

Abstract
Hantaviruses cause a persistent infection in reservoir hosts that is attributed to the upregulation of regulatory responses and downregulation of proinflammatory responses. To determine whether rat alveolar macrophages (AMs) and lung microvascular endothelial cells (LMVECs) support Seoul virus (SEOV) replication and contribute to the induction of an environment that polarizes CD4+ T cell differentiation toward a regulatory T (Treg) cell phenotype, cultured primary rat AMs and LMVECs were mock infected or infected with SEOV and analyzed for viral replication, cytokine and chemokine responses, and expression of cell surface markers that are related to T cell activation. Allogeneic CD4+ T cells were cocultured with SEOV-infected or mock-infected AMs or LMVECs and analyzed for helper T cell (i.e., Treg, Th17, Th1, and Th2) marker expression and Treg cell frequency. SEOV RNA and infectious particles in culture media were detected in both cell types, but at higher levels in LMVECs than in AMs postinfection. Expression of Ifnβ, Ccl5, and Cxcl10 and surface major histocompatibility complex class II (MHC-II) and MHC-I was not altered by SEOV infection in either cell type. SEOV infection significantly increased Tgfβ mRNA in AMs and the amount of programmed cell death 1 ligand 1 (PD-L1) in LMVECs. SEOV-infected LMVECs, but not AMs, induced a significant increase in Foxp3 expression and Treg cell frequency in allogeneic CD4+ T cells, which was virus replication and cell contact dependent. These data suggest that in addition to supporting viral replication, AMs and LMVECs play distinct roles in hantavirus persistence by creating a regulatory environment through increased Tgfβ, PD-L1, and Treg cell activity.
INTRODUCTION
Hantaviruses are negative-strand RNA viruses (family Bunyaviridae) that are maintained in the environment by causing persistent infection in rodent and insectivore hosts. Virus infection of humans results in hemorrhagic fever with renal syndrome (HFRS) or hantavirus cardiopulmonary syndrome (HCPS), both of which are caused by enhanced vascular permeability (34, 45). The absence of cytopathic effects in infected human endothelial cell cultures has led to the hypothesis that hantavirus pathogenesis in humans is due to immune-mediated mechanisms targeting the infected cells (58, 67, 71). Pathogenic hantavirus infection has been hypothesized to cause human vascular leakage by various mechanisms, including sensitizing endothelial cells to vascular endothelial growth factor (VEGF) and downregulating VE-cadherin (25, 69), recruiting quiescent platelets to infected endothelial cells (24), increasing endothelial cell surface ICAM-1 and major histocompatibility complex class I (MHC-I) levels that induce natural killer (NK) cell activation (5), or triggering viral specific CD8+ T cell responses that target infected endothelial cells (35). In addition to infecting endothelial cells, hantaviruses infect macrophages and dendritic cells (DCs) in the human host (48, 59, 62). Because the primary mode of transmission of hantaviruses to humans is through inhalation of aerosolized virus contained in rodent excreta, infection of alveolar macrophages (AMs) may contribute to viral dissemination in humans (33). Patients infected with hantaviruses have elevated levels of proinflammatory cytokines and frequencies of virus-specific CD8+ T cells but decreased regulatory T (Treg) cell activity and Treg cell numbers (14, 35, 44, 47, 49, 81). Concentrations of the regulatory cytokine transforming growth factor β1 (TGF-β1) are also reduced in HCPS patients (9) and in Syrian hamster models of HCPS (65).
In contrast to the severe diseases in humans, hantaviruses cause persistent infection in the absence of pathological disease in their reservoir hosts (15, 43). For example, in Seoul virus (SEOV)-infected Norway rats (the natural reservoir for SEOV), virus preferentially replicates in the lungs, with SEOV N protein identified in AMs and lung endothelial cells (18). Elevated regulatory responses, including the production of TGF-β1 and numbers of CD4+ CD25+ FoxP3+ Treg cells in the lungs, is associated with persistence of SEOV in Norway rats and Sin Nombre virus (SNV) in deer mice (18, 19, 68). In SEOV-infected Norway rats, the increased expression of Tgfβ occurs within 72 h of infection and precedes the induction of Treg cells in the lungs (18). The cellular source of the early production of TGF-β1 and whether this contributes to the induction of Treg cells in the lungs of infected rats are not known. In addition to the induction of regulatory responses, activity along the type I interferon (IFN) pathway and production of proinflammatory cytokines (e.g., interleukin 1β [IL-1β], IL-6, tumor necrosis factor alpha [TNF-α], and gamma interferon [IFN-γ]) remain at or below baseline throughout SEOV infection in the lungs of male rats (18, 27, 37). Conversely, in the spleen, proinflammatory and antiviral responses are elevated during acute SEOV infection (18). Infection of bone marrow-derived macrophages (BMDMs) with SEOV suppresses NF-κB-mediated inflammatory responses, including TNF-α, IL-6, and IL-10, and surface marker (i.e., MHC-II, CD80, and CD86) expression, suggesting SEOV infection suppresses the innate immune response in antigen-presenting cells (APCs) (2).
Virus-infected cells can induce Treg cells. Hepatitis C virus (HCV)-infected human hepatocytes induce activated CD4+ T cells to preferentially differentiate into Treg cells, which contribute to the maintenance of chronic HCV infection (26). Both macrophages and endothelial cells are capable of inducing naïve CD4+ T cells to differentiate into Treg cells (1, 38, 41, 72). Whether hantaviruses productively infect AMs or endothelial cells in reservoir hosts and whether infection of these cells creates a regulatory environment necessary for induction of Treg cells during persistent hantavirus infection have not been determined and formed the basis for the current studies.
MATERIALS AND METHODS
Animals and cell isolation.
Primary cells were isolated from Lewis rats, Brown Norway rats, or Sprague-Dawley rats, all of which are different inbred laboratory strains of Rattus norvegicus. Adult male Lewis rats (60 to 70 days of age) and Brown Norway rats (120 days of age) were purchased from Charles River Laboratories (Raleigh, NC) and maintained in pathogen-free facilities with a constant 14:10 light-dark cycle. AMs were isolated from Lewis rats by bronchial lavage as described previously (63) with modifications. Briefly, the rats were euthanized with CO2. The lungs and trachea were exposed, and an 18-gauge (18G) blunt needle (Small Parts Inc.) was fixed in the trachea with surgical threads. Cold phosphate-buffered saline (PBS) (5 ml) was slowly injected into the lungs using a 5-ml syringe and drawn out. The lavage step was repeated 10 times, and the fluid was pooled and centrifuged at 400 × g for 10 min at 4°C to pellet cells. Splenic CD4+ T cells were isolated from Brown Norway rats using a MagCellect rat CD4+ T cell isolation kit (R&D Systems). All procedures were performed in accordance with the guidelines of the Johns Hopkins Animal Care and Use Committee (protocol no. RA10H178).
LCM preparation.
For culturing of AMs, lung conditioned medium (LCM) was prepared using a previously described method with modifications (13). Freshly isolated lungs from Lewis rats were minced and collected after filtering through a 100-μm cell strainer. LCM was obtained by incubating minced lung pieces with complete-growth RPMI 1640 medium (10% fetal bovine serum [FBS], 2 mM l-glutamine, 1% penicillin/streptomycin) at a tissue/medium ratio of 1:4 (vol/vol) for 48 h. The LCM was filtered through a 0.2-μm filter and stored in aliquots at −80°C.
Rat LMVEC cultures.
Primary cultures of male Sprague-Dawley rat lung microvascular endothelial cells (LMVECs) (VEC Technologies, Rensselaer, NY) were grown in complete-growth RPMI 1640 medium (10% FBS, 2 mM l-glutamine, 1% penicillin/streptomycin) supplemented with a final concentration of 50 μg/ml endothelial cell growth supplement (ECGS) (BD Biosciences) and 1 μl/ml 2-mercaptoethanol (2-ME) (Invitrogen). Tissue culture flasks, plates, and transwell inserts for all LMVEC cultures were precoated with 50 μg/ml fibronectin (Sigma). Experiments were conducted between passages 3 and 9.
Virus infection and stimulation.
AMs or LMVECs were plated in 24-well tissue culture plates at 2 × 105 or 1 × 105 cells per well, respectively. The cells were mock infected or infected with 200 μl per well of diluted SEOV at a multiplicity of infection (MOI) of 0.05, 0.5, or 5 for 2 h. At the end of the incubation, the cells were washed with RPMI 1640 medium, and the infection medium was replaced with complete-growth RPMI 1640 medium supplemented with LCM for AMs or with ECGS and 2-ME for LMVECs. As specified for each experiment, cells and media were collected at 6 h or 1, 3, or 6 days postinfection (p.i.). On days 0, 2, and 5 p.i. (i.e., 6 h prior to collecting 6-h samples and 24 h prior to collecting day 1, 3, and 6 samples), recombinant rat IFN-γ (PeproTech), lipopolysaccharide (LPS) (Sigma), or poly(I·C) (pIC) (Invivogen) was added to designated wells at final concentrations of 100 U/ml, 100 ng/ml, and 1 μg/ml for AMs and 500 U/ml, 100 ng/ml, and 10 μg/ml for LMVECs, respectively. Following infection, all experiments were conducted at biosafety level 3 (BSL-3) using protocols approved by the Johns Hopkins Office of Health, Safety, and Environment (protocol no. P9902030113).
Allogeneic CD4+ T cell coculture.
For the AM/CD4+ T cell coculture and LMVEC/CD4+ T cell coculture experiments, AMs and LMVECs were plated in 24-well plates at 5 × 105 cells per well and 2 × 105 cells per well, respectively. The cells were mock infected, infected with SEOV at an MOI of 0.5 or 5, or infected with UV-inactivated SEOV at an MOI of 5. At the end of incubation, the cells were washed with RPMI medium, and the infection medium was replaced with complete-growth RPMI 1640 medium. Designated wells of AMs or LMVECs were also stimulated with LPS to induce a Th17 response. At 24 h p.i., the medium was removed, and CD4+ T cells were added to the AM culture at 5 × 106 cells per well and to the LMVEC culture at 2 × 106 cells per well in X-Vivo medium (Lonza). For controls, CD4+ T cells were either cultured alone or stimulated with 1 μg/ml plate-bound anti-rat CD3 (BD Pharmingen) and 5 μg/ml anti-rat CD28 (BD Pharmingen) in the presence or absence of 10 ng/ml recombinant TGF-β1 (R&D Systems). Cocultured cells were incubated for 4 days.
For LMVEC/CD4+ T cell coculture experiments using transwell inserts, LMVECs were plated in the lower chambers of transwell plates (24-well format) at 2 × 105 cells per well and were mock infected or infected with SEOV at an MOI of 5, as described above. On the same day as LMVEC infection, CD4+ T cells were stimulated with 1 μg/ml plate-bound anti-rat CD3 (BD Pharmingen) and 5 μg/ml anti-rat CD28 (BD Pharmingen) in X-Vivo 15 medium (Lonza). At 3 days after LMVEC infection and CD4+ T cell stimulation, the medium was removed from LMVEC cultures and replaced with 600 μl per well of X-Vivo 15 medium. Transwell inserts (6.5 mm with a 0.3-μm pore size; Thermo Scientific) were added to designated wells with or without LMVECs in the lower chamber. Activated CD4+ T cells were added at 5 × 105 cells per well in 100 μl X-Vivo 15 medium to the upper chambers of the transwell inserts to eliminate direct contact between CD4+ T cells and LMVECs in the lower chambers. As controls, activated CD4+ T cells in the upper chambers of the transwell inserts were cultured either without LMVECs present in the lower chambers or with recombinant TGF-β added to the lower chambers to a final concentration of 10 ng/ml. Cultures were incubated for 2 additional days.
Immune TCID50 assay.
SEOV-infected cell supernatants were titrated by an immune 50% tissue culture infective dose (TCID50) assay as described previously with modifications (2, 64). Vero E6 cells were plated in 96-well plates at 4 × 104 cells per well on the day before the assay. The supernatants were diluted in serial 10-fold dilutions in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) containing 2% FBS (Lonza). Vero E6 cells were infected with 100 μl of the diluted supernatants per well in sextuplicate, incubated for 12 days, and fixed with a cold solution of 95% ethanol-5% acetic acid. For immunostaining, cells were permeabilized with 0.1% Triton X-100; blocked with 3% bovine serum albumin (BSA) (Sigma-Aldrich) in PBS; and incubated with a rat anti-Andes virus (anti-ANDV) N antibody (a kind gift from Andrew Pekosz), which cross-reacts with SEOV N (2), diluted in blocking buffer (1:5,000). The cells were washed twice and incubated with a secondary horseradish peroxidase (HRP)-conjugated goat anti-rat antibody diluted in blocking buffer (1:5,000; Jackson ImmunoResearch). SEOV-positive wells were visualized by adding 3,3′,5,5′-tetramethylbenzidine (TMB) substrates (BD Biosciences), and virus titers were calculated using the Reed-Muench formula.
ELISA for TNF-α quantification.
Cell culture supernatants were exposed to UV for 3 min to inactivate virus for processing samples outside a BSL-3 facility (2). TNF-α was measured using enzyme-linked immunosorbent assay (ELISA) kits for rat TNF-α according to the manufacturer's protocol (R&D Systems).
RNA isolation and quantitative RT-PCR.
RNA was isolated using TRIzol (Invitrogen). Reverse transcription was carried out using the SuperScript III First Strand Synthesis System (Invitrogen) with SEOV S gene-specific primers for subsequent SEOV negative-strand RNA quantification or oligo(dT) for subsequent cellular mRNA quantification. For the samples in which 18S rRNA was measured by real-time PCR, random hexamer was used in the place of oligo(dT). Primers and probes for real-time PCR were either designed using Primer Express 2.0 software (Applied Biosystems) and purchased commercially (Invitrogen or Applied Biosystems) or purchased as predesigned assays (Applied Biosystems). Quantitative real-time (RT) PCR was performed in 96-well optical reaction plates using the StepOnePlus real-time PCR system (Applied Biosystems). For SEOV RNA quantification, absolute RNA copy numbers were calculated by including a set of standards composed of SR-11 S segment in plasmid pWRG7077 ranging from 2 to 2 × 106 copies/ml on each plate. For cellular mRNA quantification, serial dilutions of pools of selected samples were run on each plate to generate a standard curve, from which the expression of genes of interest was calculated. For AMs and LMVECs, host gene expression was normalized to that of Gapdh. Because Gapdh is a poor reference gene for T cell cultures (4), T cell gene expression was normalized to that of 18S rRNA.
Immunofluorescence.
AMs were grown on 4-well glass chamber slides (Nalge Nunc International Corp.) at 2 × 105 cells per well. LMVECs were grown on fibronectin-coated 6.5-mm transwell inserts (0.4-μm pore size; Corning Inc.) at 2.5 × 104 cells per transwell insert. AMs and LMVECs were infected with SEOV at an MOI of 0.5 or 5. At 3 and 6 days after infection, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with 5% normal goat serum (Thermo Scientific) for 1 h at room temperature. The cells were incubated with primary rat anti-ANDV N antibody (1:500) diluted in blocking buffer overnight, washed three times with PBS, and incubated for 1 h with Alexa Fluor 488-conjugated goat anti-rat and Alexa Fluor 594-conjugated goat anti-rabbit secondary antibodies (1:150; Invitrogen) diluted in blocking buffer. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma). A Nikon Eclipse 90i microscope and Volocity Imaging software (PerkinElmer) were used for image acquisition.
Flow cytometry.
Cells were collected and washed in cold fluorescence-activated cell sorter (FACS) buffer (PBS containing 0.5% BSA and 0.1% sodium azide) and blocked with FcR (BD Pharmingen). AMs were incubated with combinations of the following anti-rat antibodies or antibodies that cross-react with rat: MHC-I–phycoerythrin (PE) (clone OX-27; AbD Serotec), MHC-II–peridinin chlorophyll protein (PerCP) (clone OX-6; BD Pharmingen), and CD80-allophycocyanin (APC) (clone 3H5; Invitrogen). LMVECs were incubated with anti-rat MHC-I–PE (clone OX-27; AbD Serotec), MHC-II–PerCP (clone OX-6; BD Pharmingen), and ICAM-1–PE (clone 1A29; AbD Serotec). For the coculture experiments, T cells were incubated with anti-rat CD4-APC (Clone OX-35; BD Pharmingen). AMs and LMVECs were fixed in 2% paraformaldehyde for 30 min, which is sufficient for inactivating virus for processing outside a BSL-3 facility (39). AMs were permeabilized with PBS containing 0.1% Triton X-100. Intracellular CD68 was stained with anti-rat CD68-fluorescein isothiocyanate (FITC) (clone ED1; AbD Serotec) or CD68-PE (clone ED1; AbD Serotec). T cells were fixed and permeabilized using the FoxP3 staining buffer set (eBioscience) according to the manufacturer's protocol and incubated with a FoxP3-FITC antibody (clone FJK-16S; eBioscience). Cells were sorted using a BD FACSCalibur cytometer (BD Biosciences) and the CellQuest Pro software (BD Biosciences). Data were analyzed using FlowJo software (TreeStar Inc., Ashland, OR).
SDS-PAGE and Western blotting.
Mock-infected or SEOV-infected LMVECs in 24-well plates were lysed in 100 μl/well of M-PER mammalian protein extraction reagent (Thermo Scientific) supplemented with 1% (vol/vol) protease inhibitor cocktail (Sigma). Cell lysates were separated on 10% (wt/vol) SDS polyacrylamide gels and electrophoretically blotted onto polyvinylidene difluoride (PVDF) membranes (Invitrogen) according to standard procedures. The membranes were blocked with 5% nonfat milk for 1 h at room temperature and treated with rabbit anti-PD-L1 (Santa Cruz Biotechnology) diluted in 5% BSA (1:500) overnight at 4°C. After washing with phosphate-buffered saline with 0.1% Tween 20 (T-PBS), the membranes were incubated with an HRP-conjugated goat anti-rabbit secondary antibody (Santa Cruz Biotechnology) in 5% nonfat milk for 1 h at room temperature. Specific protein bands were visualized using SuperSignal West Pico Chemiluminescent Substrate and exposed to autoradiography films (Denville Scientific Inc.). For the detection of β-actin, membranes that had been blotted for PD-L1 were stripped with a stripping buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, and 100 mM β-mercaptoethanol) for 20 min, blocked with 5% milk, and reblotted with rabbit anti-β-actin (Santa Cruz Biotechnology; 1:200) in combination with a horseradish peroxidase-conjugated goat anti-rabbit secondary antibody, following the same procedures as for PD-L1 staining.
Statistical analyses.
One-way or two-way analyses of variance (ANOVAs) were used to analyze group differences in quantitative data. Significant interactions were further analyzed using Tukey pairwise comparisons. Comparison of SEOV TCID50 data was done by Student t tests. For all statistical tests, mean differences were considered significant if P was <0.05.
RESULTS
SEOV productively infects rat AMs and LMVECs.
To investigate whether AMs and LMVECs from rats support SEOV replication, cells were infected with SEOV, and in both cell types, there was a time- and dose-dependent increase in cell-associated negative-sense SEOV RNA, in which the largest amount of viral RNA was detected in cells infected with SEOV at an MOI of 5 at 6 days p.i. (Fig. 1A and B) (P < 0.001). In SEOV-infected AM cultures, virus antigen (N protein)-positive cells were detected and increased over time at all MOIs (from approximately 25% to 50% in AMs infected with SEOV at an MOI of 0.5 and from 32% to 93% in cells infected with SEOV at an MOI of 5) (Fig. 1C). In SEOV-infected LMVEC cultures, the majority of cells in the infected cultures were virus antigen positive at 3 days (approximately 90% and 98% in LMVECs infected with SEOV at an MOI of 0.5 or 5, respectively) and 6 days p.i. (97% and 98% in cells infected with SEOV at an MOI of 0.5 or 5, respectively). (Fig. 1D). Infectious SEOV particles were detected in the supernatants from AMs and LMVECs at 6 days p.i., but not 1 day p.i., with titers being higher in LMVEC than AM cultures (Fig. 1E and F) (P < 0.001). These data demonstrate that both rat AMs and LMVECs are susceptible to SEOV infection, the virus can complete its life cycle in these cells, and levels of infection are greater in LMVECs than than in AMs.
Fig 1.

SEOV productively infects AMs and LMVECs from Norway rats. AMs and LMVECs were inoculated with medium alone or with medium containing SEOV. Cells and supernatants were collected at the indicated time points. (A and B) Cell-associated negative-sense SEOV RNA in AMs (A) and LMVECs (B) was quantified by real-time PCR. The data are expressed as means ± standard errors of the mean (SEM) of three independent experiments. d, days. (C and D) SEOV N protein in AMs (C) and LMVECs (D) was identified with an antibody that cross-reacts with SEOV N protein (green), and cell nuclei were counterstained with DAPI (blue). The images were acquired using a 40× objective lens. Bars, 50 μm. (E and F) At 1 and 6 days p.i., supernatants from AMs (E) and LMVECs (F) infected with SEOV at an MOI of 5 were collected, and infectious-virus titers were quantified using an immune TCID50 assay. The dashed lines represent the lower limit of detection of the assay. The data are expressed as means and SEM of three independent experiments. The asterisks indicate significant differences relative to day 1 SEOV TCID50 titers; P < 0.001; Student's t test.
SEOV infection does not induce antiviral or proinflammatory responses in either rat AMs or LMVECs.
We next examined whether SEOV infection induces innate immune responses in these cell types. Infection at either low or high MOIs with SEOV did not induce the expression of Ifnβ, Ccl5, or Cxcl10 in AMs (Fig. 2A to C) or LMVECs (Fig. 2E to G) or the secretion of TNF-α by AMs (Fig. 2D). In contrast, both AMs and LMVECs expressed high levels of proinflammatory and antiviral cytokines and chemokines in response to inflammatory stimuli, including IFN-γ, LPS, or pIC treatment (P < 0.001 in each case), indicating that these cells are capable of mounting proinflammatory and antiviral responses but that SEOV infection does not trigger these responses in either cell type.
Fig 2.

SEOV infection does not induce antiviral or proinflammatory responses in AMs or LMVECs. AMs and LMVECs were inoculated with medium alone or medium containing SEOV. The positive controls included cells stimulated with pIC, LPS in combination with IFN-γ (AMs), or LPS (LMVECs) for 24 h prior to sample collection. Ifnβ, Ccl5, and Cxcl10 mRNA expression in AMs (A, B, and C) and LMVECs (E, F, and G) was quantified by real-time PCR and is expressed as relative gene expression (RGE) compared to mock-infected cells at 1 day (AMs) or 6 h (LMVECs). (D) TNF-α concentrations in AM supernatants were measured by ELISA. All data are expressed as means ± SEM of three independent experiments. The asterisks indicate significant differences relative to mock-infected cells at the corresponding time point; P < 0.05; 2-way ANOVAs.
SEOV infection induces Tgfβ expression but does not alter other T cell-related markers in rat AMs.
To determine whether SEOV infection alters the ability of rat AMs to induce T cell activation, the expression of T cell activation-related factors was measured at 1, 3, and 6 days p.i. with SEOV at low and high MOIs and compared with mock-infected cells and cells treated with inflammatory stimuli. In AMs, SEOV infection at an MOI of 0.5 significantly induced the expression of Tgfβ by 6 days p.i. (Fig. 3A) (P = 0.005), whereas stimulation with LPS/IFN-γ downregulated Tgfβ expression 3 days p.i. (Fig. 3A) (P = 0.024) compared with mock-treated cells. The surface expression of MHC-II on SEOV-infected AMs was comparable to that on mock-infected cells and was significantly lower than the expression on cells treated with inflammatory stimuli (Fig. 3B) (P < 0.05 in each case). The surface expression of MHC-I was not induced by either SEOV infection or LPS/IFN-γ stimulation in AMs (Fig. 3C). The surface expression of the costimulatory molecule CD80 on SEOV-infected AMs was also comparable to that of mock-infected cells and was significantly lower than the expression on cells treated with inflammatory stimuli at 3 days p.i. but significantly higher than the expression on cells treated with inflammatory stimuli at 6 days p.i. (Fig. 3D) (P < 0.05).
Fig 3.
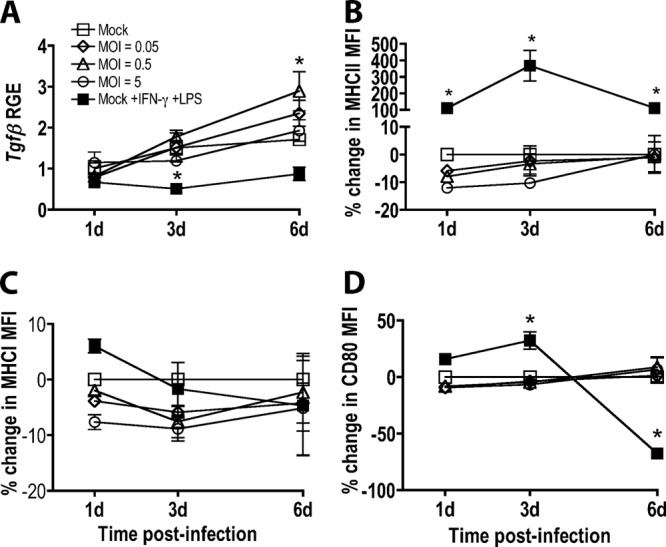
SEOV infection induces Tgfβ, but not other T cell-related marker expression, in AMs. AMs were inoculated with medium alone or medium containing SEOV. Controls included AMs stimulated with a combination of recombinant IFN-γ and LPS for 24 h prior to sample collection. (A) Tgfβ expression in AMs was quantified by real-time PCR and is expressed as RGE compared to mock-infected cells at 1 day. (B, C, and D) Cell surface MHC-II, MHC-I, and CD80 expression was quantified by flow cytometry and expressed as percent changes in median fluorescence intensity (MFI) compared to mock-infected cells at the corresponding time point. All data are expressed as means ± SEM of three independent experiments. The asterisks indicate significant differences relative to mock-infected cells at the corresponding time points; P < 0.05; 2-way ANOVAs.
SEOV infection does not increase the ability of rat AMs to induce Treg activity.
To determine whether SEOV infection of rat AMs upregulates Treg activity, AMs were mock infected or infected with SEOV at an MOI of 0.5 or 5 for 24 h and then cocultured with allogeneic CD4+ T cells for 4 days. Allogeneic CD4+ T cells cocultured with mock-infected AMs significantly upregulated Foxp3 expression compared with CD4+ T cells incubated alone (Fig. 4A) (P = 0.031), which was mitigated in T cells cocultured with SEOV-infected cells. To evaluate whether AMs induce other subsets of CD4+ T cells, the expression of genes associated with Th1, Th2, and Th17 cells was examined. The expression of Il17, Tbet, Ifnγ, Il4, and Il10 was not altered in CD4+ T cells cultured with either SEOV-infected or uninfected AMs, which were each significantly lower than the expression of these genes in CD4+ T cells stimulated with anti-CD3/CD28 (Fig. 4B to F) (P < 0.001 in each case). FoxP3+ Treg cell frequencies were significantly increased in CD4+ T cells cocultured with AMs, regardless of infection status, compared to CD4+ T cells cultured alone (Fig. 4G and H) (P < 0.001). AMs, in general, enhance frequencies of Treg cells in culture irrespective of SEOV infection.
Fig 4.

AMs induce a Treg cell phenotype in allogeneic CD4+ T cells regardless of infection status. (A to F) AMs from Lewis rats were inoculated with medium alone or with medium containing SEOV. At 1 day p.i., splenic CD4+ T cells from Brown Norway rats were added to AM cultures at a AM/T cell ratio of 1:10. After 4 days of coculture, nonadherent cells were collected, and the expression of Foxp3, Il17, Tbet, Ifnγ, Il4, and Il10 was quantified by real-time PCR and is expressed as RGE compared to CD4+ T cells cultured alone. The dashed lines represent the gene expression level of CD4+ T cells cultured alone. Data are expressed as means and SEM from three independent experiments. (G) The frequency of CD4+ FoxP3+ Treg cells among the cocultured cells was analyzed by flow cytometry. (H) The percent change in Treg cell frequencies among CD4+ T cells cultured with AMs relative to CD4+ T cells cultured alone from three independent experiments is expressed as means and SEM. The asterisks indicate significant differences relative to CD4+ T cells cultured alone; P < 0.05; one-way ANOVA.
SEOV infection selectively induces PD-L1 in rat LMVECs.
To determine whether SEOV infection alters the ability of rat LMVECs to present antigen and activate T cells, T cell-related factors, with particular attention devoted to the factors associated with endothelial cell induction of Treg cells (i.e., ICAM-1 and PD-L1) (41), were measured at 1, 3, and 6 days p.i. with SEOV at low and high MOIs and compared with mock-infected cells and cells treated with inflammatory stimuli. SEOV infection did not induce Tgfβ expression in rat LMVECs at any MOI, nor did IFN-γ stimulation alter Tgfβ expression in rat LMVECs (Fig. 5A). The surface expression of MHC-II, MHC-I, and ICAM-1 on SEOV-infected LMVECs was comparable to that on mock-infected cells and was significantly lower than the expression on cells treated with inflammatory stimuli (Fig. 5B to D) (P < 0.05 in each case). SEOV infection induced a dose-dependent increase in PD-L1 protein expression in LMVECs compared with mock-infected cells (Fig. 5E).
Fig 5.
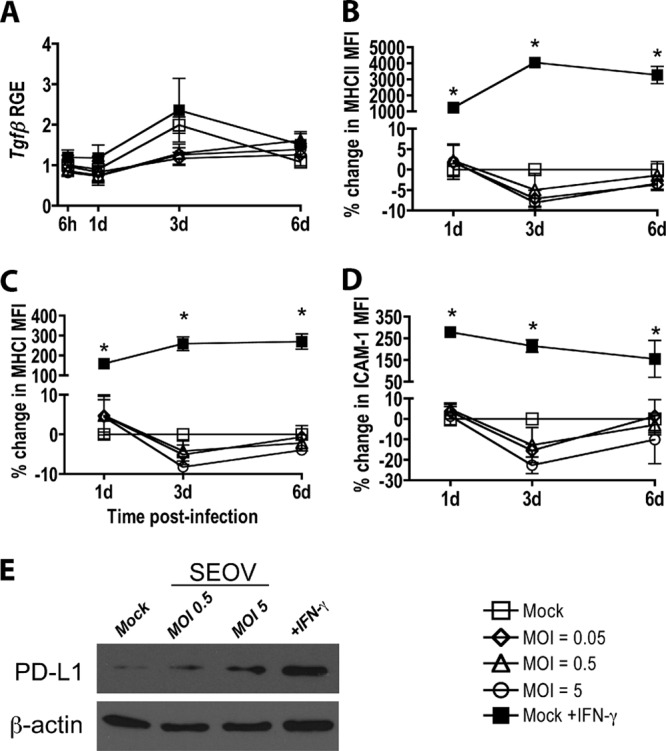
SEOV infection induces total PD-L1 protein expression but does not alter the expression of other T cell activation markers in rat LMVECs. LMVECs were inoculated with medium alone or medium containing SEOV. Controls included LMVECs stimulated with IFN-γ for 24 h prior to sample collection. (A) Tgfβ expression in LMVECs was quantified by real-time PCR and is expressed as RGE compared to mock-infected cells at 6 h. (B, C, and D) Cell surface MHC-II, MHC-I, and ICAM-1 expression was quantified by flow cytometry and expressed as percent changes in MFI compared to mock-infected cells at the corresponding time point. All data are expressed as means ± SEM of three independent experiments. The asterisks indicate significant differences relative to mock-infected cells at the corresponding time points; P < 0.05; 2-way ANOVAs. (E) PD-L1 expression was measured in mock-infected, SEOV-infected, and IFN-γ-treated LMVECs that were lysed at 3 days p.i., and PD-L1 and β-actin expression in cell lysates was detected by Western blotting. The data are representative of three independent experiments.
SEOV-infected rat LMVECs induce Treg cell activity.
To determine whether SEOV infection of rat LMVECs upregulates Treg cell activity, LMVECs were mock infected or infected with SEOV at an MOI of 0.5 or 5 for 24 h and then cocultured with allogeneic CD4+ T cells for 4 days. Coculture of allogeneic CD4+ T cells with SEOV-infected LMVECs significantly increased the expression of Foxp3 compared with CD4+ T cells cultured alone or with mock-infected LMVECs (Fig. 6A) (P < 0.05). Conversely, the expression of genes associated with Th17, Th1, and Th2 activity was not altered in CD4+ T cells cultured with either SEOV-infected or uninfected LMVECs and was significantly lower than the expression of these genes in CD4+ T cells stimulated with anti-CD3/CD28 (Fig. 6B to F) (P < 0.001 in each case). Allogeneic CD4+ T cells cocultured with SEOV-infected LMVECs had significantly higher FoxP3+ Treg cell frequencies than CD4+ T cells cultured alone or with mock-infected LMVECs (Fig. 6G and H) (P < 0.001).
Fig 6.

SEOV infection of LMVECs increases Foxp3 expression and Treg cell frequency in allogeneic CD4+ T cells. (A to F) LMVECs from Sprague-Dawley rats were inoculated with medium alone or with medium containing SEOV. At 1 day p.i., splenic CD4+ T cells from Brown Norway rats were added to LMVEC cultures at a LMVEC/T cell ratio of 1:10. After 4 days of coculture, nonadherent cells were collected, and the expression of Foxp3, Il17, Tbet, Ifnγ, Il4, and Il10 was quantified by real-time PCR and is expressed as RGE compared to CD4+ T cells cultured alone. The data are expressed as means and SEM from three independent experiments. The dashed lines represent the gene expression level of CD4+ T cells cultured alone. (G) The frequency of CD4+ FoxP3+ Treg cells among the cocultured cells was analyzed by flow cytometry. (H) The percentage change in Treg cell frequencies among CD4+ T cells cultured with LMVECs relative to CD4+ T cells cultured alone from three independent experiments are expressed as means and SEM. *, significant difference relative to CD4+ T cells cultured alone; #, significant difference relative to CD4+ T cells cocultured with mock-infected LMVECs; P < 0.05; one-way ANOVA.
The induction of Treg cells by SEOV-infected rat LMVECs is dependent on virus replication and cell contact.
To determine whether Treg cell induction by SEOV-infected LMVECs is stimulated by viral particles alone or is dependent on virus replication, LMVECs were mock infected, infected with SEOV at an MOI of 5, or infected with UV-inactivated SEOV at an MOI of 5 for 24 h and cocultured with allogeneic CD4+ T cells for 4 days. Allogeneic CD4+ T cells cocultured with SEOV-infected LMVECs showed greater expression of Foxp3 and FoxP3+ Treg cell frequencies than CD4+ T cells cultured alone or with mock-infected LMVECs (Fig. 7A and B) (P < 0.01 in each case). In contrast, allogeneic CD4+ T cells cultured with UV-inactivated SEOV-infected LMVECs had levels of Foxp3 expression and FoxP3+ Treg cell frequencies that were comparable to those of CD4+ T cells cultured alone or with mock-infected LMVECs (Fig. 7A and B).
Fig 7.
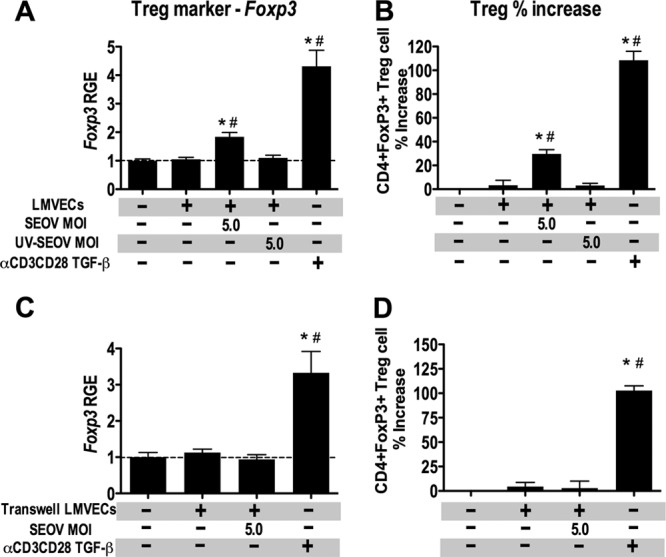
Induction of Treg cells in allogeneic CD4+ T cell cultures by SEOV-infected LMVECs requires SEOV replication and cell contact. (A) LMVECs from Sprague-Dawley rats were inoculated with medium alone, medium containing SEOV, or medium containing UV-inactivated SEOV. At 1 day p.i., splenic CD4+ T cells from Brown Norway rats were added to LMVEC cultures at a LMVEC/T cell ratio of 1:10. After 4 days of coculture, nonadherent cells were collected, and the expression of Foxp3 was quantified by real-time PCR and is expressed as RGE compared to CD4+ T cells cultured alone. The data are expressed as means and SEM from three independent experiments. (B) The frequency of CD4+ FoxP3+ Treg cells among the cocultured cells was analyzed by flow cytometry. The percent change in Treg cell frequencies among CD4+ T cells cultured with LMVECs relative to CD4+ T cells cultured alone from three independent experiments is expressed as means and SEM. (C) For transwell coculture experiments, LMVECs from Sprague-Dawley rats were cultured in the bottom chambers of transwells and inoculated with medium alone or with medium containing SEOV. Splenic CD4+ T cells from Brown Norway rats were stimulated with a combination of plate-bound anti-CD3 and soluble anti-CD28. At 3 days p.i., stimulated CD4+ T cells were added to the upper chambers of the transwells to eliminate LMVEC-CD4+ T cell contact. After 2 days of coculture, CD4+ T cells in the upper chambers were collected, and the expression of Foxp3 was quantified by real-time PCR and expressed as RGE compared to CD4+ T cells cultured alone in the upper chambers without LMVECs in the lower chamber. The data are expressed as means and SEM from three independent experiments. (D) The frequency of CD4+ FoxP3+ Treg cells among the CD4+ T cells in the upper chamber was analyzed by flow cytometry. The percent change in Treg cell frequencies relative to CD4+ T cells cultured alone from three independent experiments is expressed as means and SEM. *, significant difference relative to CD4+ T cells cultured alone; #, significant difference relative to CD4+ T cells cocultured with mock-infected LMVECs; P < 0.05; one-way ANOVA. The dashed lines represent the gene expression level of CD4+ T cells cultured alone.
To determine if increased expression of PD-L1 on SEOV-infected LMVECs contributes to the induction of Treg cells, we sought to neutralize PD-L1 on the surfaces of SEOV-infected LMVECs. The commercially available anti-rat PD-L1 antibody did not bind to undenatured PD-L1 on the surfaces of live LMVECs (data not shown), indicating that the available antibody would not work as a neutralizing antibody for directly testing whether upregulation of PD-L1 on SEOV-infected LMVECs induces Treg cells. We instead designed experiments to determine whether the induction of Treg cells by SEOV-infected LMVECs is cell contact dependent or attributed to a soluble factor(s) secreted by SEOV-infected LMVECs. Allogeneic CD4+ T cells were stimulated prior to culture in transwell upper chambers with mock or SEOV-infected (MOI, 5) LMVECs in the lower chambers to eliminate contact between CD4+ T cells and LMVECs. Coculture of stimulated allogeneic CD4+ T cells with SEOV-infected LMVECs in transwells did not increase the expression of Foxp3 compared with stimulated CD4+ T cells cultured alone or with mock-infected LMVECs in transwells, all of which were significantly lower than the Foxp3 expression of stimulated CD4+ T cells cultured in transwell inserts with TGF-β added to the lower chambers (Fig. 7C) (P < 0.001). Stimulated allogeneic CD4+ T cells cocultured with SEOV-infected LMVECs or mock-infected LMVECs in transwells also had comparable FoxP3+ Treg cell frequencies, which were all significantly lower than Treg cell frequencies of stimulated CD4+ T cells cultured with TGF-β in the lower chambers (Fig. 7D) (P < 0.001). These data indicate that induction of Treg cells by SEOV-infected LMVECs is virus replication and cell contact dependent.
DISCUSSION
Hantaviruses infect macrophages and endothelial cells in both reservoir hosts and humans (11, 43, 53, 78). The severity of hantaviral diseases in humans (i.e., HFRS and HCPS) is partially mediated by increased endothelium permeability (53, 58, 73). In contrast, reservoir hosts infected with species-specific hantaviruses exhibit no overt pathological disease, which is attributed to virus-induced upregulation of regulatory responses and inhibition of proinflammatory responses (17–19, 68); how this occurs, however, has remained elusive. The data from the present study illustrate that both AMs and endothelial cells from a rodent reservoir support hantavirus replication and contribute to the induction of an environment that polarizes CD4+ T cell differentiation toward a Treg cell phenotype.
In SEOV-infected Norway rats, the lungs, more than any other organ examined (e.g., spleen or kidney), support elevated viral replication and regulatory responses, including Tgfβ expression during the acute phase of infection followed by upregulation of Treg cell activity during the persistent phase of infection (17–19). SEOV antigen is localized to AMs and lung endothelial cells of infected rats (18). We have previously shown that rat BMDMs and BM-derived DCs support SEOV replication, with macrophages supporting significantly greater levels of SEOV replication than DCs (2). SEOV infection does not trigger antiviral or proinflammatory responses but suppresses the ability of these BM-derived APCs to respond appropriately to inflammatory stimuli (2). Embryonic fibroblasts from bank voles also do not increase Ifnβ or Mx2 mRNA expression when infected with the species-specific hantavirus Puumala virus (PUUV) (70). In contrast to reservoir host-derived cells, pathogenic hantavirus infection of human endothelial cells or DCs induces antiviral cytokine (i.e., Ifnβ) and chemokine (i.e., Ccl5 and Cxcl10) expression and increases cell surface activation markers (i.e., MHC-I and ICAM-1) that contribute to the enhanced proinflammatory response and CD8+ T cell activity in HFRS and HCPS patients (30, 40, 62, 71). Collectively, these studies suggest that hantaviruses have evolved mechanisms to evade innate immune surveillance in their reservoir hosts, but not in humans, at a cellular level.
Rat AMs and, to a greater extent, LMVECs play fundamental roles in supporting SEOV replication, with the virus completing its life cycle in both cell types. SEOV infection of rat LMVECs results in greater production of virus than with AMs or even Vero E6 cells (data not shown), a type I IFN-deficient cell line that is traditionally used to propagate hantaviruses (20, 36, 60). These data suggest that both rat AMs and LMVECs contribute to SEOV persistence in vivo. How hantaviruses replicate in reservoir host cells without inducing cellular immune responses that promote virus clearance has remained unspecified.
SEOV infection of rat AMs and LMVECs creates an environment that polarizes CD4+ T cell differentiation toward a Treg cell phenotype and provides one mechanism by which hantaviruses escape clearance and cause persistent infections in rodent reservoirs. The absence of innate proinflammatory cytokine and chemokine responses in rat AMs or LMVECs following SEOV infection minimizes the induction of Th1, Th2, and Th17 cells and shifts the CD4+ T cell population in the lungs toward a regulatory phenotype. In the lungs of SEOV-infected rats, the frequency of Treg cells significantly increases after the SEOV RNA load peaks during the acute phase of infection, suggesting the induction of Treg cells during the persistent phase of infection might be dependent on SEOV replication in cells, including AMs and lung endothelial cells, during the acute phase of infection (19). Macrophages and endothelial cells can induce CD4+ T cells into Treg cells following appropriate cell (e.g., MHC-II engagement and costimulation and PD-1–PD-L1 pathway activation) or cytokine (e.g., presence of TGF-β) stimulation (38, 41, 72). Coculturing of naïve CD4+ T cells with infected LMVECs induced CD4+ T cells to differentiate into Treg cells. SEOV-infected LMVECs induced an approximately 25% increase in Treg cell frequency in CD4+ T cell cultures, which is comparable in magnitude to the increase observed in SEOV-infected rats (19), as well as in other virus-host cell systems (12, 79, 80). The induction of Treg cells by SEOV-infected LMVECs is dependent on virus replication and not merely on the presence of virus particles. These data suggest that hantavirus replication in LMVECs contributes to the increased Treg frequency in the lungs of infected rats (19) and possibly increases Foxp3 expression in T cells from persistently Sin Nombre virus-infected deer mice (68).
Treg cell induction by endothelial cells is PD-L1 dependent (41, 72). SEOV infection of LMVECs increased PD-L1 protein expression. PD-L1 alone (i.e., PD-L1-coated beads) can induce Treg cells in vitro (22). The cell contact dependence of Treg cell induction by SEOV-infected LMVECs indicates that Treg cell induction is mediated by a LMVEC surface protein(s) rather than a LMVEC-secreted soluble factor(s). Taken together, our data suggest that PD-L1 might be one mediator of increased Treg cell activity and the absence of endothelium damage in reservoir hosts infected with species-specific hantaviruses.
The PD-1–PD-L1 pathway regulates the balance between stimulatory and inhibitory signals to maintain peripheral tolerance (23). PD-1 is expressed by activated lymphocytes and myeloid cells, and PD-L1 is expressed by IFN-γ-stimulated APCs and nonlymphoid cells in many tissues, such as the heart, lung, liver, and spleen (23, 32, 50). The engagement of PD-1 with PD-L1 inhibits lymphocyte proliferation and cytokine secretion (23). Because of its inhibitory properties, the PD-1–PD-L1 pathway is exploited by many viruses to achieve chronic infection (50, 51). For example, activation of the PD-1–PD-L1 pathway plays an important role in chronic lymphocytic choriomeningitis virus, human immunodeficiency virus (HIV), hepatitis B virus, and HCV infections (3, 6–8, 46, 50, 57, 61, 75). Whether the enhancement of PL-L1 in SEOV-infected endothelial cells is a significant factor in hantavirus persistence in vivo requires consideration. Our data suggest that the increased endothelial PD-L1 level during SEOV infection might mediate both the induction of Treg cells and possibly the inhibition of CD8+ T cell activity to cause persistent infection in reservoir hosts. It is, however, possible that other proteins in addition to PD-L1 are involved.
AMs appear to play a supportive role in the maintenance of SEOV replication and induction of an anti-inflammatory pulmonary environment. Although SEOV-infected rat AMs do not increase Treg cell frequency in CD4+ T cell cultures, these cells might be the cellular source of the early increase in Tgfβ expression in the lungs of SEOV-infected rats (18). AMs play a critical role at the interface between detection of foreign antigens, induction of immune responses, and clearance of pathogens from the lungs (16, 21, 42). AMs have evolved the ability to clear foreign antigens without extensive activation of inflammatory responses (77). The relatively long half-life (30 days) of AMs and their restrained response to infection make them an ideal niche for pathogens, such as HIV and Mycobacterium tuberculosis, that cause persistent infections in humans (28, 52, 54, 66). We hypothesize that hantaviruses may have evolved mechanisms to exploit AMs, as these cells support productive SEOV replication and are able to induce Treg cells, regardless of their infection status. SEOV-infected AMs caused a slightly mitigated induction of Treg cells that is likely attributable to SEOV-induced inhibition of MHC-II expression on APCs (2).
The goal of the current study was to investigate the possible mechanism(s) underlying the increase in Treg cell frequencies in the lungs of SEOV-infected rats (19) using an in vitro approach, because identifying cell-specific induction of Treg cells in rat lungs is currently not feasible. To achieve this goal, a mixed lymphocyte reaction assay was used to determine whether infected AMs, LMVECs, or both cell types could induce naïve T cells toward a Treg phenotype (i.e., expression of FoxP3). Although using SEOV-specific T cells might be more physiologically relevant, isolation of SEOV-specific T cells from infected rats would not serve the purpose of the current study, which was to establish the cell type involved in the induction of Treg cells from a naïve CD4+ T cell population. Allogeneic T cell cocultures have been used previously to systematically evaluate APC induction of T cell responses during virus infection (55, 56, 76). This mixed lymphocyte reaction or allogeneic T cell coculture system also has been used to study the induction of Treg cells by activated murine vascular endothelial cells (41) and human endothelial cells (72). The allogeneic coculture system enabled identification of SEOV-infected endothelial cells as a cell type responsible for Treg cell induction in rats. A limitation of the current study is that, using the allogeneic coculture system as a surrogate, it is not clear whether the induced Treg cells are SEOV specific; determining this remains a goal of future studies.
Hantavirus infection and replication in AMs and LMVECs likely contributes to making the lungs a site of persistent infection in natural reservoir hosts (10, 11, 19, 29, 31, 68). Infection of these cells from rats results in production of SEOV particles, inhibition of proinflammatory responses, induction of regulatory responses, and induction of Treg cell activity. Future studies should identify the viral proteins and cellular pathways mediating the way in which SEOV induces PD-L1 expression in rat LMVECs and Tgfβ expression in AMs. A possible effect of hantavirus infection on human endothelial cell PD-L1 expression has been proposed but not yet investigated (74). We hypothesize that differences in PD-L1 expression during hantavirus infection might underlie the drastic difference in hantavirus disease outcomes between humans and reservoir hosts.
ACKNOWLEDGMENTS
This work was supported by the W. Harry Feinstone endowment and SWHR Medtronic Award.
We thank Alan Scott, Andrew Pekosz, and members of the Virology Laboratory meeting group for helpful discussions about these data.
Footnotes
Published ahead of print 22 August 2012
REFERENCES
- 1. Alard P, Clark SL, Kosiewicz MM. 2004. Mechanisms of tolerance induced by TGF beta-treated APC: CD4 regulatory T cells prevent the induction of the immune response possibly through a mechanism involving TGF beta. Eur. J. Immunol. 34:1021–1030 [DOI] [PubMed] [Google Scholar]
- 2. Au RY, Jedlicka AE, Li W, Pekosz A, Klein SL. 2010. Seoul virus suppresses NF-kappaB-mediated inflammatory responses of antigen presenting cells from Norway rats. Virology 400:115–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barber DL, et al. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–687 [DOI] [PubMed] [Google Scholar]
- 4. Bas A, Forsberg G, Hammarstrom S, Hammarstrom ML. 2004. Utility of the housekeeping genes 18S rRNA, beta-actin and glyceraldehyde-3-phosphate-dehydrogenase for normalization in real-time quantitative reverse transcriptase-polymerase chain reaction analysis of gene expression in human T lymphocytes. Scand. J. Immunol. 59:566–573 [DOI] [PubMed] [Google Scholar]
- 5. Bjorkstrom NK, et al. 2011. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J. Exp. Med. 208:13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blackburn SD, et al. 2010. Tissue-specific differences in PD-1 and PD-L1 expression during chronic viral infection: implications for CD8 T-cell exhaustion. J. Virol. 84:2078–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blattman JN, Wherry EJ, Ha SJ, van der Most RG, Ahmed R. 2009. Impact of epitope escape on PD-1 expression and CD8 T-cell exhaustion during chronic infection. J. Virol. 83:4386–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boni C, et al. 2007. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J. Virol. 81:4215–4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Borges AA, et al. 2008. Role of mixed Th1 and Th2 serum cytokines on pathogenesis and prognosis of hantavirus pulmonary syndrome. Microbes Infect. 10:1150–1157 [DOI] [PubMed] [Google Scholar]
- 10. Botten J, et al. 2000. Experimental infection model for Sin Nombre hantavirus in the deer mouse (Peromyscus maniculatus). Proc. Natl. Acad. Sci. U. S. A. 97:10578–10583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Botten J, et al. 2003. Persistent Sin Nombre virus infection in the deer mouse (Peromyscus maniculatus) model: sites of replication and strand-specific expression. J. Virol. 77:1540–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cabrera R, et al. 2004. An immunomodulatory role for CD4(+)CD25(+) regulatory T lymphocytes in hepatitis C virus infection. Hepatology 40:1062–1071 [DOI] [PubMed] [Google Scholar]
- 13. Cavanaugh PG, Nicolson GL. 1989. Purification and some properties of a lung-derived growth factor that differentially stimulates the growth of tumor cells metastatic to the lung. Cancer Res. 49:3928–3933 [PubMed] [Google Scholar]
- 14. Chen LB, Yang WS. 1990. Abnormalities of T cell immunoregulation in hemorrhagic fever with renal syndrome. J. Infect. Dis. 161:1016–1019 [DOI] [PubMed] [Google Scholar]
- 15. Childs JE, Glass GE, Korch GW, LeDuc JW. 1989. Effects of hantaviral infection on survival, growth and fertility in wild rat (Rattus norvegicus) populations of Baltimore, Maryland. J. Wildl. Dis. 25:469–476 [DOI] [PubMed] [Google Scholar]
- 16. du Bois RM. 1985. The alveolar macrophage. Thorax 40:321–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Easterbrook JD, Klein SL. 2008. Immunological mechanisms mediating hantavirus persistence in rodent reservoirs. PLoS Pathog. 4:e1000172 doi:10.1371/journal.ppat.1000172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Easterbrook JD, Klein SL. 2008. Seoul virus enhances regulatory and reduces proinflammatory responses in male Norway rats. J. Med. Virol. 80:1308–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Easterbrook JD, Zink MC, Klein SL. 2007. Regulatory T cells enhance persistence of the zoonotic pathogen Seoul virus in its reservoir host. Proc. Natl. Acad. Sci. U. S. A. 104:15502–15507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elliott LH, et al. 1994. Isolation of the causative agent of hantavirus pulmonary syndrome. Am. J. Trop. Med. Hyg. 51:102–108 [DOI] [PubMed] [Google Scholar]
- 21. Fels AO, Cohn ZA. 1986. The alveolar macrophage. J. Appl. Physiol. 60:353–369 [DOI] [PubMed] [Google Scholar]
- 22. Francisco LM, et al. 2009. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 206:3015–3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Freeman GJ, et al. 2000. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192:1027–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gavrilovskaya IN, Gorbunova EE, Mackow ER. 2010. Pathogenic hantaviruses direct the adherence of quiescent platelets to infected endothelial cells. J. Virol. 84:4832–4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gavrilovskaya IN, Gorbunova EE, Mackow NA, Mackow ER. 2008. Hantaviruses direct endothelial cell permeability by sensitizing cells to the vascular permeability factor VEGF, while angiopoietin 1 and sphingosine 1-phosphate inhibit hantavirus-directed permeability. J. Virol. 82:5797–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hall CH, Kassel R, Tacke RS, Hahn YS. 2010. HCV+ hepatocytes induce human regulatory CD4+ T cells through the production of TGF-beta. PLoS One 5:e12154 doi:10.1371/journal.pone.0012154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hannah MF, Bajic VB, Klein SL. 2008. Sex differences in the recognition of and innate antiviral responses to Seoul virus in Norway rats. Brain Behav. Immun. 22:503–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoshino Y, et al. 2007. Mechanisms of polymorphonuclear neutrophil-mediated induction of HIV-1 replication in macrophages during pulmonary tuberculosis. J. Infect. Dis. 195:1303–1310 [DOI] [PubMed] [Google Scholar]
- 29. Hutchinson KL, Rollin PE, Peters CJ. 1998. Pathogenesis of a North American hantavirus, Black Creek Canal virus, in experimentally infected Sigmodon hispidus. Am. J. Trop. Med. Hyg. 59:58–65 [DOI] [PubMed] [Google Scholar]
- 30. Jiang H, et al. 2008. Hantaan virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon, interleukin-6 and tumor necrosis factor-alpha. Virology 380:52–59 [DOI] [PubMed] [Google Scholar]
- 31. Kariwa H, et al. 1996. Modes of Seoul virus infections: persistency in newborn rats and transiency in adult rats. Arch. Virol. 141:2327–2338 [DOI] [PubMed] [Google Scholar]
- 32. Keir ME, et al. 2006. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 203:883–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khaiboullina SF, Netski DM, Krumpe P, St Jeor SC. 2000. Effects of tumor necrosis factor alpha on sin nombre virus infection in vitro. J. Virol. 74:11966–11971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khaiboullina SF, St Jeor SC. 2002. Hantavirus immunology. Viral Immunol. 15:609–625 [DOI] [PubMed] [Google Scholar]
- 35. Kilpatrick ED, et al. 2004. Role of specific CD8+ T cells in the severity of a fulminant zoonotic viral hemorrhagic fever, hantavirus pulmonary syndrome. J. Immunol. 172:3297–3304 [DOI] [PubMed] [Google Scholar]
- 36. Kitamura T, et al. 1983. Isolation of virus causing hemorrhagic fever with renal syndrome (HFRS) through a cell culture system. Jpn. J. Med. Sci. Biol. 36:17–25 [DOI] [PubMed] [Google Scholar]
- 37. Klein SL, et al. 2004. Differential expression of immunoregulatory genes in male and female Norway rats following infection with Seoul virus. J. Med. Virol. 74:180–190 [DOI] [PubMed] [Google Scholar]
- 38. Kosiewicz MM, Alard P. 2004. Tolerogenic antigen-presenting cells: regulation of the immune response by TGF-beta-treated antigen-presenting cells. Immunol. Res. 30:155–170 [DOI] [PubMed] [Google Scholar]
- 39. Kraus AA, Priemer C, Heider H, Kruger DH, Ulrich R. 2005. Inactivation of Hantaan virus-containing samples for subsequent investigations outside biosafety level 3 facilities. Intervirology 48:255–261 [DOI] [PubMed] [Google Scholar]
- 40. Kraus AA, et al. 2004. Differential antiviral response of endothelial cells after infection with pathogenic and nonpathogenic hantaviruses. J. Virol. 78:6143–6150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Krupnick AS, et al. 2005. Murine vascular endothelium activates and induces the generation of allogeneic CD4+25+Foxp3+ regulatory T cells. J. Immunol. 175:6265–6270 [DOI] [PubMed] [Google Scholar]
- 42. Lambrecht BN. 2006. Alveolar macrophage in the driver's seat. Immunity 24:366–368 [DOI] [PubMed] [Google Scholar]
- 43. Lee PW, Yanagihara R, Gibbs CJ, Jr, Gajdusek DC. 1986. Pathogenesis of experimental Hantaan virus infection in laboratory rats. Arch. Virol. 88:57–66 [DOI] [PubMed] [Google Scholar]
- 44. Linderholm M, Ahlm C, Settergren B, Waage A, Tarnvik A. 1996. Elevated plasma levels of tumor necrosis factor (TNF)-alpha, soluble TNF receptors, interleukin (IL)-6, and IL-10 in patients with hemorrhagic fever with renal syndrome. J. Infect. Dis. 173:38–43 [DOI] [PubMed] [Google Scholar]
- 45. Maes P, Clement J, Gavrilovskaya I, Van Ranst M. 2004. Hantaviruses: immunology, treatment, and prevention. Viral Immunol. 17:481–497 [DOI] [PubMed] [Google Scholar]
- 46. Maier H, Isogawa M, Freeman GJ, Chisari FV. 2007. PD-1:PD-L1 interactions contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver. J. Immunol. 178:2714–2720 [DOI] [PubMed] [Google Scholar]
- 47. Markotic A, et al. 1999. Role of peripheral blood mononuclear cell (PBMC) phenotype changes in the pathogenesis of haemorrhagic fever with renal syndrome (HFRS). Clin. Exp. Immunol. 115:329–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marsac D, et al. 2011. Infection of human monocyte-derived dendritic cells by ANDES Hantavirus enhances pro-inflammatory state, the secretion of active MMP-9 and indirectly enhances endothelial permeability. Virol. J. 8:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mori M, et al. 1999. High levels of cytokine-producing cells in the lung tissues of patients with fatal hantavirus pulmonary syndrome. J. Infect. Dis. 179:295–302 [DOI] [PubMed] [Google Scholar]
- 50. Mueller SN, et al. 2010. PD-L1 has distinct functions in hematopoietic and nonhematopoietic cells in regulating T cell responses during chronic infection in mice. J. Clin. Invest. 120:2508–2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muhlbauer M, et al. 2006. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J. Hepatol. 45:520–528 [DOI] [PubMed] [Google Scholar]
- 52. Murphy J, Summer R, Wilson AA, Kotton DN, Fine A. 2008. The prolonged life-span of alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 38:380–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nagai T, et al. 1985. Isolation of haemorrhagic fever with renal syndrome virus from leukocytes of rats and virus replication in cultures of rat and human macrophages. J. Gen. Virol. 66:1271–1278 [DOI] [PubMed] [Google Scholar]
- 54. Nakata K, et al. 1997. Mycobacterium tuberculosis enhances human immunodeficiency virus-1 replication in the lung. Am. J. Respir. Crit. Care Med. 155:996–1003 [DOI] [PubMed] [Google Scholar]
- 55. Noone CM, et al. 2008. Natural killer cells regulate T-cell proliferation during human parainfluenza virus type 3 infection. J. Virol. 82:9299–9302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Oh S, McCaffery JM, Eichelberger MC. 2000. Dose-dependent changes in influenza virus-infected dendritic cells result in increased allogeneic T-cell proliferation at low, but not high, doses of virus. J. Virol. 74:5460–5469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Peng G, et al. 2008. PD-1 upregulation is associated with HBV-specific T cell dysfunction in chronic hepatitis B patients. Mol. Immunol. 45:963–970 [DOI] [PubMed] [Google Scholar]
- 58. Pensiero MN, Sharefkin JB, Dieffenbach CW, Hay J. 1992. Hantaan virus infection of human endothelial cells. J. Virol. 66:5929–5936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Plekhova NG, et al. 2005. Metabolic activity of macrophages infected with hantavirus, an agent of hemorrhagic fever with renal syndrome. Biochemistry 70:990–997 [DOI] [PubMed] [Google Scholar]
- 60. Prescott J, et al. 2010. New World hantaviruses activate IFNlambda production in type I IFN-deficient vero E6 cells. PLoS One 5:e11159 doi:10.1371/journal.pone.0011159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Quigley M, et al. 2010. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 16:1147–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Raftery MJ, Kraus AA, Ulrich R, Kruger DH, Schonrich G. 2002. Hantavirus infection of dendritic cells. J. Virol. 76:10724–10733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rehn B, Bruch J, Zou T, Hobusch G. 1992. Recovery of rat alveolar macrophages by bronchoalveolar lavage under normal and activated conditions. Environ. Health Perspect. 97:11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rowe RK, Suszko JW, Pekosz A. 2008. Roles for the recycling endosome, Rab8, and Rab11 in hantavirus release from epithelial cells. Virology 382:239–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Safronetz D, et al. 2011. Pathogenesis and host response in Syrian hamsters following intranasal infection with Andes virus. PLoS Pathog. 7:e1002426 doi:10.1371/journal.ppat.1002426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sanduzzi A, Fraziano M, Mariani F. 2001. Monocytes/macrophages in HIV infection and tuberculosis. J. Biol. Regul. Homeost. Agents 15:294–298 [PubMed] [Google Scholar]
- 67. Schonrich G, et al. 2008. Hantavirus-induced immunity in rodent reservoirs and humans. Immunol. Rev. 225:163–189 [DOI] [PubMed] [Google Scholar]
- 68. Schountz T, et al. 2007. Regulatory T cell-like responses in deer mice persistently infected with Sin Nombre virus. Proc. Natl. Acad. Sci. U. S. A. 104:15496–15501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shrivastava-Ranjan P, Rollin PE, Spiropoulou CF. 2010. Andes virus disrupts the endothelial cell barrier by induction of vascular endothelial growth factor and downregulation of VE-cadherin. J. Virol. 84:11227–11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stoltz M, et al. 2011. A model system for in vitro studies of bank vole borne viruses. PLoS One 6:e28992 doi:10.1371/journal.pone.0028992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sundstrom JB, et al. 2001. Hantavirus infection induces the expression of RANTES and IP-10 without causing increased permeability in human lung microvascular endothelial cells. J. Virol. 75:6070–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Taflin C, et al. 2011. Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions. Proc. Natl. Acad. Sci. U. S. A. 108:2891–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Temonen M, et al. 1993. Susceptibility of human cells to Puumala virus infection. J. Gen. Virol. 74:515–518 [DOI] [PubMed] [Google Scholar]
- 74. Terajima M, Hayasaka D, Maeda K, Ennis FA. 2007. Immunopathogenesis of hantavirus pulmonary syndrome and hemorrhagic fever with renal syndrome: do CD8+ T cells trigger capillary leakage in viral hemorrhagic fevers? Immunol. Lett. 113:117–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Urbani S, et al. 2006. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J. Virol. 80:11398–11403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Walker JD, Maier CL, Pober JS. 2009. Cytomegalovirus-infected human endothelial cells can stimulate allogeneic CD4+ memory T cells by releasing antigenic exosomes. J. Immunol. 182:1548–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wewers MD, Rennard SI, Hance AJ, Bitterman PB, Crystal RG. 1984. Normal human alveolar macrophages obtained by bronchoalveolar lavage have a limited capacity to release interleukin-1. J. Clin. Invest. 74:2208–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yanagihara R, Amyx HL, Gajdusek DC. 1985. Experimental infection with Puumala virus, the etiologic agent of nephropathia epidemica, in bank voles (Clethrionomys glareolus). J. Virol. 55:34–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yang G, et al. 2007. Association of CD4+CD25+Foxp3+ regulatory T cells with chronic activity and viral clearance in patients with hepatitis B. Int. Immunol. 19:133–140 [DOI] [PubMed] [Google Scholar]
- 80. Yoshizawa K, et al. 2010. Expansion of CD4(+)CD25(+)FoxP3(+) regulatory T cells in hepatitis C virus-related chronic hepatitis, cirrhosis and hepatocellular carcinoma. Hepatol Res. 40:179–187 [DOI] [PubMed] [Google Scholar]
- 81. Zhu LY, Chi LJ, Wang X, Zhou H. 2009. Reduced circulating CD4+CD25+ cell populations in haemorrhagic fever with renal syndrome. Clin. Exp. Immunol. 156:88–96 [DOI] [PMC free article] [PubMed] [Google Scholar]