

Background: Epithelial to mesenchymal transition (EMT) correlates with increased metastatic potential and poor prognosis.
Results: Secreted eHsp90 induces EMT, matrix metalloproteinase activity and cell motility.
Conclusion: EMT inducing activity of eHsp90 provides a mechanistic basis for its tumorigenic and metastatic function.
Significance: The requirement for eHsp90 in supporting tumorigenic events indicates that targeting eHsp90 may represent a therapeutic approach to improve prostate cancer patient survival.
Keywords: Cell Motility, Epithelial to Mesenchymal Transition, ERK, Hsp90, Matrix Metalloproteinase (MMP), Prostate, Prostate Cancer, Extracellular Hsp90
Abstract
Prostate cancer (PCa) is the most frequently diagnosed malignancy in men, and the second highest contributor of male cancer related lethality. Disease mortality is due primarily to metastatic spread, highlighting the urgent need to identify factors involved in this progression. Activation of the genetic epithelial to mesenchymal transition (EMT) program is implicated as a major contributor of PCa progression. Initiation of EMT confers invasive and metastatic behavior in preclinical models and is correlated with poor clinical prognosis. Extracellular Hsp90 (eHsp90) promotes cell motility and invasion in cancer cells and metastasis in preclinical models, however, the mechanistic basis for its widespread tumorigenic function remains unclear. We have identified a novel and pivotal role for eHsp90 in driving EMT events in PCa. In support of this notion, more metastatic PCa lines exhibited increased eHsp90 expression relative to their lineage-related nonmetastatic counterparts. We demonstrate that eHsp90 promoted cell motility in an ERK and matrix metalloproteinase-2/9-dependent manner, and shifted cellular morphology toward a mesenchymal phenotype. Conversely, inhibition of eHsp90 attenuated pro-motility signaling, blocked PCa migration, and shifted cell morphology toward an epithelial phenotype. Last, we report that surface eHsp90 was found in primary PCa tumor specimens, and elevated eHsp90 expression was associated with increased levels of matrix metalloproteinase-2/9 transcripts. We conclude that eHsp90 serves as a driver of EMT events, providing a mechanistic basis for its ability to promote cancer progression and metastasis in preclinical models. Furthermore, its newly identified expression in PCa specimens, and potential regulation of pro-metastatic genes, supports a putative clinical role for eHsp90 in PCa progression.
Introduction
One of the greatest challenges associated with the widespread and early diagnosis of prostate cancer (PCa)3 in men (1–3) is the inability to accurately distinguish which subset of patients with apparently localized disease will progress to metastatic disease. The propensity of tumor cells to metastasize to bone, rendering the disease incurable (4), is largely responsible for PCa as the second leading cause of male cancer associated lethality (5). Although rising levels of the serum protein prostate-specific antigen are indicative of tumor growth, its expression can be nonspecifically modulated by a number of benign conditions (6, 7) such that this metric alone risk cannot accurately predict risk for progression. Similarly, Gleason score cannot accurately predict disease progression, relapse, or response to treatment (8, 9). These clinical limitations illustrate the pressing need to utilize new and improved molecular indicators of PCa progression.
Activation of the evolutionarily conserved developmental epithelial to mesenchymal transition (EMT) genetic program (10, 11) is implicated as a significant contributor to PCa progression. A universal hallmark of EMT is loss of epithelial cell polarity and acquisition of elongated mesenchymal morphology, concomitant with disruption of cell adhesion, increased cell migration, invasion, and metastasis (12). The adherens junction protein E-cadherin acts as a gatekeeper in suppressing EMT events, and corresponding cell motility and dissemination, by maintaining the cuboidal phenotype and architecture that defines the morphology of normal epithelium (10, 11, 13). As such, loss of E-cadherin function is a conserved and fundamental hallmark associated with early EMT events (10, 11).
Multiple preclinical models provide strong support for EMT in mediating PCa progression. Pathological EMT events have been shown to potentiate the transition from localized prostate adenocarcinoma to invasive carcinoma and subsequent metastasis (14–19). Conversely, repression of EMT events blocks the metastatic potential of PCa cells (20). In clinical specimens, measures of cancer progression correlate with loss of E-cadherin and up-regulation of EMT-inducing transcriptional factors (18, 20–23). EMT events are correlated with metastatic recurrence following surgery (18, 24), and have recently been observed concurrently following androgen withdrawal therapy (25). Therefore, the ability to identify primary tumor cells with an increased propensity to undergo EMT-like events would improve diagnostic approaches to discriminate patients at risk for progression.
Hsp90 is a well known intracellular chaperone responsible for mediating the ATP-dependent folding and signaling function of numerous client proteins, many of which exhibit pro-tumorigenic functions (26–28). In addition to its intracellular localization, Hsp90 is also a secreted and cell surface protein. Extracellular Hsp90 (eHsp90) exhibits distinct function from the intracellular chaperone with its signal transducing activity, in tandem with its receptor low density lipoprotein-related protein (LRP1) (29, 30). eHsp90 supports cell motility and invasion in several cancer cell lines (31–34), and promotes metastatic spread in preclinical models (35–38). Clinically, eHsp90 was first reported as a tumor antigen (39, 40), and more recently found in the serum from patients afflicted with a variety of tumor types (37), including metastatic PCa (41). Although these reports strongly implicate eHsp90 in disease progression, a mechanistic basis for this putative function remains largely unknown.
The present study identifies eHsp90 as a pivotal regulator of E-cadherin function in PCa. Importantly, we demonstrate that modest, physiologically relevant expression of secreted eHsp90 reduces the expression and alters the localization of E-cadherin in phenotypically epithelial cell lines. In addition, eHsp90 modulates numerous genetic events consistent with activation of EMT, as well as promoting diminished cell adhesion, conversion to a more mesenchymal morphology, and increased cell motility. In addition to its role in initiating EMT events, eHsp90 is also essential for sustaining the mesenchymal properties and behavior of more aggressive PCa cell types. Finally, we report that surface eHsp90 is found in primary PCa tumor specimens and is correlated with markedly elevated expression of a subset of pro-tumorigenic eHsp90-regulated transcripts. This newly identified role for eHsp90 as a mediator of EMT events provides a mechanistic basis for its ability to promote cancer progression and metastasis in a variety of preclinical models. Moreover, the newly identified expression of eHsp90 in PCa specimens, coupled with its potential role in modulating gene expression, implicates a clinical role for eHsp90 in PCa progression.
MATERIALS AND METHODS
Western Blot and Antibodies
Cell extracts for Western blot analysis were prepared and performed as described (30) and all blots are representative of a minimum of two independent experiments. Antibodies for N-cadherin (ab12221), Slug (ab27568), Snail (ab63371), and Vimentin (ab8978) were purchased from Abcam. Mouse and rabbit Hsp90 antibodies (ADI-SPA-830, ADI-SPS-771) were from Enzo Lifesciences. E-cadherin (3195), ERK1/2 (4695), P-ERK1/2 (4370), MEK1/2 (9122), P-MEK1/2 (9121), and ZO-1(5406) were from Cell Signaling. FAK (AHO0502) and P-FAK (44–624G) were from Invitrogen. V5 antibody (NB600-381) was from Novus Biologicals. Twist (sc-15393) was from Santa Cruz Biotechnology, and anti-α-tubulin antibody (T6074) was from Sigma. Mouse monoclonal LRP1 antibody (11H4) was purified from a hybridoma cell line (CRL 1936) purchased from ATCC. The hybridoma supernatant was concentrated with a Vivacell 70 concentrator (Sartorius Biolab Products) and purified with an NAb protein G antibody purification kit (Thermo Scientific) according to the manufacturer's instructions, and aliquots were stored at −20 °C.
Reagents
Recombinant Hsp90α protein was obtained from Enzo Life Sciences (ADI-SPP-776). Geldanamycin was obtained from the Experimental Therapeutics Branch, National Cancer Institute, DMAG-N-oxide modified geldanamycin (or nonpermeable GA, NPGA used at 1 μm) was synthesized by Chris Lindsey and Craig Beeson (Pharmaceutical Sciences, Medical University of South Carolina). MMP inhibitors GM6001 (CC1010 used at 1 μm), MMP-2/9 inhibitor IV (444274 used at 1 μm), and MMP-3 inhibitor IV (444243 used at 5 μm) were obtained from EMD Millipore Chemicals. MEK inhibitor U0126 (V112A used at 10 μm) was obtained from Promega.
Cell Culture, Plasmids, and Transfection
The prostate cancer cell lines DU145 and LNCaP were obtained from ATCC, the ARCaP cell pair was purchased from Novicure Biotechnology, and C4–2B was obtained from Viromed. The P69/M12 cell pair was a gift from Joy Ware. The ARCaP and P69/M12 cell pair was maintained in T-media supplemented with 5% heat-inactivated fetal bovine serum, and the LNCaP and C4-2B pair was maintained in their specified media supplemented with 1% HEPES and 1% penicilin/streptomycin in a 5% CO2-humidified atmosphere. The plasmid for shLRP1 was as previously described (29, 30). The sequence for Hsp90α was cloned in-frame to a 5′ signal peptide to direct its extracellular localization, and 3′ to a V5 tag and His6 epitope, with each epitope separated by a flexible linker. The product was assembled in a Gateway entry vector and recombined (CRE) into Plenti6.3V5-Dest (Invitrogen). To obtain viral particles for LRP1 suppression and constitutive eHsp90 secretion, 293FT cells (Invitrogen) were co-transfected with the viral packaging plasmids vesicular stomatitis virus G and PΔR 8.71, along with the corresponding lentiviral vector. All plasmid transfections were performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's specifications. Following viral plasmid transfection, cell medium was harvested at 48 h, the lentiviral supernatant was concentrated by ultracentrifugation, titered, and 5 × 104 particles were used to infect the recipient cells in the presence of Polybrene (8 μg/ml). Cells transduced with eHsp90 virus were selected in blasticidin (Invivogen), whereas GFP-shLRP1-transduced cells were selected by flow cytometry to isolate the highest GFP expressing cells. Selection for eHsp90-transduced cells was performed for 2 weeks, whereupon surviving cells were pooled.
Patient Samples
Human prostate tissue was obtained via an approved research protocol with informed written consent of all participants. Adjacent tissue specimens were snap frozen in liquid nitrogen or fixed in formalin and paraffin-embedded for histological analysis. Tumor tissue specimens were mechanically and enzymatically digested as previously described (42) and dissociated tissue was filtered and passed repeatedly through a 23-gauge needle to generate single cell suspensions. Cells were resuspended and incubated (15 min at 4 °C) with either isotype-matched control or anti-Hsp90 antibody (SPS-771-PE, 1:50), and subjected to FACS analysis (BD FACS Aria II system). Cell populations corresponding to eHsp90high and eHsp90low subtypes were gated and isolated (minimum signal threshold set at 10e2).
RNA Isolation and Real Time-PCR Analysis
RNA purification from patient samples was performed according to the manufacturer's recommendations (Qiagen RNeasy Plus Micro kit; 74034). RNA purification from cells was performed following a TRIzol/chloroform extraction (Qiagen miRNeasy kit; 217004). mRNA from cells and patient material was converted into complimentary DNA (OriGene first strand cDNA synthesis kit; NP100042, and Bio-Rad iScript advanced cDNA synthesis kit; 170–8842, respectively) and amplified. Array samples were prepared according to the manufacturer's instructions (Qiagen RT2 first strand kit; 330401), and further analyzed using an EMT profiler array (Qiagen; PAHS-090A). Biological replicates were utilized for the initial array analysis, and select results were validated with additional biological replicates. Primers (supplemental Table S1) were purchased from IDT.
Cell Motility Assays
Wounding assays were performed as previously described (30). Briefly, a thin sterile pipette tip was used to create a scratch wound in confluent cell monolayers. Pictures were taken at 0 and 16–20 h with an inverted Nikon eclipse TE 2000-S microscope with ×10 magnification, and the extent of migration was calculated by measurement of the gap area using Image J software. For all motility experiments, mitomycin C (5 μg/ml) (Sigma) was added at the time of plating to suppress proliferation.
Gelatin Zymography Assay
The gelatinolytic activity of MMP-2 and MMP-9 was determined by gelatin zymography in 0.1% gelatin, 10% acrylamide gels. Samples were prepared by plating cells at 50% confluence, and conditioned medium (0.25% serum in DMEM) was collected after 36 h. 1 mm O-phenanthroline monohydrate was added to halt MMP activity. Conditioned medium was incubated with gelatin-Sepharose beads (GE Healthcare, 17-0956-01) and the eluted samples were run in nonreducing conditions. Gels were washed with 2.5% Triton X-100 renaturing buffer, incubated in a Tris-HCl/Brij 35 developing buffer (42 °C for 24 h), and subsequently stained with a 5% Coomassie solution.
Hsp90α ELISA
To detect expression of secreted Hsp90α, equivalent cell numbers (7.5 × 105) were plated overnight and replenished with complete media 24 h prior to harvest. Conditioned medium was collected, debris was removed by centrifugation (5 min, 1200 × g), and Hsp90α levels was detected by ELISA (Enzo Life Sciences). Values are presented as fold-change of Hsp90/ml of conditioned medium with the standard deviation shown.
Immunofluorescence
To evaluate cellular localization of E-cadherin and ZO-1, equivalent cell numbers (2.5 × 104) were plated overnight and replenished with complete media for 24 h. Cells were then treated with NPGA, GM6001, MMP-2/9, and MMP-3 inhibitors for the specified times. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in PBS. Immunofluorescence was performed as described (30).
Statistical Analysis
All cell motility and quantitative real time-PCR were performed in triplicate. Data shown are presented as mean ± S.D.; differences in treatment groups are defined as statistically significant at p < 0.05 value, as calculated from Student's t test.
RESULTS
An eHsp90-LRP1 Signaling Pathway Initiates Prostate Cancer Cell Motility
Although eHsp90 has been implicated in promoting cancer cell motility, invasion, and metastasis in several models (30–34, 36–38, 43), its role in PCa has not yet been explored. To investigate whether eHsp90 supports PCa motility, we examined the effects of eHsp90 inhibition in PC3 cells. To inhibit eHsp90, PC3 cells were treated with two different anti-Hsp90 antibodies, an effective approach to neutralize eHsp90 activity and diminish eHsp90 driven cell motility (30, 35–37). As an additional means to inhibit eHsp90 function, cells were treated with NPGA, a small molecule inhibitor specific for eHsp90 (30, 36, 44). Exposure of PC3 cells to either NPGA or blocking antibodies to Hsp90α and β isoforms or to Hsp90α similarly suppressed cell migration over 50% (Fig. 1A and supplemental Fig. S1A). The similar results elicited by these three distinct eHsp90 targeted approaches validate the importance of eHsp90 in PCa motility.
FIGURE 1.
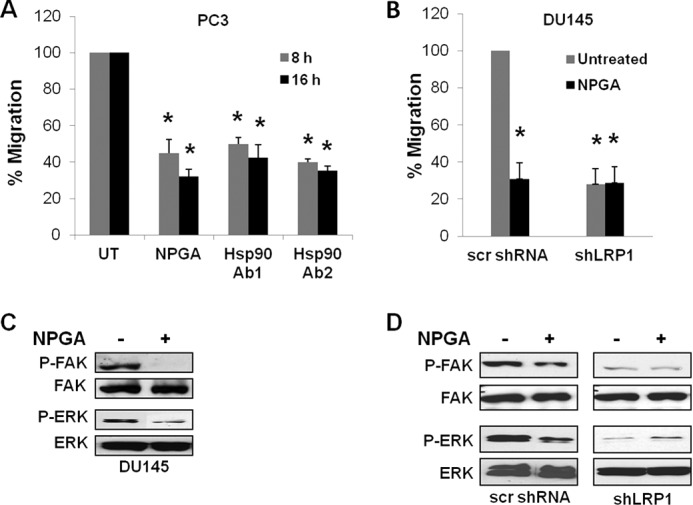
An eHsp90-LRP1 signaling pathway initiates prostate cancer cell motility. A, representative results from a scratch wound assay with PC3 prostate cancer cells following treatment with NPGA (1 μm) or either of two eHsp90 targeting antibodies (SPA-830, Ab1; SPS-771, Ab2). B, similar assay with DU145 cells transduced with either scrambled (scr) shRNA or shLRP1. C, Pharmacologic targeting of eHsp90 with NPGA (1 μm, 16 h) and effects upon FAK and ERK activity in DU145. D, comparison of shLRP1 and NPGA upon FAK and ERK activity. UT refers to untreated vehicle control. Asterisks (*) indicate significance of p ≤ 0.05.
It has been shown that eHsp90 elicits autocrine signaling through LRP1 (29, 30). We reasoned that if eHsp90 was eliciting its pro-motility effects through LRP1, then treatment of cells with either NPGA or suppression of LRP1 would similarly impair cell migration. In support of this notion, treatment of DU145 PCa cells with either NPGA or shLRP1 comparably blocked cell motility (Fig. 1B and supplemental Fig. S1B). NPGA did not further block cell motility in LRP1-suppressed cells, indicating impairment of a similar pathway.
LRP1 has been shown to be required for activation of the pro-motility proteins extracellular signal-regulated kinase (ERK) and focal adhesion kinase (FAK) (45–47). Treatment of DU145 with NPGA inhibited phosphorylation of both FAK and ERK (Fig. 1C). To provide further evidence for an eHsp90-LRP1 signaling pathway, the activation status of FAK and ERK was evaluated in LRP1 suppressed cells. As shown, LRP1 suppression in DU145 cells dramatically diminished both FAK and ERK phosphorylation, with no further effect elicited by NPGA treatment (Fig. 1D). These data indicate that eHsp90-LRP1 plays an essential role in regulating the activation status of pro-motility effectors and supporting the migratory potential of PCa cells.
eHsp90 Is Elevated in More Aggressive PCa Cell Types and Is Essential for Cell Motility
Increased cell motility is commonly associated with cells adopting a mesenchymal morphology (10, 11). We therefore next investigated whether increased eHsp90 expression was associated with more highly motile mesenchymal cell types. To examine this, we analyzed 3 sets of lineage-related prostate cancer cell pairs, with the mesenchymal derivative of each pair representing a more tumorigenic and metastatic derivative of its epithelial counterpart. The ARCaP model, consisting of ARCaPE and ARCaPM subtypes, recapitulates many of the pathological features of PCa (48), and is one of the best characterized cell pairs for investigating EMT events (14, 15, 49). The epithelial ARCaPE disseminates at low frequency, whereas its mesenchymal counterpart ARCaPM is highly aggressive and metastatic (14, 15, 17). EMT events in ARCaPE can be initiated following exposure to a variety of soluble growth factors (22, 50, 51), and sustained EMT activation increases its metastatic potential (19, 51). The cell pair represented by P69 and M12 is another useful model for monitoring EMT events. P69 is a normal nontumorigenic immortalized prostate epithelial cell line, whereas M12 a highly tumorigenic and metastatic subline (52). P69 expresses E-cadherin and is responsive to EMT inducing stimuli that confer tumorigenic properties (53), whereas M12 expresses mesenchymal markers such as vimentin (17, 53). We confirmed the epithelial and mesenchymal nature of these two cell pairs via immunoblot analysis (supplemental Fig. S2A). We included LNCaP and C4-2B as an additional lineage-related model with differential metastatic potential. LNCaP is weakly tumorigenic (54, 55), whereas C4-2B is an osteogenic derivative that is highly metastatic and efficiently forms bone metastases (56). Strikingly, we demonstrate that each of the three metastatic derivatives express severalfold higher expression of eHsp90 relative to their less metastatic epithelial counterparts (Fig. 2A).
FIGURE 2.

eHsp90 is elevated in more aggressive PCa cell types and is essential for cell motility. A, detection of secreted eHsp90α from the conditioned media of indicated cell pairs was assessed by ELISA. Each cell pair is lineage related, with the more metastatic derivative in black. B, scratch wound assay assessed the effects of either exogenous eHsp90 protein (3 day pre-treatment, 3 μg/ml) upon ARCaPE cell motility, or treatment of ARCaPM cells with NPGA. C, similar treatment of epithelial P69 cells with eHsp90, and NPGA treatment of the metastatic counterpart M12. D, effect of shLRP1 upon eHsp90-mediated cell motility in ARCaPE. UT refers to untreated vehicle control. Asterisks (*) indicate significance of p value ≤0.05.
We next evaluated whether the secretion of eHsp90 in these cell pairs influenced tumor cell motility. To assess this, exogenous Hsp90 protein (to mimic eHsp90 secretion) was added to ARCaPE, whereas eHsp90 in ARCaPM was targeted by NPGA. Addition of eHsp90 elicited a greater than 3-fold increase in ARCaPE cell motility, whereas inhibition of eHsp90 function in ARCaPM resulted in a 5-fold reduction of cell motility (Fig. 2B and supplemental Fig. S2B). Similar trends were noted in the P69/M12 pair (Fig. 2C and Fig. S2C). These findings solidify a causal relationship between eHsp90 expression and cell motility.
To establish whether eHsp90 elicited its effects via autocrine signaling through LRP1, we evaluated the impact of LRP1 suppression upon eHsp90-driven cell motility in ARCaPE. We show that down-regulation of LRP1 suppressed ARCaPE basal migration, and completely blocked eHsp90-mediated cell motility (Fig. 2D and supplemental Fig. S2D). To provide further support for eHsp90-initiated pro-motility signaling events, we evaluated the ability of eHsp90 to stimulate the MEK-ERK axis, a critical component of cell motility (57). Our results indicate that eHsp90 elicits a rapid activation of MEK-ERK signaling (supplemental Fig. S2E), supporting an autocrine eHsp90-directed signaling pathway in promoting cell motility.
eHsp90 Induces Molecular and Morphological Changes Consistent with an Epithelial to Mesenchymal Transition
Our data are consistent with the premise that increased eHsp90 expression is associated with migratory potential and mesenchymal morphology. Given this correlation, we next examined whether eHsp90 functionally regulated molecular and morphologic events consistent with activation of an EMT. We utilized ARCaPE cells as an EMT responsive model representative of early disease. Our data indicate that eHsp90 suppressed E-cadherin, while increasing the mesenchymal proteins N-cadherin and Twist (Fig. 3A). Strikingly, addition of eHsp90 also induced morphological changes consistent with a mesenchymal phenotype, such as an elongated fibroblastic morphology, and conversion from a tightly packed epithelial cobblestone pattern to a loosely packed scattered phenotype. To assess the possible role of eHsp90 in supporting the mesenchymal phenotype of ARCaPM, eHsp90 was pharmacologically targeted by NPGA. This treatment reduced the mesenchymal marker N-cadherin, concomitantly reduced the extent of cell elongation, and increased cell cohesiveness (Fig. 3B). E-cadherin is repressed in ARCaPM (50) and NPGA treatment was unable to restore this expression.
FIGURE 3.

eHsp90 induces molecular and morphological changes consistent with an epithelial to mesenchymal transition. A, ARCaPE cells treated for the indicated times (1, 3, or 5 day (d)) with exogenous Hsp90 protein, and immunoblot analysis of epithelial E-cadherin (E-cad) and mesenchymal proteins N-cadherin (N-cad) and Twist. Phase-contrast images of cell morphology. B, effects of NPGA treatment of ARCaPM upon E- and N-cadherin expression and corresponding cell morphology. C, analysis of eHsp90 treatment of P69 nontumorigenic cells as in A. D, analysis of NPGA treatment of M12 as in B.
To solidify these trends, we next examined the role of eHsp90 in the P69/M12 cell pair. Addition of eHsp90 to P69 recapitulated a similar transition toward mesenchymal-like characteristics, with a modest reduction of E-cadherin and increased expression N-cadherin and Twist (Fig. 3C). Addition of eHsp90 elicited similar EMT-like morphological changes, such as cell elongation and increased cell scattering. Targeting eHsp90 in the mesenchymal counterpart M12 resulted in decreased N-cadherin and a less dispersive phenotype, resulting in tight cellular clusters resembling epithelial cell types. These data indicate that targeting eHsp90 in mesenchymal cell types facilitates a reversal of a subset of EMT events, resulting in at least a partial mesenchymal to epithelial conversion.
Modest Elevation of eHsp90 Is Sufficient to Suppress E-cadherin Function and Promote Cell Motility
Although treatment of cells with eHsp90 consistently elicited EMT-like events, use of bacterially produced protein is a less than ideal system, due to confounding factors such as batch variation, protein instability, the potential presence of endotoxin, or additional minor protein species in the preparations. To address these concerns and to create a more physiologically relevant and reproducible model, we designed a genetic approach for eHsp90 secretion by fusing a signal peptide to the N terminus of Hsp90α. This approach has been shown to direct the extracellular localization of proteins (58), including the chaperone protein Hsp70 (59). Expression of Hsp90α was chosen due to the relative lack of activity of the Hsp90β isoform in our model (data not shown). Lentivirus expressing V5-tagged Hsp90 (V5-eHsp90) was used to infect ARCaPE cells, whereas ARCaPE cells transduced with V5-lacZ served as the control. Stably transduced ARCaPE (ARCaPE-eHsp90) exhibited a 3-fold increase in eHsp90 secretion, as determined by ELISA analysis for Hsp90α protein (Fig. 4A). The increased secretion was further confirmed by immunoblot analysis of conditioned media for Hsp90α, and by V5 detection, the latter of which is specific for exogenous V5-eHsp90 (Fig. 4A). Intracellular Hsp90 protein levels were not appreciably elevated in ARCaPE-eHsp90 (Fig. 4B). This model replicated the molecular effects of exogenously added protein, and was able to elicit the characteristic cadherin switch associated with EMT events (Fig. 4B). Furthermore, relative to control (ARCaPE-LacZ) cells, ARCaPE-eHsp90 demonstrated a sustained activation of the pro-motility kinase ERK, demonstrated a more mesenchymal morphology, and an ∼2.5-fold increase in cell motility, the latter of which was suppressed by NPGA (Fig. 4C). Consistent with a pro-EMT role, blockade of eHsp90 reduced E-cadherin to control levels (Fig. 4D).
FIGURE 4.

Modest elevation of eHsp90 is sufficient to suppress E-cadherin function and promote cell motility. A, upper panel, ELISA analysis of secreted eHsp90 protein detected from conditioned media collected from parental ARCaPE cells stably transduced with control (lacZ) or V5-tagged eHsp90α lentivirus. Lower panel, immunoblot detection of total (endogenous and exogenous) eHsp90α, or V5 detection of transduced eHsp90 protein. B, immunoblot analysis of cell lysates from ARCaPE-LacZ or ARCaPE-eHsp90 confirmed consistent levels of intracellular eHsp90α (IC Hsp90). Indicated analysis of E- and N-cadherin and ERK activity. C, representative morphology of the indicated ARCaPE cells. Analysis of cell motility of ARCaPE-eHsp90 either untreated or treated with NPGA. D, effect of NPGA upon E-cadherin expression in ARCaPE-eHsp90. E, analysis of E-cadherin localization in ARCaPE-LacZ and ARCaPE-eHsp90 untreated cells, or treated for the indicated times with NPGA. F, membrane localization of ZO1 in ARCaPE-LacZ and ARCaPE-eHsp90 untreated cells, or treated with NPGA for 3 days. Asterisks (*) indicate significance of p value ≤0.05. Scale bar is 50 μm.
We next investigated whether eHsp90 affected the integrity of cellular junctional complexes. Loss of membrane localization of the gap junction protein ZO-1 frequently accompanies the disruption of cell polarity during EMT (50, 60, 61). As clearly demonstrated, diminished expression and protein mislocalization of both E-cadherin and ZO-1 were observed in ARCaPE-eHsp90 compared with control cells (Fig. 4, E and F). A similar mislocalization of E-cadherin and ZO-1 was observed in a parallel genetic model of eHsp90-transduced P69 cells (P69-eHsp90) (supplemental Fig. S3A). To confirm the specific role of eHsp90 in modulating junctional complexes, we evaluated the ability of NPGA to normalize these structures in the ARCaPE model. As shown, both E-cadherin and ZO-1 protein localization was restored to regions of cell-cell contact, corresponding with the restoration of tightly packed cells with epithelial morphology (Fig. 4, E and F).
To explore the possible therapeutic implications of eHsp90 treatment, we next examined whether eHsp90 inhibition in more aggressive mesenchymal cell lines might restore junctional complex integrity. Although NPGA did not elicit a re-expression of E-cadherin in ARCaPM, it partially restored membrane localization of ZO-1 (supplemental Fig. S3B). Pharmacologic targeting of eHsp90 in M12 cells dramatically restored the junctional localization of E-cadherin and elicited a modest effect upon ZO-1 (supplemental Fig. S3B). Taken together, our results indicate that eHsp90 is a pivotal regulator of the junctional complexes that influence epithelial and mesenchymal morphology.
eHsp90 Modulates the Expression of Multiple Genes Associated with EMT Activation
To further strengthen the EMT-initiating role of eHsp90, we evaluated its ability to modulate additional EMT-associated transcripts. To monitor the temporal effects of eHsp90 action, RNA was harvested from ARCaPE cells following exposure to eHsp90 protein for the indicated times. The resultant heat map from a qRT-PCR array demonstrates that eHsp90 regulates a large number of EMT transcripts (Fig. 5A), including N-cadherin, as supported by our protein expression data. Longer eHsp90 protein treatments most effectively increased the core EMT mediators Snail, Slug, Zeb1, and Zeb2. Changes were also observed in cytoskeletal, integrin/ECM proteins, and proteolytic proteins.
FIGURE 5.

eHsp90 modulates the expression of multiple genes associated with EMT activation. A, a focused EMT qRT-PCR array was utilized to assess EMT-regulated genes modulated by eHsp90 in ARCaPE cells. Samples for array data were derived from two identical biological replicate experiments. B, transcript expression of E-cadherin and the indicated EMT transcriptional effectors from the array were validated by qRT-PCR in untreated (UT) ARCaPE or cells treated with eHsp90 protein for 1, 3, or 5 days (upper panel), whereas a similar analysis was performed for control or ARCaPE-eHsp90 genetically modified cells (lower panel). C, increased expression of proteolytic MMP transcripts was also validated from both protein-treated (upper panel) and ARCaPE-eHsp90-modified cells (lower panel). Quantitative PCR levels were normalized to GAPDH expression. UT refers to untreated vehicle control. Asterisks (*) indicate significance of p value ≤0.05.
To confirm these results, and to more carefully interrogate temporal effects, a subset of these targets was validated in ARCaPE in response to protein exposure for 1, 3, or 5 days. As shown, E-cadherin was progressively suppressed in a time-dependent manner (Fig. 5B). Transcriptional up-regulation of the key EMT mediators Snail, Slug, Zeb1, and Zeb2 was also validated by qRT-PCR. Our findings indicate that Slug and Zeb1 are early responders to eHsp90, whereas Snail and especially Zeb2 exhibit a delayed response. We next evaluated expression of these transcripts in our ARCaPE-eHsp90 genetic model. Whereas the suppression of E-cadherin was consistent, there was a less dramatic, but statistically significant increase in both Snail and Zeb1. Transcripts for Slug were decreased, implicating this effector as eliciting an early and possibly transient role. A robust increase in Zeb2 suggests that this EMT factor may be required for later events, a finding in accordance with the later elevation of this transcript following protein exposure. In addition to these core EMT transcripts, we also evaluated expression of MMPs, zinc-dependent endopeptidases that degrade components of the basement membrane, promote EMT events, and support metastatic spread (62, 63). We found that the MMP-9 transcript expression was up-regulated greater than 3-fold in both protein-treated ARCaPE and ARCaPE-eHsp90 cells (Fig. 5C). In contrast, MMP-3, which also plays an important role in EMT events (64), was preferentially elevated at earlier time points in the protein-treated model, whereas MMP-2 was only up-regulated in the ARCaPE-eHsp90 genetic model.
MMP and ERK Activity Are Required for eHsp90-mediated Motility and EMT Events
It has been demonstrated that eHsp90 directly interacts with MMP-2/9 to modulate proteolytic activity and subsequent cell motility (32, 35, 65, 66). We therefore utilized a standard zymogen gelatinase assay to determine whether the elevated levels of eHsp90 in ARCaPE-eHsp90 would be sufficient to increase MMP activity. As shown, MMP-2 and MMP-9 activity was increased in ARCaPE-eHsp90 cells relative to ARCaPE-LacZ control (Fig. 6A). As expected, this increase was abrogated by NPGA, demonstrating that eHsp90 is the initiating stimulus for increased MMP-2/9 activity. Given that eHsp90 activates ERK in ARCaPE-eHsp90, and the demonstrated ability of ERK to regulate MMP-2/9 activity (67, 68), we evaluated the effects of ERK upon MMP-2/9. ERK inhibition most effectively blocked this activity, supporting the premise that eHsp90-mediated ERK activation is an initiating event for MMP-2/9 activation.
FIGURE 6.

MMP and ERK activity are required for eHsp90-mediated motility and EMT events. A, a gelatin zymography assay was utilized to assay MMP-2/9 activity in control or ARCaPE-Hsp90 cells. For the indicated inhibitors, cells were treated for 2 days prior to media collection. Cells were treated with ERK inhibitor (UO126, 10 μm), MMP-2/9 inhibitor (SB-3CT, 1 μm), or NPGA (1 μm). B, transcript expression of E-cadherin was evaluated in ARCaPE-eHsp90 following a 3-day treatment with the following: NPGA (1 μm), pan-MMP inhibitor (GM6001, 1 μm), MMP-2/9 inhibitor (SB-3CT, 1 μm), MMP-3 inhibitor (inhibitor IV, 5 μm), or ERK inhibitor (UO126, 10 μm). C, immunoblot analysis of E-cadherin and ERK proteins in ARCaPE-eHsp90 following the time-dependent inhibition of the following: MMP-2/9, MMP-3, or ERK. D, the effect of 3 days of MMP and ERK inhibition upon E-cadherin and ZO-1 localization in ARCaPE-LacZ and ARCaPE-eHsp90 was assessed by confocal microscopy. Cells were treated as in A, with inclusion of the pan-MMP inhibitor (GM6001, 1 μm) and the MMP-3 inhibitor (inhibitor IV, 5 μm). E, evaluation of MMP and ERK in directing eHsp90 cell motility following a scratch wound assay. Scale bar is 50 μm. UT refers to untreated vehicle control. Asterisks (*) indicate significance p value ≤0.05.
We next evaluated the effect of MMP-2/9, MMP-3, and ERK upon E-cadherin transcript levels in ARCaPE-eHsp90. As indicated, broad spectrum targeting with GM60001 or MMP-2/9 inhibition robustly increased (∼10-fold) E-cadherin message levels, comparably to NPGA (Fig. 6B). MMP-3 inhibition weakly induced transcript expression, whereas UO126 elicited a potent (∼35-fold) increase. We further investigated whether these trends correlated with changes in E-cadherin protein expression. Inhibition of either MMP-2/9 or MMP-3 comparably restored E-cadherin protein expression by 3 days, whereas UO126 promoted the most dramatic restoration, in accordance with its effects upon message levels (Fig. 6C). It is unclear why MMP inhibition diminished E-cadherin at 5 days, as this trend was not supported via immunofluorescence microscopy (Fig. 6D). It has been shown that E-cadherin may be found in an insoluble membrane fraction (69, 70) when associated with the cytoskeletal matrix at apical junctions, offering a potential explanation. Interestingly, inhibition of either MMP-2/9 or MMP-3 attenuated ERK activity, with a more dramatic effect elicited by the former, indicating that these MMPs collaborate with eHsp90-ERK mediated suppression of E-cadherin.
Given that inhibition of MMP-2/9, MMP-3, and ERK increased E-cadherin expression, we assessed whether this effect trended with the restoration of E-cadherin at cellular junctions, a hallmark of its EMT suppressive epithelial function. As shown (Fig. 6D), pan-MMP inhibition, or targeted inhibition of MMP-3, MMP-2/9, or ERK, comparably re-established E-cadherin expression at the cell membrane in ARCaPE-eHsp90. Functional restoration of junctional complexes was further confirmed by the membrane expression ZO-1. These findings demonstrate that eHsp90-mediated activation of ERK and MMPs is required for the loss in cell polarity that accompanies the transition to a mesenchymal morphology.
We next evaluated whether MMP and ERK activity were important for eHsp90 directed pro-motility function. As shown, pan-MMP inhibition with GM6001 blocked eHsp90-mediated cell motility (Fig. 6E and supplemental Fig. 4). Interestingly, specific targeting of MMP-2/9 or MMP-3 elicited a similar inhibition, highlighting a prominent role for MMP signaling in eHsp90 directed pro-motility function. ERK inhibition comparably diminished cell migration. Therefore, MMP and ERK are critical regulators of the coordinate effects of eHsp90 upon junctional integrity and cell motility.
Detection of eHsp90 Protein and Regulated Transcripts in Human Prostatectomy Tumor Specimens
The ability of eHsp90 to initiate EMT events has important clinical ramifications. We therefore investigated the potential translational relevance of our results and determined whether eHsp90 was found in primary PCa tumors. We reasoned that tumor cells with autocrine eHsp90 function would be represented by a subpopulation exhibiting higher cell surface eHsp90. Therefore, prostatectomy specimens from high risk, locally advanced patients were subjected to FACS sorting, and tumor cell populations were isolated by either high or low surface eHsp90α expression. Interestingly, this approach reproducibly detected a subpopulation of eHsp90high cells corresponding to ∼5% of the total cell number (Fig. 7A). We next investigated whether surface eHsp90 could be utilized as a marker to define a genetically distinct subpopulation of tumor cells. A modest increase in MMP-2 and a marked induction of MMP-9 was evident in these specimens, when comparing the eHsp90high population of tumor cells, relative to each tumor matched eHsp90low population (Fig. 7B). No increases were observed in MMP-3 transcript expression. Intriguingly, these data parallel the trends observed in our constitutively expressing ARCaPE-eHsp90 genetic model (Fig. 5C). Importantly, these findings validate the clinical presence of eHsp90 in primary patient tumors, and further support the notion that eHsp90 may drive genetic events associated with an increased risk for tumor progression. A working model for eHsp90-mediated EMT events and tumor promotion is depicted in Fig. 7C.
FIGURE 7.

Detection of eHsp90 protein and regulated transcripts in human prostatectomy tumor specimens. A, prostate tissue from 2 patients was FACS sorted for eHsp90low and eHsp90high populations using a phycoerythrin-conjugated antibody specific for Hsp90α. In each instance, the subpopulation of eHsp90high cells represented ∼5% of the cell population. Patient 1 was identified as Gleason 3 + 4 (stage T3aNO), whereas Patient 2 was Gleason 4 + 5 (stage T3bNO). B, RNA was harvested from these subpopulations and transcripts for MMP-2, MMP-3, and MMP-9 were evaluated via qRT-PCR. C, proposed mechanism for eHsp90-mediated regulation of cell motility and EMT events. Tumor-secreted eHsp90 functions in an autocrine manner via its receptor LRP1 to transduce ERK phosphorylation. eHsp90-LRP1-ERK signaling subsequently initiates transcription of several pro-EMT transcription factors (Snail/Zeb/Twist), as well as MMPs. MMP activation serves to reinforce sustained ERK activation and E-cadherin suppression through several potential mechanisms (dotted arrows, see text for details). These concurrent processes deregulate junctional complexes (E-cadherin and ZO-1), resulting in a loss of cell polarity, increased migratory potential, and initiation of a subset of EMT events.
DISCUSSION
Although reports demonstrate the ability of eHsp90 to promote cell motility (30–34) and facilitate metastatic spread in preclinical models (36–38, 43), a unifying mechanistic basis for eHsp90 tumorigenic function has not yet been clearly defined. To our knowledge, we are the first to identify eHsp90 as a pivotal initiator of EMT-like events. We demonstrate that eHsp90 increases the cell motility of epithelial ARCaPE and P69 severalfold. This pro-motility function of eHsp90 is dependent upon its impairment of E-cadherin, manifest as diminished protein expression and aberrant cellular localization. Strikingly, eHsp90 elicited dramatic changes in cell morphology, converting cells from an epithelial cuboidal clustered morphology to an elongated mesenchymal morphology with loss of cell-cell contacts. Thus, eHsp90 coordinates a multitude of key events associated with cancer progression, including impaired E-cadherin function, loss of junctional integrity, initiation of EMT, and increased cell motility. Importantly, these events were achieved with relatively modest increases in eHsp90 expression comparable with levels observed in metastatic PCa tumor cells and patient sera, further underscoring that eHsp90 is a potent driver of these processes.
The EMT initiating activity of eHsp90 was further supported by its ability to modulate a wide array of genetic events consistent with this program, including up-regulation of the EMT effectors Snail, Twist, Zeb, and Slug. Not surprisingly, these factors also serve as transcriptional repressors of E-cadherin (71–74), in accordance with the diminished E-cadherin message observed in ARCaPE-eHsp90. The finding that eHsp90 increases transcript expression of the proteolytic enzymes MMP-2, MMP-3, and MMP-9 is relevant given that sustained MMP-2/9 activity increases the tumorigenic and metastatic properties of ARCaPE (19), and initiates EMT events and tumor progression in preclinical models of PCa (19, 75). Although a causal role for MMPs in tumor progression is well known, the relationship between eHsp90 and MMPs is still unfolding. Reports demonstrate that eHsp90 increases the proteolytic activity of MMP-2/9 via direct protein-protein interactions (32, 35, 65, 66). Our results indicate that eHsp90 plays a dual role in up-regulating MMP-2/9 transcript expression, as well as increasing proteolytic function. Given that the eHsp90-regulated EMT transcriptional effectors additionally contribute to MMP expression (76–78), MMP proteolytic activity may be further influenced by these factors, as well as by direct eHsp90-chaperone-mediated mechanisms (79).
Collectively, our data support a model (Fig. 7C), whereby an eHsp90-LRP1 signaling axis activates ERK and MMP activity to promote increased cell motility, impairment of E-cadherin function, and initiation of EMT events. The ability of eHsp90 to sustain ERK activation is significant, given the reported role of ERK as a potent effector of EMT, motility, malignant invasion, and metastasis (57, 80–83). Our data indicate that the relationships among ERK, MMPs, and EMT are complex. eHsp90 rapidly initiates ERK activation, which is required for increased MMP-2/9 activity, supporting ERK as an upstream regulator of MMP function. In addition, it was shown that ERK may be activated by MMP-2/9 in ARCaPE and other cancer models (19). Our data also indicate that ERK may additionally function downstream of MMP-2/9. We therefore propose that eHsp90-mediated MMP-2/9 activity is required to potentiate and sustain ERK activity, implicating a feed-forward mechanism. Our data support a contributory role for MMP-3, and MMP-3 has been implicated in promoting cell motility (64), at least in part via activation of MMP-9 (84).
Further work is required to clarify the precise contribution of these proteins to eHsp90-mediated ERK activation and E-cadherin suppression. Additional functions for MMP-2/9 may be considered (Fig. 7C). First, MMP-2/9 signaling may induce ERK activation via growth factor-initiated receptor activation (19). Although we cannot exclude this possibility, LRP1 silencing precludes ERK activation, indicating that LRP1 plays a dominant role in this process. Second, MMP-2/9 may cleave E-cadherin, thereby attenuating its tumor suppressive function (85). Although this is possible, we did not observe cleavage products,4 and MMP suppressive activity is at least partially due to its modulation of E-cadherin transcript expression. Third, MMP-9 is a ligand for LRP1 (86) and may signal through LRP1 in a nonproteolytic manner to regulate ERK activity (87). Nonetheless, our data conclusively establish that MMP-2/9 and ERK are critical regulators of the coordinate effects of eHsp90 upon junctional integrity and cell motility.
Hsp90 has been detected from patient serum and ascites fluid in a number of cancer types (37, 40, 41, 88), yet the source of this protein remains unclear. In support of a regulated secretory pathway, surface eHsp90 is detected from patients with metastatic melanoma tumors (31). Our discovery that Hsp90α is found on the surface of a subpopulation of primary tumor cells further reinforces a regulated pathway for Hsp90 secretion. A striking finding is the increased expression of MMP-2 and MMP-9 transcripts associated with the eHsp90high population of tumor cells, which supports trends from our cell-based models. Although it is possible that nontumorigenic and/or stromal cells may be present in these preparations, this does not change the interpretation of our results that eHsp90 appears to mark a distinct (presumably tumor) population of cells exhibiting transcripts associated with protumorigenic properties. Our finding that a relatively small subpopulation of eHsp90 expressing tumor cells may contribute to PCa progression also highlights the challenges in identifying a unifying genetic signature indicative of EMT events clinical samples.
Despite its EMT inducing activity, eHsp90 was unable to enforce a permanent EMT, demonstrated by the ability of NPGA to reverse EMT like events in ARCaPE-eHsp90. Within this context, it was remarkable that continued eHsp90 expression was also required to sustain the aggressive properties of metastatic PCa cell types, including cell motility, expression of N-cadherin, and mesenchymal morphology. This constitutive reliance upon eHsp90 may be widespread, supported by the reported ability of eHsp90 targeting to suppress the metastatic potential of breast and other tumor types (35–38). Although the eHsp90-mediated induction of MMP-2/9 has been implicated in several of these models (35, 37), the precise eHsp90-directed molecular events driving metastatic potential remain unresolved. A recent report has also implicated a role for N-cadherin in the invasion and metastasis of PCa (89). In light of these reports, the ability of eHsp90 targeting to attenuate a subset of EMT events may have clinical utility in blocking or delaying cancer progression. Although more mechanistic details need to be elucidated, our data conclusively position eHsp90 as a novel and pivotal effector of tumor cell EMT plasticity.
Supplementary Material
Acknowledgments
We thank Joy Ware for the P69 and M12 cells, Chris Lindsey and Craig Beeson for synthesis of DMAG-N-oxide (NPGA), and Enzo Life Sciences for generous reagent support. Imaging facilities for this research were supported, in part, by Cancer Center Support Grant P30 CA138313 to the Hollings Cancer Center, Medical University of South Carolina. We also thank Phillip Howe and Simon Hayward for critical comments on this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 CA135297 (to J. S. I.) and Training Grant T32 CA119945 (to M. W. H.) and Institutional Research and Academic Career Development Award Training Grant K12 GM081265 (to J. E. B.).

This article contains supplemental Figs. S1–S4 and Table S1.
M. W. Hance, K. Dole, U. Gopal, J. E. Bohonowych, A. Jezierska-Drutel, C. A. Neumann, H. Liu, I. P. Garraway, and J. S. Isaacs, unpublished data.
- PCa
- prostate cancer
- eHsp90
- extracellular Hsp90
- ERK
- extracellular signal-regulated kinase
- MMP
- matrix metalloprotease
- NPGA
- nonpermeable geldanamycin
- EMT
- epithelial to mesenchymal transition
- FAK
- focal adhesion kinase
- LRP1
- low density lipoprotein-related protein
- qRT-PCR
- quantitative reverse transcriptase PCR.
REFERENCES
- 1. Arya M., Bott S. R., Shergill I. S., Ahmed H. U., Williamson M., Patel H. R. (2006) The metastatic cascade in prostate cancer. Surg. Oncol. 15, 117–128 [DOI] [PubMed] [Google Scholar]
- 2. Crawford E. D. (2009) Understanding the epidemiology, natural history, and key pathways involved in prostate cancer. Urology 73, S4–10 [DOI] [PubMed] [Google Scholar]
- 3. Jemal A., Siegel R., Ward E., Hao Y., Xu J., Thun M. J. (2009) Cancer statistics, 2009. CA Cancer J. Clin. 59, 225–249 [DOI] [PubMed] [Google Scholar]
- 4. Efstathiou E., Logothetis C. J. (2010) A new therapy paradigm for prostate cancer founded on clinical observations. Clin. Cancer Res. 16, 1100–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Siegel R., Naishadham D., Jemal A. (2012) Cancer statistics, 2012. CA Cancer J. Clin. 62, 10–29 [DOI] [PubMed] [Google Scholar]
- 6. Culp S., Porter M. (2009) The effect of obesity and lower serum prostate-specific antigen levels on prostate-cancer screening results in American men. BJU Int. 104, 1457–1461 [DOI] [PubMed] [Google Scholar]
- 7. Strope S. A., Andriole G. L. (2010) Prostate cancer screening. Current status and future perspectives. Nat. Rev. Urol. 7, 487–493 [DOI] [PubMed] [Google Scholar]
- 8. Andriole G. L. (2009) Pathology. The lottery of conventional prostate biopsy. Nat. Rev. Urol. 6, 188–189 [DOI] [PubMed] [Google Scholar]
- 9. Markert E. K., Mizuno H., Vazquez A., Levine A. J. (2011) Molecular classification of prostate cancer using curated expression signatures. Proc. Natl. Acad. Sci. U.S.A. 108, 21276–21281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Polyak K., Weinberg R. A. (2009) Transitions between epithelial and mesenchymal states. Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 9, 265–273 [DOI] [PubMed] [Google Scholar]
- 11. Thiery J. P., Acloque H., Huang R. Y., Nieto M. A. (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 [DOI] [PubMed] [Google Scholar]
- 12. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer. The next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 13. Onder T. T., Gupta P. B., Mani S. A., Yang J., Lander E. S., Weinberg R. A. (2008) Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 68, 3645–3654 [DOI] [PubMed] [Google Scholar]
- 14. Zhau H. E., Li C. L., Chung L. W. (2000) Establishment of human prostate carcinoma skeletal metastasis models. Cancer 88, 2995–3001 [DOI] [PubMed] [Google Scholar]
- 15. Xu J., Wang R., Xie Z. H., Odero-Marah V., Pathak S., Multani A., Chung L. W., Zhau H. E. (2006) Prostate cancer metastasis. Role of the host microenvironment in promoting epithelial to mesenchymal transition and increased bone and adrenal gland metastasis. Prostate 66, 1664–1673 [DOI] [PubMed] [Google Scholar]
- 16. Acevedo V. D., Gangula R. D., Freeman K. W., Li R., Zhang Y., Wang F., Ayala G. E., Peterson L. E., Ittmann M., Spencer D. M. (2007) Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell 12, 559–571 [DOI] [PubMed] [Google Scholar]
- 17. Zhang X., Fournier M. V., Ware J. L., Bissell M. J., Yacoub A., Zehner Z. E. (2009) Inhibition of vimentin or β1-integrin reverts morphology of prostate tumor cells grown in laminin-rich extracellular matrix gels and reduces tumor growth in vivo. Mol. Cancer Ther. 8, 499–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mak P., Leav I., Pursell B., Bae D., Yang X., Taglienti C. A., Gouvin L. M., Sharma V. M., Mercurio A. M. (2010) ERβ impedes prostate cancer EMT by destabilizing HIF-1α and inhibiting VEGF-mediated snail nuclear localization. Implications for Gleason grading. Cancer Cell 17, 319–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lue H. W., Yang X., Wang R., Qian W., Xu R. Z., Lyles R., Osunkoya A. O., Zhou B. P., Vessella R. L., Zayzafoon M., Liu Z. R., Zhau H. E., Chung L. W. (2011) LIV-1 promotes prostate cancer epithelial-to-mesenchymal transition and metastasis through HB-EGF shedding and EGFR-mediated ERK signaling. PLoS One 6, e27720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xie D., Gore C., Liu J., Pong R. C., Mason R., Hao G., Long M., Kabbani W., Yu L., Zhang H., Chen H., Sun X., Boothman D. A., Min W., Hsieh J. T. (2010) Role of DAB2IP in modulating epithelial-to-mesenchymal transition and prostate cancer metastasis. Proc. Natl. Acad. Sci. U.S.A. 107, 2485–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kwok W. K., Ling M. T., Lee T. W., Lau T. C., Zhou C., Zhang X., Chua C. W., Chan K. W., Chan F. L., Glackin C., Wong Y. C., Wang X. (2005) Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res. 65, 5153–5162 [DOI] [PubMed] [Google Scholar]
- 22. Graham T. R., Zhau H. E., Odero-Marah V. A., Osunkoya A. O., Kimbro K. S., Tighiouart M., Liu T., Simons J. W., O'Regan R. M. (2008) Insulin-like growth factor I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 68, 2479–2488 [DOI] [PubMed] [Google Scholar]
- 23. Contreras H. R., Ledezma R. A., Vergara J., Cifuentes F., Barra C., Cabello P., Gallegos I., Morales B., Huidobro C., Castellón E. A. (2010) The expression of syndecan-1 and -2 is associated with Gleason score and epithelial-mesenchymal transition markers, E-cadherin and β-catenin, in prostate cancer. Urol. Oncol. 28, 534–540 [DOI] [PubMed] [Google Scholar]
- 24. Zhang Q., Helfand B. T., Jang T. L., Zhu L. J., Chen L., Yang X. J., Kozlowski J., Smith N., Kundu S. D., Yang G., Raji A. A., Javonovic B., Pins M., Lindholm P., Guo Y., Catalona W. J., Lee C. (2009) Nuclear factor-κB-mediated transforming growth factor-β-induced expression of vimentin is an independent predictor of biochemical recurrence after radical prostatectomy. Clin. Cancer Res. 15, 3557–3567 [DOI] [PubMed] [Google Scholar]
- 25. Sun Y., Wang B. E., Leong K. G., Yue P., Li L., Jhunjhunwala S., Chen D., Seo K., Modrusan Z., Gao W. Q., Settleman J., Johnson L. (2012) Androgen deprivation causes epithelial-mesenchymal transition in the prostate. Implications for androgen-deprivation therapy. Cancer Res. 72, 527–536 [DOI] [PubMed] [Google Scholar]
- 26. Isaacs J. S., Xu W., Neckers L. (2003) Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 3, 213–217 [DOI] [PubMed] [Google Scholar]
- 27. Whitesell L., Lindquist S. L. (2005) HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772 [DOI] [PubMed] [Google Scholar]
- 28. Bohonowych J. E., Gopal U., Isaacs J. S. (2010) Hsp90 as a gatekeeper of tumor angiogenesis. Clinical promise and potential pitfalls. J. Oncol. 2010, 412985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheng C. F., Fan J., Fedesco M., Guan S., Li Y., Bandyopadhyay B., Bright A. M., Yerushalmi D., Liang M., Chen M., Han Y. P., Woodley D. T., Li W. (2008) Transforming growth factor α (TGFα)-stimulated secretion of HSP90α. Using the receptor LRP-1/CD91 to promote human skin cell migration against a TGFβ-rich environment during wound healing. Mol. Cell. Biol. 28, 3344–3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gopal U., Bohonowych J. E., Lema-Tome C., Liu A., Garrett-Mayer E., Wang B., Isaacs J. S. (2011) A novel extracellular Hsp90 mediated co-receptor function for LRP1 regulates EphA2-dependent glioblastoma cell invasion. PLoS One 6, e17649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Becker B., Multhoff G., Farkas B., Wild P. J., Landthaler M., Stolz W., Vogt T. (2004) Induction of Hsp90 protein expression in malignant melanomas and metastases. Exp. Dermatol. 13, 27–32 [DOI] [PubMed] [Google Scholar]
- 32. Eustace B. K., Sakurai T., Stewart J. K., Yimlamai D., Unger C., Zehetmeier C., Lain B., Torella C., Henning S. W., Beste G., Scroggins B. T., Neckers L., Ilag L. L., Jay D. G. (2004) Functional proteomic screens reveal an essential extracellular role for hsp90α in cancer cell invasiveness. Nat. Cell Biol. 6, 507–514 [DOI] [PubMed] [Google Scholar]
- 33. Sidera K., Gaitanou M., Stellas D., Matsas R., Patsavoudi E. (2008) A critical role for HSP90 in cancer cell invasion involves interaction with the extracellular domain of HER-2. J. Biol. Chem. 283, 2031–2041 [DOI] [PubMed] [Google Scholar]
- 34. Yang Y., Rao R., Shen J., Tang Y., Fiskus W., Nechtman J., Atadja P., Bhalla K. (2008) Role of acetylation and extracellular location of heat shock protein 90α in tumor cell invasion. Cancer Res. 68, 4833–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stellas D., El Hamidieh A., Patsavoudi E. (2010) Monoclonal antibody 4C5 prevents activation of MMP2 and MMP9 by disrupting their interaction with extracellular HSP90 and inhibits formation of metastatic breast cancer cell deposits. BMC Cell Biol. 11, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsutsumi S., Scroggins B., Koga F., Lee M. J., Trepel J., Felts S., Carreras C., Neckers L. (2008) A small molecule cell-impermeant Hsp90 antagonist inhibits tumor cell motility and invasion. Oncogene 27, 2478–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X., Song X., Zhuo W., Fu Y., Shi H., Liang Y., Tong M., Chang G., Luo Y. (2009) The regulatory mechanism of Hsp90α secretion and its function in tumor malignancy. Proc. Natl. Acad. Sci. U.S.A. 106, 21288–21293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sahu D., Zhao Z., Tsen F., Cheng C. F., Park R., Situ A. J., Dai J., Eginli A., Shams S., Chen M., Ulmer T. S., Conti P., Woodley D. T., Li W. (2012) A potentially common peptide target in secreted heat shock protein-90α for hypoxia-inducible factor-1α-positive tumors. Mol. Biol. Cell 23, 602–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ullrich S. J., Robinson E. A., Law L. W., Willingham M., Appella E. (1986) A mouse tumor-specific transplantation antigen is a heat shock-related protein. Proc. Natl. Acad. Sci. U.S.A. 83, 3121–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luo L. Y., Herrera I., Soosaipillai A., Diamandis E. P. (2002) Identification of heat shock protein 90 and other proteins as tumor antigens by serological screening of an ovarian carcinoma expression library. Br. J. Cancer 87, 339–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burgess E. F., Ham A. J., Tabb D. L., Billheimer D., Roth B. J., Chang S. S., Cookson M. S., Hinton T. J., Cheek K. L., Hill S., Pietenpol J. A. (2008) Prostate cancer serum biomarker discovery through proteomic analysis of α2-macroglobulin protein complexes. Proteomics Clin. Appl. 2, 1223–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garraway I. P., Sun W., Tran C. P., Perner S., Zhang B., Goldstein A. S., Hahm S. A., Haider M., Head C. S., Reiter R. E., Rubin M. A., Witte O. N. (2010) Human prostate sphere-forming cells represent a subset of basal epithelial cells capable of glandular regeneration in vivo. Prostate 70, 491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stellas D., El Hamidieh A., Patsavoudi E. (2010) Monoclonal antibody 4C5 prevents activation of MMP2 and MMP9 by disrupting their interaction with extracellular HSP90 and inhibits formation of metastatic breast cancer cell deposits. BMC Cell Biol. 11, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qin Z., DeFee M., Isaacs J. S., Parsons C. (2010) Extracellular Hsp90 serves as a co-factor for MAPK activation and latent viral gene expression during de novo infection by KSHV. Virology 403, 92–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Orr A. W., Pedraza C. E., Pallero M. A., Elzie C. A., Goicoechea S., Strickland D. K., Murphy-Ullrich J. E. (2003) Low density lipoprotein receptor-related protein is a calreticulin coreceptor that signals focal adhesion disassembly. J. Cell Biol. 161, 1179–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shi Y., Mantuano E., Inoue G., Campana W. M., Gonias S. L. (2009) Ligand binding to LRP1 transactivates Trk receptors by a Src family kinase-dependent pathway. Sci. Signal 2, ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Langlois B., Perrot G., Schneider C., Henriet P., Emonard H., Martiny L., Dedieu S. (2010) LRP-1 promotes cancer cell invasion by supporting ERK and inhibiting JNK signaling pathways. PLoS One 5, e11584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhau H. Y., Chang S. M., Chen B. Q., Wang Y., Zhang H., Kao C., Sang Q. A., Pathak S. J., Chung L. W. (1996) Androgen-repressed phenotype in human prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 93, 15152–15157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. He H., Yang X., Davidson A. J., Wu D., Marshall F. F., Chung L. W., Zhau H. E., Wang R. (2010) Progressive epithelial to mesenchymal transitions in ARCaP E prostate cancer cells during xenograft tumor formation and metastasis. Prostate 70, 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhau H. E., Odero-Marah V., Lue H. W., Nomura T., Wang R., Chu G., Liu Z. R., Zhou B. P., Huang W. C., Chung L. W. (2008) Epithelial to mesenchymal transition (EMT) in human prostate cancer. Lessons learned from ARCaP model. Clin. Exp. Metastasis 25, 601–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Josson S., Nomura T., Lin J. T., Huang W. C., Wu D., Zhau H. E., Zayzafoon M., Weizmann M. N., Gururajan M., Chung L. W. (2011) β2-Microglobulin induces epithelial to mesenchymal transition and confers cancer lethality and bone metastasis in human cancer cells. Cancer Res. 71, 2600–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bae V. L., Jackson-Cook C. K., Maygarden S. J., Plymate S. R., Chen J., Ware J. L. (1998) Metastatic sublines of an SV40 large T antigen immortalized human prostate epithelial cell line. Prostate 34, 275–282 [DOI] [PubMed] [Google Scholar]
- 53. Rojas A., Liu G., Coleman I., Nelson P. S., Zhang M., Dash R., Fisher P. B., Plymate S. R., Wu J. D. (2011) IL-6 promotes prostate tumorigenesis and progression through autocrine cross-activation of IGF-IR. Oncogene 30, 2345–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fu X., Herrera H., Hoffman R. M. (1992) Orthotopic growth and metastasis of human prostate carcinoma in nude mice after transplantation of histologically intact tissue. Int. J. Cancer 52, 987–990 [DOI] [PubMed] [Google Scholar]
- 55. Stephenson R. A., Dinney C. P., Gohji K., Ordóñez N. G., Killion J. J., Fidler I. J. (1992) Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J. Natl. Cancer Inst. 84, 951–957 [DOI] [PubMed] [Google Scholar]
- 56. Thalmann G. N., Anezinis P. E., Chang S. M., Zhau H. E., Kim E. E., Hopwood V. L., Pathak S., von Eschenbach A. C., Chung L. W. (1994) Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 54, 2577–2581 [PubMed] [Google Scholar]
- 57. Smolen G. A., Zhang J., Zubrowski M. J., Edelman E. J., Luo B., Yu M., Ng L. W., Scherber C. M., Schott B. J., Ramaswamy S., Irimia D., Root D. E., Haber D. A. (2010) A genome-wide RNAi screen identifies multiple RSK-dependent regulators of cell migration. Genes Dev. 24, 2654–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Janda C. Y., Li J., Oubridge C., Hernández H., Robinson C. V., Nagai K. (2010) Recognition of a signal peptide by the signal recognition particle. Nature 465, 507–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Figueiredo C., Wittmann M., Wang D., Dressel R., Seltsam A., Blasczyk R., Eiz-Vesper B. (2009) Heat shock protein 70 (HSP70) induces cytotoxicity of T-helper cells. Blood 113, 3008–3016 [DOI] [PubMed] [Google Scholar]
- 60. Thiery J. P. (2003) Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 15, 740–746 [DOI] [PubMed] [Google Scholar]
- 61. Polette M., Gilles C., Nawrocki-Raby B., Lohi J., Hunziker W., Foidart J. M., Birembaut P. (2005) Membrane-type 1 matrix metalloproteinase expression is regulated by zonula occludens-1 in human breast cancer cells. Cancer Res. 65, 7691–7698 [DOI] [PubMed] [Google Scholar]
- 62. Chambers A. F., Matrisian L. M. (1997) Changing views of the role of matrix metalloproteinases in metastasis. J. Natl. Cancer Inst. 89, 1260–1270 [DOI] [PubMed] [Google Scholar]
- 63. Nistico P., Bissell M. J., Radisky D. C. (2012) Cold Spring Harbor Perspect. Biol. 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Radisky D. C., Levy D. D., Littlepage L. E., Liu H., Nelson C. M., Fata J. E., Leake D., Godden E. L., Albertson D. G., Nieto M. A., Werb Z., Bissell M. J. (2005) Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 436, 123–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lagarrigue F., Dupuis-Coronas S., Ramel D., Delsol G., Tronchère H., Payrastre B., Gaits-Iacovoni F. (2010) Matrix metalloproteinase-9 is up-regulated in nucleophosmin-anaplastic lymphoma kinase-positive anaplastic lymphomas and activated at the cell surface by the chaperone heat shock protein 90 to promote cell invasion. Cancer Res. 70, 6978–6987 [DOI] [PubMed] [Google Scholar]
- 66. Song X., Wang X., Zhuo W., Shi H., Feng D., Sun Y., Liang Y., Fu Y., Zhou D., Luo Y. (2010) The regulatory mechanism of extracellular Hsp90α on matrix metalloproteinase-2 processing and tumor angiogenesis. J. Biol. Chem. 285, 40039–40049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ito H., Duxbury M., Benoit E., Clancy T. E., Zinner M. J., Ashley S. W., Whang E. E. (2004) Prostaglandin E2 enhances pancreatic cancer invasiveness through an Ets-1-dependent induction of matrix metalloproteinase-2. Cancer Res. 64, 7439–7446 [DOI] [PubMed] [Google Scholar]
- 68. Yoshida T., Hisamoto T., Akiba J., Koga H., Nakamura K., Tokunaga Y., Hanada S., Kumemura H., Maeyama M., Harada M., Ogata H., Yano H., Kojiro M., Ueno T., Yoshimura A., Sata M. (2006) Spreds, inhibitors of the Ras/ERK signal transduction, are dysregulated in human hepatocellular carcinoma and linked to the malignant phenotype of tumors. Oncogene 25, 6056–6066 [DOI] [PubMed] [Google Scholar]
- 69. Hinck L., Näthke I. S., Papkoff J., Nelson W. J. (1994) Dynamics of cadherin/catenin complex formation. Novel protein interactions and pathways of complex assembly. J. Cell Biol. 125, 1327–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liwosz A., Lei T., Kukuruzinska M. A. (2006) N-Glycosylation affects the molecular organization and stability of E-cadherin junctions. J. Biol. Chem. 281, 23138–23149 [DOI] [PubMed] [Google Scholar]
- 71. Batlle E., Sancho E., Francí C., Domínguez D., Monfar M., Baulida J., García De Herreros A. (2000) The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumor cells. Nat. Cell Biol. 2, 84–89 [DOI] [PubMed] [Google Scholar]
- 72. Cano A., Pérez-Moreno M. A., Rodrigo I., Locascio A., Blanco M. J., del Barrio M. G., Portillo F., Nieto M. A. (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2, 76–83 [DOI] [PubMed] [Google Scholar]
- 73. Grooteclaes M. L., Frisch S. M. (2000) Evidence for a function of CtBP in epithelial gene regulation and anoikis. Oncogene 19, 3823–3828 [DOI] [PubMed] [Google Scholar]
- 74. Yang J., Mani S. A., Donaher J. L., Ramaswamy S., Itzykson R. A., Come C., Savagner P., Gitelman I., Richardson A., Weinberg R. A. (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939 [DOI] [PubMed] [Google Scholar]
- 75. Johnson T. R., Koul S., Kumar B., Khandrika L., Venezia S., Maroni P. D., Meacham R. B., Koul H. K. (2010) Loss of PDEF, a prostate-derived Ets factor is associated with aggressive phenotype of prostate cancer. Regulation of MMP-9 by PDEF. Mol. Cancer 9, 148. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 76. Jordà M., Olmeda D., Vinyals A., Valero E., Cubillo E., Llorens A., Cano A., Fabra A. (2005) Up-regulation of MMP-9 in MDCK epithelial cell line in response to expression of the Snail transcription factor. J. Cell Sci. 118, 3371–3385 [DOI] [PubMed] [Google Scholar]
- 77. Sun L., Diamond M. E., Ottaviano A. J., Joseph M. J., Ananthanarayan V., Munshi H. G. (2008) Transforming growth factor-β1 promotes matrix metalloproteinase-9-mediated oral cancer invasion through snail expression. Mol. Cancer Res. 6, 10–20 [DOI] [PubMed] [Google Scholar]
- 78. Zhao X. L., Sun T., Che N., Sun D., Zhao N., Dong X. Y., Gu Q., Yao Z., Sun B. C. (2011) Promotion of hepatocellular carcinoma metastasis through matrix metalloproteinase activation by epithelial-mesenchymal transition regulator Twist1. J. Cell Mol. Med. 15, 691–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sims J. D., McCready J., Jay D. G. (2011) Extracellular heat shock protein (Hsp)70 and Hsp90α assist in matrix metalloproteinase-2 activation and breast cancer cell migration and invasion. PLoS One 6, e18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ellenrieder V., Hendler S. F., Boeck W., Seufferlein T., Menke A., Ruhland C., Adler G., Gress T. M. (2001) Transforming growth factor-β1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. 61, 4222–4228 [PubMed] [Google Scholar]
- 81. Vial E., Sahai E., Marshall C. J. (2003) ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell 4, 67–79 [DOI] [PubMed] [Google Scholar]
- 82. Doehn U., Hauge C., Frank S. R., Jensen C. J., Duda K., Nielsen J. V., Cohen M. S., Johansen J. V., Winther B. R., Lund L. R., Winther O., Taunton J., Hansen S. H., Frödin M. (2009) RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol. Cell 35, 511–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shin S., Dimitri C. A., Yoon S. O., Dowdle W., Blenis J. (2010) ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol. Cell 38, 114–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Johnson J. L., Dwivedi A., Somerville M., George S. J., Newby A. C. (2011) Matrix metalloproteinase (MMP)-3 activates MMP-9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arterioscler. Thromb. Vasc. Biol. 31, e35–44 [DOI] [PubMed] [Google Scholar]
- 85. David J. M., Rajasekaran A. K. (2012) Dishonorable discharge. The oncogenic roles of cleaved E-cadherin fragments. Cancer Res. 72, 2917–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hahn-Dantona E., Ruiz J. F., Bornstein P., Strickland D. K. (2001) The low density lipoprotein receptor-related protein modulates levels of matrix metalloproteinase 9 (MMP-9) by mediating its cellular catabolism. J. Biol. Chem. 276, 15498–15503 [DOI] [PubMed] [Google Scholar]
- 87. Mantuano E., Inoue G., Li X., Takahashi K., Gaultier A., Gonias S. L., Campana W. M. (2008) The hemopexin domain of matrix metalloproteinase-9 activates cell signaling and promotes migration of Schwann cells by binding to low-density lipoprotein receptor-related protein. J. Neurosci. 28, 11571–11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vidal C. I., Mintz P. J., Lu K., Ellis L. M., Manenti L., Giavazzi R., Gershenson D. M., Broaddus R., Liu J., Arap W., Pasqualini R. (2004) An HSP90-mimic peptide revealed by fingerprinting the pool of antibodies from ovarian cancer patients. Oncogene 23, 8859–8867 [DOI] [PubMed] [Google Scholar]
- 89. Tanaka H., Kono E., Tran C. P., Miyazaki H., Yamashiro J., Shimomura T., Fazli L., Wada R., Huang J., Vessella R. L., An J., Horvath S., Gleave M., Rettig M. B., Wainberg Z. A., Reiter R. E. (2010) Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat. Med. 16, 1414–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.