

Abstract
Background
Cold storage of platelets reduces bacterial growth and preserves their hemostatic properties better than current procedures do. However, storage at 0°C induces [14-3-3ζ-glycoprotein Ibα] association, 14-3-3ζ release from phospho-Bad, Bad activation and apoptosis.
Design and Methods
We investigated whether arachidonic acid, which also binds 14-3-3ζ, contributes to coldinduced apoptosis.
Results
Cold storage activated P38-mitogen-activated protein kinase and released arachidonic acid, which accumulated due to cold inactivation of cyclooxygenase-1/thromboxane synthase. Accumulated arachidonic acid released 14-3-3ζ from phospho-Bad and decreased the mitochondrial membrane potential, which are steps in the induction of apoptosis. Addition of arachidonic acid did the same and its depletion made platelets resistant to cold-induced apoptosis. Incubation with biotin-arachidonic acid revealed formation of an [arachidonic acid-14-3-3ζ-glycoprotein Ibα] complex. Indomethacin promoted complex formation by accumulating arachidonic acid and released 14-3-3ζ from cyclo-oxygenase-1. Arachidonic acid depletion prevented the cold-induced reduction of platelet survival in mice.
Conclusions
We conclude that cold storage induced apoptosis through an [arachidonic acid-14-3-3ζ-glycoprotein Ibα] complex, which released 14-3-3ζ from Bad in an arachidonic acid-dependent manner. Although arachidonic acid depletion reduced agonist-induced thromboxane A2 formation and aggregation, arachidonic acid repletion restored these functions, opening ways to reduce apoptosis during storage without compromising hemostatic functions post-transfusion.
Key words: platelets, 14-3-3ζ, arachidonic acid, Glycoprotein Ibα, apoptosis, cold storage
Introduction
Current protocols for the storage of platelet concentrates recommend a temperature of 22-24°C and a maximum of 7 days.1 Problems associated with the relatively high temperature are the growth of bacteria which occasionally contaminate platelet concentrates and the gradual loss of hemostatic functions. Improvements are sought by cooling the concentrates to 0-4°C, but this introduces new problems as it induces redistribution of the von Willebrand factor (VWF) receptor, glycoprotein (GP) Ibα. The change in GPIbα starts apoptosis,2 platelet destruction by macrophages3 and generation of thromboxane A2 (TxA2) upon rewarming.4
We previously described that the cold-induced change makes GPIbα a “sink” for the adapter protein 14-3-3ζ.2 Formation of a [14-3-3ζ-GPIbα] complex is accompanied by dissociation of [14-3-3ζ-phosphoBad], inducing Bad activation, a fall in mitochondrial membrane potential (ΔΨm) and caspase-9 activation. These reactions drive the surface exposure of phosphatidylserine (PS) which together with GPIbα and surface-expressed P-selectin mediate binding to macrophages and platelet destruction. Cold storage followed by rewarming starts TxA2 formation, which initiates a second wave of apoptosis induction, as do most platelet activating agents.5
At a physiological temperature, VWF binds to GPIbα and activates cytosolic phospholipase A2 (cPLA2), arachidonic acid (AA) release from membrane phospholipids and TxA2 formation.6 cPLA2 is activated through phosphorylation of Ser505 by P38-mitogen-activated protein kinase (P38MAPK).7,8 The major part of released AA is metabolized by cyclo-oxygenase-1 (COX-1) to endoperoxides and further converted by thromboxane synthase to TxA2. Released AA is also a substrate for 12-lipo-oxygenase, which generates hydro (pero)xy-eicosatetraenoic acid9 and possibly for cytochrome P450 mono-oxygenase, which catalyzes formation of 14,15-epoxyeicosatrienoic acid.10 COX-1 is the target of aspirin, which acetylates Ser530 blocking access of AA to the active site.11
The caspase-9 induction followed by phosphatidylserine surface exposure and binding/phagocytosis by macrophages are major responses to the relatively small effect that association with GPIbα might have on 14-3-3ζ localization. We, therefore, searched for other pathways that might contribute to cold-induced apoptosis, acting either in parallel or in synergy with the pathway initiated by GPIbα. A candidate is the release of AA. In U937 phagocytic cells, interference with reacylation/deacylation of membrane phospholipids induces accumulation of free AA and apoptosis.12 Neurons stimulated with AA respond with depolarization of the inner mitochondrial membrane and caspase-3 activation.13 In tumor cells, over-expression of COX-2 to increase AA removal blocks apoptosis. The reduction in cell death correlates inversely with the cellular level of free AA. Conversely, COX-2 inhibition restores the apoptotic response.13
In this report, we describe a novel role for AA in apoptosis induction during cold storage of platelets.
Design and Methods
Materials
The materials used in this study are described in detail in the Online Supplementary Design and Methods.
Platelet isolation and incubations
Human platelets were isolated14 while maximally preventing their activation using free-flow blood collection and discarding the first 2 mL of blood and all collections that showed microaggregates as determined by particle sizing. The procedures were approved by the Medical Ethical Committee of our hospital; the laboratory is certified for ISO-9001:2008. Platelets were resuspended in Hepes-Tyrode (2×1011 cells/L, pH 7.2) and incubated without stirring for 10 min at room temperature (defined as fresh platelets) and 4 h at 0°C followed by 1 h at 37°C to mimic cold storage and post-transfusion conditions,4 unless stated otherwise. Inhibitors used were SQ30741 (25 μM) for TPα, SB203580 (10 μM) for P38MAPK, indomethacin (30 μM) for COX-1, ETI (25 μM) for lipooxygenase and SK&F96365 (30 μM) for cytochrome P450 mono-oxygenase, added 15 min before the incubation at 0°C. To deplete platelets of AA, FAFBSA was present (75 g/L in Hepes-Tyrode, pH 7.2) during the 4 h of incubation at 0°C with concurrent incubation with normal albumin as a control, as applied earlier to platelets.15-17 The platelets were then washed in the presence of PGI2 and left at room temperature for 30 min to restore responsiveness.
Eight-week old strain- and sex-matched C57Bl/6 wild-type mice from Harlan (Boxmeer, the Netherlands) were used for isolation of murine platelets. The experimental protocols were approved by the local ethics committees for animal experiments. Mice were anesthetized with isoflurane and blood was collected by cardiac puncture into a 0.1 volume of 130 mM trisodium citrate and centrifuged (420g, 3 min, 22°C, no brake). The pellet, together with one-third of the red blood cell fraction, was collected and centrifuged again (960g, 1 min, 22°C). Platelets were collected and resuspended in Hepes-Tyrode (pH 6.5), washed in a 0.1 volume of ACD and PGI2 (2700g, 2 min, 22°C) and resuspended in Hepes-Tyrode (pH 7.2) to a final concentration of 2×1011 platelets/L.
Western blots and immunoprecipitation
Platelet suspensions were added to lysis buffer. Proteins were separated by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE). After blocking with Odyssey Blocking buffer, membranes were incubated with primary antibodies (1 μg/mL) and protein bands visualized with an Odyssey Imaging system (LI-COR Biosciences, Lincoln, NE, USA). Quantification was performed with Image-J software (NIH, Bethesda, MD, USA). For immunoprecipitation, 450 μL washed platelets (5×1011 platelets/L) were lysed (15 min, 0°C), centrifuged (10 000g, 10 mins, 4°C) to remove cell debris and mixed with 55 μL (10% vol/vol) protein G beads together with antibody (1 μg/mL, 30 min, 4°C, rotating). Data are expressed as percentages of fresh platelets. Control studies by FACS analysis with inhibitors of ADAM17 (TAPI-2, GM6001) confirmed that GPIbα ectodomain shedding was absent (data not shown).
Flow cytometric analysis
For determination of the mitochondrial membrane potential ΔΨm, a 100 μL platelet suspension was incubated with JC-1 (0.5 μM, 30 min, 37°C) and 10 000 platelets were measured on a FACS Calibur (BD Biosciences, San Jose, CA, USA). In viable cells, the high ΔΨm promotes a directional uptake of JC-1 into the matrix where JC-1 forms J-aggregates (λex 490 nm, λem 570-610 nm). In apoptotic cells, the low ΔΨm preserves the monomeric form (λex 490 nm, λem 535 nm).18 Changes in ΔΨm were calculated from the ratio of platelets in lower- over upperright quartiles and expressed as the ratio of treated to fresh platelets.
P38-mitogen-activated protein kinase activity assays
P38MAPK phosphorylation was measured as described elsewhere.14 The catalytic activity was measured in immunoprecipitates, resuspended in 15 μL kinase buffer containing 50 μM ATP and 2 μg activating transcription factor-2 (ATF-2)-fusion protein. After incubation (30 min, 30 °C), 15 μL sample buffer with DDT was added and phospho-ATF-2 (Thr171) was measured on western blots.
Measurement of free arachidonic acid, conversion of arachidonic acid to thromboxane A2 and cyclo-oxygenase-1 activity
To estimate free AA formation at 0°C, platelet suspensions were incubated for the indicated periods in the presence of SQ30741 (25 μM). Samples were then transferred to an environment at 37°C and incubated for exactly 60 min. The suspensions were put on ice to halt COX-1 activity, centrifuged (15 000xg, 30 s, 22°C) and supernatants were collected for TxA2 analysis. Data were expressed as AA equivalents formed/min/1011 platelets during the 60 min incubation period. To measure the activity of COX-1/Txsynthase at different temperatures, platelets in Hepes-Tyrode preincubated with SB203580 (30 min, 37°C) to block release of endogenous AA were incubated at the indicated temperatures. Then, exogenous AA (50 μM final concentration) was added and 60 min later TxA2 formation was measured. The catalytic activity of COX-1 was measured in platelet lysates by luminal luminescence in a Spectramax-L (MDS Analytic Technology, Sunnyvale, CA, USA) as described by Hohlfeld et al.19
Platelet survival in vivo
Before transfusion, platelets were isolated from donor mice, resuspended in Tyrode pH 6.5 and labeled with CMFDA (2.5 μM, 1 h, 22°C). After washing in the presence of PGI2, platelets were resuspended in Tyrode pH 7.2 and incubated for 4 h at room temperature (control), in buffer containing either 75 g/L FAF-BSA or normal BSA at 0°C and in Tyrode buffer containing 1 μM AA at 0°C. The platelets were then washed in the presence of PGI2 and resuspended to a concentration of 1012 cells/L. After incubation for 30 min at 22°C to inactivate PGI2, 1×108 CMFDA-labeled platelets were injected into the tail vein of syngeneic recipient mice. For recovery and survival determinations, blood samples were collected at 2 min and 2, 24, 48, and 72 h after injection into small vacuum EDTA tubes by mandibular puncture. Then, 1 μL of whole blood was diluted in Tyrode (1/250 by volume), analyzed by FACS, and the percentage of CMFDA-positive platelets was determined in a total of 50 000 platelets per sample. Initial recovery was determined 2 min after injection and survival times were calculated as described previously.20
Statistical analysis
Data are means ± SEM of the results from three mice in each group. Statistical analyses were performed using GraphPad InStat (San Diego, CA, USA) software. Differences were considered statistically significant when the P value was less than 0.05 [between fresh and treated samples (*) and between incubations (*)] Blots are representative examples of three experiments.
Results
Changes in mitochondrial membrane potential induced by arachidonic acid
We and others previously reported that cold storage starts platelet apoptosis through activation of the proapoptotic Bcl-2 protein Bad, depolarization of the mitochondrial mitochondrial membrane (ΔΨm change), and caspase-9 mediated exposure of surface phosphatidylserine.2,21 Figure 1A confirms these observations for the ΔΨm change and shows a 7-fold increase after incubation for 4 h at 0°C. Cold/rewarming initiates formation of TxA2.4 To investigate whether AA metabolites contribute to the induction of apoptosis, platelets were incubated with the COX-1 inhibitor indomethacin, but this treatment did not inhibit the ΔΨm change. Instead, indomethacin induced a significant increase in this apoptosis parameter. Subsequent rewarming (1 h, 37°C) further increased the ΔΨm change This response was inhibited by indomethacin and was, therefore, caused by TxA2, a known platelet apoptosis inducer.5 To identify steps that contributed to apoptosis induction at 0°C, samples were incubated with metabolic inhibitors (Figure 1B). P38MAPK is an upstream regulator of cPLA2 and AA release.8 Blockade of P38MAPK by SB203580 completely suppressed the cold-induced ΔΨm change and, in addition, abolished the effect of indomethacin. Inhibition of the TxA2 receptor, TPα, (SQ30741) had no effect, in agreement with the low TxA2 formation found at 0°C.4 Apart from being a precursor of TxA2 formation through COX-1/Tx-synthase, AA is converted to hydro (pero)xy-eicosatetraenoic acids by 12-lipooxygenase and, possibly, to 14,15-epoxyeicosatrienoic acids by cytochrome P450 mono-oxygenase.9,10 Blockade of the respective pathways with ETI and SK&F96365 (SK&F) in combination with indomethacin did not further increase the ΔΨm change. Apparently, AA triggers apoptosis without being metabolized and indomethacin enhances this response by blocking a residual conversion of AA to TxA2 precursors.
Figure 1.
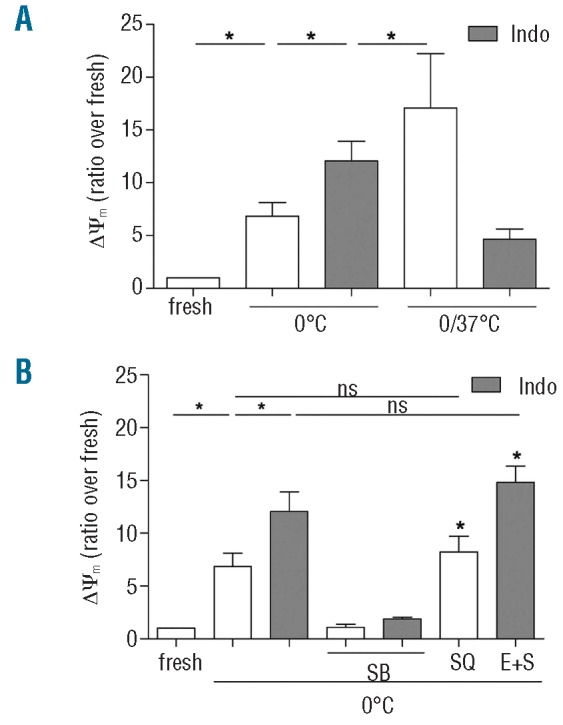
Role of arachidonic acid in cold-induced platelet apoptosis. Apoptosis induction in platelets incubated at 0°C (4 h) and 0/37°C (4 h/1 h) was analyzed by measuring the change in mitochondrial membrane potential (ΔΨm) and compared with that of fresh platelets. (A) Platelets in the absence (open bars) and presence of indomethacin (indo, 30 μM f.c., gray bars). (B) Platelets incubated at 0°C (4 h) without and with indomethacin, the P38MAPK-blocker SB203580 (SB; 10 μM), the TPα-blocker SQ30741 (SQ; 25 μM) and the combined (E+S) lipo-oxygenase- (ETI; 25 μM) and cytochrome P450 blockers (SK&F96365, 30 μM). Data are means ± SEM (n=3) with significant difference indicated by an asterisk (P<0.05) compared with fresh platelets and between treatments.
These findings suggest that 0°C incubation activates upstream regulators of AA release and inactivates downstream regulators of AA degradation inducing AA accumulation and ΔΨm change. Analysis of P38MAPK phosphorylation confirmed that the enzyme was activated upon cooling to 0°C and inhibited by SB203580; subsequent rewarming suppressed enzyme phosphorylation (Figure 2A). To understand the temperature dependence of P38MAPK activation, platelets were incubated at different temperatures and the catalytic activity was inferred from the phosphorylation of ATF-2. After incubation for 4 h, optimal activation was found at 10°C. Conversely, the capacity of COX-1 to convert added AA to TxA2 and the catalytic activity of COX-1 were low at 0°C and increased at higher temperature (Figure 2B). A time course at 0°C showed rapid P38MAPK activation after 10 min which was accompanied by accumulation of free AA (Figure 2C). To confirm that free AA has apoptotic properties, ΔΨm was measured in fresh platelets incubated with exogenous AA. There was a dose-dependent increase in ΔΨm change, both at 0 and 37°C (Figure 2D). Apoptosis induction by endogenous AA was measured in cold-stored platelets with normal and lowered AA content (AA-depleted platelets, in short). These platelets were prepared by prior incubation at 0°C in normal BSA and FAF-BSA containing buffer respectively. AA-depleted platelets showed a 60% lower ΔΨm change than controls (Figure 2E). As expected, incubation with FAF-BSA lowered TRAP-induced TxA2 formation and aggregation (Online Supplementary Figure S1A-C). Subsequent repletion of AA stores restored these functions. Importantly, recovery of these functions was not accompanied by the induction of apoptosis (Online Supplementary Figure S1D).
Figure 2.

Free arachidonic acid triggers platelet apoptosis. (A, left panel) Cold storage activates P38MAPK. Platelets without and with the P38MAPK inhibitor SB203580 (SB) were incubated for 10 min at room temperature (fresh), at 0°C (0 and 4 h) and 0/37°C (4 h/1 h), and phosphorylated P38MAPK was measured. (A, right panel) P38MAPK catalytic activity shown by the phosphorylation of ATF-2 after 4 h incubation at the indicated temperatures. (B) Temperature dependence of P38MAPK activity (catalytic assay, ■ - ■), COX-1 activity (bars) and the COX-1/Tx-synthase reaction (O - - O). COX-1 activity measured as relative luminescence units19 showed a 6-fold difference between platelets stored at 0°C or 37°C. Bars show % activity in platelet lysates at 37°C and 0°C (set at 100%). For measurement of the COX-1/Txsynthase reaction, platelets with blocked P38MAPK (SB203580), to prevent release of endogenous AA, were incubated for 60 min with 50 μM exogenous AA at the indicated temperatures and TxA2 was measured. (C) Time course of P38MAPK catalytic activity (■ - ■), and accumulation of free AA (O - - O) during incubation at 0°C. Platelets with blocked TPα-receptor to prevent TxA2-signaling were incubated for the indicated times at 0°C. Then, samples were collected for P38MAPK measurement and for a standard incubation for 60 min at 37°C to allow conversion of free AA to TxA2. (D) Exogenous AA triggers apoptosis. The change in mitochondrial membrane potential (ΔΨm) was measured in platelets incubated with 100 nM-100 μM AA for 10 min at 0°C (■ - ■), and 37°C (O - - O). AA concentrations up to 100 μM did not compromise cell integrity. (E) AA depletion decreases apoptosis. Platelets were incubated (4 h, 0°C) in buffer with normal BSA (open bars) and with FAF-BSA to lower platelet AA content (gray bar), and the ΔΨm change was measured.
At a physiological temperature, AA release contributes to aggregation and secretion through TxA2 formation and extracellular feed-back signaling. To investigate whether at 37°C, AA preserves apoptotic properties, normal and AA-depleted platelets were stimulated with the Ca2+-ionophore A23187, the PAR-1 activator TRAP and the TxA2-mimetic U46619. In all conditions, stimulation led to a change in ΔΨm (Online Supplementary Figure S2A). In AAdepleted platelets, the ΔΨm change was much smaller (A23187) or virtually absent (TRAP, U46619). Thus, apoptosis induction by activators of platelet aggregation occurs mainly through AA. Blockade of the TPα receptor reduced the ΔΨm change by TRAP (Online Supplementary Figure S2B). Apoptosis induction by U46619 was virtually absent, confirming complete blockade of TPα. AA depletion further reduced the TRAP-induced ΔΨm change, but the ionophore response remained unchanged.
Regulation of the [14-3-3ζ-Bad] association by arachidonic acid
A key step in apoptosis induction is the release of 14-3-3ζ adapter protein from [14-3-3ζ-phospho-Bad] complex, inducing dephosphorylation of phospho-Bad, Bad activation and further signaling to pro-apoptotic Bax and Bak. We showed earlier that the ΔΨm change in cold-stored platelets is accompanied by a fall in [14-3-3ζ-Bad].2 To investigate whether AA contributes to this process, platelets with blocked TPα were incubated at 0°C (4 h) and subsequently incubated at 37°C (1 h) and the complex was measured in immunoprecipitates of Bad. Cold/rewarming induced a fall in [14-3-3ζ-Bad] (Figure 3A). At 0°C, the addition of AA decreased the complex (Figure 3B) and AA depletion prevented the decrease (Figure 3C). The 0/37°C-induced dephosphorylation of total Ser reported earlier2 was confirmed for phospho-Ser112 and also induced by added AA (Figure 3D). These data show that AA induces dissociation of [14-3-3ζ-Bad] complex and Bad dephosphorylation.
Figure 3.

Arachidonic acid dissociates the [14-3-3ζ-Bad] complex. Measurement of [14-3-3ζ-Bad] in (A) fresh, 0°C- and 0/37°C-treated platelets, (B) fresh platelets and platelets incubated with 1, 10 and 50 μM AA for 10 min at 0°C, and (C) fresh and 0°C-treated platelets incubated with normal BSA (open bar) and FAF-BSA (AA-depleted, gray bar). (The shape of the spots is disturbed by the high albumin content of the medium). (D) Bad-Ser112 dephosphorylation in coldrewarmed platelets and platelets treated with AA.
Regulation of the [14-3-3ζ-cyclo-oxygenase-1] association by arachidonic acid
14-3-3 proteins are phospho-Ser/phospho-Thr binding proteins through specific interactions with Arg-X-X-X-Ser-X-Pro and Arg-Ser-X-Ser-X-Pro sequences.22 COX-1 contains two potential 14-3-3ζ binding sites (Arg60-Thr-Gly-Tyr-Ser-Gly-Pro66 and Arg149-Ile-Leu-Pro-Ser-Val-Pro155; UniProt: KB-P23219). Immunoprecipitates of 14-3-3ζ of fresh platelets with blocked TPα confirmed the association of 14-3-3ζ with COX-1 (Online Supplementary Figure S3A). Cold storage induced a 40% fall and rewarming induced re-association. Addition of AA (0°C) induced a dose-dependent decrease in [14-3-3ζ-COX-1], and AA depletion prevented dissociation (Online Supplementary Figure S3B, C). These findings show that cold storage induced a fall in [14-3-3ζ-COX-1] through formation of free-AA, in parallel with the fall in [14-3-3ζ-Bad]. Rewarming restored the complex, to levels above those in fresh platelets, probably as a result of P38MAPK deactivation and removal of AA by TxA2 formation. The 14-3-3 bindings site Arg149-Ile-Leu-Pro-Ser-Val-Pro155 of COX-1 is located in the catalytic site of COX-1 (amino acids 120-385). Indomethacin binds COX-1 at Tyr355, forming a salt bridge between the carboxylate site of the drug and Arg120, preventing binding of AA at Tyr385.23 Addition of indomethacin to cold-stored platelets should, therefore, displace 14-3-3ζ from COX-1. In fresh platelets, indomethacin induced a 30% fall in [14-3-3ζ-COX-1]. Incubation with AA (0°C) lowered the complex and again indomethacin induced a further decrease (Online Supplementary Figure S3D).
Cold-induced platelet apoptosis by arachidonic acid-mediated [14-3-3ζ−glycoprotein Ibα] association
Earlier studies in our laboratory showed that cold storage increases the 14-3-3ζ association with GPIbα and decreases the 14-3-3ζ association with Bad enabling Bad dephosphorylation and induction of apoptosis.2 To clarify the relation between the GPIbα pathway and the AA pathway, platelets were pretreated with OSGE to remove the GPIbα ectodomain and with SB203580 to arrest P38MAPK-mediated production of free AA. The separate treatments (Figure 4A) and the combination (data not shown) induced complete blockade of cold-induced ΔΨm change. Apparently, GPIbα and free AA are both essential components in the induction of apoptosis. Cold-induced P38MAPK activation and production of AA equivalents were insensitive to OSGE treatment and, therefore, independent of GPIbα (Figure 4B and data not shown).
Figure 4.
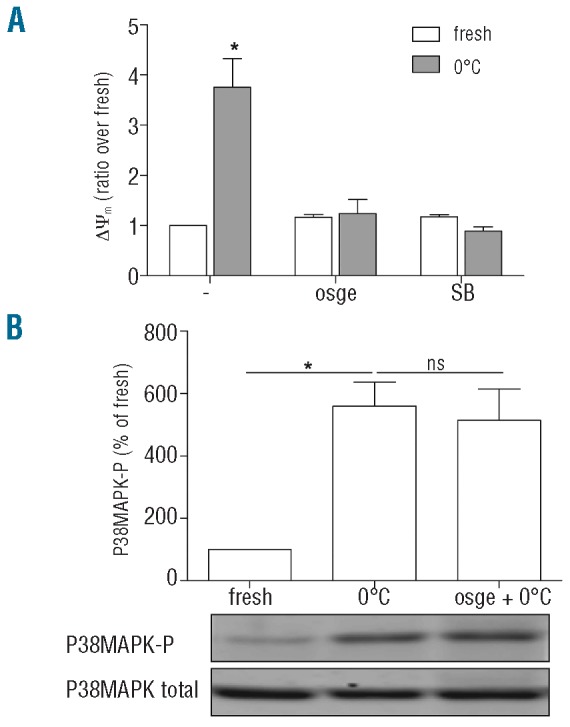
Contribution of GPIbα and arachidonic acid signaling in apoptosis induction. (A) Both GPIbα and free-AA are essential for cold-induced apoptosis. Fresh and cold-treated platelets (4 h, 0°C) were incubated without and with OSGE (to remove GPIbα ectodomain) and SB203580 (to block P38MAPK), and the ΔΨm change was measured. (B) Cold-induced P38MAPK activation does not depend on GPIbα. Phosphorylated P38MAPK was measured in fresh and cold-stored platelets (4 h, 0°C) without and with OSGE treatment.
To clarify the contribution of GPIbα and AA in more detail, platelets were treated with 1, 10 and 50 μM biotinlabeled AA (biotin-AA). This treatment induced a dose-dependent association of 14-3-3ζ (Figure 5A). The fall in [14-3-3ζ-Bad] and [14-3-3ζ-COX-1] found earlier might, therefore, reflect transfer of 14-3-3ζ to AA. Interestingly, GPIbα also bound biotin-AA (Figure 5B) as did COX-1 (Figure 5C). In contrast, there was no binding of Bad to biotin-AA (data not shown). Together with 14-3-3ζ binding to GPIbα at 0°C2 and confirmed in AA-treated platelets (Figure 5D), these data suggest formation of an [AA-14-3-3ζ-GPIbα] complex upon cold storage.
Figure 5.

Free arachidonic acid binds 14-3-3ζ, GPIbα and COX-1. Formation of a [AA-14-3-3ζ-GPIbα] complex. Platelets were incubated with 1, 10 and 50 μM biotin-AA (10 min, 0°C). Lysates from the same incubations were incubated with streptavidin-beads, centrifuged and analyzed for association with (A) 14-3-3ζ, (B) GPIbα and (C) COX-1. (D) AA-induced [14-3-3ζ-GPIbα] association. Platelets were incubated with 50 μM AA (10 min, 0°C) and 14-3-3ζ, was measured in immunoprecipitates of GPIbα.
Arachidonic acid depletion enhances in vivo survival of cold-stored platelets
Apoptosis induction by added AA was also observed in murine platelets and AA depletion inhibited the coldinduced ΔΨm change (Online Supplementary Figure S4A, B). The question of whether AA depletion would improve the survival of cold-stored platelets was addressed by incubating murine platelets for 4 h at room temperature (control) and for 4 h at 0°C in buffer with fatty acid free BSA (FAFBSA platelets), normal BSA (BSA platelets) and in buffer containing 1 μM AA. The platelets were then washed with protection by PGI2 and injected into recipient mice (Figure 6A). The recovery at 2 min after injection was ~80% for the controls stored at room temperature and the cold-treated FAF-BSA platelets (Figure 6B). For cold-treated BSA platelets and AA-treated platelets, recoveries were ~71% and ~56% respectively. The survival of FAF-BSA platelets (~88 hours) was better than that of platelets stored at room temperature (~79 hours) whereas BSAplatelets (~70 hours) and AA-treated platelets (~60 hours) showed much shorter survival. Thus, AA depletion prevents the cold-induced reduction of platelet survival.
Figure 6.

Platelet survival in C57Bl/6 mice. (A-B) Mice platelets labeled with CMFDA were incubated for 4 h at room temperature (control) and for 4 h at 0°C either in buffer with fatty acid free BSA (FAF-BSA platelets), normal BSA (BSA platelets) and in buffer containing 1 μM AA. Then, platelets were washed with protection by PGI2 and injected into recipient mice. Blood was collected 2 min, 2, 24, 48 and 72 h after injection. (A) Recovery of platelets incubated at room temperature (RT) for 2 min (80.8±5.9) was set at 100%; other data points are presented relative to this value. (B) Recoveries and survival times of CMFDA-labeled platelets (means ± SEM of three mice in each group). Data statistically compared to either RT platelets (*) or BSA platelets [(*)]. (C) Schematic representation of apoptosis induction in cold-stored platelets. Cold releases free-AA from membrane phospholipids initiating (i) 14-3-3ζ translocation from phospho-Bad to GPIbα, (ii) dephosphorylation/activation of pro-apoptotic Bad, (iii) displacement of Bak and Bax from pro-survival Bcl-xL by Bad, (iv) Bak/Bax-induced permeabilization of the mitochondrial membrane, (v) cytochrome C (Cyt c) release and (vi) apoptosis.
Discussion
The main findings of this study are: (i) cold storage releases AA from membrane phospholipids, which accumulates since COX-1/Tx-synthase have little activity at low temperature; (ii) released AA induces formation of an [AA-14-3-3ζ-GPIbα] complex, inducing [14-3-3ζ-Bad] dissociation, Bad dephosphorylation and ΔΨm change; (iii) indomethacin increases AA accumulation enhancing apoptosis and releases 14-3-3ζ from COX-1; and (iv) AA depletion reduces apoptosis in vitro and improves the survival of cold-stored platelets in vivo (Figure 6C).
Cold storage activates P38MAPK, an upstream activator of cPLA2 and AA release, and inhibits COX-1/Txsynthase, suppressing the metabolism of AA to TxA2. P38MAPK is a member of the stress-activated protein kinase family and is especially sensitive to thermal stress.24 Chilling induces P38MAPK phosphorylation/activation when the temperature falls below 10°C. At this temperature, the platelet plasma membrane undergoes a phase transition which might be a trigger for P38MAPK phosphorylation. 25 P38MAPK phosphorylation by cold has been found in other types of cells, including hepatocytes,26 endothelial cells,26 brain,27 and in plants in which it is the result of phosphatase PP2A inhibition.28 P38MAPK is an upstream regulator of cPLA2 and mediates TxA2 formation in platelets stimulated with collagen,7 lipopolysaccharide,29 VWF30 and low density lipoprotein at 37°C.31 The Ca2+ increase in cold-stored platelets32 together with the P38MAPK-induced phosphorylation of cPLA2-Ser505/Ser727 translocates the enzyme to the plasma membrane,33,34 inducing the release of AA. Induction of ΔΨm change, but not P38MAPK activation, depends on GPIbα. Chilling triggers desialylation of the receptor, exposing galactose and β-N-acetyl-D-glucosamine residues which become recognition sites for receptors on macrophages and hepatocytes. 35 Apparently, cold storage also makes GPIbα a participant in apoptosis induction.
Released AA induces an [AA-14-3-3ζ-GPIbα] complex by trapping 14-3-3ζ from [14-3-3ζ-Bad]. Dissociation of [14-3-3ζ-Bad] removes the constraint that prevents Bad dephosphorylation and activation.36 The role of AA as a Bad activator is supported by the ΔΨm change upon AA addition and its reduction in AA-depleted platelets. These data support earlier correlations found between free AA and cell death and formation of labeled 14-3-3 in [3H]AAneurons. 37 The introduction of biotin-AA confirmed that platelet 14-3-3ζ binds AA. An unexpected result was that GPIbα also binds biotin-AA. Apparently, cold-induced apoptosis by a GPIbα change2 and by accumulated free-AA (present study) act together in activating Bad. Earlier, Li et al. showed that at 37°C, VWF induced a ΔΨm change, caspase-3 activation and phosphatidylserine exposure in platelets and that a mutated 14-3-3-ζ binding site in GPIbα site abolished apoptosis induction in CHO cells.38 Thus, Bad activation in cold-stored platelets might have properties in common with VWF-stimulated platelets. Its importance in apoptosis induction is illustrated by the prolonged platelet lifespan in Bad-/- mice.39
At 37°C, thrombin and a TxA2-analogue (U46619) affect the downstream target of Bad, the pro-apoptotic Bax, which translocates to the mitochondria and induces a ΔΨm change.5,40 Our studies show that free AA might mediate apoptosis induction by these agents. Without blockade of TPα receptor signaling, the ΔΨm change by TRAP and TxA2-analog was lowered by AA depletion, indicating the contribution of TxA2 formation and TPα signaling to ΔΨm. With TPα receptor blockade, a slight TRAP induction remained present, which completely disappeared in AAdepleted cells. The difference reflects the contribution of free AA in TRAP-induced apoptosis. The Ca2+-ionophore A23187 triggers entry of extracellular Ca2+ and is a potent apoptosis inducer independently of receptor activation. Without TPα blockade, the ΔΨm change was suppressed by AA depletion, but with TPα blockade, a mechanism for apoptosis induction independent of free AA and TxA2 signaling became apparent. Studies by Arachiche et al. confirmed that a high cytosolic Ca2+ concentration is an independent inducer of platelet apoptosis.41
The COX-1 inhibitor indomethacin increased the ΔΨm change at 0°C in the absence of P38MAPK blocker but not with SB203580 present. Thus, at low temperature indomethacin facilitates the accumulation of free AA. Cold storage induced formation of [14-3-3ζ-COX-1]. Complex formation was optimal in fresh platelets, fell after cold storage and was restored to pre-treatment values and more after incubation at 37°C. These changes parallel variations in [14-3-3ζ-Bad].42 The capacity to bind 14-3-3ζ might be important, since indomethacin releases 14-3-3ζ from COX-1. Binding of 14-3-3 proteins depends on the phosphorylation status of specific Ser-residues on target molecules. Factors that control COX-1 Ser-phosphorylation might, therefore, contribute to 14-3-3ζ translocation. Apart from COX-1, 14-3-3 proteins are known to bind pro-apoptotic Bax,43 members of the GPIb-V-IX complex,2,44 GTPase-activating protein Rap1GAP2,45 phosphoinositide 3-kinase,46 c-Raf-1 and insulin receptor substrate-1.47 Trapping of 14-3-3ζ by accumulated AA might, therefore, affect many steps that control platelet functions.
Apoptosis induction by added AA was also observed in mouse platelets and AA depletion inhibited the coldinduced ΔΨm change. In vivo experiments showed that AA depletion improved survival of cold-stored platelets bringing it into the range of survival of platelets stored at room temperature. Phosphatidylserine exposure and platelet binding/phagocytosis by macrophages are downstream steps in cold-induced Bad activation.2 The improved survival of AA-depleted platelets might, therefore, result from decreased Bad activation. A problem of AA depletion is that it also impairs TRAP-induced TxA2 formation and aggregation, which would jeopardize hemostatic functions after transfusion. The finding that this treatment is fully reversible is important, since it opens ways to suppress apoptotic and hemostatic functions during storage with an expected normalization in the recipient. Equally important is the fact that during the recovery phase, the addition of AA does not start apoptosis, indicating that AA-depleted platelets give priority to restoration of AA stores in the plasma membrane.
The combination of cold and AA-depletion might significantly improve preservation conditions for platelet transfusion. Apart from suppressing bacterial growth, cold storage will decrease platelet energy metabolism extending the time the suspension medium supports metabolic ATP regeneration. Cold also increases the chance that artificial media can fully replace plasma and plasma-buffer combinations, lowering risks of virus infections and transfusion reactions.48 Furthermore, cold storage better preserves the platelets' capacity to aggregate and secrete their granule contents upon later stimulation at 37°C.49,50 AA depletion will reduce platelet activation during preparation of platelet concentrates, since mechanical disturbances can activate this metabolic pathway.51 Since this process is reversible, addition of AA prior to transfusion - or when transfused platelets bind plasma AA-albumin in the circulation - will fully restore hemostatic functions. Under in vitro conditions, normal platelets incorporate exogenous AA at a rate of 0.7 μmol/min/1011 platelets, reaching saturation after 90 min (37°C).52 The uptake might be faster in AA-depleted platelets, but the kinetics of this process in vitro and especially in circulating blood and its implications for the arrest of bleeding remain subjects for further studies.
Supplementary Material
Acknowledgments
the authors thank Dr. R.T. Urbanus at the UMCU for discussions.
Funding: this study was supported by a grant from the Landsteiner Foundation of Blood Transfusion Research (LSBR grant n. 0510). Prof. Dr. J.W.N. Akkerman is supported by the Netherlands Thrombosis Foundation.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures: The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Shrivastava M. The platelet storage lesion. Transfus Apher Sci. 2009;41(2):105-13 [DOI] [PubMed] [Google Scholar]
- 2.van der Wal DE, DU VX, Lo KS, Rasmussen JT, Verhoef S, Akkerman JW. Platelet apoptosis by cold-induced glycoprotein Ibalpha clustering. J Thromb Haemost. 2010;8(11):2554-62 [DOI] [PubMed] [Google Scholar]
- 3.Hoffmeister KM, Felbinger TW, Falet H, Denis CV, Bergmeier W, Mayadas TN, et al. The clearance mechanism of chilled blood platelets. Cell. 2003;112(1):87-97 [DOI] [PubMed] [Google Scholar]
- 4.van der Wal DE, Verhoef S, Schutgens RE, Peters M, Wu Y, Akkerman JW. Role of glycoprotein Ibalpha mobility in platelet function. Thromb Haemost. 2010;103(5):1033-43 [DOI] [PubMed] [Google Scholar]
- 5.Tonon G, Luo X, Greco NJ, Chen W, Shi Y, Jamieson GA. Weak platelet agonists and U46619 induce apoptosis-like events in platelets, in the absence of phosphatidylserine exposure. Thromb Res. 2002;107(6):345-50 [DOI] [PubMed] [Google Scholar]
- 6.Garcia A, Quinton TM, Dorsam RT, Kunapuli SP. Src family kinase-mediated and Erk-mediated thromboxane A2 generation are essential for VWF/GPIb-induced fibrinogen receptor activation in human platelets. Blood. 2005;106(10):3410-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saklatvala J, Rawlinson L, Waller RJ, Sarsfield S, Lee JC, Morton LF, et al. Role for p38 mitogen-activated protein kinase in platelet aggregation caused by collagen or a thromboxane analogue. J Biol Chem. 1996;271(12):6586-9 [DOI] [PubMed] [Google Scholar]
- 8.Kramer RM, Roberts EF, Um SL, Borsch-Haubold AG, Watson SP, Fisher MJ, et al. p38 mitogen-activated protein kinase phosphorylates cytosolic phospholipase A2 (cPLA2) in thrombin-stimulated platelets. Evidence that proline-directed phosphorylation is not required for mobilization of arachidonic acid by cPLA2. J Biol Chem. 1996;271(44):27723-9 [DOI] [PubMed] [Google Scholar]
- 9.Maskrey BH, Bermudez-Fajardo A, Morgan AH, Stewart-Jones E, Dioszeghy V, Taylor GW, et al. Activated platelets and monocytes generate four hydroxyphosphatidylethanolamines via lipoxygenase. J Biol Chem. 2007;282(28):20151-63 [DOI] [PubMed] [Google Scholar]
- 10.Zhu Y, Schieber EB, McGiff JC, Balazy M. Identification of arachidonate P-450 metabolites in human platelet phospholipids. Hypertension. 1995;25(4 Pt 2):854-9 [DOI] [PubMed] [Google Scholar]
- 11.Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367(6460):243-9 [DOI] [PubMed] [Google Scholar]
- 12.Perez R, Matabosch X, Llebaria A, Balboa MA, Balsinde J. Blockade of arachidonic acid incorporation into phospholipids induces apoptosis in U937 promonocytic cells. J Lipid Res. 2006;47(3):484-91 [DOI] [PubMed] [Google Scholar]
- 13.Cao Y, Pearman AT, Zimmerman GA, McIntyre TM, Prescott SM. Intracellular unesterified arachidonic acid signals apoptosis. Proc Natl Acad Sci USA. 2000;97(21):11280-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Korporaal SJ, van Eck M, Adelmeijer J, Ijsseldijk M, Out R, Lisman T, et al. Platelet activation by oxidized low density lipoprotein is mediated by CD36 and scavenger receptor-A. Arterioscler Thromb Vasc Biol. 2007;27(11):2476-83 [DOI] [PubMed] [Google Scholar]
- 15.Yoshida N, Aoki N. Release of arachidonic acid from human platelets. A key role for the potentiation of platelet aggregability in normal subjects as well as in those with nephrotic syndrome. Blood. 1978;52(5):969-77 [PubMed] [Google Scholar]
- 16.Ramesha CS, Taylor LA. Measurement of arachidonic acid release from human polymorphonuclear neutrophils and platelets: comparison between gas chromatographic and radiometric assays. Anal Biochem. 1991;192(1):173-80 [DOI] [PubMed] [Google Scholar]
- 17.Surya II, Gorter G, Akkerman JW. Arachidonate transfer between platelets and lipoproteins. Thromb Haemost. 1992;68(6):719-26 [PubMed] [Google Scholar]
- 18.Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, et al. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA. 1991;88(9):3671-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hohlfeld T, Zimmermann N, Weber AA, Jessen G, Weber H, Schror K, et al. Pyrazolinone analgesics prevent the antiplatelet effect of aspirin and preserve human platelet thromboxane synthesis. J Thromb Haemost. 2008;6(1):166-73 [DOI] [PubMed] [Google Scholar]
- 20.Baker GR, Sullam PM, Levin J. A simple, fluorescent method to internally label platelets suitable for physiological measurements. Am J Hematol. 1997;56(1):17-25 [DOI] [PubMed] [Google Scholar]
- 21.Liu Q, Xu L, Jiao SX, Wang TX, Song Y, Wen ZK. Trehalose inhibited the phagocytosis of refrigerated platelets in vitro via preventing apoptosis. Transfusion. 2009;49(10):2158-66 [DOI] [PubMed] [Google Scholar]
- 22.van Hemert MJ, Steensma HY, van Heusden GP. 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. Bioessays. 2001;23(10):936-46 [DOI] [PubMed] [Google Scholar]
- 23.Mancini JA, Riendeau D, Falgueyret JP, Vickers PJ, O'Neill GP. Arginine 120 of prostaglandin G/H synthase-1 is required for the inhibition by nonsteroidal anti-inflammatory drugs containing a carboxylic acid moiety. J Biol Chem. 1995;270(49):29372-7 [DOI] [PubMed] [Google Scholar]
- 24.Cowan KJ, Storey KB. Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. J Exp Biol. 2003;206(Pt 7):1107-15 [DOI] [PubMed] [Google Scholar]
- 25.Gousset K, Tsvetkova NM, Crowe JH, Tablin F. Important role of raft aggregation in the signaling events of cold-induced platelet activation. Biochim Biophys Acta. 2004;1660(1-2):7-15 [DOI] [PubMed] [Google Scholar]
- 26.Laszlo V, Rauen U. Evidence for the involvement of ERK in cold-induced injury [abstract]. Cryobiology 2010; Abstract Annual Meeting Society for Cryobiology. [Google Scholar]
- 27.Zheng G, Chen Y, Zhang X, Cai T, Liu M, Zhao F, et al. Acute cold exposure and rewarming enhanced spatial memory and activated the MAPK cascades in the rat brain. Brain Res. 2008;1239:171-80 [DOI] [PubMed] [Google Scholar]
- 28.Monroy AF, Sangwan V, Dhindsa RS. Low temperature signal transduction during cold acclimation: protein phosphatase 2A as an early target for cold-inactivation. Plant J. 1998;13(5):653-60 [Google Scholar]
- 29.Brooks AC, Menzies-Gow NJ, Wheeler-Jones C, Bailey SR, Cunningham FM, Elliott J. Endotoxin-induced activation of equine platelets: evidence for direct activation of p38 MAPK pathways and vasoactive mediator production. Inflamm Res. 2007;56(4):154-61 [DOI] [PubMed] [Google Scholar]
- 30.Canobbio I, Reineri S, Sinigaglia F, Balduini C, Torti M. A role for p38 MAP kinase in platelet activation by von Willebrand factor. Thromb Haemost. 2004;91(1):102-10 [DOI] [PubMed] [Google Scholar]
- 31.Hackeng CM, Relou IA, Pladet MW, Gorter G, van Rijn HJ, Akkerman JW. Early platelet activation by low density lipoprotein via p38MAP kinase. Thromb Haemost. 1999;82(6):1749-56 [PubMed] [Google Scholar]
- 32.Hoffmeister KM, Falet H, Toker A, Barkalow KL, Stossel TP, Hartwig JH. Mechanisms of cold-induced platelet actin assembly. J Biol Chem. 2001;276(27):24751-9 [DOI] [PubMed] [Google Scholar]
- 33.Borsch-Haubold AG, Bartoli F, Asselin J, Dudler T, Kramer RM, pitz-Castro R, et al. Identification of the phosphorylation sites of cytosolic phospholipase A2 in agonist-stimulated human platelets and HeLa cells. J Biol Chem. 1998;273(8):4449-58 [DOI] [PubMed] [Google Scholar]
- 34.Das S, Rafter JD, Kim KP, Gygi SP, Cho W. Mechanism of group IVA cytosolic phospholipase A(2) activation by phosphorylation. J Biol Chem. 2003;278(42):41431-42 [DOI] [PubMed] [Google Scholar]
- 35.Rumjantseva V, Grewal PK, Wandall HH, Josefsson EC, Sorensen AL, Larson G, et al. Dual roles for hepatic lectin receptors in the clearance of chilled platelets. Nat Med. 2009;15(11):1273-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xing H, Zhang S, Weinheimer C, Kovacs A, Muslin AJ. 14-3-3 proteins block apoptosis and differentially regulate MAPK cascades. EMBO J. 2000;19(3):349-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brock TG. Arachidonic acid binds 14-3-3zeta, releases 14-3-3zeta from phosphorylated BAD and induces aggregation of 14-3-3zeta. Neurochem Res. 2008;33(5):801-7 [DOI] [PubMed] [Google Scholar]
- 38.Li S, Wang Z, Liao Y, Zhang W, Shi Q, Yan R, et al. The glycoprotein Ibalpha-von Willebrand factor interaction induces platelet apoptosis. J Thromb Haemost. 2010;8(2):341-50 [DOI] [PubMed] [Google Scholar]
- 39.Kelly PN, White MJ, Goschnick MW, Fairfax KA, Tarlinton DM, Kinkel SA, et al. Individual and overlapping roles of BH3-only proteins Bim and Bad in apoptosis of lymphocytes and platelets and in suppression of thymic lymphoma development. Cell Death Differ. 2010;17(10):1655-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lopez JJ, Salido GM, Pariente JA, Rosado JA. Thrombin induces activation and translocation of Bid, Bax and Bak to the mitochondria in human platelets. J Thromb Haemost. 2008;6(10):1780-8 [DOI] [PubMed] [Google Scholar]
- 41.Arachiche A, Kerbiriou-Nabias D, Garcin I, Letellier T, Chary-Prigent J. Rapid procoagulant phosphatidylserine exposure relies on high cytosolic calcium rather than on mitochondrial depolarization. Arterioscler Thromb Vasc Biol. 2009;29(11):1883-9 [DOI] [PubMed] [Google Scholar]
- 42.Masters SC, Yang H, Datta SR, Greenberg ME, Fu H. 14-3-3 inhibits Bad-induced cell death through interaction with serine-136. Mol Pharmacol. 2001;60(6):1325-31 [DOI] [PubMed] [Google Scholar]
- 43.Nomura M, Shimizu S, Sugiyama T, Narita M, Ito T, Matsuda H, et al. 14-3-3 Interacts directly with and negatively regulates proapoptotic Bax. J Biol Chem. 2003;278(3):2058-65 [DOI] [PubMed] [Google Scholar]
- 44.Mangin PH, Receveur N, Wurtz V, David T, Gachet C, Lanza F. Identification of five novel 14-3-3 isoforms interacting with the GPIb-IX complex in platelets. J Thromb Haemost. 2009;7(9):1550-5 [DOI] [PubMed] [Google Scholar]
- 45.Hoffmeister M, Riha P, Neumuller O, Danielewski O, Schultess J, Smolenski AP. Cyclic nucleotide-dependent protein kinases inhibit binding of 14-3-3 to the GTPase-activating protein Rap1GAP2 in platelets. J Biol Chem. 2008;283(4):2297-306 [DOI] [PubMed] [Google Scholar]
- 46.Munday AD, Berndt MC, Mitchell CA. Phosphoinositide 3-kinase forms a complex with platelet membrane glycoprotein Ib-IXV complex and 14-3-3zeta. Blood. 2000;96(2):577-84 [PubMed] [Google Scholar]
- 47.Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, et al. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 1997;91(7):961-71 [DOI] [PubMed] [Google Scholar]
- 48.Stroncek DF, Rebulla P. Platelet transfusions. Lancet. 2007;370(9585):427-38 [DOI] [PubMed] [Google Scholar]
- 49.Choi JW, Pai SH. Influence of storage temperature on the responsiveness of human platelets to agonists. Ann Clin Lab Sci. 2003;33(1):79-85 [PubMed] [Google Scholar]
- 50.Babic AM, Josefsson EC, Bergmeier W, Wagner DD, Kaufman RM, Silberstein LE, et al. In vitro function and phagocytosis of galactosylated platelet concentrates after long-term refrigeration. Transfusion. 2007;47(3):442-51 [DOI] [PubMed] [Google Scholar]
- 51.Stevens DE, Joist JH, Sutera SP. Role of platelet-prostaglandin synthesis in shearinduced platelet alterations. Blood. 1980;56(5):753-8 [PubMed] [Google Scholar]
- 52.Bakken AM, Farstad M. The activities of acyl-CoA:1-acyl-lysophospholipid acyltransferase(s) in human platelets. Biochem J. 1992;288(Pt 3):763-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.