

Background: Macrophage migration inhibitory factor (MIF) family members promote AMP-activated protein kinase (AMPK) activation in nontransformed cells.
Results: MIF family members cooperatively inhibit AMPK in lung adenocarcinoma cell lines.
Conclusion: Inhibition of AMPK activation in lung adenocarcinoma cells by MIF family members promotes efficient mTOR activation.
Significance: Therapeutic targeting of MIF family members may benefit lung cancer patients.
Keywords: AMP-activated Kinase (AMPK), Cancer Biology, Cytokine Action, LKB, Lung Cancer, GLUT4, Glucose, Glutathione, Reactive Oxygen Species
Abstract
AMP-activated protein kinase (AMPK) is a nutrient- and metabolic stress-sensing enzyme activated by the tumor suppressor kinase, LKB1. Because macrophage migration inhibitory factor (MIF) and its functional homolog, d-dopachrome tautomerase (d-DT), have protumorigenic functions in non-small cell lung carcinomas (NSCLCs) but have AMPK-activating properties in nonmalignant cell types, we set out to investigate this apparent paradox. Our data now suggest that, in contrast to MIF and d-DTs AMPK-activating properties in nontransformed cells, MIF and d-DT act cooperatively to inhibit steady-state phosphorylation and activation of AMPK in LKB1 wild type and LKB1 mutant human NSCLC cell lines. Our data further indicate that MIF and d-DT, acting through their shared cell surface receptor, CD74, antagonize NSCLC AMPK activation by maintaining glucose uptake, ATP production, and redox balance, resulting in reduced Ca2+/calmodulin-dependent kinase kinase β-dependent AMPK activation. Combined, these studies indicate that MIF and d-DT cooperate to inhibit AMPK activation in an LKB1-independent manner.
Introduction
Nutrient stress-induced AMP-activated protein kinase (AMPK)3 facilitates a cellular stress response that serves to reduce ATP-consuming processes and enhance ATP-generating processes (1). In malignant cells, AMPK activation is thought to promote tumor suppression by facilitating inhibition of protein synthesis, cell cycle arrest, and programmed cell death (2). Given the restricted nutrient and metabolic microenvironments of solid tumors, it is not surprising that some tumors have developed mechanisms to evade AMPK activation. For example, in non-small cell lung carcinomas (NSCLCs), the AMPK-activating tumor suppressor kinase, LKB1, is mutated in >30% of malignant lesions (3). LKB1 is an environmental nutrient and oxygen stress sensor, and when activated, it serves to phosphorylate the activation loop of AMPK resulting in tumor suppression (2). In a mouse model of K-RasG12D-initiated lung adenocarcinoma, LKB1 deletion cooperates with K-RasG12D more potently than p53 knock-out mice (4). LKB1-deficient K-RasG12D lung adenocarcinomas exhibit metabolic stress resistance, shorter latency, and have more frequent metastases than LKB1-competent lesions (4). Importantly, LKB1 wild type, but not LKB1 kinase-deficient, allelic reconstitution into LKB1 mutant A549 NSCLC cells significantly reactivates A549 AMPK-dependent signaling, resulting in reduced anchorage independence and lung metastases (4).
Macrophage migration inhibitory factor (MIF) is a hypoglycemia- and hypoxia-inducible gene product that is overexpressed in a large majority of NSCLC lesions (5–8). Prior studies from our laboratory demonstrate that MIF is an important contributor to anchorage independence, cell motility, and invasive potential of A549 lung adenocarcinoma cells (9, 10). MIF signals as an extracellular autocrine and paracrine-acting cytokine and is necessary for maximal tumor-associated neovascularization (7, 11), evasion from cell senescence (12), and inhibition of the tumor suppressor, p53 (13–15). Recent studies revealed that the only other known MIF family member, d-dopachrome tautomerase (d-DT), functionally cooperates with, and compensates for, MIF in dictating neoangiogenic potential in human NSCLC cell lines (11).
Recent studies show that both MIF and d-DT activate AMPK in nonmalignant cell types (16–18). Because AMPK-activating phenotypes in malignant lung adenocarcinoma are tumor-suppressive (4) and MIF/d-DT-associated NSCLC phenotypes are generally tumor-promoting (9–11), we sought to evaluate MIF and d-DT individual and combined actions on AMPK activation in lung adenocarcinoma cell lines. We now describe that MIF and d-DT act cooperatively to inhibit AMPK phosphorylation and activity in both LKB1 wild type and LKB1 mutant cell lines. Our data indicate that MIF and d-DT signal through their shared cell surface receptor, CD74, to regulate glucose uptake, ATP generation, and steady-state inhibition of CaMKKβ-dependent AMPK activation. Because the two most commonly used AMPK activators, metformin and 5-amino-1-β-d-ribofuranosyl-imidazole-4-carboxamide (AICAR), have anti-cancer activities (19, 20) but are ineffective at inducing AMPK activation and subsequent tumor suppression in LKB1 mutant cells (21, 22), the discovery of unique LKB1-independent AMPK activators would be expected to have a significant impact on NSCLC disease management for clinicians.
EXPERIMENTAL PROCEDURES
Materials
2-Deoxy-d-glucose (2-DG), methyl pyruvate, N-acetylcysteine, N-2-mercaptopropionyl glycine, BAPTA, EGTA, and STO-609 were all obtained from Sigma.
Cell Culture
LKB1 mutant A549 cells were cultured in DMEM supplemented with 10% fetal calf serum, 2 mm glutamine, and 50 μg/ml gentamicin. LKB1 wild type H1299 and LKB1 mutant H460 cells were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, 2 mm glutamine, and 50 μg/ml gentamicin. Cells were subjected to hypoxic conditions as indicated using a Modular Incubator Chamber purged with 1% oxygen.
Immunoblotting
Whole cell extracts were prepared from cells after the indicated treatments. Cells were lysed in 1× lysis buffer (20 mm Tris, 137 mm NaCl, 1 mm EGTA, 1% Triton X-100, 10% glycerol, 1.5 mm MgCl2, 1 mm NaVO4, 2 mm NaF, and 1× protease inhibitor) by repeated passages through a 27-gauge needle. Equal amounts of cellular protein were fractionated on SDS-polyacrylamide gels (Bio-Rad) and transferred to polyvinylidene difluoride membranes (PVDF; Millipore). Immunoblotting was performed with antibodies directed against AMPK, phospho-AMPK (Thr-172), acetyl-CoA carboxylase, phosphoacetyl-CoA carboxylase, S6 kinase (S6K), phospho-S6K, phospho-rpS6, phospho-TSC2 (Cell Signaling Technology), GAPDH, MIF, CaMKKβ (Santa Cruz Biotechnology), GLUT4 (Millipore), Na+/K+ ATPase (Sigma), Golgin-97 (Molecular Probes), and d-DT (prepared by our laboratory) (11). Densitometric analysis of Western blots was performed using Bio-Rad Quantity One analysis software.
RNA Interference
Cells were transfected with MIF, d-DT, or nonspecific scrambled siRNA oligonucleotides using Oligofectamine reagent (Invitrogen) as described previously (11). Commercially available siRNA oligonucleotides for human CD74, TSC2, and CaMKKβ were purchased from Santa Cruz Biotechnology. Cells were incubated after siRNA transfection for the times indicated.
Adenovirus Preparation and Cell Infection
Adenovirus for human MIF and d-DT was prepared as described previously (11). Dominant negative AMPKα recombinant adenovirus was purchased from Eton Bioscience and was used to infect cells at ∼2 × 107 virus particles/ml. Cells were infected at ∼ 70–80% confluence as indicated.
Glucose Uptake Assay
Fresh medium was provided 24 h after siRNA transfection of cells. After an additional 24-h incubation period, spent cell medium was analyzed for remaining glucose using the Amplex Red glucose assay kit (Invitrogen) according to the manufacturer's protocol. Glucose uptake was measured by subtracting from glucose concentration in fresh medium.
GLUT4 Translocation Analysis by Differential Centrifugation
Cells were harvested 48 h after siRNA transfection and lysed in HES buffer (20 mm HEPES, 1 mm EDTA, 250 mm sucrose, 1 mm Na3VO4, 50 mm) using a ball-bearing homogenizer. Differential centrifugation was used to isolate plasma membrane (PM) and low-density microsome (LDM) fractions, as described previously (23). Unbroken cells and nuclei were pelleted from the lysate by centrifugation for 10 min at 600 rpm. The lysate was then centrifuged for 20 min at 19,000 × g to generate a supernatant to be processed at a later step and to pellet mitochondria and PM. The pellet was resuspended in HES buffer and layered onto 1.12 m sucrose and centrifuged for 60 min at 100,000 × g, which resulted in a band at the interface enriched in PM, whereas the mitochondria was pelleted and discarded. The PM was isolated from the interface fraction by dilution of the sucrose and pelleting at 40,000 × g for 20 min. The supernatant yielded from the centrifugation of the lysate was centrifuged at 41,000 × g for 20 min to pellet and discard of the high density microsome fraction. The resultant supernatant was then centrifuged for 75 min at 180,000 × g, pelleting the LDM fraction. The PM and LDM pellets were resuspended in 1× sample buffer. Equal volumes of lysate, PM, and LDM were assessed as indicated by Western analysis. Na+/K+ ATPase and Golgin-97 expression were used to normalize loading of PM and LDM fractions, respectively.
ATP Assay
Cell medium was replaced with fresh medium 24 h after siRNA transfection. Cells were lysed after an additional 24-h incubation period. Lysates were normalized to protein content and were assessed for intracellular ATP levels using the Cell Titer-Glow assay kit (Promega).
Measurement of Intracellular ROS and GSH
siRNA-transfected cells were lifted by trypsinization and washed with PBS. Cells were incubated with either the ROS indicator H2DCF-DA (5 μm, Invitrogen) or the reduced-glutathione detection reagent, ThiolTracker (10 μm, Invitrogen), in PBS supplemented with CaCl2 and MgCl2. After a 30-min incubation at 37 °C, cells were washed in PBS, and fluorescence was analyzed using a FACSCalibur flow cytometer (BD Biosciences). Data were analyzed and prepared using FlowJo software.
Statistics
Results are expressed as means ± S.D. Data comparisons were derived by one-way ANOVA, and p values < 0.01 were considered significant.
RESULTS
MIF and d-DT Cooperatively Inhibit AMPK Activity in NSCLC
MIF (16), and very recently, d-DT (18), were shown to activate AMPK in nonmalignant cell types. Because AMPK has tumor-suppressive activities in malignant cells (2, 4) and MIF is induced by the metabolic stressors, hypoxia and hypoglycemia (5, 6, 12), we set out to determine whether MIF and d-DT functionally regulate AMPK activity in human lung adenocarcinoma cell lines. Surprisingly, simultaneous siRNA knockdown of MIF and d-DT resulted in a significant increase in AMPK phosphorylation and activity as assessed by phosphorylation of acetyl-CoA carboxylase, the prototypical AMPK protein substrate (Fig. 1A) (25). Reminiscent of MIF and d-DT redundant functions in promoting angiogenic potential in human NSCLC cell lines, individual silencing of MIF or d-DT resulted in only a modest activation of AMPK, whereas simultaneous knockdown of both proteins resulted in a strong induction of AMPK phosphorylation/activation (Fig. 1B). Importantly, reintroduction of MIF and/or d-DT expression by adenoviral delivery efficiently reversed AMPK activation in MIF/d-DT-deficient cells (Fig. 1C).
FIGURE 1.

MIF and d-DT cooperate to antagonize AMPK activation. Scrambled (Scr), MIF, and/or d-DT siRNAs were transfected into A549 cells using Oligofectamine as indicated for 72 h (A and B), and lysates were examined by immunoblotting. C, A549 cells were siRNA-transfected as indicated for 48 h and then infected with GFP (used as a control for adenoviral infection/overexpression), MIF, d-DT, or MIF + d-DT (M+D) adenovirus overnight. Lysates were examined by immunoblotting. D, A549 cells were siRNA-transfected as indicated for 72 h, and lysates were examined by immunoblotting. Bio-Rad Quantity One software was used for densitometry and P-AMPK/GAPDH (top panels, n = 2) densitometry values are depicted in the graph. *, p < 0.05 by one-way ANOVA analysis is shown for individual group comparisons. Data shown are representative of two independent experiments. Error bars, S.E. MIF, d-DT, and CD74 were reduced by ∼85%, 70%, and 80%, respectively (not shown).
The MIF cell surface receptor, CD74, is overexpressed in a significant proportion of NSCLC lesions (7). Importantly, CD74 is responsible for signaling to AMPK activation by MIF and d-DT (16, 26). To determine the role of CD74 in NSCLC AMPK regulation, we evaluated the relative phosphorylation state of AMPK in CD74 siRNA-transfected cells. As shown in Fig. 1D, CD74 silencing phenocopies MIF/d-DT silencing in inducing AMPK phosphorylation in A549 lung adenocarcinoma cells. Combined, these findings suggest that MIF and d-DT, through CD74, cooperate to inhibit AMPK phosphorylation and activity in human NSCLC cell lines.
MIF and d-DT Suppress AMPK-dependent Inhibition of the mTOR Pathway
Because a central target of activated AMPK is mTORC1 inhibition mediated by AMPK-dependent phosphorylation of TSC2 (27, 28), we next determined whether MIF and d-DT regulate the activation of S6K, the primary and prototypical substrate of mTOR kinase activity (27, 29). Overexpression of MIF, d-DT, and combined MIF/d-DT strongly induced the phosphorylation of S6K and its immediate substrate, rpS6. Importantly, MIF overexpression also results in a concurrent inhibition of AMPK phosphorylation and activation under both hyper- (25 mm) and hypoglycemic (1 mm) conditions (Fig. 2B).
FIGURE 2.

MIF and d-DT suppress AMPK-dependent inhibition of mTOR signaling. A549 cells were infected with GFP (G), MIF (M), d-DT (D), or MIF+d-DT (M+D) adenovirus for 24 h. A, lysates were analyzed by immunoblotting. B and C, after 24 h, cells were challenged with high (25 mm) or low (1 mm) glucose medium (B), 10 mm 2-DG (C), or 1% oxygen (C) for an additional 6 h before lysing and immunoblotting as indicated. D and E, H1299 cells were transfected with siRNA as indicated for 72 h (D) or for 48 h and then infected with GFP or dominant negative AMPK adenovirus for an additional 24 h (E). Lysates were examined by immunoblotting. F, A549 cells were transfected with siRNA as indicated for 72 h, and lysates were examined by immunoblotting.
Activated AMPK contributes to the well documented inhibition of mTOR that accompanies metabolic stress induction by acute hypoxia or glucose restriction (30, 31). As such, we next tested whether forced overexpression of MIF or d-DT can rescue mTOR activation under conditions of low glucose (induced by 2-DG) or low oxygen tensions. As shown in Fig. 2C, MIF and d-DT overexpression independently restored the phosphorylation of S6 kinase under conditions of acute hypoxia or 2-DG treatment.
We next investigated whether endogenous MIF and/or d-DT is necessary for maximal, steady-state S6K activation in lung adenocarcinoma cell lines. As shown in Fig. 2D, individual siRNA knockdown of MIF or d-DT only moderately reduced S6K phosphorylation whereas simultaneous silencing of MIF and d-DT resulted in a much more pronounced reduction of phosphorylated S6K. Corresponding closely with decreases in S6K phosphorylation were commensurate increases in phosphorylated AMPK (Fig. 2D) consistent with a role for AMPK in inhibiting S6K activation. To determine whether the dysregulated activation of AMPK observed in MIF/d-DT-deficient cells was responsible for the observed loss of phosphorylated/activated S6K in the same cells, an adenoviral dominant negative mutant allele of AMPK was employed (32). As shown in Fig. 2E, adenoviral mediated delivery and expression of dominant negative AMPK resulted in significantly enhanced phosphorylation of S6K and rpS6 in control siRNA transfected cells (Scr), essentially phenocopying MIF and d-DT-induced activation of S6K/inhibition of AMPK (Fig. 2, A and B), and, more importantly, partially restored defective phosphorylation of S6K and rpS6 in MIF/d-DT-deficient cells (Fig. 2E).
Because inhibition of mTOR by AMPK is primarily mediated by AMPK-dependent phosphorylation of the mTOR-inhibiting protein TSC2 (27), we next investigated the potential involvement of TSC2 in mediating defective mTOR/S6K activation observed in MIF/d-DT-deficient cells (Fig. 2, D and E). As shown in Fig. 2F, simultaneous siRNA knockdown of MIF and d-DT increased the phosphorylation of TSC2 on the AMPK phosphorylation site, Ser-1387 (27). Furthermore, siRNA knockdown of TSC2 in MIF/d-DT-deficient cells effectively rescued the phosphorylation and activation of S6K. Combined, these findings suggest that MIF and d-DT cooperatively promote mTOR activation, at least in part, by antagonizing AMPK-dependent activation of TSC.
MIF and d-DT Promote Glucose Uptake and ATP Generation
As a metabolic stress sensor, AMPK activity is tightly regulated by changes in ATP/AMP (22) and ATP/ADP (33). Because MIF has previously been shown to regulate glucose uptake and catabolism (34, 35), we investigated whether loss of MIF and/or d-DT influences glucose uptake properties in human lung adenocarcinoma cells. As shown in Fig. 3A, individual loss of either MIF or d-DT resulted in modest reductions of glucose uptake into cells whereas simultaneous MIF/d-DT-deficiency resulted in a substantially larger reduction in uptake (Fig. 3A). We next evaluated the possibility that alterations in glucose transporter expression or plasma membrane translocation may be involved in the observed defective glucose uptake in MIF/d-DT-deficient cells. Using differential centrifugation and normalizing, we found that GLUT4 is distributed preferentially away from PM fractions and into LDM fractions in MIF/d-DT-deficient cells (Fig. 3B), consistent with prior studies on MIF (16). Expression levels of Na+/K+ ATPase and Golgin-97 were used as markers to normalize distribution of GLUT4 in the PM and LDM fractions, respectively (Fig. 3B). Consistent with suboptimal glucose uptake and metabolism, total ATP levels were reduced (Fig. 3C) in a pattern and manner similar to the observed reductions in glucose uptake in MIF, d-DT, and combined MIF/d-DT siRNA knockdown cells (Fig. 3A). To investigate whether increasing cellular ATP levels was sufficient to reverse the observed increase in phosphorylated AMPK in MIF/d-DT-deficient cells, a cell-permeable form of the TCA cycle entry substrate, pyruvate (methyl pyruvate (36)), was added exogenously to MIF/d-DT-deficient cells. As shown in Fig. 3D, methyl pyruvate dose-dependently reduced AMPK phosphorylation in MIF/d-DT-deficient cells while having little to no effect on control siRNA-transfected cell (Scr) phosphorylated AMPK. These findings suggest that MIF and d-DT contribute to lung adenocarcinoma glucose uptake and energy balance that, in turn, contributes to an inhibition of steady-state AMPK activity.
FIGURE 3.

MIF and d-DT inhibit AMPK activation by regulating glucose levels and ATP/AMP ratios. H460 cells (LKB1 mutant; p53 wild type) were siRNA transfected as indicated, and 24 h later medium was replaced. 24 h after medium change, supernatants were analyzed for remaining glucose. A, uptake was determined by subtracting from glucose concentration in fresh medium. B, A549 cells were siRNA-transfected as indicated for 48 h, followed by isolation of total cell lysate, PM, and LDM fractions by differential centrifugation. Levels of GLUT4 and the appropriate controls (Na+/K+ ATPase for plasma membrane and Golgin-97 for LDM) were assessed by immunoblotting in each fraction. GLUT4 protein levels were determined using control Na+/K+ ATPase (lysate and PM fractions) and Golgin-97 (LDM fraction) protein band intensities from two independent experiments. **, p < 0.01 by one-way ANOVA analysis is shown for individual group comparisons. C, H460 cells were transfected with siRNA as indicated and, 48 h later, lysed and examined for ATP levels. Trypan blue staining of parallel cell transfectants revealed no differences in cell viability (not shown). D, MIF and d-DT were silenced as indicated in H460 cells and cultured for 40 h, followed by addition of methyl pyruvate at the indicated concentrations for an additional 8 h. Lysates were examined by immunoblotting. Error bars, S.E.
CaMKKβ Is Required for Maximal MIF/d-DT-dependent Regulation of AMPK
Lung adenocarcinomas are unique among human malignancies in that they have, by a considerable margin, the highest frequency of LKB1 tumor suppressor mutations compared with other cancers (4, 37, 38). LKB1 mutation/deficiency results in metabolic stress resistance, shorter latency, and increased metastases, and these phenotypes are primarily considered to be due to an inability to efficiently activate AMPK-dependent tumor suppression (4). Intriguingly, increased phosphorylation and activation of AMPK observed in MIF/d-DT knockdown NSCLC cells were observed in both LKB1 mutant cell lines (A549, Fig. 1, A and B; H460, Fig. 3D) and an LKB1 wild type line (H1299, Fig. 2D) suggesting an LKB1-independent mechanism of AMPK regulation by MIF and d-DT in lung adenocarcinoma.
CaMKKβ has been identified as an alternative upstream activating kinase of AMPK (39). Because AMPK activation requires not only reduced ATP/AMP or ATP/ADP ratios but also phosphorylation on Thr-172 in its activation loop (1), we next determined whether CaMKKβ participates in MIF/d-DT-associated AMPK modulation. As shown in Fig. 4A, the CaMKKβ-specific inhibitor, STO-609, completely attenuated both steady-state and MIF/d-DT depletion-associated phosphorylation of AMPK in A549 cells. To validate the hypothesized requirement for CaMKKβ in mediating AMPK phosphorylation in MIF/d-DT-deficient cells, siRNA silencing of CaMKKβ was employed. As shown in Fig. 4B, siRNA targeting CaMKKβ very efficiently reduced CaMKKβ expression and significantly attenuated the aberrant phosphorylation and activation of AMPK observed in MIF/d-DT-deficient cells.
FIGURE 4.

CaMKKβ is required for maximal MIF/d-DT-dependent regulation of AMPK. A, A549 cells were siRNA-transfected as indicated for 40 h followed by addition of vehicle or the CaMKK inhibitor, STO-609 (5 μm), for an additional 8 h. Lysates were examined by immunoblotting. B and C, A549 cells were siRNA-transfected as indicated for 72 h (B) and treated with and without 10 μm BAPTA (C) or 5 mm EGTA (C) for an additional 6 h. Lysates were examined by immunoblotting.
As their names imply, Ca2+/calmodulin-dependent protein kinase kinases are regulated by free calcium. To investigate whether the enhanced CaMKKβ-dependent AMPK activation observed in MIF/d-DT-deficient cells (Fig. 4, A and B) requires free calcium, BAPTA, an intracellular calcium chelator, and EGTA, an extracellular calcium chelator, were tested for their respective abilities to block MIF/d-DT deficiency-induced, CaMKKβ-dependent, AMPK activation. As shown in Fig. 4C, BAPTA (top panel) strongly reduced, and EGTA (bottom panel) reduced but to a lesser extent, AMPK activation in MIF/d-DT-deficient cells, suggesting that increased CaMKKβ-mediated AMPK phosphorylation requires free calcium. This further supports the observation that CaMKKβ activity is necessary for the aberrant AMPK activation induced by simultaneous siRNA silencing of MIF and d-DT.
MIF and d-DT Regulate AMPK in a Redox-dependent Fashion
Two independent studies recently described an important role for ROS in facilitating intracellular calcium increases and subsequent induction of CaMKKβ-dependent phosphorylation of AMPK (40, 41). Because increased glucose uptake in transformed cells enhances pentose phosphate pathway flux-mediated generation of NADPH that, in turn, is necessary for reducing glutathione (GSH) and maintaining low ROS levels (36), we hypothesized that loss of MIF and d-DT may result in inefficient glutathione reduction as a result of defective glucose uptake (Fig. 3A). To test this, the reduced thiol indicator, ThiolTracker (36), was used to determine levels of free intracellular thiols in MIF/d-DT silenced cells. As shown in Fig. 5A, simultaneous knockdown of MIF and d-DT results in significant decreases in reduced GSH levels. Because reduced GSH represents a critically important cellular antioxidant that serves to minimize oxidative damage caused by a variety of intracellular oxidative species (42), we next used H2DCF-DA to detect cellular oxidative species (43) in MIF/d-DT-deficient cells. As shown in Fig. 5B, MIF/d-DT-deficient cells exhibited significantly higher DCF-detectable ROS when assessed by flow cytometry than that of control siRNA-transfected cells.
FIGURE 5.

MIF and d-DT regulate AMPK in a redox-dependent fashion. A and B, A549 cells were siRNA-transfected as indicated for 72 h, and intracellular GSH levels (A) and intracellular ROS levels (B) were assessed by flow cytometry after incubation with ThiolTracker or DCF-DA, respectively. The left panel shows a histogram overlay, whereas the right panels show the individual histograms with an arbitrary gate representing the percentage of cells with increased GSH (A) or ROS levels (B). Results are also graphically expressed as relative geometric means of triplicate samples. **, p < 0.01; ***, p < 0.001. C, A549 cells were siRNA-transfected as indicated. 48 h later, increasing concentrations of N-acetylcysteine or N-2-mercaptopropionyl glycine (MPG) were added to the cells for an additional 24 h. Lysates were analyzed by immunoblotting. Error bars, S.E.
Because we observed increased ROS levels in MIF/d-DT-deficient cells, consistent with decreased reduced GSH levels (Fig. 5A), we next assessed the relative requirements for ROS in mediating aberrant AMPK activation in MIF/d-DT-knockdown cells using ROS scavengers. As shown in Fig. 5C, N-acetylcysteine and 2-mercaptopropionyl glycine both attenuated, in a dose-dependent manner, the aberrant AMPK phosphorylation observed in MIF/d-DT-deficient cells consistent with their ability to reduce DCF-detectable ROS (supplemental Fig. S1).
DISCUSSION
MIF, and more recently d-DT, are novel, extracellular activators of AMPK signaling in nontransformed cells (16, 18). Our current studies now indicate that MIF and d-DT, through their shared, cell surface receptor, CD74 (11, 26, 44), actively inhibit the phosphorylation and activation of AMPK resulting in enhanced mTOR/S6K signaling in human lung adenocarcinoma cell lines. Our data further suggest that MIF and d-DT act cooperatively to promote glucose uptake and maintain both ATP and reduced glutathione levels resulting, ultimately, in the negative regulation of CaMKKβ-dependent phosphorylation of AMPK. The aberrant activation of CaMKKβ-mediated AMPK phosphorylation observed in MIF/d-DT-deficient cells was also found to require free intracellular calcium and reactive oxygen species. Combined, we propose a model where MIF and d-DT promote glucose uptake into malignant NSCLC cells resulting in enhanced ATP and increased GSH leading to reduced ROS-mediated, CaMKKβ-dependent AMPK phosphorylation (Fig. 6).
FIGURE 6.
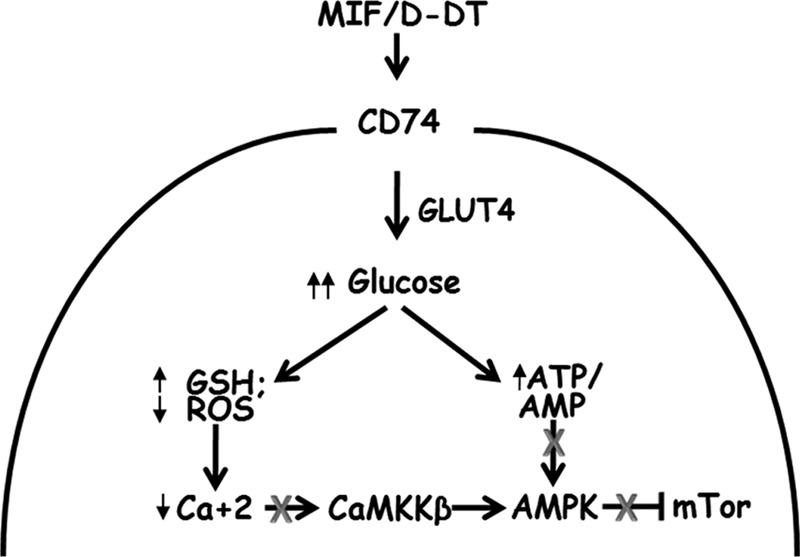
Hypothesized pathway for MIF and d-DT cooperativity in regulating AMPK activation.
Perhaps the most notable and intriguing aspect of these findings is the fact that MIF and d-DT negatively regulate AMPK activity in an LKB1-independent manner. Mutated in >30% of all human NSCLCs (4, 21), the LKB1 tumor suppressor is one of the most important determinants of metabolic stress adaptation. Given that ∼70% of NSCLC lesions have functional LKB1 tumor suppressor alleles, it is highly likely that other, as yet unidentified, AMPK regulatory pathways exist in NSCLC tumors. Our findings indicate that MIF and d-DT functionally promote a unique metabolic stress regulatory pathway that is usurped by malignant NSCLC cells to counter AMPK-dependent mTOR inhibition. Both MIF and CD74 are overexpressed in a large majority of human NSCLC lesions (7), and this overexpression correlates closely with increased angiogenic growth factor expression and microvascular density (7, 8). MIF and d-DT cooperate, in a CD74-dependent manner, to promote VEGF expression and angiogenic potential in human lung adenocarcinoma cell lines (11). Because TSC2 regulates VEGF expression using both mTOR-dependent and -independent mechanisms (45), it is not unreasonable to speculate that MIF and d-DT may promote NSCLC angiogenic potential by negatively regulating AMPK activation. It is also interesting to note that MIF deficiency or inhibition renders A549 cells defective in both anchorage independence and cell motility/invasion (9, 10) in a manner that essentially phenocopies wild type LKB1 allelic reconstitution into LKB1 mutant A549 cells (4). Although further studies are needed, it is conceivable that this phenocopy is due to the aberrant activation of AMPK observed in MIF-deficient cells (albeit not as pronounced as combined MIF/d-DT deficiency) which would be similarly expected upon reintroduction of a functional LKB1 allele (4).
An important question raised by these findings is how do MIF and d-DT activate AMPK in some nontransformed cell types (16, 18) and inhibit AMPK in malignant lung adenocarcinoma cell lines? The fact that the MIF/d-DT receptor, CD74, appears to be necessary for both activation (16, 18) and inhibition (Fig. 1D) of phenotypes indicates that other, as yet unidentified, factors dictate MIF/d-DT-dependent activation or inhibition of AMPK. Given the requirement for CD74 in both AMPK activating and inhibiting cell types, coupled with the fact that MIF:CD74 signaling can occur through several different co-receptor complexes (24, 46, 47), it is not unreasonable to speculate that malignant cells differentially coordinate MIF/d-DT co-receptor expression/activity as a means of evading AMPK-dependent tumor suppression. Studies are currently underway to examine this possibility.
In conclusion, we report for the first time that the cytokine/growth factor family members, MIF and d-DT, functionally inhibit steady-state phosphorylation and activation of AMPK in an LKB1-independent manner. Because traditional AMPK activating drugs such as metformin and AICAR are unable to induce AMPK-dependent tumor suppression in LKB1 mutant NSCLC lesions (21), MIF and d-DT dual targeting small molecules or neutralizing antibodies may represent a novel chemotherapeutic approach to human NSCLC clinical disease management.
Supplementary Material
Acknowledgment
We thank Dr. John W. Eaton for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant CA129967 (to R. A. M).

This article contains supplemental Fig. S1.
- AMPK
- AMP-activated protein kinase
- BAPTA
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- CaMKKβ
- Ca2+/calmodulin-dependent kinase kinase β
- d-DT
- d-dopachrome tautomerase
- 2-DG
- 2-deoxy-d-glucose
- GLUT4
- glucose transporter 4
- H2DCF-DA
- 2′,7′-dichlorodihydrofluorescein diacetate
- LDM
- low density microsomal
- LKB1
- liver kinase B1
- MIF
- macrophage migration inhibitory factor
- mTOR
- mammalian target of rapamycin
- NSCLC
- non-small cell lung carcinoma
- PM
- plasma membrane
- ROS
- reactive oxygen species
- rpS6
- ribosomal protein S6
- S6K
- S6 kinase
- TSC2
- tuberous sclerosis protein 2.
REFERENCES
- 1. Hardie D. G. (2007) AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8, 774–785 [DOI] [PubMed] [Google Scholar]
- 2. Luo Z., Zang M., Guo W. (2010) AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol. 6, 457–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Makowski L., Hayes D. N. (2008) Role of LKB1 in lung cancer development. Br. J. Cancer 99, 683–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ji H., Ramsey M. R., Hayes D. N., Fan C., McNamara K., Kozlowski P., Torrice C., Wu M. C., Shimamura T., Perera S. A., Liang M. C., Cai D., Naumov G. N., Bao L., Contreras C. M., Li D., Chen L., Krishnamurthy J., Koivunen J., Chirieac L. R., Padera R. F., Bronson R. T., Lindeman N. I., Christiani D. C., Lin X., Shapiro G. I., Jänne P. A., Johnson B. E., Meyerson M., Kwiatkowski D. J., Castrillon D. H., Bardeesy N., Sharpless N. E., Wong K. K. (2007) LKB1 modulates lung cancer differentiation and metastasis. Nature 448, 807–810 [DOI] [PubMed] [Google Scholar]
- 5. Bacher M., Schrader J., Thompson N., Kuschela K., Gemsa D., Waeber G., Schlegel J. (2003) Up-regulation of macrophage migration inhibitory factor gene and protein expression in glial tumor cells during hypoxic and hypoglycemic stress indicates a critical role for angiogenesis in glioblastoma multiforme. Am. J. Pathol. 162, 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Winner M., Koong A. C., Rendon B. E., Zundel W., Mitchell R. A. (2007) Amplification of tumor hypoxic responses by macrophage migration inhibitory factor-dependent hypoxia-inducible factor stabilization. Cancer Res. 67, 186–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McClelland M., Zhao L., Carskadon S., Arenberg D. (2009) Expression of CD74, the receptor for macrophage migration inhibitory factor, in non-small cell lung cancer. Am. J. Pathol. 174, 638–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. White E. S., Flaherty K. R., Carskadon S., Brant A., Iannettoni M. D., Yee J., Orringer M. B., Arenberg D. A. (2003) Macrophage migration inhibitory factor and CXC chemokine expression in non-small cell lung cancer: role in angiogenesis and prognosis. Clin. Cancer Res. 9, 853–860 [PubMed] [Google Scholar]
- 9. Rendon B. E., Roger T., Teneng I., Zhao M., Al-Abed Y., Calandra T., Mitchell R. A. (2007) Regulation of human lung adenocarcinoma cell migration and invasion by macrophage migration inhibitory factor. J. Biol. Chem. 282, 29910–29918 [DOI] [PubMed] [Google Scholar]
- 10. Winner M., Meier J., Zierow S., Rendon B. E., Crichlow G. V., Riggs R., Bucala R., Leng L., Smith N., Lolis E., Trent J. O., Mitchell R. A. (2008) A novel, macrophage migration inhibitory factor suicide substrate inhibits motility and growth of lung cancer cells. Cancer Res. 68, 7253–7257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coleman A. M., Rendon B. E., Zhao M., Qian M. W., Bucala R., Xin D., Mitchell R. A. (2008) Cooperative regulation of non-small cell lung carcinoma angiogenic potential by macrophage migration inhibitory factor and its homolog, d-dopachrome tautomerase. J. Immunol. 181, 2330–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Welford S. M., Bedogni B., Gradin K., Poellinger L., Broome Powell M., Giaccia A. J. (2006) HIF1α delays premature senescence through the activation of MIF. Genes Dev. 20, 3366–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hudson J. D., Shoaibi M. A., Maestro R., Carnero A., Hannon G. J., Beach D. H. (1999) A proinflammatory cytokine inhibits p53 tumor suppressor activity. J. Exp. Med. 190, 1375–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fingerle-Rowson G., Petrenko O., Metz C. N., Forsthuber T. G., Mitchell R., Huss R., Moll U., Müller W., Bucala R. (2003) The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc. Natl. Acad. Sci. U.S.A. 100, 9354–9359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martin J., Duncan F. J., Keiser T., Shin S., Kusewitt D. F., Oberyszyn T., Satoskar A. R., VanBuskirk A. M. (2009) Macrophage migration inhibitory factor (MIF) plays a critical role in pathogenesis of ultraviolet-B (UVB)- induced nonmelanoma skin cancer (NMSC). FASEB J. 23, 720–730 [DOI] [PubMed] [Google Scholar]
- 16. Miller E. J., Li J., Leng L., McDonald C., Atsumi T., Bucala R., Young L. H. (2008) Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature 451, 578–582 [DOI] [PubMed] [Google Scholar]
- 17. Heinrichs D., Knauel M., Offermanns C., Berres M. L., Nellen A., Leng L., Schmitz P., Bucala R., Trautwein C., Weber C., Bernhagen J., Wasmuth H. E. (2011) Macrophage migration inhibitory factor (MIF) exerts antifibrotic effects in experimental liver fibrosis via CD74. Proc. Natl. Acad. Sci. U.S.A. 108, 17444–17449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iwata T., Taniguchi H., Kuwajima M., Taniguchi T., Okuda Y., Sukeno A., Ishimoto K., Mizusawa N., Yoshimoto K. (2012) The action of d-dopachrome tautomerase as an adipokine in adipocyte lipid metabolism. PLoS ONE 7, e33402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rocha G. Z., Dias M. M., Ropelle E. R., Osório-Costa F., Rossato F. A., Vercesi A. E., Saad M. J., Carvalheira J. B. (2011) Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth. Clin. Cancer Res. 17, 3993–4005 [DOI] [PubMed] [Google Scholar]
- 20. Swinnen J. V., Beckers A., Brusselmans K., Organe S., Segers J., Timmermans L., Vanderhoydonc F., Deboel L., Derua R., Waelkens E., De Schrijver E., Van de Sande T., Noël A., Foufelle F., Verhoeven G. (2005) Mimicry of a cellular low energy status blocks tumor cell anabolism and suppresses the malignant phenotype. Cancer Res. 65, 2441–2448 [DOI] [PubMed] [Google Scholar]
- 21. Shaw R. J., Kosmatka M., Bardeesy N., Hurley R. L., Witters L. A., DePinho R. A., Cantley L. C. (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. U.S.A. 101, 3329–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sim A. T., Hardie D. G. (1988) The low activity of acetyl-CoA carboxylase in basal and glucagon-stimulated hepatocytes is due to phosphorylation by the AMP-activated protein kinase and not cyclic AMP-dependent protein kinase. FEBS Lett. 233, 294–298 [DOI] [PubMed] [Google Scholar]
- 23. Marsh B. J., Alm R. A., McIntosh S. R., James D. E. (1995) Molecular regulation of GLUT4 targeting in 3T3-L1 adipocytes. J. Cell Biol. 130, 1081–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tarnowski M., Grymula K., Liu R., Tarnowska J., Drukala J., Ratajczak J., Mitchell R. A., Ratajczak M. Z., Kucia M. (2010) Macrophage migration inhibitory factor is secreted by rhabdomyosarcoma cells, modulates tumor metastasis by binding to CXCR4 and CXCR7 receptors, and inhibits recruitment of cancer-associated fibroblasts. Mol. Cancer Res. 8, 1328–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Munday M. R., Campbell D. G., Carling D., Hardie D. G. (1988) Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur. J. Biochem. 175, 331–338 [DOI] [PubMed] [Google Scholar]
- 26. Merk M., Mitchell R. A., Endres S., Bucala R. (2012) d-Dopachrome tautomerase (d-DT or MIF-2): doubling the MIF cytokine family. Cytokine 59, 10–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Inoki K., Zhu T., Guan K. L. (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590 [DOI] [PubMed] [Google Scholar]
- 28. Shaw R. J., Bardeesy N., Manning B. D., Lopez L., Kosmatka M., DePinho R. A., Cantley L. C. (2004) The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6, 91–99 [DOI] [PubMed] [Google Scholar]
- 29. Burnett P. E., Barrow R. K., Cohen N. A., Snyder S. H., Sabatini D. M. (1998) RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. U.S.A. 95, 1432–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu L., Cash T. P., Jones R. G., Keith B., Thompson C. B., Simon M. C. (2006) Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 21, 521–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jones R. G., Plas D. R., Kubek S., Buzzai M., Mu J., Xu Y., Birnbaum M. J., Thompson C. B. (2005) AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293 [DOI] [PubMed] [Google Scholar]
- 32. Woods A., Azzout-Marniche D., Foretz M., Stein S. C., Lemarchand P., Ferré P., Foufelle F., Carling D. (2000) Characterization of the role of AMP-activated protein kinase in the regulation of glucose-activated gene expression using constitutively active and dominant negative forms of the kinase. Mol. Cell. Biol. 20, 6704–6711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oakhill J. S., Steel R., Chen Z. P., Scott J. W., Ling N., Tam S., Kemp B. E. (2011) AMPK is a direct adenylate charge-regulated protein kinase. Science 332, 1433–1435 [DOI] [PubMed] [Google Scholar]
- 34. Benigni F., Atsumi T., Calandra T., Metz C., Echtenacher B., Peng T., Bucala R. (2000) The proinflammatory mediator macrophage migration inhibitory factor induces glucose catabolism in muscle. J. Clin. Invest. 106, 1291–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Waeber G., Calandra T., Roduit R., Haefliger J. A., Bonny C., Thompson N., Thorens B., Temler E., Meinhardt A., Bacher M., Metz C. N., Nicod P., Bucala R. (1997) Insulin secretion is regulated by the glucose-dependent production of islet beta cell macrophage migration inhibitory factor. Proc. Natl. Acad. Sci. U.S.A. 94, 4782–4787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schafer Z. T., Grassian A. R., Song L., Jiang Z., Gerhart-Hines Z., Irie H. Y., Gao S., Puigserver P.., Brugge J. S. (2009) Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carretero J., Medina P. P., Pio R., Montuenga L. M., Sanchez-Cespedes M. (2004) Novel and natural knockout lung cancer cell lines for the LKB1/STK11 tumor suppressor gene. Oncogene 23, 4037–4040 [DOI] [PubMed] [Google Scholar]
- 38. Matsumoto S., Iwakawa R., Takahashi K., Kohno T., Nakanishi Y., Matsuno Y., Suzuki K., Nakamoto M., Shimizu E., Minna J. D., Yokota J. (2007) Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene 26, 5911–5918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hawley S. A., Pan D. A., Mustard K. J., Ross L., Bain J., Edelman A. M., Frenguelli B. G., Hardie D. G. (2005) Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2, 9–19 [DOI] [PubMed] [Google Scholar]
- 40. Gusarova G. A., Trejo H. E., Dada L. A., Briva A., Welch L. C., Hamanaka R. B., Mutlu G. M., Chandel N. S., Prakriya M., Sznajder J. I. (2011) Hypoxia leads to Na,K-ATPase down-regulation via Ca2+ release-activated Ca2+ channels and AMPK activation. Mol. Cell Biol. 31, 3546–3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mungai P. T., Waypa G. B., Jairaman A., Prakriya M., Dokic D., Ball M. K., Schumacker P. T. (2011) Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol. Cell. Biol. 31, 3531–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ballatori N., Krance S. M., Notenboom S., Shi S., Tieu K., Hammond C. L. (2009) Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 390, 191–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang H., Joseph J. A. (1999) Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic. Biol. Med. 27, 612–616 [DOI] [PubMed] [Google Scholar]
- 44. Xin D., Rendon B. E., Zhao M., Winner M., McGhee Coleman A., Mitchell R. A. (2010) The MIF homolog, d-dopachrome tautomerase (d-DT), promotes COX-2 expression through β-catenin-dependent and independent mechanisms. Mol. Cancer Res. 8, 1601–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brugarolas J. B., Vazquez F., Reddy A., Sellers W. R., Kaelin W. G., Jr. (2003) TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 4, 147–158 [DOI] [PubMed] [Google Scholar]
- 46. Bernhagen J., Krohn R., Lue H., Gregory J. L., Zernecke A., Koenen R. R., Dewor M., Georgiev I., Schober A., Leng L., Kooistra T., Fingerle-Rowson G., Ghezzi P., Kleemann R., McColl S. R., Bucala R., Hickey M. J., Weber C. (2007) MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 13, 587–596 [DOI] [PubMed] [Google Scholar]
- 47. Weber C., Kraemer S., Drechsler M., Lue H., Koenen R. R., Kapurniotu A., Zernecke A., Bernhagen J. (2008) Structural determinants of MIF functions in CXCR2-mediated inflammatory and atherogenic leukocyte recruitment. Proc. Natl. Acad. Sci. U.S.A. 105, 16278–16283 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.