

The FLT3 receptor tyrosine kinase is constitutively activated by internal tandem duplication (ITD) in its juxtamembrane domain or tyrosine kinase domain in 30% of acute myeloid leukemia (AML) cases.1-3 Alternatively, FLT3 can also be activated by mutations in the kinase domain (such as the D835Y mutation, which are observed in 7% of AML cases.1,2 Both in vitro and in vivo data have demonstrated that FLT3-ITD and FLT3-D835Y encode constitutively activated kinases that drive the proliferation and survival of hematopoietic cells. Furthermore, FLT3-ITD mutations confer a bad prognosis in AML,1,4,5 and FLT3 inhibitors are expected to improve the outcome of AML patients in this subgroup. These data provide the rationale for the use of FLT3 kinase inhibitors for the treatment of AML.2
While pre-clinical experiments with cell-based assays and mouse models documented a potent effect of various FLT3 kinase inhibitors, first clinical trials showed only moderate efficacy.2 One complicating factor for the treatment of AML is the heterogeneity of the disease, with FLT3 mutations being only one of the many mutations present in human AML samples. Molecular data also suggest that FLT3 mutations tend to occur late in the multi-step development of AML, limiting the effect of FLT3 inhibitors to the subset of AML cells within the leukemia that harbor FLT3 mutations.
In an ongoing multinational phase II trial of the FLT3 inhibitor AC220 as monotherapy, a complete remission rate of 45% was reported at an interim report at the EHA meeting in 2011.6,7 Despite these promising results, patients treated with AC220 develop resistance to this inhibitor and Smith et al. have demonstrated that this is due to the acquisition of FLT3 kinase mutations.8 In that study, mutations at 3 different positions in the kinase domain of FLT3 were identified that confer resistance to AC220: F691, D835 and Y842.8 These mutations were identified by an in vitro resistance screen, and in AML samples obtained from patients who developed resistance to AC220 treatment. Although these mutations conferred resistance to AC220, defined by a clear shift in the IC50 values compared to FLT3-ITD and by their identification as acquired mutations in relapsed patients during AC220 treatment, they could still be inhibited by higher concentrations of the drug.
Eight years ago, we predicted resistance mutations in the FLT3 kinase domain using an in vitro mutagenesis technology in FLT3-ITD transformed Ba/F3 cells.9 In this way, we identified 3 positions in FLT3 (N676, F691 and G697) that upon mutation conferred high level resistance to PKC412. One of these mutations (N676D) was later also identified as a resistance mutation in an AML patient treated with PKC412, indicating the validity of the in vitro predictions.10 The F691L mutation in FLT3 was recently identified as a resistance mutation in AML patients treated with AC220.8
Here we show that also the G697R and N676D mutations confer resistance to AC220. Ba/F3 cells dependent on the expression of FLT3-ITD or FLT3-ITD with additional N676D, F691I, F691L or G697R mutation were treated with increasing concentrations of AC220. We measured the proliferation of these cells over a period of 24 h and we determined the level of autophosphorylation of FLT3 as a measurement of its activity after 90 min of inhibitor treatment. All mutations conferred resistance to AC220, with G697R and F691L/I mutations being the most resistant, and N676D conferring a lower level of resistance (Figure 1). In addition, we also used the Ba/F3 cells expressing FLT3-D835Y, and we observed that also these cells were highly resistant to AC220 (Figure 1).
Figure 1.
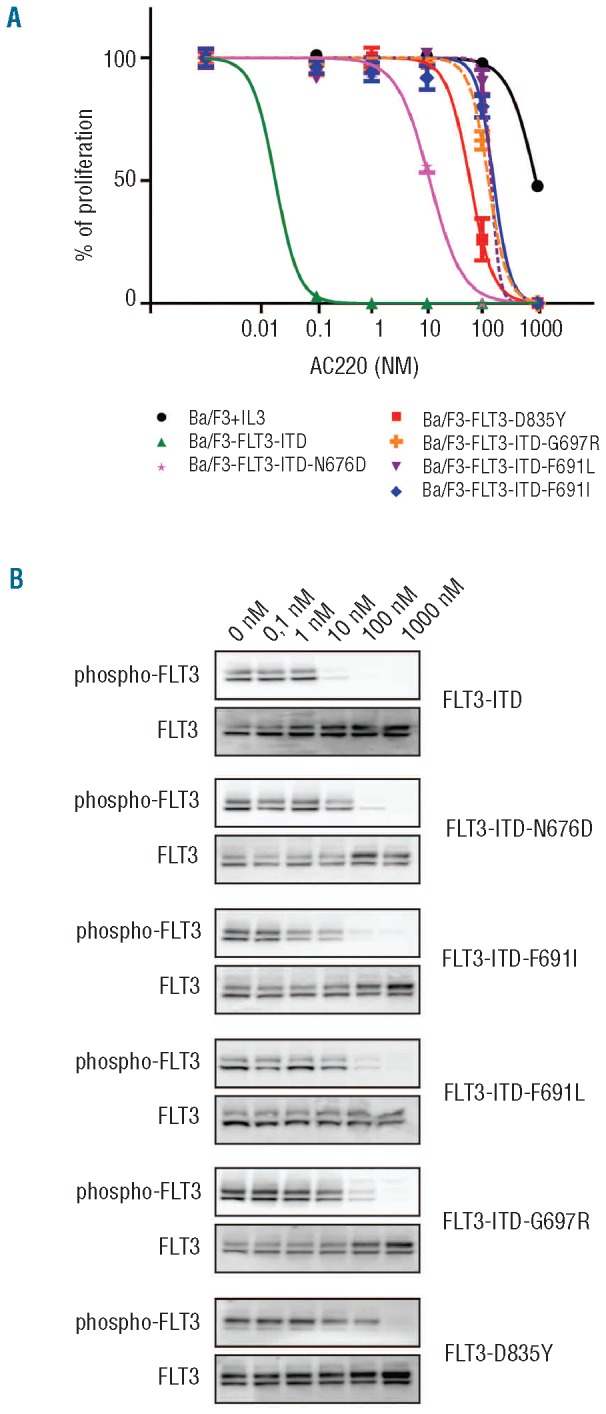
Sensitivity of FLT3-ITD mutants to the AC220 inhibitor. Ba/F3 cells expressing FLT3-ITD (W51 mutation as described by Kelly et al.11), FLT3-D835Y and different FLT3-ITD mutants were treated with increasing concentrations of AC220 and their proliferation was measured over a period of 24 h (A). (B) The same cells were also harvested for Western blot analysis after treatment for 90 min with different concentrations of AC220. The phosphorylation of the different FLT3 proteins was determined using a phospho-FLT3 antibody (Tyr591/3466, Cell Signaling Technology).
Our data show that there are additional mutations that confer resistance to AC220; in particular, the G697R mutation that confers resistance similar to the recently described F691L mutation. All these mutations can be easily acquired by a single nucleotide change in the FLT3 gene sequence. Notably, the F691L, G697R and N676D mutations that confer resistance to AC220 remain sensitive to ponatinib or sorafenib, other known FLT3 inhibitors currently being tested in clinical trials. These results point out that combinations of AC220 and ponatinib or sorafenib may be useful to overcome resistance to single agents.12,13
Acknowledgments
Funding: this study was supported by grants from the KU Leuven (JC), Stichting tegen Kanker (JC), ERC-Starting grant (JC). D.P. is supported by a Ph.D. grant of the Agency for Innovation by Science and Technology (IWT), Flanders, Belgium.
Footnotes
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
Reference
- 1.Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010;116(24):5089-102 [DOI] [PubMed] [Google Scholar]
- 3.Kayser S, Schlenk RF, Londono MC, Breitenbuecher F, Wittke K, Du J, et al. ; German-Austrian AML Study Group (AMLSG). Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009;114(12):2386-92 [DOI] [PubMed] [Google Scholar]
- 4.Wagner K, Damm F, Thol F, Göhring G, Görlich K, Heuser M, et al. FLT3-internal tandem duplication and age are the major prognostic factors in patients with relapsed acute myeloid leukemia with normal karyotype. Haematologica. 2011;96(5):681-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker A, Marcucci G. Impact of molecular prognostic factors in cytogenetically normal acute myeloid leukemia at diagnosis and relapse. Haematologica. 2011;96(5):640-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114(14):2984-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cortes J, et al. A phase ii open-label, ac220 monotherapy efficacy (ace) Study in patients with acute myeloid leukemia (aml) with flt3-itd activating mutations: interim results in 16th Congress of the European Hematology Association (Haematologica, 2011). [Google Scholar]
- 8.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cools J, Mentens N, Furet P, Fabbro D, Clark JJ, Griffin JD, et al. Prediction of resistance to small molecule FLT3 inhibitors: implications for molecularly targeted therapy of acute leukemia. Cancer Res. 2004;64(18):6385-9 [DOI] [PubMed] [Google Scholar]
- 10.Heidel F, Solem FK, Breitenbuecher F, Lipka DB, Kasper S, Thiede MH, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107(1):293-300 [DOI] [PubMed] [Google Scholar]
- 11.Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99(1):310-8 [DOI] [PubMed] [Google Scholar]
- 12.Zirm E, Spies-Weisshart B, Heidel F, Schnetzke U, Böhmer FD, Hochhaus A, et al. Ponatinib may overcome resistance of FLT3-ITD harbouring additional point mutations, notably the previously refractory F691I mutation. Br J Haematol. 2012;157(4):483-92 [DOI] [PubMed] [Google Scholar]
- 13.Lierman E, Lahortiga I, Van Miegroet H, Mentens N, Marynen P, Cools J. The ability of sorafenib to inhibit oncogenic PDGFRbeta and FLT3 mutants and overcome resistance to other small molecule inhibitors. Haematologica. 2007;92(1):27-34 [DOI] [PubMed] [Google Scholar]