

Abstract
Many viruses express inhibitors of programmed cell death (apoptosis), thereby countering host defenses that would otherwise rapidly clear infected cells. To counter this, viruses such as adenoviruses and herpesviruses express recognizable homologs of the mammalian prosurvival protein Bcl-2. In contrast, the majority of poxviruses lack viral Bcl-2 (vBcl-2) homologs that are readily identified by sequence similarities. One such virus, myxoma virus, which is the causative agent of myxomatosis, expresses a virulence factor that is a potent inhibitor of apoptosis. In spite of the scant sequence similarity to Bcl-2, myxoma virus M11L adopts an almost identical 3-dimensional fold. We used M11L as bait in a sequence similarity search for other Bcl-2-like proteins and identified six putative vBcl-2 proteins from poxviruses. Some are potent inhibitors of apoptosis, in particular sheeppox virus SPPV14, which inhibited cell death induced by multiple agents. Importantly, SPPV14 compensated for the loss of antiapoptotic F1L in vaccinia virus and acts to directly counter the cell death mediators Bax and Bak. SPPV14 also engages a unique subset of the death-promoting BH3-only ligands, including Bim, Puma, Bmf, and Hrk. This suggests that SPPV14 may have been selected for specific biological roles as a virulence factor for sheeppox virus.
INTRODUCTION
Viruses employ diverse strategies to subvert the death of infected host cells by apoptosis (23). The process of determining whether a cell lives or dies is critically dependent on the actions of Bcl-2 protein family members, which are key regulators of the mitochondrial or intrinsic pathway of apoptosis (68). Their central role is reinforced by the realization that certain viruses express sequence, structural, or functional homologs of mammalian Bcl-2 (13). Such members of the wider Bcl-2 family are readily recognizable, as they share one to four regions of sequence similarity, the Bcl-2 homology (BH) domains. In higher organisms, the prosurvival family members, such as Bcl-2, Bcl-xL, and Mcl-1, keep cells viable unless they are bound and inactivated by the proapoptotic BH3-only proteins, such as Bim or Bad (44). BH3-only proteins are activated by damage signals triggered after cellular insults, such as growth factor deprivation or exposure to cytotoxic drugs, to initiate the cell death machinery (55). Unlike the prosurvival Bcl-2 proteins, which contain multiple BH domains, the BH3-only proteins contain only an α-helical BH3 domain, which binds a receptor-like groove on the prosurvival proteins (44). The key consequence is to activate the multidomain death mediators Bax and Bak to drive mitochondrial outer membrane permeabilization (MOMP), which results ultimately in the activation of proteolytic enzymes (caspases) that drive cellular demolition (27).
A number of viruses, including adenovirus (66), Kaposi sarcoma-associated herpesvirus (KSHV) (52), Epstein-Barr virus (EBV) (30), fowlpox virus FPV039 (1), and orf virus ORFV125 (64, 65), encode recognizable homologous Bcl-2-like proteins that were recognized by their sequence similarities indicating the presence of one or more BH domains (13). Subsequent structure determination of KSHV Bcl-2 (35) and EBV BHRF1 (36, 41) confirmed that both adopt the classical Bcl-2 fold encompassing the canonical hydrophobic binding groove observed in their cellular counterparts. Notably, viral Bcl-2-like proteins are often required for successful viral propagation, emphasizing the critical role inhibition of host cell apoptosis plays during viral infections (23). However, other viruses express antiapoptotic proteins that appear unrelated by sequence to known cell death regulators. They include myxoma virus-encoded M11L (19, 20, 25), human cytomegalovirus-encoded vMIA (24), murine cytomegalovirus-encoded m38 (38), and vaccinia virus-encoded F1L (21, 48, 49, 61, 63) and N1L (2, 7, 12). Despite the lack of sequence similarity to the Bcl-2 protein family, both F1L and M11L adopt a Bcl-2 fold (16, 40, 42), and the structure of M11L in complex with the BH3 peptide from Bak revealed that M11L binds BH3 ligands via the canonical BH3 binding groove (40). Similar to mammalian cell death antagonists, M11L acts to directly counter the cell death mediators Bax and Bak expressed by the host (40).
Overall, these studies suggest that there may be other yet-to-be-identified viral Bcl-2 proteins, and we embarked on a search for other viral gene products that bear sequence similarity to M11L to sample a different region of sequence space compared to cellular Bcl-2 proteins. Six candidate poxvirus genes were identified using a BLAST search, and one originating from sheeppox virus was selected for further analysis, since it appeared to be as potent an inhibitor of apoptosis as M11L in an initial cell death assay. Sheeppox virus is a member of the genus Capripoxvirus within the subfamily Chordopoxvirus of the Poxviridae. Sheeppox is endemic in Southwest and Central Asia, India, and North Africa and is a significant economic factor due to high mortality rates (50 to 70%), particularly in young animals. In cellular assays, we showed that sheeppox virus SPPV14 potently protects against diverse apoptotic stimuli, and here, we also report studies to define the molecular mechanism of action, and in particular, how it compares with other related proteins, such as myxoma virus M11L. Even though SPPV14 directly inhibits Bax and Bak, similar to M11L, it also binds a unique subset of the BH3-only proteins, suggesting that it may be functionally selected for a specific biological role(s) during the sheeppox virus life cycle.
MATERIALS AND METHODS
Expression and retroviral constructs.
The cDNAs of deerpox virus (DPV83gp022 and DPV84gp022), swinepox virus (SPV12), shope fibroma virus (gp011L), lumpy skin disease virus (LD17), and sheeppox virus (SPPV14) were synthesized (GeneScript) and verified by sequencing. All mammalian expression vectors for hemagglutinin (HA)-tagged BH3-only proteins, HA-Bax and HA-Bak, subcloned into pEF PGKhygro have been described previously (9, 34, 46, 67). Similar constructs, made by subcloning into pEF PGKpuro, were generated for all viral proteins. A retroviral expression construct (BimS2A) was generated by subcloning into pMIG (murine stem cell virus [MSCV]-internal ribosome entry site [IRES]-green fluorescent protein [GFP], where the GFP sequence is that of enhanced GFP [EGFP]), as previously described (9, 67). The GFP selection cassette was replaced with the hygromycin resistance gene (58) for the constructs expressing viral proteins. Yeast constructs were made by subcloning into the pGALL(TRP) vector, as previously described (22). All cDNAs were of human or viral origin.
Details of all oligonucleotides and constructs used are available on request.
Tissue culture, cell death induction, retroviral infections, and apoptosis assays.
All cell lines except Jurkat cells (HEK293T immortalized human embryonal kidney cell line, Phoenix Ecotropic packaging cells [39], and mouse embryonic fibroblasts [MEFs]) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) and in some cases with 250 mM l-asparagine and 50 mM 2-mercaptoethanol. HuTK−-143B and Jurkat cells were obtained from ATCC and maintained as previously described (62). Jurkat cells overexpressing Bcl-2 were generated and cultured as previously described (6); Bak- and Bax-deficient Jurkat cells were a gift from H. Rabinowich (University of Pittsburgh School of Medicine, Pittsburgh, PA) (60).
All MEFs (9, 67) were generated from embryonic day 13 (E13) to E14.5 embryos and immortalized at passages 2 to 4 with simian virus 40 (SV40) large T antigen. All mice used were of C57BL/6 origin or had been backcrossed (>10 generations), and their genotypes were determined as previously described (details are available on request).
Cell death was induced with 10 to 40 μM etoposide, UV irradiation (100 J/m2), cytosine arabinoside (AraC), ABT-737 (1 μM; Abbott) (47), or FasL (Alexis) or by retroviral infection. pMIG retroviral constructs encoding BH3-only proteins were transiently transfected into Phoenix Ecotropic packaging cells, and viral supernatants were used to infect cells as described previously (9). Cell viability in both short-term assays and long-term assays of colony formation was determined as described previously (9). Long-term survival is expressed as a percentage of the number of colonies obtained relative to the number of colonies obtained after retroviral infection with empty parental retrovirus.
Bak activation was assessed by infecting 1 × 106 Jurkat cells, Jurkat cells expressing Bcl-2, or Bak- and Bax-deficient Jurkat cells at a multiplicity of infection (MOI) of 10 with VVEGFP (control wild-type vaccinia virus), VVΔF1L (F1L-deficient virus), or VVΔF1L-FLAG-SPPV14 (F1L-deficient virus expressing FLAG-tagged SPPV14). Six hours postinfection, the cells were fixed in 0.25% paraformaldehyde, permeabilized with 500 μg/ml (0.05%) digitonin (Sigma-Aldrich), and stained with a conformation-specific anti-Bak Ab-1 antibody (Oncogene Research Products) (28). Phycoerythrin-conjugated anti-mouse antibody was used to counterstain the cells (Jackson ImmunoResearch) before analysis by flow cytometry (FACScan; Becton Dickinson) using the FL-2 channel equipped with a 585-nm filter (42-nm band-pass). Data were analyzed using CellQuest software.
Protein production.
The cDNA of SPPV14 was used to amplify the region coding for residues 1 to 145 of SPPV14 (deleting the C-terminal 31 amino acids, referred to as SPPV14ΔC31), which was cloned into the pET DUET vector (Invitrogen) using an introduced 5′ BamHI restriction site and 3′ EcoRI site, followed by a stop codon, and expressed in Escherichia coli BL21(DE3) pLysS cells. After homogenization in lysis buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10 mM 2-mercaptoethanol), the cell lysate was centrifuged and filtered prior to loading onto a 1-ml His-Trap column (Amersham). The protein was eluted in lysis buffer supplemented with 250 mM imidazole and subjected to gel filtration chromatography in 20 mM HEPES, pH 7.5, 150 mM NaCl, and 10 mM dithiothreitol (DTT) using a Superdex 200 column (Amersham), where it eluted as a single peak.
Immunoprecipitation and immunoblotting.
The transfection and metabolic labeling of the human embryonic kidney (HEK293T cells with [35S]methionine/[35S]cysteine (NEN) have been described previously (33, 46). Immunoprecipitation of SPPV14 with Bak or Bax was performed using mouse monoclonal anti-FLAG (M2; Sigma) or anti-HA (HA11; Covance) antibody in buffer containing 20 mM Tris-HCl, pH 7.4, 135 mM NaCl, 1% Triton X-100, 10% glycerol in the presence of protease inhibitor (Roche). Control immunoprecipitations were performed using an anti-mouse Glu-Glu (MMS-115R; CRP) antibody. Proteins were resolved by SDS-PAGE (Novex gels; Invitrogen), transferred onto nitrocellulose membranes, and detected with X-ray film (Hyperfilm; GE Healthcare).
Vaccinia virus production.
SPPV14 cDNA was subcloned into pSC66, which places the gene under the control of a poxvirus promoter to generate VVΔF1L-FLAG-SPPV14 (17). Recombinant VVΔF1L-FLAG-SPPV14 was generated by homologous recombination of pSC66-FLAG-SPPV14 into the thymidine kinase locus of VVΔF1L, as described previously (17). In brief, 1 × 106 baby green monkey kidney (BGMK) cells were transfected with 5 μg of pSC66-Flag-SPPV14 and infected with VVΔF1L at an MOI of 0.05. Recombinant viruses were selected by growth on HuTK−-143B cells in the presence of 5-bromodeoxyuridine (Sigma-Aldrich) and plaque purification using 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (Rose Scientific Ltd.) to visualize β-galactosidase-positive viruses. All viruses were grown on BGMK cells.
Cytochrome c release.
Jurkat cells (1 × 106) were infected with VVEGFP, VVΔF1L-SPPV014, and VVΔF1L at an MOI of 5 at 4, 8, and 12 h of infection. The cells were permeabilized in lysis buffer containing 75 mM NaCl, 1 mM NaH2PO4, 8 mM Na2HPO4, 250 mM sucrose, and 190 μg/ml of digitonin (Sigma-Aldrich), and the lysates were incubated on ice for 10 min (62). Mitochondrial and cytoplasmic fractions were separated by ultracentrifugation at 10,000 × g for 5 min. The mitochondrial pellet was resuspended in Triton X-100 lysis buffer containing 25 mM Tris, pH 8.0, and 0.1% Triton X-100 (Fisher Scientific). Samples were subjected to SDS-PAGE and immunoblotted with mouse anti-cytochrome c (BD PharMingen) and anti-Bak NT as a control.
PARP cleavage assay.
To detect cleavage of poly-ADP ribose polymerase (PARP), Jurkat cells (1 × 106) were mock infected or infected with VVEGFP, VVΔF1L-SPPV014, and VVΔF1L at an MOI of 5. Cells were harvested 4, 8, 12, and 16 h postinfection and lysed in SDS-PAGE sample buffer containing 8 M urea. Samples were subjected to SDS-PAGE and immunoblotted with anti-PARP (BD PharMingen), anti-β-tubulin (EMC Bioscience), and anti-I3L to detect virus infection.
Solution competition assays.
Solution competition assays using the Biacore optical biosensor were performed as described previously (9), using identical BH3 domain peptides and 10 nM SPPV14ΔC31.
Yeast colony assays.
Saccharomyces cerevisiae W303α cells were cotransformed with pGALL(TRP) vector only, pGALL(TRP)-Bcl-xL, or pGALL(TRP)-DPV022 and pGALL(Leu)-Bak or pGALL(Leu)-Bax. pGALL(TRP) and pGALL(Leu) place genes under the control of a galactose-inducible promoter (29). Cells were spotted as 5-fold serial dilutions onto medium containing 2% (wt/vol) galactose (inducing [ON]), which induces protein expression, or 2% (wt/vol) glucose (repressing [OFF]), which prevents protein expression, as previously described (37). Plates were incubated for 48 h at 30°C and then photographed.
RESULTS
Identifying putative viral Bcl-2 homologs.
Although proteins such as Epstein-Barr virus BHRF1 (30) and adenovirus E1B19K (66) were first isolated due to their roles in modulating host-pathogen interactions during infection and in extending the life span of infected cells, they were later recognized as viral homologs of Bcl-2 (vBcl-2) by virtue of their sequence similarity to mammalian Bcl-2 (11). In contrast, herpesvirus saimiri-encoded ORF16 (54) and fowlpox virus-encoded FPV039 (1) were specifically recognized due to sequence similarity to mammalian Bcl-2. Aside from these readily identifiable viral Bcl-2-related proteins, the recent molecular and structural characterization of proteins such as M11L in myxoma virus (40, 59) and F1L in vaccinia virus (21, 42, 63), which appear to be unrelated to Bcl-2 by conventional sequence comparisons, has opened up another approach to identifying vBcl-2 proteins.
By searching for putative proteins that are related by sequence to M11L instead of mammalian Bcl-2, we circumvented the need for conventional genetic or functional screens in order to identify putative antiapoptotic factors encoded by other organisms. In a BLASTP search of the NCBI database (40), the top hits included six sequences that encode putative open reading frames (ORFs) from deerpox virus (DPV83gp022 and DPV84gp022), swinepox virus (SPV12), shope fibroma virus (gp011L), lumpy skin disease virus (LD17), and sheeppox virus (SPPV14) (Fig. 1A). Even though the overall sequence similarity to M11L was low, the critical residues forming the canonical binding groove in M11L (40) were conserved in all six proteins, strongly suggesting the presence of an analogous BH3 binding grove in these proteins (Fig. 1B and 2A and B).
Fig 1.

Identifying putative viral Bcl-2 (vBcl-2) homologs. (A) Using myxoma virus M11L as the query sequence in a BLASTP search of the NCBI database, six putative Bcl-2 family proteins encoded by viruses were identified (40). The pairwise identities (percent) in the amino acid sequences between the proteins were determined using ClustalW (57). Note the low sequence homology (<15%) of all the putative vBcl-2 proteins to human Bcl-xL. (B) The amino acid sequence of a vBcl-2, SPPV14, was structurally aligned with those of myxoma virus M11L (40), mouse Bcl-xL (44), and KSHV Bcl-2 (35). Based on this alignment, the residues making up α-helices 1 to 8 are marked “H.” The colored shading indicates the BH1 (magenta), BH2 (orange), BH3 (green), and BH4 (yellow) Bcl-2 homology regions in Bcl-xL and KSHV Bcl-2. The residues marked in cyan are those in the BH3 binding groove of M11L and conserved between it and SPPV14 (Fig. 2A). TM indicates the putative transmembrane region as taken from M11L.
Fig 2.

Some putative vBcl-2 proteins block apoptosis. (A) Cartoon of the binding interface formed by the M11L-Bak complex (40). The M11L surface is shown in gray, and residues conserved between SPPV14 and M11L (Fig. 1B) that form part of the canonical binding groove in M11L are shaded in cyan. The Bak BH3 peptide is shown in orange. (B) Cartoon of the binding interface formed by the M11L-Bak complex. The colors are as in panel A, and the Bak BH3 peptide is removed for clarity. (C) The expression of FLAG-tagged viral proteins in MEFs was detected by flow cytometry. MEFs expressing novel vBcl-2 proteins were fixed and stained with an anti-FLAG antibody (red lines). The staining of MEFs expressing the empty vector is shown as the negative control (black lines). (D) The viability of MEFs expressing the empty vector, M11L, or poxvirus vBcl-2 proteins treated with etoposide (2.5 μM) for 24 h was determined by propidium iodide (PI) exclusion. The relative viability was determined by normalizing to that of M11L-expressing cells treated with etoposide. The data represent means ± standard deviations (SD) of two experiments performed with each cell line.
To test if these ORFs encode antiapoptotic proteins, we expressed tagged versions of all six proteins in MEFs (Fig. 2C) and assessed their abilities to counter killing induced by the cytotoxic drug etoposide (Fig. 2D). DPV83gp022, LD17, and SPPV14 potently inhibited etoposide-induced killing, whereas DPV84gp022, swinepox virus (SPV12), and shope fibroma virus (gp011L) did not afford protection against etoposide, even though they were abundantly expressed. Interestingly, we noted that four residues (M52, T67, L68, and A71) in the binding groove of M11L are likely to be important for function, since the identified DPV84gp022, SPV12, and shope (rabbit) fibroma virus gp011L, which do not protect against apoptosis, have critical substitutions for these residues (see Fig. S1 in the supplemental material), possibly pointing to a biological function that does not involve apoptosis. We elected to focus further studies on SPPV14, since it was the most potent (Fig. 2D) out of all 6 identified putative vBcl-2 proteins and afforded protection against etoposide that appeared superior to that of M11L. Notably, SPPV14 shows 22% sequence identity to M11L. However, significantly higher conservation was apparent in the region corresponding to the central α5 helix that constitutes the BH1 region in M11L (10/21 amino acids; ∼50%) (Fig. 1B).
Sheeppox virus SPPV14 is a potent inhibitor of mitochondrially mediated cell death.
To extend the functional analysis, we examined the dose response following etoposide treatment. SPPV14 proved to be as efficacious as M11L (Fig. 3A), and its ability to counter killing induced by other agents, such as AraC or UV irradiation, was also comparable (Fig. 3B). Furthermore, SPPV14 appears to act at the mitochondrial checkpoint and not at the level of caspases, since it potently inhibits cytochrome c release (Fig. 3C).
Fig 3.

SPPV14 inhibits apoptosis, but not NF-κB activation. (A) Sheeppox virus SPPV14 is functionally comparable to myxoma virus M11L. Shown is the viability of wild-type (WT) MEFs expressing M11L or SPPV14, or the vector control, 24 h after treatment with 0 to 40 μM etoposide. (B) SPPV14 blocks apoptosis induced by diverse agents. Shown is the viability of MEFs (described in the legend to Fig. 1B) 24 h after treatment with 10 μM AraC or exposure to UV irradiation (100 J/m2). The bars on the right show the viability of similarly treated Bax/Bak-doubly deficient MEFs. (C) SPPV14 prevents cytochrome c release. Jurkat cells were infected with VVEGFP, VVΔF1L-SPPV014, and VVΔF1L at an MOI of 10 for 4, 8, and 12 h. Mitochondrial pellets and cytoplasmic supernatants were separated via ultracentrifugation after treatment with digitonin. The mitochondrial pellets were resuspended in lysis buffer containing 0.1% Triton X-100, and 20% of the mitochondrial fractions and 50% of the cytoplasmic fractions were subjected to SDS-PAGE and immunoblotted with anti-cytochrome c and anti-Bak NT antibody as a control. WB, Western blotting. (D) SPPV14 does not block NF-κB activation induced by IL-1β or TNF-α. HEK293 cells were transiently transfected with an NF-κB luciferase reporter plasmid and the empty vector, dominant-negative (DN) IκBα, Bcl-2, B14, or SPPV14. The relative luciferase activity was measured 8 h after treatment with IL-1β (100 ng/ml) or TNF-α (100 ng/ml). The data represent means ± SD from 2 independent experiments.
As M11L was previously reported to protect against Fas-induced cell killing (59), we assessed the abilities of various virus-encoded antiapoptotic proteins to inhibit the extrinsic pathway of apoptosis. To induce CD95 (Fas) killing, FLAG-tagged Fas ligand was aggregated with the anti-FLAG antibody (32, 53). Notably, in cells treated with Fas ligand and cycloheximide, which is required for Fas-induced killing of MEFs, potent killing was observed (see Fig. S2 in the supplemental material). Expression of M11L, SPPV14, or vaccinia virus B14, a vaccinia virus virulence factor that inhibits IκB kinase (IKK) (26), had no impact upon Fas-induced killing (see Fig. S2). In MEFs, killing by Fas did not appear to be primarily regulated by the Bcl-2 protein family, since the loss of the essential death mediators Bax and Bak made little difference (see Fig. S2).
Having established that SPPV14 is a bone fide antiapoptotic protein (Fig. 2 and 3), we next determined if SPPV14 also interfered with NF-κB signaling, since some vBcl-2 proteins, such as vaccinia virus B14, A52, and N1L (2, 15, 26), fold like Bcl-2 but inhibit NF-κB. As expected, a dominant interfering mutant of IκBα effectively blocked activation of NF-κB induced by two cytokines, interleukin 1β (IL-1β) and tumor necrosis factor alpha (TNF-α), when HEK293T cells were cotransfected with a NF-κB reporter plasmid. Although we were able to confirm the ability of B14 to block NF-κB activation in this assay, neither Bcl-2 nor SPPV14 could (Fig. 3D), even though they were expressed. Overall, our results demonstrate that sheeppox virus SPPV14 can effectively inhibit multiple forms of apoptosis induced by the intrinsic (mitochondrial) pathway (Fig. 2 and 3) but does not appear to interfere with death through the extrinsic pathway or with NF-κB activation.
Sheeppox virus SPPV14 inhibits apoptosis during viral infection.
Since SPPV14 can effectively inhibit killing triggered by experimentally applied stimuli, but not Fas, we next investigated its ability to inhibit apoptosis during viral infection (Fig. 4 and 5). Wild-type vaccinia virus infects and propagates in a wide range of mammalian cell lines, including Jurkat and HeLa cells. Deletion of the antiapoptotic protein F1L (63) from vaccinia virus results in apoptosis of infected cells, indicated by the activation of the key cell death mediators Bak (Fig. 4A) and Bax (Fig. 5). To determine if SPPV14 could functionally replace F1L and inhibit vaccinia virus-induced apoptosis, we inserted SPPV14 into a F1L-deficient vaccinia virus, and remarkably, SPPV14 was able to completely block Bak (Fig. 4A) and Bax (Fig. 5B and C) activation, thereby maintaining cell viability during infection with vaccinia virus. These findings are further supported by the observation that SPPV14 in the context of vaccinia virus infection also inhibits PARP cleavage (Fig. 5D).
Fig 4.

SPPV14 can functionally replace F1L during vaccinia virus infection. (A to C) SPPV14 inhibits Bak activation during vaccinia virus infection. Wild-type (A), Bax/Bak-doubly deficient (B), or Bcl-2-overexpressing (C) Jurkat cells were mock infected or infected with VVEGFP, VVΔF1L, or VVΔF1L-FLAG-SPPV14 (F1L-deficient virus expressing FLAG-tagged SPPV14) vaccinia virus. Bak activation was detected 6 h later by flow cytometric analyses of cells stained with the conformation-specific anti-Bak Ab-1 antibody (28). Positive-control mock-infected cells were treated with the broad-spectrum kinase inhibitor staurosporine (+STS) (A, a), known to induce apoptosis in many cell types. The bars mark the populations that contained activated Bak. Experiments were performed in triplicate. Fractions of Bak-activated cells are given as percentages.
Fig 5.

SPPV14 inhibits Bax activation during vaccinia virus infection. (A) HeLa cells were infected with VVEGFP, VVΔF1L, or VVΔF1L-FLAG-SPPV14 and incubated for 24 h. Infected cells or UV-C-irradiated HeLa cells were fixed and stained with the conformation-specific anti-Bax antibody 6A7, which specifically recognizes activated Bax (14, 31). (B) HeLa cells infected with VVEGFP, VVΔF1L, or VVΔF1L-FLAG-SPPV14 were lysed with Triton X-100 or CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate}-based buffers and immunoprecipitated (IP) using the conformation-specific anti-Bax antibody 6A7. The immunoprecipitants were subjected to SDS-PAGE and blotted with the anti-Bax antibody 2D2, an antibody that recognizes all forms of Bax in immunoblots. (C) SPPV14 inhibits Bax activation induced by VVΔF1L. HeLa cells infected with VVEGFP, VVΔF1L, or VVΔF1L-FLAG-SPPV14 were incubated for 24 h and fixed, and Bax activation was measured by staining with anti-Bax 6A7 antibody (31) that specifically recognizes activated Bax. The data were quantified by counting 200 cells per experiment; means and SD of three replicate experiments are shown. UV-C, positive-control UV-irradiated HeLa cells. (D) SPPV14 inhibits PARP cleavage induced by VVΔF1L. Jurkat cells were mock infected or infected with VVEGFP, VVΔF1L-SPPV14, or VVΔF1L at an MOI of 5. Samples were collected 4, 8, 12, and 16 h postinfection, and the cells were lysed in SDS-PAGE sample buffer containing 8 M urea. Samples were subjected to SDS-PAGE and immunoblotted for PARP to determine the trigger of apoptosis; β-tubulin was used as a loading control and I3L as a sign of infection.
In summary, we have demonstrated that sheeppox virus SPPV14 counters multiple experimentally applied inducers of apoptosis and appears fully functionally competent even in the context of infection with a distantly related virus, such as vaccinia virus.
SPPV14 interacts with a selective subset of mammalian Bcl-2 proteins.
The capacity of most vBcl-2 proteins to inhibit apoptosis depends on the ability to bind and interfere with the action of mammalian proapoptotic proteins (4, 23, 40, 41, 61). Since SPPV14 potently prevented the activation of proapoptotic Bax and Bak (Fig. 4 and 5), we further investigated the molecular basis of Bax and Bak inhibition by testing the ability of SPPV14 to interact with mammalian proapoptotic Bcl-2 proteins. Initially, we evaluated the ability of recombinant SPPV14 to bind the BH3 domains of eight BH3-only proteins, as well as Bax and Bak, via solution competition assays (Fig. 6A and Table 1) (9). Bak and Bax BH3 domains were bound strongly with a 50% inhibitory concentration (IC50) of <50 nM. For the BH3-only proteins, strong binding (IC50 < 50 nM) to Bim was observed, whereas other BH3-only proteins, such as Puma, Bmf, Hrk, and Bid, bound more weakly to SPPV14 (Table 1). No binding was detected with Noxa, Bik, or Bad when tested at the highest peptide concentration (2 μM), and these findings were confirmed in vivo in immunoprecipitation assays (Fig. 7A) for Bim, Bid, Puma, Bad, and Bmf.
Fig 6.
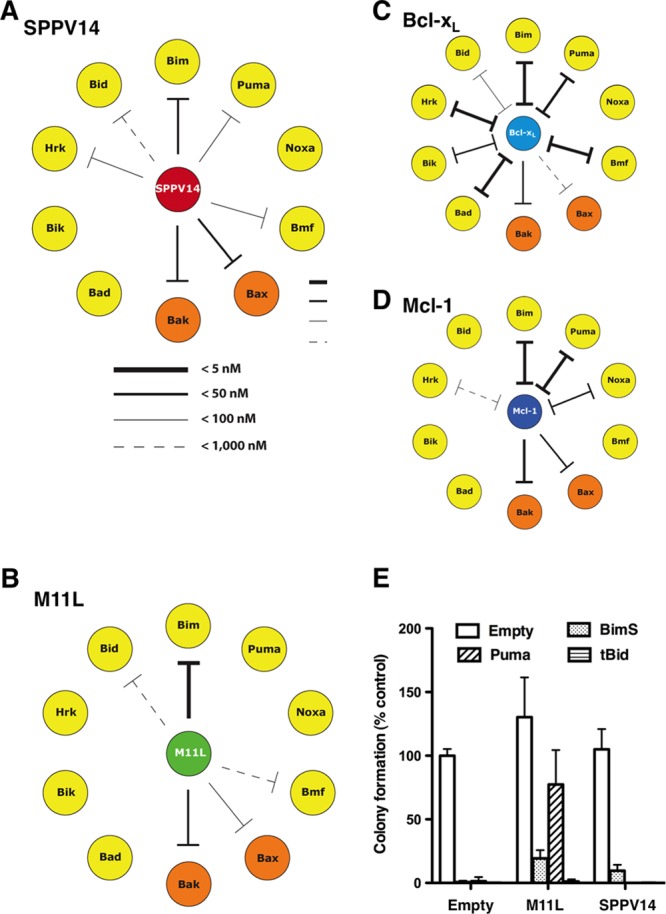
SPPV14 binds a unique set of mammalian proapoptotic proteins. (A to D) SPPV14 binds the BH3 domains of Bak, Bax, and certain BH3-only proteins. The relative binding affinities (IC50) of recombinant SPPV14 (lacking its C terminus) for long BH3 peptides were determined by solution competition assays using the Biacore optical biosensor (9) (Table 1). The relative affinities of the interactions for SPPV14 (A), M11L (40) (B), Bcl-xL (9) (C), and Mcl-1 (9) (D) are indicated by the thickness of the lines. Absence of a line indicates no binding. (E) Overexpression of BimS, Puma, and tBid counters the protection afforded by SPPV14. Shown is colony formation of M11L- or SPPV14-expressing MEFs infected with viruses expressing BimS, Puma, tBid, or an empty control. The number of colonies formed is expressed as a proportion of the colonies formed with the empty vector. The data represent means and SD from 2 independent experiments.
Table 1.
Binding affinities of selected prosurvival Bcl-2 proteins for BH3 ligands
| BH3 ligand | Binding affinity (IC50a in nM) |
|||||||
|---|---|---|---|---|---|---|---|---|
| SPPV14 | M11L | BHRF1 | Bcl-2 | Bcl-w | Bcl-xL | Mcl-1 | A1 | |
| Bad | >2,000 | >1,000 | >2,000 | 16 | 30 | 5.3 | >100,000 | 15,000 |
| Bid | 341 ± 16 | 100 | 42 | 6,800 | 40 | 82 | 2,100 | 1 |
| Bik | >2,000 | >1,000 | >2,000 | 850 | 12 | 43 | 1,700 | 58 |
| Bim | 26 ± 4 | 5 | <50 | 2.6 | 4.3 | 4.6 | 2.4 | 1 |
| Bmf | 67 ± 6 | 100 | >2,000 | 3 | 9.8 | 9.7 | 1,100 | 180 |
| Hrk | 63 ± 6 | >1,000 | >1,000 | 320 | 49 | 3.7 | 370 | 46 |
| Noxa | >2,000 | >1,000 | >2,000 | >100,000 | >100,000 | >100,000 | 24 | 20 |
| Puma | 65 ± 1 | >1,000 | <50 | 3.3 | 5.1 | 6.3 | 5 | 1 |
| Bak | 46 ± 3 | 50 | NA | >1,000 | 500 | 50 | 10 | NA |
| Bax | 32 ± 5 | 75 | NA | 100 | 58 | 130 | 12 | NA |
| Bim2A | >1,000 | NA | NA | >10,000 | >10,000 | >10,000 | 19 | 520 |
| ABT-737 | >1,000 | NA | >20,000 | 3.5 | 9.5 | 5.7 | >2,000 | >2,000 |
Fig 7.

SPPV14 binds both Bak and Bax in mammalian cells and counters growth suppression of yeast by Bax and Bak. (A) Interactions between overexpressed FLAG-SPPV14 and HA-tagged BimS, tBid, Puma, Bad, and Bmf were evaluated by coimmunoprecipitation. Equivalent CHAPS-containing 293T cell lysates were immunoprecipitated with antibodies to the HA tag, subjected to SDS-PAGE, and analyzed by Western blotting using anti-FLAG antibody. (B) Interactions between overexpressed FLAG-SPPV14 and HA-tagged Bak or Bax were evaluated by coimmunoprecipitation. Equivalent 35S-labeled CHAPS-containing 293T cell lysates were immunoprecipitated with antibodies to FLAG (F), HA (H), or control (C) tags. (C) SPPV14 counters growth suppression of yeast by Bak and Bax. Yeasts cotransformed with plasmids encoding the indicated prosurvival proteins and Bak or Bax, each under the control of an inducible (GAL) promoter, were spotted onto repressing glucose (OFF) or inducing galactose (ON) plates as 5-fold serial dilutions. The images are representative of 2 independent experiments.
Compared to the binding profiles observed for other prosurvival Bcl-2 proteins (Fig. 6 and Table 1), SPPV14 was found to be unique. Whereas both M11L and SPPV14 engaged Bim, Bak, and Bax, SPPV14 also showed significant affinity for Puma, Hrk, and Bmf (Table 1). In accordance with the in vitro binding assay, the BH3-only proteins Bim, tBid, and Puma, which could bind SPPV14, also countered its prosurvival effect when overexpressed in MEFs (Fig. 6E). Unlike SPPV14, M11L cannot bind Puma, and its prosurvival effect was thus not countered by Puma overexpression in MEFs. Moreover, the pattern observed for SPPV14 is distinct from that observed with the two broad classes of mammalian prosurvival Bcl-2 proteins exemplified by Bcl-xL and Mcl-1 (Fig. 6). The unique pattern observed with SPPV14 may well reflect the specific functional requirements for such an inhibitor in the context of a viral infection.
SPPV14 can functionally antagonize cell death mediated by Bax or Bak.
Since SPPV14 can bind both Bax and Bak in vitro (Fig. 6) and these findings were confirmed in vivo in immunoprecipitation assays (Fig. 7B), we next determined if SPPV14 could counter Bax and Bak directly. We utilized a yeast-based assay, where overexpression of Bax or Bak leads to yeast growth arrest (37) that can be overcome by overexpression of mammalian prosurvival Bcl-2 proteins. SPPV14 can counter the growth suppression in yeast cells when either Bax or Bak is overexpressed (Fig. 7C). Thus, we surmise that this most likely reflects direct binding, since yeast cells do not express recognizable Bcl-2 family members. Furthermore, SPPV14 specifically blocks Bax- or Bak-mediated cell death, since expressing SPPV14 in cells deficient in either of the cell death mediators Bak or Bax counters apoptosis (Fig. 8A). Furthermore, when all the endogenous mammalian prosurvival proteins in MEFs were inactivated (Fig. 8B) by a combination of the Bim variant BimS2A, which selectively targets Mcl-1 (43), and the BH3 mimetic compound ABT-737 (to target Bcl-2, Bcl-xL, and Bcl-w and spare SPPV14 [Table 1]) (58), expression of SPPV14 maintained cell viability (Fig. 8C), consistent with the notion that it can directly inhibit Bax or Bak. This capacity to restrain Bax and Bak is presumably dependent on how much SPPV14 is sequestered by those BH3-only proteins that target it (Fig. 6 and Table 1), since overexpression of Bim, tBid, and Puma neutralizes the antiapoptotic effect of SPPV14 (Fig. 6E) in colony-forming assays.
Fig 8.

SPPV14 inhibits both Bax and Bak and functionally compensates for endogenous prosurvival Bcl-2. (A) SPPV14 inhibits both Bak- and Bax-mediated apoptosis. Shown is the viability of SPPV14-expressing Bak- or Bax-deficient MEFs treated for 24 h or 48 h with etoposide (10 mM). (B) Selectivity of BH3-only proteins for mammalian prosurvival Bcl-2 proteins (9, 58). (C) SPPV14 potently inhibits apoptosis even if all endogenous mammalian prosurvival Bcl-2 proteins are neutralized. Shown is colony formation of parental or SPPV14-expressing wild-type or Bax- or Bak-deficient MEFs infected with the Mcl-1-selective ligand BimS2A (43) and cotreated with ABT-737 (1 μM), a combination that inactivates all endogenous mammalian prosurvival proteins expressed in MEFs (9, 67). The data in panels A and C represent means and SD from 2 independent experiments using a clone of each genotype.
In summary, our studies identified SPPV14 as a novel direct inhibitor of Bax and Bak. It also has a highly distinctive binding profile for the proapoptotic Bcl-2 family proteins expressed in mammals.
DISCUSSION
Apoptosis is a potent defense mechanism deployed by higher organisms against viruses, and in turn, viruses have evolved an astonishing array of strategies to ensure their successful proliferation, propagation, and survival (23, 51). Bcl-2-like proteins from numerous viruses, including structural homologs recently recognized in poxviruses (40, 42), have been shown to be of critical importance for subverting host apoptotic defenses. Consequently, loss of viral Bcl-2 proteins often results in apoptosis upon viral infection, as observed for myxoma virus M11L (59) and vaccinia virus F1L (63).
Here, we report the identification and characterization of a novel subgroup of vBcl-2 proteins, the founding members being myxoma virus M11L (25, 40) and vaccinia virus F1L (42, 63). Like the M11L gene, all the novel genes we identified encode putative proteins that bear little primary sequence resemblance to mammalian prosurvival Bcl-2 proteins. Even though the overall sequence similarity to M11L is also low, the critical residues forming the canonical binding grove in M11L (40) were conserved in all six proteins, strongly suggesting the presence of an analogous BH3 binding grove in these proteins (Fig. 1 and 2). Despite the presence of these critical residues, unexpectedly, not all 6 identified M11L orthologs harbored antiapoptotic activity (Fig. 2D). SPV12 (from swinepox virus), gp011L (from shope fibroma virus), and DPV084 (from deerpox virus) did not inhibit etoposide-induced apoptosis when expressed in MEFs. Close inspection of the residues in the putative binding grooves of all six orthologs suggests that four residues, M52, T67, L68, and A71, as a group may determine antiapoptotic activity (see Fig. S1 in the supplemental material). Notably, the only sequence differences between DPV83 and -84 are T25M, A71S, and A101T. With A71 located in the putative binding grove, it is tempting to speculate that the A71S substitution is responsible for the observed difference in antiapoptotic activity. Although the finding that not all six identified M11L orthologs are antiapoptotic was unexpected, it is well established that simple fold conservation in the case of Bcl-2-like proteins is not sufficient for maintaining function. As shown for the vaccinia virus Bcl-2-like proteins A52 and B14 (26), the presence of a Bcl-2 fold does not automatically result in an ability to inhibit Bcl-2-mediated apoptosis. In contrast, both A52 (26) and B14 (10) act on the NF-κB pathway. Similarly, N1L from vaccinia virus also adopts a Bcl-2 fold (2, 12) and acts on NF-κB (7). Importantly, N1L was also shown to inhibit Bcl-2-mediated apoptosis (12), thus displaying dual functionality, which is mediated by two independent sites on the protein (45). It is possible that the three M11L orthologs that were inactive in our apoptosis assays act on NF-κB; however, this remains to be tested experimentally.
In addition to the structure-based sequence analysis, our detailed characterization of sheeppox virus SPPV14 and the previously reported deerpox virus DPV022 (4) support the idea that these viral proteins adopt a Bcl-2-like fold. Like the mammalian prosurvival proteins and myxoma virus M11L, a number of BH3-only proteins can bind SPPV14 (Fig. 6 and Table 1). The binding pattern we observed is unique: Puma can bind and antagonize SPPV14, but not M11L (Fig. 6). Furthermore, SPPV14 can act to directly counter the cell death mediators Bax and Bak, when tested directly (Fig. 7 and 8), in accordance with its ability to bind them (Fig. 6 and Table 1). Considering the similarities in the BH3 domain binding profile and activity against Bax and Bak, the additional ability of SPPV14 to engage Puma, which is absent in M11L, is striking. Among the currently characterized Bcl-2-like proteins from poxviruses, only ORFV125 (65) appears to engage Puma, in addition to Bim, Bik, Hrk, Noxa, and Bax, whereas with F1L (21, 42), N1L (2, 12), and FWPV039 (3, 5), a Puma interaction is not observed. Although the importance of the SPPV14-Puma interaction is currently not established, it is tempting to speculate that, based on the high IC50 of 63 nM, the interaction may be functionally relevant.
Functionally, SPPV14 appears to be equipotent to M11L in apoptosis inhibition (Fig. 2 and 3), despite the observed differences in BH3 domain binding. In addition to interactions with Bcl-2 family members (40, 59), M11L has also been reported to form part of the mitochondrial permeability transition pore complex (20) and to intersect with death receptor-mediated apoptosis signaling (59). While it is unclear if SPPV14 also harbors such additional functionality with respect to the mitochondrial permeability transition pore complex, we did not observe an effect of either SPPV14 or M11L on Fas ligand-mediated death signaling (see Fig. S2 in the supplemental material).
In the context of vaccinia virus infection, SPPV14 is able to replace F1L (Fig. 4 and 5) to prevent apoptosis. F1L has been shown to have a highly restricted BH3 binding profile and engages only Bim, Bak, and Bax BH3 domains in biosensor assays (21, 42). Although an interaction with Bax was detected using recombinant protein and peptides, no evidence of a similar interaction in the cellular context has been reported. Functionally, F1L has been shown to engage Bak (48, 61) and Bim (56) and to replace the antiapoptotic activity of Mcl-1 (8), although the precise molecular basis for F1L-mediated apoptosis inhibition remains unclear, with evidence both for (56) and against (18, 50) a role for Bim being reported. Although SPPV14 is able to replace F1L in the context of vaccinia virus, it remains to be established if the mechanism utilized by SPPV14 is similar to the one used by F1L. This is particularly pertinent when considering three issues: first, SPPV14 and F1L ligand binding profiles are substantially different, and second, F1L does not appear to inhibit Bax directly, whereas SPPV14 appears to be M11L-like and to act by neutralizing both Bax and Bak. Third, SPPV14 expression in our experimental system was driven by the strong synthetic poxviral thymidine kinase (17) locus early/late promoter, leading to expression levels and patterns for SPPV14 that are likely to be different from those for endogenous F1L.
Taken together and considering that both vaccinia virus F1L and myxoma virus M11L play key roles for these viruses, we speculate that SPPV14 performs a similar role in sheeppox virus. Key sequence regions in the Bcl-2 family that regulate apoptosis are typically highly conserved. In sheep, Bim harbors two amino acid substitutions at the periphery of the BH3 domain, whereas the Bax BH3 domain is fully conserved, suggesting that at least the SPPV14 interactions with Bim and Bax are likely to be relevant in the context of the sheeppox virus natural host.
By using M11L as bait, we identified a family of novel antiapoptotic factors expressed by poxviruses. Our functional and biophysical characterization of sheeppox virus SPPV14 strongly suggested that SPPV14 adopts a Bcl-2 fold in spite of scant sequence similarity. Interestingly, while SPPV14 seems to play a prominent role in apoptosis inhibition, it does not impact upon the activity of NF-κB, unlike certain other vBcl-2 proteins expressed by vaccinia virus (26). Thus, we anticipate that the detailed characterization of vBcl-2 proteins, such as SPPV14, will continue to unravel the processes that control essential cellular functions in higher organisms.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Fletcher, M. Hinds, S. L. Khaw, B. Smith, and M. Yabal for discussions; Abbott Laboratories for ABT-737; F. Battye, J. Blyth, A. Georgiou, H. Ierino, and A. Wardak for excellent technical assistance; and P. Bouillet, D. Fairlie, S. Gerondakis, E. Lee, H. Rabinowich, and A. Strasser for reagents.
Our work is supported by the ARC (fellowship to T.O.), the Cancer Council of Victoria (fellowships to P.M.C.), the Australian National Health and Medical Research Council (program grant 461221; fellowships to P.M.C., D.C.S.H., and M.K.; and IRIISS grant 361646), the Leukemia and Lymphoma Society (SCOR grant 7413; fellowship to M.K.), the Australian Cancer Research Foundation, the Victorian State Government (OIS grant), the Natural Sciences and Engineering Research Council of Canada (graduate scholarship to S.C.), the Alberta Heritage Foundation for Medical Research (studentship to S.C.), the Canadian Institutes of Health Research, the Howard Hughes Medical Institute, and Alberta Innovates Health Solutions. M.B. is a Tier I Canada Research Chair, a Senior Scholar of Alberta Innovates Health Solutions, and a Howard Hughes Medical Institute Scholar in Infection and Parasitology.
Footnotes
Published ahead of print 15 August 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Afonso CL, et al. 2000. The genome of fowlpox virus. J. Virol. 74:3815–3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aoyagi M, et al. 2007. Vaccinia virus N1L protein resembles a B cell lymphoma-2 (Bcl-2) family protein. Protein Sci. 16:118–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Banadyga L, Gerig J, Stewart T, Barry M. 2007. Fowlpox virus encodes a Bcl-2 homologue that protects cells from apoptotic death through interaction with the proapoptotic protein Bak. J. Virol. 81:11032–11045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Banadyga L, et al. 2010. Deerpox virus encodes an inhibitor of apoptosis that regulates Bak and Bax. J. Virol. 85:1922–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Banadyga L, Veugelers K, Campbell S, Barry M. 2009. The fowlpox virus BCL-2 homologue, FPV039, interacts with activated Bax and a discrete subset of BH3-only proteins to inhibit apoptosis. J. Virol. 83:7085–7098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barry M, et al. 2000. Granzyme B short-circuits the need for caspase 8 activity during granule-mediated cytotoxic T-lymphocyte killing by directly cleaving Bid. Mol. Cell. Biol. 20:3781–3794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bartlett N, Symons JA, Tscharke DC, Smith GL. 2002. The vaccinia virus N1L protein is an intracellular homodimer that promotes virulence. J. Gen. Virol. 83:1965–1976 [DOI] [PubMed] [Google Scholar]
- 8. Campbell S, Hazes B, Kvansakul M, Colman P, Barry M. 2010. Vaccinia virus F1L interacts with Bak using highly divergent Bcl-2 homology domains and replaces the function of Mcl-1. J. Biol. Chem. 285:4695–4708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen L, et al. 2005. Differential targeting of pro-survival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17:393–403 [DOI] [PubMed] [Google Scholar]
- 10. Chen RA, Ryzhakov G, Cooray S, Randow F, Smith GL. 2008. Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 4:e22 doi:10.1371/journal.ppat.0040022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cleary ML, Smith SD, Sklar J. 1986. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell 47:19–28 [DOI] [PubMed] [Google Scholar]
- 12. Cooray S, et al. 2007. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J. Gen. Virol. 88:1656–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cuconati A, White E. 2002. Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes Dev. 16:2465–2478 [DOI] [PubMed] [Google Scholar]
- 14. Dewson G, Snowden RT, Almond JB, Dyer MJ, Cohen GM. 2003. Conformational change and mitochondrial translocation of Bax accompany proteasome inhibitor-induced apoptosis of chronic lymphocytic leukemic cells. Oncogene 22:2643–2654 [DOI] [PubMed] [Google Scholar]
- 15. DiPerna G, et al. 2004. Poxvirus protein N1L targets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by the tumor necrosis factor superfamily of receptors, and inhibits NF-kappaB and IRF3 signaling by Toll-like receptors. J. Biol. Chem. 279:36570–36578 [DOI] [PubMed] [Google Scholar]
- 16. Douglas AE, Corbett KD, Berger JM, McFadden G, Handel TM. 2007. Structure of M11L: a myxoma virus structural homolog of the apoptosis inhibitor, Bcl-2. Protein Sci. 16:695–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Earl PL, Moss B, Wyatt LS, Carroll MW. 1998. Generation of recombinant vaccinia viruses, p 16.17.11–16.17.19 In Ausubel FM, et al. (ed), Current protocols in molecular biology. Wiley Interscience, New York, NY: [DOI] [PubMed] [Google Scholar]
- 18. Eitz Ferrer P, et al. 2011. Induction of Noxa-mediated apoptosis by modified vaccinia virus Ankara depends on viral recognition by cytosolic helicases, leading to IRF-3/IFN-beta-dependent induction of pro-apoptotic Noxa. PLoS Pathog. 7:e1002083 doi:10.1371/journal.ppat.1002083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Everett H, et al. 2000. M11L: a novel mitochondria-localized protein of myxoma virus that blocks apoptosis of infected leukocytes. J. Exp. Med. 191:1487–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Everett H, et al. 2002. The myxoma poxvirus protein, M11L, prevents apoptosis by direct interaction with the mitochondrial permeability transition pore. J. Exp. Med. 196:1127–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fischer SF, et al. 2006. Modified vaccinia virus Ankara protein F1L is a novel BH3-domain binding protein and acts together with the early viral protein E3L to block virus-associated apoptosis. Cell Death Differ. 13:109–118 [DOI] [PubMed] [Google Scholar]
- 22. Fletcher JI, et al. 2008. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc. Natl. Acad. Sci. U. S. A. 105:18081–18087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. 2008. Viral control of mitochondrial apoptosis. PLoS Pathog. 4:e1000018 doi:10.1371/journal.ppat.1000018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goldmacher VS, et al. 1999. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to bcl-2. Proc. Natl. Acad. Sci. U. S. A. 96:12536–12541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Graham KA, Opgenorth A, Upton C, McFadden G. 1992. Myxoma virus M11L ORF encodes a protein for which cell surface localization is critical in manifestation of viral virulence. Virology 191:112–124 [DOI] [PubMed] [Google Scholar]
- 26. Graham SC, et al. 2008. Vaccinia virus proteins A52 and B14 share a Bcl-2-like fold but have evolved to inhibit NF-kappaB rather than apoptosis. PLoS Pathog. 4:e1000128 doi:10.1371/journal.ppat.1000128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Green DR, Kroemer G. 2004. The pathophysiology of mitochondrial cell death. Science 305:626–629 [DOI] [PubMed] [Google Scholar]
- 28. Griffiths GJ, et al. 1999. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 144:903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hawkins CJ, Wang SL, Hay BA. 1999. A cloning method to identify caspases and their regulators in yeast: identification of Drosophila IAP1 as an inhibitor of the Drosophila caspase DCP-1. Proc. Natl. Acad. Sci. U. S. A. 96:2885–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Henderson S, et al. 1993. Epstein virus-coded BHRF 1 protein, a viral homologue of Bcl-2 protects human B cells from programmed cell death. Proc. Natl. Acad. Sci. U. S. A. 90:8479–8483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hsu Y-T, Youle RJ. 1998. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J. Biol. Chem. 273:10777–10783 [DOI] [PubMed] [Google Scholar]
- 32. Huang DC, et al. 1999. Activation of Fas by FasL induces apoptosis by a mechanism that cannot be blocked by Bcl-2 or Bcl-xL. Proc. Natl. Acad. Sci. U. S. A. 96:14871–14876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang DCS, Cory S, Strasser A. 1997. Bcl-2, Bcl-XL and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene 14:405–414 [DOI] [PubMed] [Google Scholar]
- 34. Huang DCS, O'Reilly LA, Strasser A, Cory S. 1997. The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J. 16:4628–4638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang Q, Petros AM, Virgin HW, Fesik SW, Olejniczak ET. 2002. Solution structure of a Bcl-2 homolog from Kaposi sarcoma virus. Proc. Natl. Acad. Sci. U. S. A. 99:3428–3433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang Q, Petros AM, Virgin HW, Fesik SW, Olejniczak ET. 2003. Solution structure of the BHRF1 protein from Epstein-Barr virus, a homolog of human Bcl-2. J. Mol. Biol. 332:1123–1130 [DOI] [PubMed] [Google Scholar]
- 37. Jabbour AM, et al. 2006. Human Bcl-2 cannot directly inhibit the Caenorhabditis elegans Apaf-1 homologue CED-4, but can interact with EGL-1. J. Cell Sci. 119:2572–2582 [DOI] [PubMed] [Google Scholar]
- 38. Jurak I, Schumacher U, Simic H, Voigt S, Brune W. 2008. Murine cytomegalovirus m38.5 protein inhibits Bax-mediated cell death. J. Virol. 82:4812–4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kinsella TM, Nolan GP. 1996. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum. Gene Ther. 7:1405–1413 [DOI] [PubMed] [Google Scholar]
- 40. Kvansakul M, et al. 2007. A structural viral mimic of prosurvival Bcl-2: a pivotal role for sequestering proapoptotic Bax and Bak. Mol. Cell 25:933–942 [DOI] [PubMed] [Google Scholar]
- 41. Kvansakul M, et al. 2010. Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS Pathog. 6:e1001236 doi:10.1371/journal.ppat.1001236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kvansakul M, et al. 2008. Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ. 15:1564–1571 [DOI] [PubMed] [Google Scholar]
- 43. Lee EF, et al. 2008. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J. Cell Biol. 180:341–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu X, Dai S, Zhu Y, Marrack P, Kappler JW. 2003. The structure of a Bcl-xL/Bim fragment complex: implications for Bim function. Immunity 19:341–352 [DOI] [PubMed] [Google Scholar]
- 45. Maluquer de Motes C, et al. 2011. Inhibition of apoptosis and NF-kappaB activation by vaccinia protein N1 occur via distinct binding surfaces and make different contributions to virulence. PLoS Pathog. 7:e1002430 doi:10.1371/journal.ppat.1002430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. O'Connor L, et al. 1998. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 17:384–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Oltersdorf T, et al. 2005. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435:677–681 [DOI] [PubMed] [Google Scholar]
- 48. Postigo A, Cross JR, Downward J, Way M. 2006. Interaction of F1L with the BH3 domain of Bak is responsible for inhibiting vaccinia-induced apoptosis. Cell Death Differ. 13:1651–1662 [DOI] [PubMed] [Google Scholar]
- 49. Postigo A, Martin MC, Dodding MP, Way M. 2009. Vaccinia-induced epidermal growth factor receptor-MEK signalling and the anti-apoptotic protein F1L synergize to suppress cell death during infection. Cell Microbiol. 11:1208–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Postigo A, Way M. 2012. The vaccinia virus-encoded Bcl-2 homologues do not act as direct Bax inhibitors. J. Virol. 86:203–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Roulston A, Marcellus RC, Branton PE. 1999. Viruses and apoptosis. Annu. Rev. Microbiol. 53:577–628 [DOI] [PubMed] [Google Scholar]
- 52. Sarid R, Sato T, Bohenzky RA, Russo JJ, Chang Y. 1997. Kaposi's sarcoma-associated herpesvirus encodes a functional Bcl-2 homologue. Nat. Med. 3:293–298 [DOI] [PubMed] [Google Scholar]
- 53. Schneider P, et al. 1998. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J. Exp. Med. 187:1205–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smith CA. 1995. A novel viral homologue of Bcl-2 and Ced-9. Trends Cell Biol. 5:344. [DOI] [PubMed] [Google Scholar]
- 55. Strasser A. 2005. The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 5:189–200 [DOI] [PubMed] [Google Scholar]
- 56. Taylor JM, Quilty D, Banadyga L, Barry M. 2006. The vaccinia virus protein F1L interacts with Bim and inhibits activation of the pro-apoptotic protein Bax. J. Biol. Chem. 281:39728–39739 [DOI] [PubMed] [Google Scholar]
- 57. Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. van Delft MF, et al. 2006. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10:389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang G, et al. 2004. Myxoma virus M11L prevents apoptosis through constitutive interaction with Bak. J. Virol. 78:7097–7111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang GQ, et al. 2001. Resistance to granzyme B-mediated cytochrome c release in Bak-deficient cells. J. Exp. Med. 194:1325–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wasilenko ST, Banadyga L, Bond D, Barry M. 2005. The vaccinia virus F1L protein interacts with the proapoptotic protein Bak and inhibits Bak activation. J. Virol. 79:14031–14043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wasilenko ST, Meyers AF, Vander Helm K, Barry M. 2001. Vaccinia virus infection disarms the mitochondrion-mediated pathway of the apoptotic cascade by modulating the permeability transition pore. J. Virol. 75:11437–11448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wasilenko ST, Stewart TL, Meyers AF, Barry M. 2003. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc. Natl. Acad. Sci. U. S. A. 100:14345–14350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Westphal D, et al. 2007. A novel Bcl-2-like inhibitor of apoptosis is encoded by the parapoxvirus ORF virus. J. Virol. 81:7178–7188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Westphal D, et al. 2009. The orf virus inhibitor of apoptosis functions in a Bcl-2-like manner, binding and neutralizing a set of BH3-only proteins and active Bax. Apoptosis 14:1317–1330 [DOI] [PubMed] [Google Scholar]
- 66. White E, et al. 1992. The 19-kilodalton adenovirus E1B transforming protein inhibits programmed cell death and prevents cytolysis by tumor necrosis factor α. Mol. Cell. Biol. 12:2570–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Willis SN, et al. 2005. Pro-apoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 19:1294–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Youle RJ, Strasser A. 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9:47–59 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.