

Background: The toxin VgrG1 actin cross-linking domain (ACD) cross-links G-actin, leading to F-actin depolymerization. It is homologous to the MARTX toxin family.
Results: We solved the crystal structure of active VgrG1 ACD.
Conclusion: Complexes with ADP/ATP and divalent cations help in understanding ACD active site function.
Significance: The structural characterization of ACDs provides opportunities to design compounds targeting their active site.
Keywords: Actin; ATP; Cholera Toxin; Crystal Structure; Secretion; MARTX; Type VI Secretion,; VgrG; Actin Cross-linking; G-actin
Abstract
Vibrio cholerae is the cause of the diarrheal disease cholera. V. cholerae produces RtxA, a large toxin of the MARTX family, which is targeted to the host cell cytosol, where its actin cross-linking domain (ACD) cross-links G-actin, leading to F-actin depolymerization, cytoskeleton rearrangements, and cell rounding. These effects on the cytoskeleton prevent phagocytosis and bacterial engulfment by macrophages, thus preventing V. cholerae clearance from the gut. The V. cholerae Type VI secretion-associated VgrG1 protein also contains a C-terminal ACD, which shares 61% identity with MARTX ACD and has been shown to covalently cross-link G-actin. Here, we purified the VgrG1 C-terminal domain and determined its crystal structure. The VgrG1 ACD exhibits a V-shaped three-dimensional structure, formed of 12 β-strands and nine α-helices. Its active site comprises five residues that are conserved in MARTX ACD toxin, within a conserved area of ∼10 Å radius. We showed that less than 100 ACD molecules are sufficient to depolymerize the actin filaments of a fibroblast cell in vivo. Mutagenesis studies confirmed that Glu-16 is critical for the F-actin depolymerization function. Co-crystals with divalent cations and ATP reveal the molecular mechanism of the MARTX/VgrG toxins and offer perspectives for their possible inhibition.
Introduction
The life-threatening disease cholera is caused by the Gram-negative bacterium Vibrio cholerae. The main symptom of cholera is severe, profuse watery diarrhea. Humans are usually poisoned by absorbing water contaminated by the stools of infected people associated with poor sanitation (1, 2). Cholera pandemics have spread around the world, and the most recent outbreak started in Haiti a few months after the 2010 earthquake (2, 3). During its life in water, V. cholerae has to resist predation by amoebas. It also kills competing bacteria to colonize the niche and target human host cells (4). A large repertoire of toxins and virulence factors has been linked to V. cholerae pathogenesis. The O1 and O139 V. cholerae serogroups produce an enterotoxin, the cholera toxin, which is internalized and induces ADP-ribosylation of G protein. The resulting constitutive activation of adenylate cyclase provokes massive loss of water and electrolytes (5).
A second toxin is RtxA, the prototype of the multifunctional autoprocessing repats-in-toxins (MARTX)5 family (6). Upon secretion by a dedicated Type I secretion system, the catalytic actin cross-linking domain (ACD) of RtxA is delivered into host cells, where it mediates the covalent cross-linking of G-actin, leading to cytoskeleton rearrangements and cell rounding (7). ACDs are enzyme ligases that catalyze isopeptide bond formation between residues Glu-270 and Lys-50 of two actin monomers (6). By curbing actin assembly and dynamics, RtxA prevents phagocytosis and bacterial engulfment (6, 8). MARTX toxins carrying an ACD are found in several Vibrio species as well as closely related pathogens, such as Aeromonas (8).
Interestingly, the ACD domain is also found combined with the V. cholerae VgrG1 protein (7, 9). VgrG is a core component of the Type VI secretion system, a versatile macromolecular machine dedicated to the secretion of toxins toward eukaryotic or prokaryotic target cells (10–15). The Type VI secretion system shares structural similarities with tailed bacteriophage, and recent data have demonstrated that a sheath-like structure acts as a contractile machine to deliver the extracellular portion of the secretion apparatus to target cells (16). This extracellular portion is thought to be composed of a tail-like structure formed by the polymerization of the Hcp protein concluded by the VgrG protein. The trimer of the VgrG protein shares similarities with the trimeric gp27-gp5 complex (i.e. the bacteriophage tail spike required to puncture the bacterial cell) (17, 18). A number of VgrG proteins, called specialized VgrGs, carry an additional C-terminal domain (19). By analogy with the bacteriophage spike, it has been proposed that upon host cell puncturing, the C-terminal domain is delivered into the cytosol. Indeed, the C-terminal ACD domain of the V. cholerae VgrG1 protein is translocated into target cells, where it covalently cross-links G-actin (20, 21). The resulting cross-linked dimers or polymers are in a conformation not compatible with F-actin filament formation. Therefore, the depletion of the pool of G-actin displaces the equilibrium between F-actin and G-actin toward G-actin and leads to F-actin depolymerization, thus disabling phagocytic functions.
Despite the critical importance of the MARTX and VgrG1 ACD in bacterial pathogenesis, we still lack structural information to better understand the catalytic mechanism of actin cross-linking. Here, we report the crystal structure and the activity of the V. cholerae VgrG1 ACD. The V-shaped VgrG1 ACD crystal structure harbors an active site composed of five residues, conserved among all MARTX ACDs (8). We show that, as for MARTX ACD, the VgrG1 ACD E16Q variant is impaired for in vitro and in vivo actin cross-linking activity. The catalytic site is blocked by the N-terminal segment in the apo-form. Interestingly, this segment is displaced by ATP/ADP and Mg2+/Mn2+ in the holo-forms. We confirm that the purified VgrG1 ACD cross-links G-actin in vitro. This activity requires ATP and Mg2+ or Mn2+. Using microinjection in fibroblasts (22), we demonstrate that less than 100 molecules of VgrG1 ACD suffice to induce F-actin depolymerization. Finally, modeling of the MARTX ACD and comparison with the VgrG1 ACD reveals the complete conservation of the active site and of the probable G-actin binding interface between both molecules.
EXPERIMENTAL PROCEDURES
Protein Cloning and Production
The sequence encoding the vgrG1 effector domain (residues 701–1095) of V. cholerae strain O395 was cloned into pETG-20A expression vector (a kind gift from Dr. Arie Geerlof, EMBL, Hamburg) according to standard GatewayTM protocols. The final construct allows the production of the soluble cytoplasmic VgrG1-Ct fragment fused to an N-terminal hexahistidine-tagged thioredoxin followed by a tobacco etch virus protease cleavage site. The plasmid was transformed into the Escherichia coli T7 Iq pLysS expression strain (New England Biolabs). Cells were grown at 37 °C in terrific broth until the A600 reached 0.9, and vgrG1 expression was inducted with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside overnight at 17 °C. After cell harvesting, cells were resuspended in 50 mm Tris-HCl, pH 7.5, 300 mm NaCl, and lysozyme was added (0.25 mg/ml final concentration). Cells were broken by sonication. The soluble proteins were separated from inclusion bodies and cell debris by centrifugation for 30 min at 20,000 × g. The VgrG1-Ct fusion was purified using Ni2+ affinity chromatography (HisTrap, 5 ml (GE Healthcare) on an AKTA FPLC system) and eluted with a step gradient of 250 mm imidazole. The VgrG1-Ct fusion protein was digested overnight at 4 °C by a hexahistidine-tagged tobacco etch virus protease using a 1:20 (w/w) protease/protein ratio. The tobacco etch virus protease and contaminants were bound on a second Ni2+ affinity chromatography, and the native VgrG1-Ct was collected in the flow-through and then separated on a preparative Superdex 200 gel filtration column (GE Healthcare) equilibrated in 10 mm Tris-HCl, pH 7.5, 150 mm NaCl. The final concentration of the VgrG1 solution was 13 mg/ml. The selenomethionine derivative of VgrG1-Ct was produced in minimal medium supplemented with 100 mg/liter lysine, phenylalanine, and threonine, 50 mg/liter isoleucine, leucine, valine, and selenomethionine. Protein production was performed overnight at 17 °C after the addition of 0.5 mm isopropyl 1-thio-β-d-galactopyranoside, and the selenomethionine-VgrG1-Ct protein was purified as described above. The E16Q variant was produced at levels comparable with the native protein (∼70 mg/liter), and the E16A variant could not be expressed at all.
Characterization of the VgrG1 ACD
Bioinformatics analysis indicated the similarity between the ACD of the V. cholerae RtxA toxin and the C-terminal effector domain of VgrG1 (8) (supplemental Fig. S1A). Sequence alignment of both domains yielded 61% amino acid sequence identity and no insertions or deletions (supplemental Fig. S1B). The puncturing device of VgrG1 is linked to the effector domain by a linker of ∼100 amino acids. Analysis of this linker using the metaserver MeDor (23) indicates that most of it is intrinsically disordered (24) (supplemental Fig. S1A). Sequence comparison and disorder analysis suggested that the effector ACD domain of VgrG1 is comprised between residue Ala-701 and the C-terminal residue Val-1095. This 395-amino acid domain (called hereafter VgrG1 ACD) was produced in high yields (∼70 mg/liter) and behaved as a monomer in solution, and CD spectra indicated a high content of secondary structures.
Activity in Vitro on Purified G-actin
Recombinant VgrG1-ACD (WT or E16Q mutant; 1.8 μm) was incubated at 37 °C with purified rabbit skeletal monomeric G-actin (cytoskeleton; 4 μm) in a buffer containing 20 mm Hepes, pH 7.5, 2 mm MgCl2 or MnCl2 or CaCl2, 20 mm KCl, and 0.5 mm ATP or AMP-PNP. Reactions were allowed to proceed for 2 h. Reaction mixtures were separated by 8% SDS-PAGE, and actin was visualized by silver staining or by Western blot analysis with actin antiserum (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Quantification of the relative abundances of multimers of actin was performed using the software ImageJ.
Activity in Vivo Using Microinjection
The human fibroblast HEL299 cells (ATCC number CCL-137) were grown in minimum Eagle's medium, 10% FBS. Microinjections were performed as described previously (22) with the following modifications. Cells were microinjected using a FemtoJet microinjector that was controlled by an InjectMan NI 2 and coupled to Femtotips needles (Eppendorf AG, Hamburg, Germany). The injection pressure of 120 hectopascals for 0.3 s allowed us to microinject ∼10 fl/cell, corresponding to ∼75 microinjected molecules (0.5 μg/ml) or ∼700 molecules (5 μg/ml) in the cell viability test. After injection, the cells were returned for 60 min at 37 °C in a 5% CO2 atmosphere, except where indicated. At this point, the cells were either prepared for live observation or fixed and prepared for immunofluorescence. The F-actin cytoskeleton was stained with BODIPY-FL (Molecular Probes, Inc., Eugene, OR), and cytotoxicity was measured by 6-carboxyfluorescein diacetate staining as recommended by the manufacturer (Sigma-Aldrich). The resulting effects of the microinjection process in live cells were directly visualized using time lapse imaging. Knowing the volume and the molarity of the injected solution and the molecular weight of VgrG-ACD, we calculated the number of injected VgrG1-ACDs as being 75 ± 25.
Crystallization and Structure Determination
VgrG1-Ct crystallization trials were carried out by the sitting drop vapor diffusion method in 96-well Greiner crystallization plates at 20 °C, using a nanodrop-dispensing robot (Cartesian Inc.) (25). The crystal of the selenomethionine VgrG1-Ct grew in a few days after mixing 300 nl of protein at 13 mg/ml with 100 nl of 2.17 m AmSO4, 0.1 m BisTris, pH 7.0. Crystal of the native VgrG1-Ct grew in a few days after mixing 300 nl of protein at 13 mg/ml with 100 nl of 2.4 m AmSO4, 0.1 m BisTris, pH 6.3. Crystals were cryoprotected with 4.0 m trimethylamine N-oxide and flash frozen in liquid nitrogen. A single anomalous dispersion data set was collected on the crystal of selenomethionine VgrG1-Ct at the selenium edge wavelength was collected at the SOLEIL Proxima 1 beamline with a 30º offset reorientation of the crystal using the κ axis of the Proxima1 goniometer. After processing the data sets using the XDS (26) program, the scaling was done using SCALA (27) (Table 1). The selenium sites were determined using SHELXD (28), and phases were calculated using autoSHARP (29). This led to an interpretable map from which an initial model comprising more than 85% of the final structure was built using ARP/wARP (30). VgrG1-ACD derivatives were obtained by soaking native protein crystals in a synthetic solution of 2.4 m AmSO4, 0.1 m BisTris, pH 6.3, with 2 mm AMP-PNP, ADP, or ATP and 5 mm Mg2+ or Mn2+. The crystals were cryocooled as for the native protein. Structure refinement was performed with AutoBUSTER (31) alternated with model rebuilding using COOT (32). Figures were made with PyMOL (33) or Chimera (34).
TABLE 1.
Data collection and refinement statistics
| Native selenium | NAT/AMP-PNP/Mg | NAT/ATP/Mg | NAT/ADP/Mg | NAT/ATP/Mn | NAT/ADP/Mn | E16Q/ADP/Mn | |
|---|---|---|---|---|---|---|---|
| Data collection | |||||||
| Protein Data Bank code | 4DTD | 4DTF | 4DTH | 4E1C | 4DTL | 4E1D | 4E1F |
| Source | Soleil Proxima 1 | Rotating anode | Soleil Proxima 1 | Rotating anode | Rotating anode | Rotating anode | Rotating anode |
| Wavelength (Å) | 0.97918 | 1.5418 | 0.97918 | 1.5418 | 1.5418 | 1.5418 | 1.5418 |
| Resolution limitsa (Å) | 49.1–2.5 (2.63–2.50) | 50–2.12 (2.17–2.12) | 50–1.78 (1.82–1.78) | 50–2.25 (2.3–2.25) | 50–2.39 (2.46–2.39) | 50–2.49 (2.56–2.49) | 50–2.1 (2.15–2.1) |
| Rmergea (%) | 7 (56.7) | 6.8 (48) | 4.1 (51.4) | 5.3 (43.6) | 12.9 (65) | 10.6 (41.6) | 5.2 (28.1) |
| No. of observationsa | 392,359 (55172) | 235,146 (15,055) | 398,821 (28,852) | 121,386 (8384) | 109,346 (7867) | 178,370 (8101) | 738,783 (53,822) |
| No. of unique reflectionsa | 22,710 (3235) | 36,963 (2616) | 62,298 (4533) | 28,824 (2096) | 25,641 (1858) | 42,340 (3008) | 72,859 (5448) |
| Mean ((I)/σ(I))a | 27.2/5.0 | 26.1 (4.3) | 26.5 (3.6) | 21.4 (3.7) | 11.5/2.2 | 10.2 (3.0) | 36.3 (10.1) |
| Completenessa (%) | 99.9 (99.5) | 99.6 (96.4) | 99.9 (99.6) | 92.7 (93.2) | 99.7 (99.4) | 98.6 (94.8) | 100 (100) |
| Multiplicitya | 17.3 (17.1) | 15.6 (14) | 6.4 (6.4) | 4.2 (4) | 4.2 (4.2) | 4.2 (2.7) | 10.1 (9.9) |
| Refinement | |||||||
| Resolutiona (Å) | 45.9–2.50 (2.62–2.50) | 45.4–2.12 (2.18–2.12) | 46.1–1.78 (1.83–1.78) | 40.6–2.25 (2.33–2.25) | 45.9–2.39 (2.49–2.39) | 45.4–2.49 (2.61–2.49) | 41.3–2.1 (2.16–2.1) |
| No. of reflectionsa | 22,605 (2810) | 36,813 (2941) | 62,135 (4531) | 28,721 (3061) | 25,603 (2675) | 22,761 (2799) | 38,592 (2951) |
| No. of proteins/waters/others | 2785/165/6 | 2777/523/60 | 2818/578/65 | 2771/478/55 | 2772/405/81 | 2772/155/50 | 2762/510/63 |
| No. of test set reflections | 1154 | 1284 | 1883 | 1019 | 1307 | 951 | 1210 |
| Rwork/Rfreea (%) | 18.8/21.3 (21–29) | 17.8/19.9 (20.5–23.7) | 18.1/19.2 (21.5/24.7) | 17.5/19.6 (20.8/22.4) | 19.2/20.9 (22.8/24.6) | 22.3/24.1 (23/26.1) | 17.3/18.7 (19.1/23.3) |
| Root mean square deviation, bonds (Å)/angles (degrees) | 0.01/1.18 | 0.009/1.06 | 0.009/1.01 | 0.010/1.08 | 0.008/1.08 | 0.008/1.06 | 0.01/1.10 |
| BWilson/Baverage | 64.05/62.24 | 29.0/30.7 | 27.7/32.8 | 39.5/37.2 | 39.3/33.9 | 43.4/30.9 | 27.5/29.4 |
| Ramachandran preferred/allowed (%) | 97.4/2.6 | 97.4/2.6 | 97.3/2.7 | 97.4/2.6 | 97.4/2.6 | 96.6/3.4 | 97.4/2.6 |
a Numbers in parentheses refer to the highest resolution bin.
Transient Intermediate Modelization
Transient intermediate modelization was performed, taking in consideration that the Glu-270 position in the active site is mimicked by an active site sulfate ion. Using this pivotal position, the G-actin 1 in the first transient intermediate was modeled, taking into account the activity results in the presence of actin-binding protein and shape complementarity. The G-actin 2, in the second transient intermediate, was modeled, taking into account the position of Lys-50 NH2 close to Glu-270 of G-actin 1 and shape complementarity with the first transient intermediate. These models have been established using COOT (32) and Chimera (34).
RESULTS
G-actin Cross-linking Activity of Native VgrG1-ACD in Vitro
Bioinformatics analysis indicated the similarity between the ACD of the V. cholerae RtxA toxin and the C-terminal effector domain of VgrG1 (8) (supplemental Fig. S1A). It has been shown previously that the MARTX ACD domain efficiently cross-links G-actin in vitro between Lys-50 and Glu-270 in the presence of ATP and divalent cations, such as Mg2+ (or Mn2+) (7, 35–39). The cross-linking activity of the full-length VgrG1 protein was also demonstrated in vitro using cell extracts or purified G-actin in the presence of ATP/Mg2+ (17).
In order to confirm the activity of our VgrG1 ACD domain, we assayed the in vitro activity of VgrG ACD on purified rabbit muscle G-actin (Fig. 1, A and B). The reconstituted reactions using purified G-actin were supplemented with ATP and one of the divalent cations, Mg2+, Mn2+, Zn2+, or Ca2+. They showed that ATP and Mg2+ or Mn2+, but not Zn2+ or Ca2+, are required for VgrG1 ACD activity (Fig. 1, A and B). No activity was observed when ATP was replaced by a non-cleavable analog, AMP-PNP. It is noteworthy that the cross-linking activity observed in the presence of Mn2+ was higher than with Mg2+, especially for cross-linked species of higher molecular weight.
FIGURE 1.
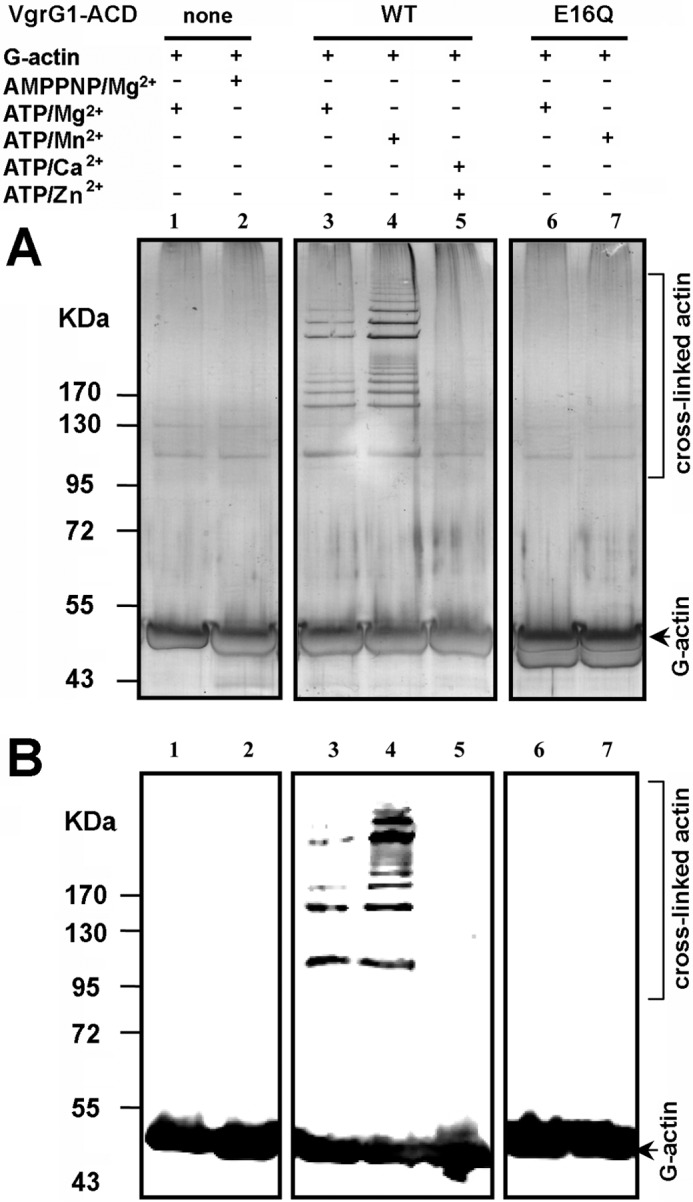
In vitro cross-linking activity of the V. cholerae VgrG1 ACD. VgrG1 WT or E16Q ACD was incubated with purified rabbit skeletal monomeric G-actin (Cytoskeleton, Denver, CO) at 37 °C for 2 h. Reactions were supplemented with ATP-Mg, ATP-Mn, or ATP-Ca, as indicated. Reaction mixtures were separated by SDS-PAGE, and actin was visualized by silver staining (A) or Western blot analysis using actin antiserum (B).
It was previously reported that G-actin cross-linking activity was lost for MARTX when residue Glu-16 (Satchell numbering is used throughout this report (8)) was substituted by Ala (8, 39). Because of the large similarity between VgrG1 ACD and MARTX sequences, we constructed a VgrG1 ACD variant in which the Glu-16 residue was replaced by Gln. The VgrG1-ACD E16Q variant was totally inactive, demonstrating the critical importance of Glu-16 for activity (Fig. 1).
VgrG1 ACD Is Highly Active in Vivo after Cytosolic Delivery
Upon cell contact, the VgrG1 ACD domain is delivered into the host cell cytosol by the Type VI secretion system using a dynamic contractile mechanism (20). We developed in vivo functional tests mimicking the bacterial secretion system by using a microinjector process. This approach has proved to be efficient in a previous study on toxin/antitoxin (22). To confirm that VgrG1-ACD is active in vivo, we examined the organization of actin filaments in VgrG1 ACD-microinjected cells using FITC-BODIPY staining. The ability of the VgrG1 ACD to affect the F-actin network was confirmed using the HEL299 human fibroblast endothelial cell line. The cells injected with buffer or an E. coli chaperonin displayed an intact F-actin network (Fig. 2, A and B). Upon microinjection, VgrG1 ACD disrupted the organized cytoskeleton (Fig. 2D), whereas cells microinjected with VgrG-E16Q exhibited an intact cytoskeleton with an intricate network of filamentous F-actin and stress fibers, comparable with control cells microinjected with buffer chaperonin (Fig. 2C). These experiments demonstrate that VgrG1 ACD, when targeted to the cytosol, causes irreversible collapse of the F-actin cytoskeleton and cell rounding. In these experiments with HEL299 fibroblast endothelial cells, ∼75 single molecules of VgrG1 ACD were injected. This number is compatible with firing of ∼25 T6S machineries from a phagocyted bacterium to a surrounding eukaryotic cell.
FIGURE 2.

Inside cell delivery of VgrG1 ACD by microinjection drastically disturbs actin skeleton. Cells were microinjected with rhodamine dextran sulfate (red fluorescence) as a tracer of microinjected proteins. F-actin was visualized with BODIPY-FL staining (green fluorescence), and the nucleus was visualized by DAPI staining (blue fluorescence). HEL299 fibroblast cells (A–D) were microinjected with buffer (A), E. coli chaperone (B), VgrG1-EA16Q variant (C), or VgrG1-ACD (D). The number of injected molecules was ∼75/cell. Scale bar, 20 μm.
Crystal Structure of Native and Complexed VgrG1 ACD
VgrG1-ACD contains 395 amino acids, between Ala-701 and the C-terminal residue Val-1095. Crystals of VgrG1-ACD appeared in nanodrops with ammonium sulfate, and optimized crystals diffracted to 2.5 Å at the Soleil synchrotron (Saint-Aubin, France). The structure was solved from selenomethionine crystals by single anomalous dispersion using the selenium edge (Table 1).
The native VgrG1 ACD electron density map covers the sequence between residues Ala-708 (residue 0) and Glu-1065 (residue 355). Mass spectroscopy footprint analysis indicated that the N-terminal and the C-terminal residues, not visible in the electron density map, were present in the purified protein and are therefore disordered in the present structure. It is noteworthy that the first eight residues in the structure (residues 0–7) have a weaker electron density than the rest of the protein.
VgrG1 ACD has an overall V-shape, with the putative active site in a cleft located within the two arms of the V (Fig. 3, A and B). The first domain, forming the left arm of the V, is mainly composed of β-strands, whereas the second domain, forming the right arm of the V, is helical. The first domain is composed of a central anti-parallel β-sheet of eight β-strands (β7,4,5,6,2,9,12,10), decorated by two adjacent small β-sheets, one above the central β-sheet (β8,3,6) and the second below (β1,11,12) (Fig. 3, A and B). A long helix (α1) packs against strands β7-β4-β5 from the central β-sheet, and strand β8 from the upper β-sheet (Fig. 3, A and B). Finally, the terminal helix α9 packs against strands β2 and β9. The second domain is composed of seven α-helices (α2–α8) and does not contain any β-strands (Fig. 3, A and B).
FIGURE 3.

Three-dimensional structure of VgrG1 ACD (residues 701–1095 and −11 to 384 (8)). A, topological representation of the secondary structures colored in rainbow mode. B, ribbon view of the native protein colored in rainbow mode. The side chains of five catalytic residues in the active site are shown as sticks. The β-sheets are boxed and numbered Sh1 to -3. C, ribbon view of the complex with AMP-PNP, Mg2+ (green) and a sulfate ion shown in sphere mode. D, surface representation of the complex with ATP and Mg2+ displayed as spheres. The N-terminal segment (residues 0–5; Nt-complex) is colored in blue. For comparison, the N-terminal segment in the native protein is represented in stick mode (green; Nt-native) and is in part masked by the ATP, which occupies the same position in the complex. The rotation of the N terminus upon ATP binding (red arrow) renders the active site accessible to catalysis.
We performed a quaternary structure analysis with the PISA server (40). The results indicated a possible VgrG1 ACD dimer formation (PISA coefficient of 0.83), and a buried water-accessible surface was determined to be 1200 Å2, ∼8% of the total surface. Together with the observation of VgrG1 ACD being a monomer in solution, the PISA analysis points only to an eventual possibility of dimerization.
We used the DALI server (41) to find possible similar structures in the Protein Data Bank. DALI returned a large number of hits with low similarity, displaying Z-score values between 6 and 3.8 and root mean square distances between 5.5 and 3.5 Å (∼150 residues included in the alignment on a total of ∼400–500 on average). The DALI best scores all pointed to a class of synthetases/ligases, such as the glutamine ligases/γ-glutamylcysteine synthetase (GCS; Protein Data Bank code 1VA6 (42)). It is noteworthy that such similarities between MARTX ACD and ligases had already been reported from local sequence comparisons (8). Using the results proposed by DALI, we superimposed a few members of this class, largely represented in the Protein Data Bank, on VgrG1-ACD. Such a superposition of GCS in complex with ATP and a suicide inhibitor (Protein Data Bank code 1VA6 (42)) and VgrG1 ACD is presented in supplemental Fig. S2A. Based on these comparisons, we concluded that VgrG1 ACD, although belonging to a novel fold, shares a few β-strands (three or four) with the ligase/synthetase family. Importantly, most of the catalytic residues of the GCS ligase structure (Protein Data Bank code 1VA6) superposed well with identical residues in the VgrG1 ACD native structure (supplemental Fig. S2B). The local similarity could further be improved by superposing the structures using the active site residues. The crystal structure of GCS was reported in complex with ATP, Mg2+, and a sulfoximine suicide inhibitor (42). In this structure, ATP provides its γ-phosphate to sulfoximine, to yield a sulfoximine phosphate stably bound to the active site, thus blocking the reaction. Surprisingly, we noticed that the ATP binding site of native VgrG1 ACD, identified by homology with that of GCS, is occupied by the N-terminal segment (residues −2 to 5).
To test whether the presence of VgrG1 ACD N terminus in the active site blocks the reaction, we crystallized VgrG1 ACD in the presence of AMP-PNP, a non-hydrolyzable analog of ATP, and Mg2+. Crystals were obtained readily, and diffraction data were collected on the laboratory rotating anode (Table 1). AMP-PNP was easily fitted in the electron density map and was well ordered except for the two last phosphate moieties, which displayed high B-factors (Fig. 3C). AMP-PNP occupies a position similar to that of ADP in complexed GCS (Protein Data Bank code 1VA6 (42)). As a consequence, the N-terminal segment is displaced from its position in the native protein structure, the bifurcation occurring at amino acid Pro-5 (Fig. 3, C and D). It is noteworthy that the N terminus borders the AMP-PNP binding site and participates in its shape, in particular Phe-4, which projects its side chain to establish a ring/ring contact with the AMP adenine moiety. We have analyzed the interaction of the N terminus with the core of the apo- and holo-form of VgrG1-ACD. The results presented in supplemental Table S1 clearly indicate a stronger interaction of the N terminus of VgrG1-ACD apo with the core, as compared with the holo-form because four strong hydrogen bonds are observed as well as a larger buried surface area.
We also generated a mutated VgrG1-ACD lacking the residues not visible in the electron density and starting at residue 1. The construct has been tentatively expressed, but only minor amounts of unfolded inclusion bodies have been obtained, indicating the importance of this stretch of residues in stability.
The Active Site of Native and E16Q Variant VgrG1-ACD
The catalytic mechanism of ligases, such as GCS (42) or MARTX-ACD (6, 35, 38), involves a two-step mechanism. In the case of the GCS, a glutamic acid is coupled to a cysteine (42). In the first step, the γ-phosphate of ATP is transferred to the glutamic acid carboxylic group. This transfer is catalyzed by three divalent cations, which weaken the β-phosphate γ-phosphate bond. Once the transfer has occurred, the phosphate of the phosphate-carboxylic group aduct is stabilized by the two cations during a time long enough for the second step to occur. Further stabilization is provided by positively charged amino acids, such as Arg-330. In the second step, the cysteine SH group performs a nucleophilic attack (SN2) on the carboxyl group of the aduct, expelling the phosphate, leading to the formation of a thioester bond.
Such a mechanism has been proposed to occur in a similar way for MARTX-ACD (6, 35, 38). In this case, the glutamic acid is replaced by the Glu-270 of actin, and the nucleophile is the amino group of Lys-50 of actin. The resulting product is an actin dimer cross-linked by a amide bond between side chains of Glu-270 and Lys-50 (Fig. 4A) (38).
FIGURE 4.
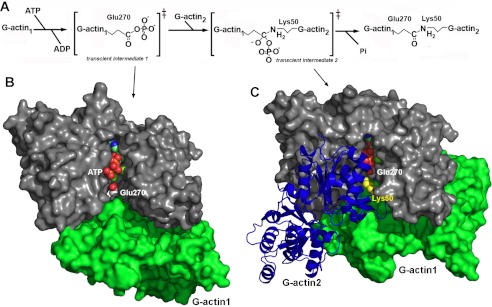
The catalytic mechanism catalyzed by VgrG1-ACD. A, schematic representation of the two-step mechanism (adapted from Satchell (8)). B, model of the first transient intermediate of VgrG1-ACD with G-actin1. G-actin 1 Glu-270 is close enough to the ATP γ-phosphate for transfer. C, model of the second transient intermediate of VgrG1-ACD with G-actin 1 and G-actin 2. G-actin 2 Lys-50 is in favorable position for nucleophilic attack on phosphorylated Glu-270 to occur, leading to a covalent amide bond between Glu-270 and Lys-50 side chains.
In order to characterize structurally and better understand the VgrG1-ACD catalytic mechanism, we grew crystals of VgrG1 ACD in the presence of ADP or ATP and Mg2+. In the ATP-bound form (Fig. 5A), ATP occupies a position strictly similar to that of AMP-PNP, with the last phosphate moieties exhibiting larger B-factors. ADP adopts also a conformation similar to its counterpart in the ATP complex (Fig. 5B). Two Mg2+ ions could be assigned in both structures, at 5.33 ± 0.1 Å from each other. A sulfate ion is observed in contact with one of the Mg2+ ions, tightly bound to Arg-341, with an identical position in all of the structures. Electrostatics at the binding site surface indicates that ATP (or ADP) is located in a positively charged surface, whereas the cations face a highly negatively charged area (supplemental Fig. S3).
FIGURE 5.

Three-dimensional structure of the VgrG1 ACD active site. A, native complex with ATP and Mg2+. B, native complex with ADP and Mg2+. C, native complex with ATP and Mn2+ compared with the complex with ATP and Mg2+ (gray sticks). D, E16Q variant complex with ADP and Mn2+ compared with the native complex with ATP and Mn2+ (gray sticks). E, E16Q variant complex with ADP and Mn2+ compared with the native complex with ATP and Mn2+ (blue sticks) and with the native complex with ADP and Mn2+ (yellow sticks). F, native VgrG1 ACD complex with ATP and Mn2+ (sticks) compared with the superposed structure of GCS (Protein Data Bank code 1VA6; side chains colored by atom type with violet carbons) in complex with ADP (green), Mg2+ (violet spheres), and the phosphosulfoximine inhibitor (blue sticks). The labels of the pairs of corresponding residues have identical colors, but GCS labels are italicized.
We then investigated the structures of co-crystals obtained with ATP or ADP (Fig. 5, C and E) in the presence of Mn2+. Because Mn2+ has a significant anomalous contribution (∼3 e−) at the x-ray wavelength of the rotating anode (1.54 Å), we identified two peaks at 3.95 ± 0.08 Å from each other in the anomalous map at ∼5 σ threshold, corresponding to the two Mn2+ ion positions. These positions are slightly displaced compared with those of the two Mg2+ ions in the ATP/Mg2+ and ADP/Mg2+ structures (Fig. 5, A–C). In contrast, the conformations of ATP in the Mg2+ and the Mn2+ structures are significantly different, the latter being more extended toward the sulfate ion. In the Mn2+ structure, the closest γ-oxygen is at 2.1 Å from the cation and 2.3 Å from the sulfate, whereas in the Mg2+ structure, the closest γ-oxygen is at 4.8 Å from the cation and 4.8 Å from the sulfate.
We submitted the E16Q inactive variant to crystallization assays with ATP and ADP in the presence of Mn2+. The anomalous map made it possible to identify a unique Mn2+ ion at a different position, peripheral to the active site, compared with those of the two cations observed in the native structure (Fig. 5E). The Mn2+ ion is only bound to a phosphate oxygen of the ADP and remotely by Glu-78 and not at all by Gln-16. The E16Q substitution is therefore sufficient to disrupt the catalytic site (the divalent cations) and therefore to abolish the ACD activity.
Comparison of VgrG1-ACD with the GCS (Protein Data Bank code 1VAT) Structure
The presence of three Mg2+ ions has been reported in the GCS structure (42). The positions of two of these cations coincide quite well those of the VgrG1 ACD Mn2+ ions (Fig. 5F, 0.6-Å shift) but less with those of the Mg2+ ions. Notably, VgrG1 ACD lacks a third divalent cation (Fig. 5F). When examining the environment of the third GCS Mg2+ ion, we noticed that it is firmly bound to GCS Glu-67 (∼Glu-78 in VgrG1 ACD), Asp-60 (∼Asp-51), and Glu-29 (∼Glu-18). Interestingly, we noticed in the VgrG1 ACD structures with ATP/ADP that the side chain of the third ligand, Glu-18 is rotated by 120°, bringing the carboxylic group to a non-binding distance of a putative divalent cation (5.9 Å; Fig. 5F). This Glu-18 conformation is stabilized by a hydrogen bond with the OH group of Thr-76 (2.7 Å). In GCS, Thr-76 is replaced by Leu-65, which is not available for establishing such a hydrogen bond. Leu-65 instead is more bulky and therefore pushes Glu-29 at a position accommodating the third Mg2+ ion. Finally, it should be mentioned that the γ-phosphate of ATP in the native VgrG1 ACD/ATP/Mn2+ structure is close to the position of the transferred γ-phosphate of the sulfoximine inhibitor in the GCS (Protein Data Bank code 1VA6) structure. Their relative positions and conformations are compatible with a perfect positioning of the ATP for a productive nucleophilic attack by the G-actin Glu-270 carboxylic group (Fig. 5F).
Similarity between VgrG1 and MARTX Actin Cross-linking Domains
The VgrG1 and MARTX ACD sequences share 61% identity, without insertions or deletions (8). Notably, when projecting this sequence similarity onto the VgrG1 ACD surface, we observed that the ATP/cations/ligand binding area was conserved (supplemental Fig. S2C). Furthermore, large patches of conserved residues were observed at the surface of VgrG1 ACD, located mainly around the active site side and between the two arms of the V-shape. These surfaces are likely to accommodate the two monomers of G-actins in both ACD.
Because of the high sequence identity and the lack of insertions/deletions, we could readily model the MARTX ACD domain. Examination of the energy-refined model reveals that the conformations of all of the residues of the active site are conserved, suggesting that binding of cofactors and divalent cations is probably identical to that determined for the VgrG1 ACD. We can therefore conclude that the G-actin cross-linking requirements, the cations involved, and the catalytic mechanism of both enzymatic domains should be identical. The MARTX ACD domain being part of a larger multidomain protein, like the VgrG1 ACD, we hypothesize that the N-terminal fragment might be similarly involved in the folding process and/or in the catalytic mechanism.
DISCUSSION
In this study, we report the crystal structure of the actin cross-linking domain of an evolved VgrG1 actin cross-linking domain from V. cholerae. Toxins of the MARTX family are important virulence factors because they affect formation of F-actin filament and disable eukaryotic host cells. Both MARTX and VgrG1 domains have a protein ligase activity by catalyzing isopeptide bond formation between two actin monomers (43). The in vivo and in vitro activity experiments reported here confirm that the VgrG1 ACD domain is sufficient for G-actin cross-linking. Conversely, the N terminus of VgrG1 might be devoted to puncturing the target cell membrane (20), consistent with the function of the homologuous bacteriophage T4 (gp27-gp5)3 tail spike complex (18). The ∼100-amino acid linker domain located between the (gp27-gp5)3 domain and the ACD is predicted as naturally disordered (24). One may hypothesize that this region is targeted by host cell proteases upon injection to deliver the ACD in the cytosol, a hypothesis that remains to be tested. Our in vivo assays, using a microinjection device, made it possible to quantify VgrG1 ACD activity in fibroblast cells. They indicated that the VgrG1 ACD is active upon microinjection of a small number of ACDs (50 < n < 100), a value compatible with the concomitant firing of 15–30 type 6 secretion machineries.
The comparison of the crystal structures of the VgrG1 ACD in the presence and absence of ATP shows that ATP binding induces a conformational modification of the N-terminal segment. In the absence of ATP, this fragment is inserted in the cleft between the two arms of the ACD V shape and therefore fills in the active site and stabilizes it. In the presence of ATP, this segment is dislodged from the cleft and displaced toward the left part of the ACD. In this conformation, it (i) liberates the active site and (ii) participates in the stabilization of the ATP molecule in the active site (supplemental Table S1).
The activity of the ACD domain requires both ATP and a divalent cation (Mg2+ or Mn2+). Mutagenesis studies have been performed by Satchell's group (8, 39) on MARTX-ACD. The results indicate that Glu-16 is the only essential residue for activity (Table 2). Mutations of other residues lead to a decrease of the cross-linking activity (Table 2). We confirmed the critical role of the invariant Glu-16 residue in the cross-linking activity, in vitro and in vivo. We showed that in the E16Q structure, only one Mg2+/Mn2+ cation is present in the structure, at a position incompatible with activity. Because Glu-16 is bridging the two Mg2+/Mn2+ cations (Fig. 6), its mutation disrupts the cation position and hence the catalytic activity. In contrast, mutations of Glu-78 or Glu-339 (8, 39), which bind only one cation (Fig. 6), Mn2 and Mn1, respectively, lead only to an activity decrease, indicating thus that the cation positions are modified but not enough to cancel catalysis. Some variations in the postions and the nature of the cations seems to be compatible with activity because both Mn2+ and Mg2+ are able to catalyze the phosphate transfer, although with different efficiencies, despite significant differences in their respective positions.
TABLE 2.
Residues mutated in MARTX-ACD and the functional result of the mutations
Numbering equivalence is presented between ACD domains, VgrG1 and MARTX numbering.
| VgrG1/MARTX-ACD | VgrG1 | MARTX | GCS (1VA6) | Mutations affecting cross-linking in MARTX-ACD (9, 39) | Function in ACD |
|---|---|---|---|---|---|
| Glu-16 | 727 | 1990 | Glu-27 | Abolished | Binds both cations |
| Glu-18 | 729 | 1992 | Glu-29 | Decreased | Far from active site; actin-1 binding? |
| Asp-51 | 762 | 2025 | Asp-60 | Decreased | Far from cations; water-mediated binding? |
| Glu-78 | 789 | 2052 | Glu-67 | Decreased | Binds a cation (Mg2) |
| His-109 | Decreased | Far from cations; actin-1 binding? | |||
| Glu-339 | 1050 | 2313 | Glu-328 | Decreased | Binds a cation (Mg1) |
| Arg-341 | 1052 | 2315 | Arg-330 | Decreased | Binds SO4; putative carboxyl binding site |
FIGURE 6.
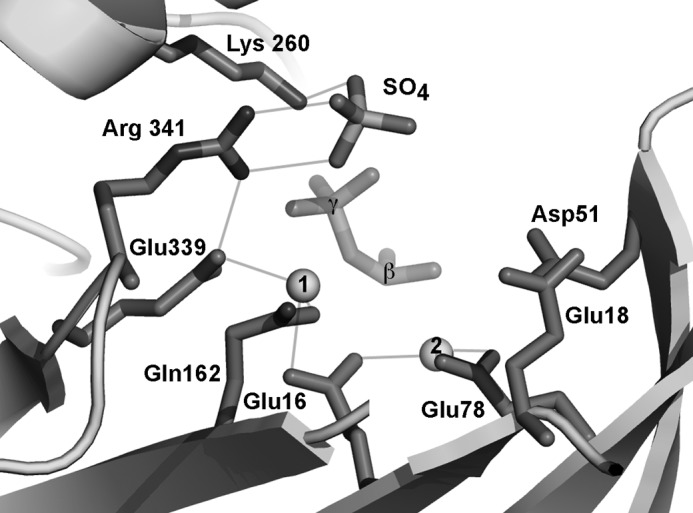
Synthetic representation of the three-dimensional structure of the VgrG1 ACD active site residues, cation-conserved sulfate anion, and the β- and γ-phosphates of ATP.
The mutation of Arg-341 leads also to an activity reduction (8, 39). In our structures, Arg-341 binds to a sulfate ion in the active site, close to the ATP γ-phosphate (Fig. 6). We think that this sulfate ion occupies the position where Glu-270 binds in the first transient intermediate (Fig. 4, A and B). Its mutation probably diminishes the stablilization of the intermediate and decreases the enzyme's activity. Mutations of residues Glu-18, Asp-51, and His-109 also lead to an activity decrease. However, these residues do not directly interact with the cations. The only possible explanation for their function would be a significant stabilizing interaction with actins in step 1 or 2.
It has been shown that actin-binding proteins modulate the cross-linking activity of MARTX-ACD (37). Profilin, thymosin-β4, and gelsolin are compatible with actin cross-linking activity, whereas cofilin and DNAseI inhibit it. The observation of the available actin·actin-binding protein complexes makes it possible to interpret these results. The first set of proteins bind in a area far from both Glu-270 and Lys-50, whereas DNase I binds the loop carrying Lys-50, thus inhibiting the second step of the reaction, the nucleophilic attack by actin-2 (Fig. S4).
These results together with consideration of the Glu-270 position in the active site led us to suggest models of the first and second transient intermediates (Fig. 4, B and C). The model of the first transient binary intermediate indicates that accessibility is left for the second G-actin to perform the SN2 reaction. The model of the second transient ternary intermediate indicates that such a compact reacting intermediate is possible and that only the modeled position is realistic (within a small positional uncertainty). Indeed, it is not possible to crystallize such labile species, but we hope that these models may help in the design of mutants aimed at refining them.
Finally, the structural characterization of the VgrG1 ACD toxin provides unique opportunities to design compounds targeting the active site. The close relationship between the active sites of the MARTX/VgrG1 ACD and that of the Gly-Cys synthetases suggests that sulfoximines might be good candidates to perform this task.
Supplementary Material
Acknowledgments
We thank Laure Journet and members of the E. C. and C. C. Type VI secretion system groups for fruitful discussions and Ella Danloss for encouragement. We gratefully acknowledge the help of Pierre Legrand (Proxima 1) for data collection and the Soleil synchrotron for beamtime allocation. We thank Michel R. Popoff for the kind gift of toxin B of Clostridium difficile.

This article contains supplemental Table S1 and Figs. S1–S4.
- MARTX
- multifunctional autoprocessing repeats-in-toxins
- ACD
- actin cross-linking domain
- AMP-PNP
- 5′-adenylyl-β,γ-imidodiphosphate
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- GCS
- γ-glutamylcysteine synthetase.
REFERENCES
- 1. Charles R. C., Ryan E. T. (2011) Cholera in the 21st century. Curr. Opin. Infect. Dis. 24, 472–477 [DOI] [PubMed] [Google Scholar]
- 2. Piarroux R., Barrais R., Faucher B., Haus R., Piarroux M., Gaudart J., Magloire R., Raoult D. (2011) Understanding the cholera epidemic, Haiti. Emerging Infect. Dis. 17, 1161–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weil A. A., Ivers L. C., Harris J. B. (2012) Cholera. Lessons from Haiti and beyond. Curr. Infect. Dis. Rep. 14, 1–8 [DOI] [PubMed] [Google Scholar]
- 4. Nelson E. J., Harris J. B., Morris J. G., Jr., Calderwood S. B., Camilli A. (2009) Cholera transmission. The host, pathogen, and bacteriophage dynamic. Nat. Rev. Microbiol. 7, 693–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sanchez J., Holmgren J. (2011) Cholera toxin. A foe and a friend. Ind. J. Med. Res. 133, 153–163 [PMC free article] [PubMed] [Google Scholar]
- 6. Satchell K. J. (2011) Structure and function of MARTX toxins and other large repetitive RTX proteins. Annu. Rev. Microbiol. 65, 71–90 [DOI] [PubMed] [Google Scholar]
- 7. Sheahan K. L., Cordero C. L., Satchell K. J. (2004) Identification of a domain within the multifunctional Vibrio cholerae RTX toxin that covalently cross-links actin. Proc. Natl. Acad. Sci. U.S.A. 101, 9798–9803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Satchell K. J. (2009) Actin cross-linking toxins of Gram-negative bacteria. Toxins 1, 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pukatzki S., Ma A. T., Sturtevant D., Krastins B., Sarracino D., Nelson W. C., Heidelberg J. F., Mekalanos J. J. (2006) Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc. Natl. Acad. Sci. U.S.A. 103, 1528–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cascales E. (2008) The type VI secretion toolkit. EMBO Rep. 9, 735–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schwarz S., Hood R. D., Mougous J. D. (2010) What is type VI secretion doing in all those bugs? Trends Microbiol. 18, 531–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schwarz S., West T. E., Boyer F., Chiang W. C., Carl M. A., Hood R. D., Rohmer L., Tolker-Nielsen T., Skerrett S. J., Mougous J. D. (2010) Burkholderia type VI secretion systems have distinct roles in eukaryotic and bacterial cell interactions. PLoS Pathog. 6, e1001068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Russell A. B., Hood R. D., Bui N. K., LeRoux M., Vollmer W., Mougous J. D. (2011) Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475, 343–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hood R. D., Singh P., Hsu F., Güvener T., Carl M. A., Trinidad R. R., Silverman J. M., Ohlson B. B., Hicks K. G., Plemel R. L., Li M., Schwarz S., Wang W. Y., Merz A. J., Goodlett D. R., Mougous J. D. (2010) A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 7, 25–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cascales E., Cambillau C. (2012) Structural biology of type VI secretion systems. Phil. Trans. R. Soc. B 367, 1102–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Basler M., Pilhofer M., Henderson G. P., Jensen G. J., Mekalanos J. J. (2012) Type VI secretion requires a dynamic contractile phage tail-like structure. Nature 483, 182–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pukatzki S., Ma A. T., Revel A. T., Sturtevant D., Mekalanos J. J. (2007) Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc. Natl. Acad. Sci. U.S.A. 104, 15508–15513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leiman P. G., Basler M., Ramagopal U. A., Bonanno J. B., Sauder J. M., Pukatzki S., Burley S. K., Almo S. C., Mekalanos J. J. (2009) Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc. Natl. Acad. Sci. U.S.A. 106, 4154–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pukatzki S., McAuley S. B., Miyata S. T. (2009) The type VI secretion system. Translocation of effectors and effector domains. Curr. Opin. Microbiol. 12, 11–17 [DOI] [PubMed] [Google Scholar]
- 20. Ma A. T., McAuley S., Pukatzki S., Mekalanos J. J. (2009) Translocation of a Vibrio cholerae type VI secretion effector requires bacterial endocytosis by host cells. Cell Host Microbe 5, 234–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Satchell K. J. (2009) Bacterial martyrdom. Phagocytes disabled by type VI secretion after engulfing bacteria. Cell Host Microbe 5, 213–214 [DOI] [PubMed] [Google Scholar]
- 22. Audoly G., Vincentelli R., Edouard S., Georgiades K., Mediannikov O., Gimenez G., Socolovschi C., Mège J. L., Cambillau C., Raoult D. (2011) Effect of rickettsial toxin VapC on its eukaryotic host. PLoS One 6, e26528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lieutaud P., Canard B., Longhi S. (2008) MeDor. A metaserver for predicting protein disorder. BMC Genomics 9, Suppl. 2, S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Receveur-Bréchot V., Bourhis J. M., Uversky V. N., Canard B., Longhi S. (2006) Assessing protein disorder and induced folding. Proteins 62, 24–45 [DOI] [PubMed] [Google Scholar]
- 25. Sulzenbacher G., Gruez A., Roig-Zamboni V., Spinelli S., Valencia C., Pagot F., Vincentelli R., Bignon C., Salomoni A., Grisel S., Maurin D., Huyghe C., Johansson K., Grassick A., Roussel A., Bourne Y., Perrier S., Miallau L., Cantau P., Blanc E., Genevois M., Grossi A., Zenatti A., Campanacci V., Cambillau C. (2002) A medium-throughput crystallization approach. Acta Crystallogr. D Biol. Crystallogr. 58, 2109–2115 [DOI] [PubMed] [Google Scholar]
- 26. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Collaborative Computational Project, Number 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 28. Schneider T. R., Sheldrick G. M. (2002) Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 58, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 29. Bricogne G., Vonrhein C., Flensburg C., Schiltz M., Paciorek W. (2003) Generation, representation, and flow of phase information in structure determination. Recent developments in and around SHARP 2.0. Acta Crystallogr. D Biol. Crystallogr. 59, 2023–2030 [DOI] [PubMed] [Google Scholar]
- 30. Morris R. J., Perrakis A., Lamzin V. S. (2003) ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol. 374, 229–244 [DOI] [PubMed] [Google Scholar]
- 31. Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., Bricogne G. (2004) Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D Biol. Crystallogr. 60, 2210–2221 [DOI] [PubMed] [Google Scholar]
- 32. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 33. DeLano W. The PyMOL Molecular Graphics System, version 1.3r1, Schrodinger, LLC, New York [Google Scholar]
- 34. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera. A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 35. Cordero C. L., Kudryashov D. S., Reisler E., Satchell K. J. (2006) The actin cross-linking domain of the Vibrio cholerae RTX toxin directly catalyzes the covalent cross-linking of actin. J. Biol. Chem. 281, 32366–32374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cordero C. L., Sozhamannan S., Satchell K. J. (2007) RTX toxin actin cross-linking activity in clinical and environmental isolates of Vibrio cholerae. J. Clin. Microbiol. 45, 2289–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kudryashov D. S., Cordero C. L., Reisler E., Satchell K. J. (2008) Characterization of the enzymatic activity of the actin cross-linking domain from the Vibrio cholerae MARTX Vc toxin. J. Biol. Chem. 283, 445–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kudryashov D. S., Durer Z. A., Ytterberg A. J., Sawaya M. R., Pashkov I., Prochazkova K., Yeates T. O., Loo R. R., Loo J. A., Satchell K. J., Reisler E. (2008) Connecting actin monomers by iso-peptide bond is a toxicity mechanism of the Vibrio cholerae MARTX toxin. Proc. Natl. Acad. Sci. U.S.A. 105, 18537–18542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Geissler B., Bonebrake A., Sheahan K. L., Walker M. E., Satchell K. J. (2009) Genetic determination of essential residues of the Vibrio cholerae actin cross-linking domain reveals functional similarity with glutamine synthetases. Mol. Microbiol. 73, 858–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 41. Holm L., Rosenström P. (2010) Dali server. Conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hibi T., Nii H., Nakatsu T., Kimura A., Kato H., Hiratake J., Oda J. (2004) Crystal structure of γ-glutamylcysteine synthetase. Insights into the mechanism of catalysis by a key enzyme for glutathione homeostasis. Proc. Natl. Acad. Sci. U.S.A. 101, 15052–15057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sheahan K. L., Satchell K. J. (2007) Inactivation of small Rho GTPases by the multifunctional RTX toxin from Vibrio cholerae. Cell. Microbiol. 9, 1324–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.