulation of α1C and α1G currents by coexpression with α2δ-2. Since expression of the cloned T-type channels is highly variable between batches of oocytes, each batch was injected with both α1 and α1α2δ-2 and stimulation by α2δ-2 was measured for each batch then averaged.
Stimulation of N-type current expression by α2δ-1 and α2δ-2 subunits (Fig. 3, A–C) is similar to the previously described effect of α2δ-1 on P/Q-type Ca2+ channels formed by α1A and β subunits (36, 37), which has been shown to depend on α2δ-1 subunit glycosylation (35). Thus, it is likely that α2δ-2 subunit is glycosylated when expressed in Xenopus oocytes, as is expected from biochemical and sequence analysis. Noticeably, the α2δ-1 but not the α2δ-2 subunit was able to hasten N-type Ca2+ channel inactivation. Indeed, at the end of a 50-ms test pulse to +20 mV, the size of the current was reduced to 33 ± 10% (n = 8) of the peak current for α1B+β3+α2δ-1, to 59 ± 5% (n = 24) of the peak current for α1B+β3+α2δ-2, and to 51 ± 4% (n = 15) of the peak current for α1B+β3 subunit combinations.
To test for an α2δ-2 effect on L-type channels, either α1C cRNA alone or α1C plus α2δ-2 cRNA were injected into oocytes. Peak currents measured during a series of test potentials were averaged (Fig. 3E). When peak current amplitudes measured at +30 mV were compared, α1C/α2δ-2 currents were significantly larger than α1C by 201% (t test, p < 0.001). However, there were no significant differences in the position of the current-voltage curves, which peaked at ~+35 mV. Similar to the α2δ-2 effect on α1C channels, coinjection of α2δ-2 cRNA with α1G cRNA increased T-type current amplitudes by 176% (t test, p < 0.05), compared with α1G alone (Fig. 3F). There were no significant differences in their biophysical properties including activation threshold, position of their current-voltage curves, reversal potentials, and activation and inactivation kinetics. We conclude from these experiments that the cloned α2δ-2 protein is able to function as an auxiliary subunit of all three subfamilies of voltage-gated Ca2+ channels.
DISCUSSION
This study describes the cloning and functional properties of a novel α2δ subunit of voltage-gated Ca2+ channels. The gene (CACNA2D2) was discovered by positional cloning while searching for a lung cancer tumor suppressor gene. GenBank™ deposits AF040709 and AF042792 represent alternatively spliced forms (in the 5′-untranslated region) with the same conceptual 1,145-amino acid sequence. GenBank™ deposit AF042793 represents another 5′ alternatively spliced form uncommonly found in lung cDNA clones resulting in an open reading frame beginning at the second 5′ methionine at codon 70 and, thus, resulting in a deletion of the 70 N-terminal amino acids found in the common α2δ-2 form studied here. Sequences were deposited in the GenBank™ to stimulate research on its function. Klugbauer et al. (7) cloned another related α2δsubunit, then proposed the following nomenclature: α2δ-1, for the original α2δ cloned from skeletal muscle; α2δ-2, for the protein described herein, and α2δ-3, for their novel sequence. Similarly the genes will be referred to as CACNA2D1, CACNA2D2, and CACNA2D3, respectively (38). While this paper was in preparation, an α2δ-2 clone (KIAA0558, GenBank™ number AB011130) was independently isolated by the Kazusa DNA Research Institute from human brain as part of large scale anonymous cDNA sequencing efforts (39). The present study reports on the expression of the CACNA2D2 gene in human tissues and on electrophysiological studies that show it can modulate the expression of functional Ca2+ channels.
Expression of the CACNA2D2 gene was determined by Northern analysis. It was most highly expressed in lung and testis, well expressed in brain, heart, and pancreas, and expressed to a lower extent in skeletal muscle and prostate. Our results do not agree with those of Klugbauer et al. (7), who found abundant cross-reactive material from what they reported to be α2δ-2 in mRNA from skeletal muscle, pancreas, and heart, with hardly any signal from lung. We feel our expression pattern is the correct one since we had performed four independent Northern blot analysis using four probes including one (nt 2729 –3293) that is very similar to the probe that Klugbauer et al. (7) used (nucleotides 2877–3249). The result of our cDNA screening also supports the high expression of α2δ-2 in lung, since we obtained 120 α2δ-2 clones from a screening of 1 million clones of a lung cDNA library. A possible explanation for the discrepancy could be that their probe cross-reacted with α2δ-1, since it has an expression pattern very similar to what they reported for α2δ-2 (40). Furthermore, it is unlikely that α2δ-2 is highly expressed in skeletal muscle, because α2δ proteins were purified from that tissue, and only the sequence of α2δ-1 was detected (13).
The tissue distribution of mRNA for the three α2δ subunits is very different (7, 40). All three genes are expressed in brain, which is the only tissue that expresses α2δ-3. The α2δ-1 gene is highly expressed in skeletal muscle, where we find little or no expression of α2δ-2. Both α2δ-1 and -2 are expressed in heart. The α2δ-2 gene is highly expressed in lung where the expression of α2δ-1 is low. It will be important to determine what cells in the lung express α2δ-2; however, we have shown that several lung cancers representing different lung epithelial types can express α2δ-2, so that presumably some normal lung epithelial cells also express α2δ-2. In this regard, it is also interesting to note that α1C was cloned from lung cDNA libraries (19), and L-type currents have been characterized from tracheal smooth muscle (41). The only β subunit detected in lung mRNA is β2 (3). Therefore, the minimum subunit composition of lung L-type channels can be deduced as α1Cα2δ-2β2.
The possible role of α2δ-2 as a Ca2+ channel subunit was examined using the Xenopus oocyte expression system. We tested for an effect on currents using three α1 subunits. The α1 subunits were chosen to represent each of the three subfamilies of Ca2+ channels: Cav1.2 or α1C, Cav2.2 or α1B, and a low voltage-activated channel Cav3.1 or α1G. In each case, α2δ-2 was able to stimulate functional expression. No effect was observed on the biophysical properties of the current, suggesting that α2δ-2 simply increased the number of functional channels at the plasma membrane. Similar results were obtained with α2δ-1 on the expression of α1G in both COS cells and Xenopus oocytes (4).
Coexpression studies of α2δ-2 plus α1B also included the β3 subunit. In these experiments we observed the largest stimulatory effect on expression. Some studies report a synergistic action of α2 and β on α1B expression (17). The experiments with α1C did not include a β subunit because they stimulate current so much already that it has been difficult to see any effect of α2δ at the whole cell level (18).
Interest in the physiological roles of Ca2+ channels has increased due to findings that mutations in their genes can lead to human diseases (42). In addition, defects in the auxiliary subunits of Ca2+ channels have been described in mouse models of absence epilepsy. These include mouse strains that have lost the expression of β4 and the recently discovered γ2 subunit (5, 43). In this regard, after we cloned CACNA2D2 we noted with great interest that the syntenic region in the mouse (mouse chromosome 9, 59.0 – 60.0 centimorgan) contains the mouse mutant ducky and also 4 other flanking genes (CISH, GNAI2, GNAT, and HYAL1) that we have identified in our ~600-kb region (25) and deposited as GenBank™ numbers AF132297 for CISH and U03056 for HYAL1. Our partial mouse cDNA sequence is 92% identical to the human α2δ-2 sequence (GenBank™ number AF169633.1). In fact, preliminary evidence suggests that loss of α2δ-2 expression leads to the epileptic phenotype, ducky (44). Histological examination of mouse ducky mutants reveals atrophy of the cerebellum, medulla oblongata, and spinal cord (45). These mice develop a spike-and-wave phenotype in the electroencephalogram, which is similar to that observed in absence epilepsy patients. Thus, it will be of great interest to see if inherited defects in CACNA2D2 also occur in humans (46). It remains to be determined how these Ca2+ channel defects lead to these epileptic phenotypes.
We began these studies searching for a human lung cancer tumor suppressor gene. The specific 600-kb 3p21.3 chromosome region within which the CACNA2D2 gene resides is a site of homozygous deletions occurring in lung and breast cancer and is a frequent target region for allele loss occurring very early in the pathogenesis of lung and other cancers (25, 47– 49). Thus, we are also studying CACNA2D2 for mutations, expression alterations, and functional characteristics of a tumor suppressor gene in these cancers. In this regard we were interested to see its high expression in normal lung tissue and in some but not all lung cancer cell lines. A clinical connection between voltage-dependent calcium channels and lung cancer is well established by the Lambert-Eaton myasthenic syndrome, seen in some small cell lung cancer patients (50). Lambert-Eaton myasthenic syndrome is a human autoimmune disorder that impairs neuromuscular transmission such that patients with this syndrome have a defect in the Ca2+-dependent quantal release of acetylcholine from motor nerve terminals (51). In this syndrome patients develop antibodies (presumably initiated by expression of the channel proteins in their small cell lung cancer) that react with voltage-gated calcium channel polypeptides that block depolarization-induced Ca2+ influx, leading to the myasthenia (52–54). In this report we have seen α2δ-2 to functionally interact with the T-type channel subunit α1G. Thus, it was of great interest to us when Toyota et al. (55) reported that CACNA1G encoding this subunit could have its expression inactivated by aberrant methylation of its 5′ CpG island in human tumors such as colorectal cancers, gastric cancers, and acute myelogenous leukemias. CACNA1G maps to chromosome region 17q21, another site of frequent allele loss in human cancer. Such acquired CpG island methylation in promoter regions of cancer cells as an acquired abnormality silencing genes such as tumor suppressor genes is well described (56, 57). Ca2+ influx via voltage-gated calcium channels including T-type channels and intracellular calcium signaling plays a role in apoptosis (58). In addition, platelet-derived growth factor-stimulated calcium influx changed during transformation of mouse C3H 10T1/2 fibroblasts accompanied by a marked reduction in expression of T-type calcium channels (59). Thus, the inactivation of voltage-gated calcium channel subunits such as CACNA2D2 and CACNA1G by any of several means merit serious consideration as an important step in cancer pathogenesis.
Acknowledgments
We thank Meena Viswanathan, Yang Song, and David Burbee for assistance in this research.
Footnotes
This work was supported by National Institutes of Health (NCI) Grants CA71618, P50-CA70907, NS38691, and NO1-CO-56000 and by the Hibino Memorial Medical Fund.
The abbreviations used are: contig, group of overlapping clones; kb, kilobase(s).
References
- 1.Perez-Reyes E, Schneider T. Kidney Int. 1995;48:1111–1124. doi: 10.1038/ki.1995.395. [DOI] [PubMed] [Google Scholar]
- 2.Randall A, Benham C. Mol Cell Neurosci. 1999;14:255–272. doi: 10.1006/mcne.1999.0795. [DOI] [PubMed] [Google Scholar]
- 3.Castellano A, Perez-Reyes E. Biochem Soc Trans. 1994;22:483–488. doi: 10.1042/bst0220483. [DOI] [PubMed] [Google Scholar]
- 4.Dolphin AC, Wyatt CN, Richards J, Beattie RE, Craig P, Lee JH, Cribbs LL, Volsen SG, Perez-Reyes E. J Physiol (Lond) 1999;519:35–45. doi: 10.1111/j.1469-7793.1999.0035o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Letts VA, Felix R, Biddlecome GH, Arikkath J, Mahaffey CL, Valenzuela A, Bartlett FS, II, Mori Y, Campbell KP, Frankel WN. Nat Genet. 1998;19:340–347. doi: 10.1038/1228. [DOI] [PubMed] [Google Scholar]
- 6.Black JL, III, Lennon VA. Mayo Clin Proc. 1999;74:357–61. doi: 10.4065/74.4.357. [DOI] [PubMed] [Google Scholar]
- 7.Klugbauer N, Lacinova L, Marais E, Hobom M, Hofmann F. J Neurosci. 1999;19:684–691. doi: 10.1523/JNEUROSCI.19-02-00684.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharp AH, Campbell KP. J Biol Chem. 1989;264:2816–2825. [PubMed] [Google Scholar]
- 9.Suh-Kim H, Wei X, Klos A, Pan S, Ruth P, Flockerzi V, Hofmann F, Perez-Reyes E, Birnbaumer L. Receptors & Channels. 1996;4:217–225. [PubMed] [Google Scholar]
- 10.Singer D, Biel M, Lotan I, Flockerzi V, Hofmann F, Dascal N. Science. 1991;253:1553–1557. doi: 10.1126/science.1716787. [DOI] [PubMed] [Google Scholar]
- 11.Schmid A, Barhanin J, Coppola T, Borsotto M, Lazdunski M. Biochemistry. 1986;25:3492–3495. doi: 10.1021/bi00360a002. [DOI] [PubMed] [Google Scholar]
- 12.McEnery MW, Snowman AM, Sharp AH, Adams ME, Snyder SH. Proc Natl Acad Sci U S A. 1991;88:11095–11099. doi: 10.1073/pnas.88.24.11095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellis SB, Williams ME, Ways NR, Brenner R, Sharp AH, Leung AT, Campbell KP, McKenna E, Koch WJ, Hui A, et al. Science. 1988;241:1661–1664. doi: 10.1126/science.2458626. [DOI] [PubMed] [Google Scholar]
- 14.De Jongh KS, Warner C, Catterall WA. J Biol Chem. 1990;265:14738–14741. [PubMed] [Google Scholar]
- 15.Williams ME, Feldman DH, McCue AF, Brenner R, Velicelebi G, Ellis SB, Harpold MM. Neuron. 1992;8:71–84. doi: 10.1016/0896-6273(92)90109-q. [DOI] [PubMed] [Google Scholar]
- 16.Brown JP, Gee NS. J Biol Chem. 1998;273:25458–25465. doi: 10.1074/jbc.273.39.25458. [DOI] [PubMed] [Google Scholar]
- 17.Brust PF, Simerson S, Mccue AF, Deal CR, Schoonmaker S, Williams ME, Velicelebi G, Johnson EC, Harpold MM, Ellis SB. Neuropharmacology. 1993;32:1089–1102. doi: 10.1016/0028-3908(93)90004-m. [DOI] [PubMed] [Google Scholar]
- 18.Wei X, Pan S, Lang W, Kim H, Schneider T, Perez-Reyes E, Birnbaumer L. J Biol Chem. 1995;270:27106–27111. doi: 10.1074/jbc.270.45.27106. [DOI] [PubMed] [Google Scholar]
- 19.Biel M, Ruth P, Bosse E, Hullin R, Stuhmer W, Flockerzi V, Hofmann F. FEBS Lett. 1990;269:409–412. doi: 10.1016/0014-5793(90)81205-3. [DOI] [PubMed] [Google Scholar]
- 20.Shirokov R, Ferreira G, Yi J, Rios E. J Gen Physiol. 1998;111:807–823. doi: 10.1085/jgp.111.6.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bangalore R, Mehrke G, Gingrich K, Hofmann F, Kass RS. Am J Physiol. 1996;270:H1521–H1528. doi: 10.1152/ajpheart.1996.270.5.H1521. [DOI] [PubMed] [Google Scholar]
- 22.Jones LP, Wei SK, Yue DT. J Gen Physiol. 1998;112:125–143. doi: 10.1085/jgp.112.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qin N, Olcese R, Stefani E, Birnbaumer L. Am J Physiol. 1998;274:C1324–C1331. doi: 10.1152/ajpcell.1998.274.5.C1324. [DOI] [PubMed] [Google Scholar]
- 24.Stefani A, Spadoni F, Bernardi G. Neuropharmacology. 1998;37:83–91. doi: 10.1016/s0028-3908(97)00189-5. [DOI] [PubMed] [Google Scholar]
- 25.Wei MH, Latif F, Bader S, Kashuba V, Chen JY, Duh FM, Sekido Y, Lee CC, Geil L, Kuzmin I, Zabarovsky E, Klein G, Zbar B, Minna JD, Lerman MI. Cancer Res. 1996;56:1487–1492. [PubMed] [Google Scholar]
- 26.Fondon JW, III, Mele GM, Brezinschek RI, Cummings D, Pande A, Wren J, O’Brien KM, Kupfer KC, Wei MH, Lerman M, Minna JD, Garner HR. Proc Natl Acad Sci U S A. 1998;95:7514–7519. doi: 10.1073/pnas.95.13.7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellinor PT, Zhang JF, Horne WA, Tsien RW. Nature. 1994;372:272–275. doi: 10.1038/372272a0. [DOI] [PubMed] [Google Scholar]
- 28.Tanabe T, Takeshima H, Mikami A, Flockerzi V, Takahashi H, Kangawa K, Kojima M, Matsuo H, Hirose T, Numa S. Nature. 1987;328:313–318. doi: 10.1038/328313a0. [DOI] [PubMed] [Google Scholar]
- 29.Hullin R, Singer-Lahat D, Freichel M, Biel M, Dascal N, Hofmann F, Flockerzi V. EMBO J. 1992;11:885–890. doi: 10.1002/j.1460-2075.1992.tb05126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudy B, Iverson LE. In: Methods in Enzymology. Abelson JN, Simon MI, editors. Vol. 207. Academic Press, Inc; San Diego: 1992. pp. 225–390. [Google Scholar]
- 31.Bezprozvanny I, Scheller RH, Tsien RW. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- 32.Wei X, Neely A, Olcese R, Lang W, Stefani E, Birnbaumer L. Receptors & Channels. 1996;4:205–215. [PubMed] [Google Scholar]
- 33.Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, Lee JH. Nature. 1998;391:896–900. doi: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- 34.Chuang RSI, Jaffe H, Cribbs LL, Perez-Reyes E, Swartz KJ. Nat Neurosci. 1998;1:668–674. doi: 10.1038/3669. [DOI] [PubMed] [Google Scholar]
- 35.Gurnett CA, De Waard M, Campbell KP. Neuron. 1996;16:431–440. doi: 10.1016/s0896-6273(00)80061-6. [DOI] [PubMed] [Google Scholar]
- 36.De Waard M, Campbell KP. J Physiol (Lond) 1995;485:619–634. doi: 10.1113/jphysiol.1995.sp020757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walker D, De Waard M. Trends Neurosci. 1998;21:148–154. doi: 10.1016/s0166-2236(97)01200-9. [DOI] [PubMed] [Google Scholar]
- 38.Lory P, Ophoff RA, Nahmias J. Hum Genet. 1997;100:149–150. doi: 10.1007/s004390050481. [DOI] [PubMed] [Google Scholar]
- 39.Nagase T, Ishikawa K, Miyajima N, Tanaka A, Kotani H, Nomura N, Ohara O. DNA Res. 1998;5:31–39. doi: 10.1093/dnares/5.1.31. [DOI] [PubMed] [Google Scholar]
- 40.Angelotti T, Hofmann F. FEBS Lett. 1996;397:331–337. doi: 10.1016/s0014-5793(96)01205-7. [DOI] [PubMed] [Google Scholar]
- 41.Welling A, Felbel J, Peper K, Hofmann F. Am J Physiol. 1992;262:L351–L359. doi: 10.1152/ajplung.1992.262.3.L351. [DOI] [PubMed] [Google Scholar]
- 42.Lehmann-Horn F, Jurkat-Rott K. Physiol Rev. 1999;79:1317–1372. doi: 10.1152/physrev.1999.79.4.1317. [DOI] [PubMed] [Google Scholar]
- 43.Burgess DL, Jones JM, Meisler MH, Noebels JL. Cell. 1997;88:385–392. doi: 10.1016/s0092-8674(00)81877-2. [DOI] [PubMed] [Google Scholar]
- 44.Barclay J, Kusumi K, Lander E, Perez-Reyes E, Frankel W, Gardiner M, Rees M. Epilepsia. 1999;40:137. [Google Scholar]
- 45.Meier H. Acta Neuropathol. 1968;11:15–28. doi: 10.1007/BF00692792. [DOI] [PubMed] [Google Scholar]
- 46.Noebels JL. In: Idiopathic Generalized Epilepsies: Clinical, Experimental, and Genetic Aspects. Malafosse A, Genton P, Hirsch E, Marescaux D, Broglin D, Bernasconi R, editors. John Libbey & Co. Ltd; London: 1994. pp. 215–225. [Google Scholar]
- 47.Sekido Y, Ahmadian M, Wistuba II, Latif F, Bader S, Wei MH, Duh FM, Gazdar AF, Lerman MI, Minna JD. Oncogene. 1998;16:3151–3157. doi: 10.1038/sj.onc.1201858. [DOI] [PubMed] [Google Scholar]
- 48.Sekido Y, Fong K, Minna J. Biochim Biophys Acta. 1998;1378:F21–F59. doi: 10.1016/s0304-419x(98)00010-9. [DOI] [PubMed] [Google Scholar]
- 49.Wistuba I, Behrens C, Milchgrub S, Bryant D, Hung J, Minna JD, Gazdar AF. Oncogene. 1999;18:643–650. doi: 10.1038/sj.onc.1202349. [DOI] [PubMed] [Google Scholar]
- 50.Takamori M. Intern Med. 1999;38:86–96. doi: 10.2169/internalmedicine.38.86. [DOI] [PubMed] [Google Scholar]
- 51.O’Neill JH, Murray NM, Newsom-Davis J. Brain. 1988;111:577–596. doi: 10.1093/brain/111.3.577. [DOI] [PubMed] [Google Scholar]
- 52.Lennon VA, Kryzer TJ, Griesmann GE, O’Suilleabhain PE, Windebank AJ, Woppmann A, Miljanich GP, Lambert EH. N Engl J Med. 1995;332:1467–1474. doi: 10.1056/NEJM199506013322203. [DOI] [PubMed] [Google Scholar]
- 53.Raymond C, Walker D, Bichet D, Iborra C, Martin-Moutot N, Seagar M, De Waard M. Neuroscience. 1999;90:269–277. doi: 10.1016/s0306-4522(98)00378-9. [DOI] [PubMed] [Google Scholar]
- 54.Voltz R, Carpentier AF, Rosenfeld MR, Posner JB, Dalmau J. Muscle Nerve. 1999;22:119–122. doi: 10.1002/(sici)1097-4598(199901)22:1<119::aid-mus19>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 55.Toyota M, Ho C, Ohe-Toyota M, Baylin SB, Issa JP. Cancer Res. 1999;59:4535–4541. [PubMed] [Google Scholar]
- 56.Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Adv Cancer Res. 1998;72:141–196. [PubMed] [Google Scholar]
- 57.Schmutte C, Jones PA. Biol Chem Hoppe-Seyler. 1998;379:377–88. doi: 10.1515/bchm.1998.379.4-5.377. [DOI] [PubMed] [Google Scholar]
- 58.Berridge MJ, Bootman MD, Lipp P. Nature. 1998;395:645–648. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- 59.Estacion M, Mordan LJ. Cell Signal. 1997;9:363–366. doi: 10.1016/s0898-6568(96)00184-2. [DOI] [PubMed] [Google Scholar]