

Background: Factor XIa proteolytically activates factor IX.
Results: XIa cleaves IX after Arg145, forming IXα, and then after Arg180, forming IXaβ. Both reactions require substrate binding to the XIa A3 domain.
Conclusion: XIa activates IX by an exosite-mediated release-rebind mechanism. Efficiency of the second cleavage is enhanced by changes resulting from the first cleavage.
Significance: The data support a new model for IX activation by XIa.
Keywords: Coagulation Factors, Enzyme Catalysis, Enzyme Kinetics, Enzyme Mechanisms, Enzyme Structure, Exosite, Factor IX, Factor XIa
Abstract
During blood coagulation, the protease factor XIa (fXIa) activates factor IX (fIX). We describe a new mechanism for this process. FIX is cleaved initially after Arg145 to form fIXα, and then after Arg180 to form the protease fIXaβ. FIXα is released from fXIa, and must rebind for cleavage after Arg180 to occur. Catalytic efficiency of cleavage after Arg180 is 7-fold greater than for cleavage after Arg145, limiting fIXα accumulation. FXIa contains four apple domains (A1–A4) and a catalytic domain. Exosite(s) on fXIa are required for fIX binding, however, there is lack of consensus on their location(s), with sites on the A2, A3, and catalytic domains described. Replacing the A3 domain with the prekallikrein A3 domain increases Km for fIX cleavage after Arg145 and Arg180 25- and ≥90-fold, respectively, and markedly decreases kcat for cleavage after Arg180. Similar results were obtained with the isolated fXIa catalytic domain, or fXIa in the absence of Ca2+. Forms of fXIa lacking the A3 domain exhibit 15-fold lower catalytic efficiency for cleavage after Arg180 than for cleavage after Arg145, resulting in fIXα accumulation. Replacing the A2 domain does not affect fIX activation. The results demonstrate that fXIa activates fIX by an exosite- and Ca2+-mediated release-rebind mechanism in which efficiency of the second cleavage is enhanced by conformational changes resulting from the first cleavage. Initial binding of fIX and fIXα requires an exosite on the fXIa A3 domain, but not the A2 or catalytic domain.
Introduction
Factor IX (fIX)2 is the zymogen of a protease, factor IXaβ (fIXaβ), which is required for proper formation and maintenance of blood clots at sites of vascular injury (1, 2). Congenital fIX deficiency causes the severe bleeding disorder hemophilia B (1). Human fIX is a 57-kDa protein composed of an N-terminal calcium-binding Gla domain, two epidermal growth factor domains, an activation peptide, and a trypsin-like catalytic domain (1, 2). It shares this structure with coagulation factors VII and X (3). The Gla domains of these proteins facilitate binding to phospholipid membranes.
Conversion of fIX to fIXaβ requires cleavage of the Arg145–Ala146 and Arg180–Val181 peptide bonds to release the activation peptide (2, 4, 5). The physiologic mediators of this process are the serine proteases factor VIIa (fVIIa) and factor XIa (fXIa). FIX activation by fVIIa requires Ca2+ and phospholipid (6–8). In the presence of the cofactor tissue factor, fVIIa cleaves fIX first after Arg145, forming the inactive intermediate fIXα. FIXα is released from fVIIa and must rebind to the protease to be cleaved after Arg180 to form fIXaβ. As the second cleavage is rate-limiting, fIXα accumulates during fIX activation by fVIIa.
FXIa appears to activate fIX by a different process than fVIIa. FXIa, is a dimer of identical subunits, each containing a trypsin-like catalytic domain and a heavy chain comprised of four apple domains (A1–A4) (9–12). In contrast to fVIIa, fXIa lacks a Gla domain. Perhaps as a result, fIX activation by fXIa, although Ca2+-dependent, does not require phospholipid. During fIX activation by fXIa an intermediate does not accumulate (13, 14). This is unrelated to the dimeric structure of the protease, as isolated fXIa subunits convert fIX to fIXaβ without intermediate accumulation (14, 15). FXIa readily cleaves fIX with an R180A substitution after Arg145, whereas fIX with an R145A substitution undergoes insignificant cleavage after Arg180 (14). This indicates that the structure of fIX facilitates ordered bond cleavage, with the Arg145–Ala146 bond presented first to the fXIa active site. Changes in conformation resulting from cleavage after Arg145 may then facilitate cleavage after Arg180. A similar process was reported previously for prothrombin activation (16).
There are conflicting hypotheses regarding the manner in which fXIa is able to activate fIX without fIXα accumulation. It has been proposed that fIXα may not be released from fXIa prior to conversion to fIXaβ (13, 17). However, at least a portion of the intermediate must be released, as active site-blocked fXIa competes with fXIa for fIXα binding during fIX activation (14). Active site-blocked fIXaβ is a competitive inhibitor of fIX cleavage by fXIa (18), and fIXα may have similar inhibitory properties. If fIXα is released from fXIa, the lack of fIXα accumulation implies that cleavage after Arg145 is rate-limiting, although this has yet to be demonstrated.
There is compelling evidence that one or more exosites (substrate binding sites distinct from the protease active site) on fXIa are required for normal fIX activation (17–23), but there is a lack of agreement on their locations. It is clear that Ca2+-dependent fIX activation requires the fXIa heavy chain (19, 20). One report indicated that the A2 domain contains a fIX binding site (21), whereas others point to A3 (22, 23). More recent work suggests a distinct fIX binding site may be located on the fXIa catalytic domain (17). Km for fIX activation by the isolated fXIa catalytic domain (a species lacking the heavy chain) was reported to be similar to Km for fIX activation by full-length fXIa, implying the catalytic domain was largely responsible for recognition and specificity of fIX binding. These data are not compatible with those showing that A3 domain substitution results in a marked increase in Km for fIX activation (18, 22, 23).
We conducted a kinetic analysis of fIX and fIXα activation to fIXaβ by fXIa and fXIa variants, to address the conflicting information regarding the mechanism of fIX activation and the locations of substrate binding exosites on fXIa. Binding studies were used to support the kinetic data, which show that fIX activation by fXIa is a largely sequential process in which fIX is converted to fIXα and then to fIXaβ, with a substantial increase in catalytic efficiency of the second cleavage explaining the absence of fIXα accumulation. The A3 domain serves as the major binding site for both fIX and fIXα. The dramatic decrease in catalytic efficiency of forms of fXIa lacking the functional exosite illustrates the critical role of Ca2+-dependent fIX and fIXα binding to fXIa.
EXPERIMENTAL PROCEDURES
Materials
Human fIX, fIXaβ, glutamyl-glycyl-arginyl-chloromethyl ketone (EGR-CK), Phe-Pro-Arg-chloromethyl ketone (FPR-CMK), and biotinylated EGR-CK were from Hematologic Technologies (Essex Junction, VT). Factor XIIa (fXIIa) and goat anti-human factor IX IgG-HRP were from Enzyme Research (South Bend, IN). l-Pyroglutamyl-l-prolyl-l-arginine p-nitroaniline S-(2366) was from Diapharma (West Chester, OH). Soybean trypsin inhibitor-agarose and bovine serum albumin (BSA) were from Sigma. Strepavidin-agarose was from Pierce. Anti-fXI IgG O1A6 (24) and anti-fIX IgG SB249417 (25) have been described.
Recombinant FXIa
The system for expressing human wild type fXI (fXI-WT) and fXI variants has been reported (14, 22, 23). HEK293 fibroblasts (ATCC-CRL1573) were transfected with 40 μg of pJVCMV containing a fXI cDNA and 2 μg of pRSVneo encoding a neomycin resistance marker using an Electrocell Manipulator 600, (BTX, San Diego, CA). Cells were initially grown in Dulbecco's modified Eagle's medium with 5% fetal bovine serum and 500 μg/ml of G418, then switched to serum-free medium (Cellgro Complete, Mediatech, Herndon, VA). FXI was purified from conditioned media by affinity chromatography using anti-human fXI-IgG 1G5.12 (22, 23).
FXI chimeras in which apple domains are replaced with prekallikrein (PK) apple domains have been described (22, 26). This study used fXI with the PK A2 or A3 domain (fXI/PKA2 or fXI/PKA3) (22), and fXI in which the entire heavy chain is replaced with the PK heavy chain (FXI-CD/PK-HC) (26). In fXI/PKA2, Lys140 was changed to Ser to prevent autoproteolysis.
FXI was converted to fXIa by incubation with fXIIa (100:1 substrate to enzyme molar ratio) at 37 °C for 24 h in 50 mm Tris-HCl, pH 7.4, 100 mm NaCl (TBS). Conversion of the 80-kDa zymogen fXI subunits to the 45-kDa heavy chain and 35-kDa catalytic domain of fXIa was confirmed by SDS-PAGE. In studies of fIX activation by fXIa or fXIa variants, removal of fXIIa, or inhibition of fXIIa, had no effect on fIX or fIXα cleavage.
To prepare isolated fXIa heavy chain (fXIa-HC) and isolated fXIa catalytic domain (fXIa-CD) (18), fXI with Cys362 and Cys482 changed to Ser (fXI-Ser362/Ser482) was activated and passed over a soybean trypsin inhibitor-agarose column. FXIa-CD binds to the column and is eluted with TBS containing 200 mm benzamidine, whereas fXIa-HC flows through the column.
Preparation of FIXα
FIX (14.3 μm) was incubated with 50 nm fXIa in TBS, pH 7.4, 20 mm EDTA for 12 min at 37 °C. FIXaβ was inhibited by incubating 90 min at 37 °C with biotinylated EGR-CK (10-fold molar excess over fIX). After dialysis against PBS, strepavidin immobilized on agarose resin was added, and incubated for 1 h at room temperature with mixing. Resin was removed by centrifugation, and the supernatant containing fIXα was dialyzed against 50 mm HEPES, pH 7.4, 125 mm NaCl, 1 mg/ml of polyethylene glycol (PEG) 8000.
Hydrolysis of S-2366 by FXIa
FXIa (6 nm) was incubated with S-2366 (50–2000 μm) in TBS at room temperature. Free p-nitroaniline formation was followed by continuous monitoring of absorbance at 405 nm on a SpectraMax 340 plate reader (Molecular Devices, Sunnyvale, CA). Rates of p-nitroaniline generation (nm/s) were determined using an extinction coefficient of 9920 OD units (405 nm) per mol per cm of p-nitroaniline. Km and kcat for S-2366 cleavage were determined by nonlinear least squares fitting performed with MicroMath Scientific Software.
Activation of FIX and FIXα by FXIa
FIX or fIXα (25 nm to 5 μm) in assay buffer (50 mm HEPES, pH 7.4, 125 mm NaCl, 5 mm CaCl2 (or 25 mm EDTA), 1 mg/ml of PEG 8000) was incubated at room temperature with fXIa (1 to 240 nm active sites, depending on the fXIa species and substrate) in tubes coated with PEG 20,000. At various times (0 to 640 min), aliquots were removed into nonreducing SDS-sample buffer, size fractionated on 17% polyacrylamide-SDS gels, and stained with GelCode Blue (Pierce). Gels were imaged on an Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE) using an excitation λ of 685 nm, and emission λ of 720 nm. Conversion of fIX to fIXα and fIXaβ, and fIXα to fIXaβ was assessed by densitometry. To determine the amount of protein per band, standards were run on a separate gel, with one standard also run on the gel with the time course samples. Full progress curves were constructed from data for disappearance of fIX, and appearance of fIXα and fIXaβ. In some reactions, antibodies to fXIa or fIX were included.
Kinetic Analyses
Steady-state kinetic parameters for fIX or fIXα cleavage were obtained by numerical integration fitting of full progress curves of substrate depletion, and intermediate and product formation at substrate concentrations from 25 to 5000 nm; and by analysis of initial rate dependence of substrate depletion as a function of substrate concentration. Rates of cleavage of the Arg145–Ala146 and Arg180–Val181 bonds were analyzed simultaneously with KinTek Explorer Version 2.5 software (27) using the reaction mechanism shown in Equations 1 and 2. IXα* indicates the intermediate bound to fXIa in the correct orientation for cleavage after Arg180.
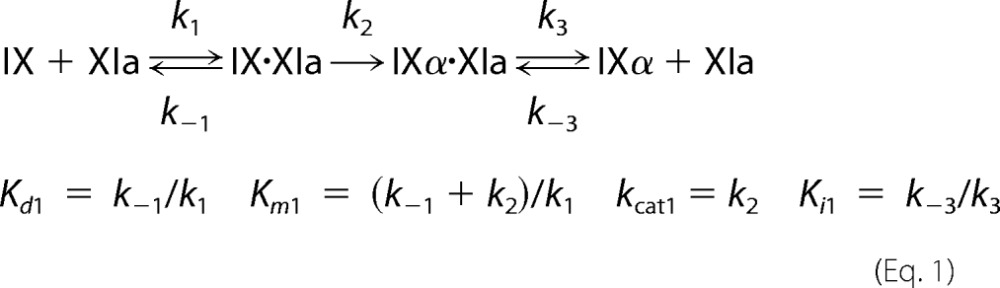 |
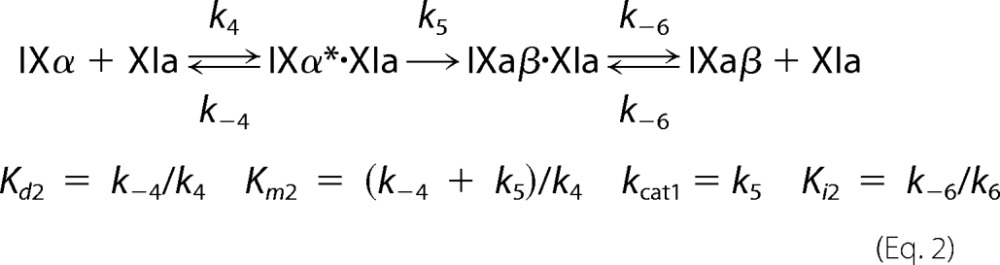 |
We reported that the product fIXaβ binds to fXIa and is a competitive inhibitor of fIX activation by fXIa (18). A similar assumption was made for product inhibition by fIXα. Kd and Ki values were initially constrained to those obtained from surface plasmon resonance studies (below) and full progress curve analysis of fIXaβ formation, and no rapid equilibrium assumptions were imposed on association and dissociation rates. By fitting full progress curves for fIX disappearance, and fIXα and fIXaβ appearance, Km, kcat, Kd, Ki, and catalytic efficiency (kcat/Km) were determined for both cleavages in fIX. The same method was used with fIXα as substrate. As fIXα preparations have ∼20% fIX contamination, analyses of conversion of fIXα to fIXaβ included the cleavage of the fIX.
Results of numerical analysis were compared with those obtained by conventional Michaelis-Menten analysis of both cleavages. With fIX as substrate, initial velocities (v0) of cleavage after Arg145 were determined from the initial slopes of progress curves for disappearance of fIX, normalized to 1 nm fXIa active sites. Values for v0 were analyzed with the Michaelis-Menten equation, and values for Km and kcat were obtained from direct nonlinear least squares analysis using Scientist Software. With fIXα as substrate, Km and kcat for cleavage after Arg180 were determined in a similar manner. Competitive binding of fXIa to fIX in the fIXα preparation was calculated by a cubic equation, and was taken into account for determining the fXIa concentration available for the reaction with fIXα.
Surface Plasmon Resonance
Binding studies were performed on a Biacore T100 flow biosensor (Biacore, Uppsala, Sweden) at 25 °C. FIX, fIXα, or fIXaβ were immobilized on carboxymethyl-dextran flow cells (CM5 sensor chips, GE Healthcare) using amine-coupling chemistry. To prevent cleavage of bound fIX or fIXα, fXIa active sites were blocked with FPR-CMK. FIXaβ active sites were blocked with EGR-CK. Flow cell surfaces were activated with a mixture of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and N-hydroxysulfosuccinimide for 5 min (flow rate 10 μl/min), after which the protein (30 μg/ml in sodium acetate, pH 4.0) was injected onto the surface. Unreacted sites were blocked for 5 min with 1 m ethanolamine. Analytes (fXIa species, 1 to 5000 nm) were perfused through flow cells in HBS-P buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 2 mm CaCl2, 1 mm benzamidine, 0.005% (v/v) P20) at 10 μl/min for 6 min. After changing to HBS-P buffer without fXIa, analyte dissociation was monitored for 10 min. Flow cells were regenerated with HBS-P containing 30 mm EDTA (no CaCl2). FXIa binding to fIX species was also tested in the absence of Ca2+ (10 mm EDTA). Data were corrected for nonspecific binding by subtracting signals obtained with analytes infused through a flow cell without coupled protein.
Binding was analyzed with BIAevaluation software (Biacore) using a bivalent binding model for dimers (fXIa-WT, fXIa/PKA2, fXIa/PKA3) and a 1:1 binding model for monomers (fXIa-CD, fXIa-CD/PK-HC). Kd values were calculated from the quotient of the derived dissociation (kd) and association (ka) rate constants. In addition, a steady state affinity model was used in which Kd was derived from nonlinear regression fitting of the response at equilibrium to fXIa concentration.
FIX Activation in the Presence of FXIa-HC
FIX (100 nm) was incubated with fXIa-WT (1 nm) with or without fXIa-HC (1 μm) in assay buffer at room temperature. At various times, aliquots were removed into nonreducing SDS sample buffer, size fractionated on 12% polyacrylamide-SDS gels, and transferred to a nitrocellulose membrane. Western blots were performed using an HRP-conjugated goat polyclonal anti-human fIX IgG and chemiluminescence for detection.
RESULTS
Recombinant FXIa
All fXI species migrate at ∼160 kDa on nonreducing SDS-PAGE, except for fXI-CD/PK-HC, which is an 80-kDa monomer (Fig. 1A). FXI-Ser362/Ser482 lacks the disulfide bond that links the A4 and catalytic domains after conversion to fXIa. Activation of this variant results in an ∼90 kDa heavy chain dimer and 35-kDa catalytic domains that dissociate (Fig. 1B). Under reducing conditions the heavy chain dimer dissociates into 45-kDa monomers (Fig. 1B). Chromatography on soybean trypsin inhibitor-agarose separates the heavy chains and catalytic domains. All species of fXIa cleaved the tripeptide S-2366 (18–20) with similar Km (190–290 μm) and kcat (140–150 s−1), indicating domain manipulations did not significantly alter active site conformation.
FIGURE 1.
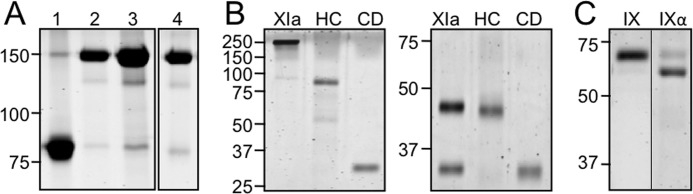
Purified proteins. A, nonreducing SDS-polyacrylamide gel stained with GelCode blue of recombinant (1) fXI-CD/PK-HC), (2) fXI-WT, (3) fXI/PKA3, and (4) fXI/PKA2-Ser140. FXI-CD/PK-HC is an 80-kDa monomer, whereas other fXI species are 160-kDa dimers. B, purified fXIa heavy chain (HC) and catalytic domain (CD) isolated from fXI-Ser362/Ser482 are compared with fXIa-WT (XIa) on nonreducing (left) and reducing (right) SDS-polyacrylamide gels. C, fIX and fIXα on a nonreducing SDS-polyacrylamide gel. Note that fIXα migrates more rapidly than fIX, despite having the same molecular mass. Positions of molecular mass standards are indicated on the left of each panel in kilodaltons.
FIXα
FIXα was prepared from fIX by limited digestion with fXIa in the absence of Ca2+ (Fig. 1C). A disulfide bond connects the polypeptides comprising fIXα, which has essentially the same molecular mass as fIX. FIXα migrates slightly faster than fIX on nonreducing PAGE, likely due to conformational changes that accompany cleavage (Fig. 1C). FIXα preparations contain ∼10–20% fIX (Fig. 1C), and this is taken into account as an initial substrate in kinetic analyses.
Cleavage of FIX and FIXα by FXIa-WT
FIX activation can be followed by measuring cleavage of a chromogenic substrate by fIXaβ (18); however, this method lacks sensitivity, making it difficult to measure initial activation rates. Sensitivity can be improved by using a coupled assay in which fIXaβ converts fX to fXa, which is then detected with a chromogenic substrate (17, 22). Chromogenic substrate-based techniques share a limitation. They are uninformative about fIXα generation. To address this, we monitored fIX activation by densitometry of Coomassie Blue-stained gels imaged at infrared wavelengths. FIX, fIXα, and fIXaβ migrate differently under nonreducing conditions on SDS-PAGE, and the low signal to noise ratio facilitated studying reactions with substrate concentrations down to 25 nm.
Initially, we assessed fIX conversion to fIXaβ by fXIa-WT in the presence of Ca2+ ions (Fig. 2, A–C). As expected, little fIXα accumulation was observed. Although fIXα is usually not detected when fIX is activated by plasma-derived fXIa, traces may be seen with recombinant fXIa. Results of the kinetic analysis by numerical integration and by the conventional Michaelis-Menten approach are summarized in Tables 1 and 2. The catalytic efficiency (kcat/Km) is ∼7-fold higher for cleavage after Arg180 compared with Arg145 (400 ± 100 and 60 ± 6 μm−1 min−1, respectively), with an estimated ∼2.5-fold lower Km and ∼3-fold higher kcat. The catalytic efficiency of cleavage after Arg180 in purified fIXα (130 ± 20 μm−1 min−1, Fig. 2, A, D, and E, and Tables 1 and 2) was ∼3-fold lower than for fIXα formed in situ during fIX activation, mainly due to a lower kcat, suggesting conformational differences between the nascent intermediate and purified fIXα bound to fXIa. Dynamic simulation indicated that kcat for cleavage after Arg145 is very sensitive to changes in v0 for disappearance of fIX, and good agreement was found with the kinetic parameters determined by Michaelis-Menten analysis of fIX activation (Fig. 2C). Taken as a whole, the results are consistent with a mechanism in which Ca2+-dependent cleavage of Arg145–Ala146 increases the efficiency of cleavage of Arg180–Val181, without fIXα accumulation.
FIGURE 2.

FIX and fIXα cleavage by fXIa-WT. A, nonreducing 17% polyacrylamide SDS gels of 100 nm fIX (top) or fIXα (bottom) in assay buffer with Ca2+ incubated at room temperature with 3 nm (active sites) fXIa-WT. Positions of standards for fIX, fIXα, and fIXaβ are indicated at the right of each panel. B, progress curves of fIX disappearance (●), and fIXα (○), and fIXaβ (▴) generation from panel A (top). Lines represent the least-squares fits to the data. C, initial velocities of cleavage after Arg145 (conversion of fIX to fIXα) by 1 μm fXIa-WT active sites, as a function of fIX concentration. Initial rates were obtained from the slopes of the linear portions of progress curves documenting the disappearance of fIX with time. D, progress curves of fIXα cleavage by fXIa-WT determined from panel A (bottom). E, initial velocities of cleavage after Arg180 (conversion of fIXα to fIXaβ) by 1 μm fXIa-WT as a function of fIXα concentration.
TABLE 1.
Kinetic parameters for cleavage of fIX and fIXα to fIXaβ by fXIa
Fitting of full-progress experimental traces was performed with KinTek software (KinTek Explorer version 2.5) using the reaction equations shown under “Experimental Procedures.” Km and kcat for activation were calculated from individual rate constants for each step. Ki values were fixed to lower limits obtained by surface plasmon resonance. Values are the mean ± S.D. for each experiment.
| Protease | Substrate | kcat | Kd | Km | Catalytic efficiency | Ki |
|---|---|---|---|---|---|---|
| min−1 | μm | μm−1 min−1 | μm | |||
| Cleavage of fIX after Arg145 | ||||||
| fXIa-WT | fIX | 12 ± 1 | 0.10 ± 0.01 | 0.20 ± 0.01 | 60 ± 6 | 0.14 |
| fXIa-WT no Ca2+ | fIX | 6.5 ± 0.4 | 4.9 ± 0.4 | 4.9 ± 0.4 | 1.3 ± 0.3 | 5 |
| fXIa-CD | fIX | 3.2 ± 0.1 | 4.9 ± 0.2 | 4.9 ± 0.2 | 0.7 ± 0.1 | 3 |
| fXIa/PKA3 | fIX | 4.9 ± 0.1 | 4.9 ± 0.2 | 4.9 ± 0.2 | 1.0 ± 0.1 | 3 |
| fXIa/PKA2-Ser140 | fIX | 16 ± 1 | 0.03 ± 0.01 | 0.10 ± 0.01 | 160 ± 20 | 0.15 |
| fXIa-CD/PK-HC | fIX | 1.6 ± 0.1 | 2.4 ± 0.2 | 2.4 ± 0.2 | 0.7 ± 0.1 | 5 |
| Cleavage of fIX after Arg180a | ||||||
| fXIa-WT | fIX | 35 ± 6 | 0.06 ± 0.01 | 0.08 ± 0.01 | 400 ± 100 | 0.06 |
| fXIa-WT | fIXα | 12 ± 1 | 0.08 ± 0.01 | 0.09 ± 0.01 | 130 ± 20 | 0.06 |
| fXIa-WT no Ca2+ | fIX | 3.6 ± 0.2 | 15 ± 1 | 15 ± 1 | 0.24 ± 0.04 | 5 |
| fXIa-CD | fIX | 0.5 ± 0.1 | 11 ± 1 | 11 ± 1 | 0.05 ± 0.01 | 5 |
| fXIa-CD | fIXα | 0.5 ± 0.1 | 13 ± 2 | 13 ± 2 | 0.04 ± 0.01 | 5 |
| fXIa/PKA3 | fIX | 0.6 ± 0.1 | 7 ± 1 | 7 ± 1 | 0.09 ± 0.02 | 3 |
| fXIa/PKA3 | fIXα | 0.6 ± 0.1 | 7 ± 1 | 7 ± 1 | 0.09 ± 0.02 | 3 |
| fXIa/PKA2-Ser140 | fIX | 40 ± 10 | 0.04 ± 0.01 | 0.08 ± 0.02 | 500 ± 200 | 0.06 |
| fXIa-CD/PK-HC | fIX | 0.4 ± 0.1 | 10 ± 1 | 10 ± 1 | 0.04 ± 0.01 | 5 |
| fXIa-CD/PK-HC | fIXα | 0.4 ± 0.1 | 10 ± 1 | 10 ± 1 | 0.04 ± 0.01 | 5 |
a Due to the low affinity between the enzyme and substrate in certain reactions, we were not able to reach saturation for reactions with fXIa-CD, fXIa/PKA3, fXIa-CD/PK-HC, or fXIa-WT in the absence of Ca2+. Fits for kcat and Km are linked, with the listed Km values representing lower estimates for the reaction. kcat/Km is the appropriate parameter for comparing these reactions.
TABLE 2.
Kinetic parameters for cleavage of fIX and fIXα by fXIa determined from initial velocities
The initial velocities (v0) of cleavage after Arg145 in fIX, and cleavage after Arg180 in fIXα were plotted against initial substrate concentration, and analyzed using the Michaelis-Menten equation. Km and kcat were obtained from direct nonlinear least squares analysis using Scientist Software. Due to the low affinity between the enzyme and substrate, we were not able to reach saturation reactions with fXIa-CD, fXIa/PKA3, and fXIa-CD/PK-HC. Thus, values for Km are approximate and values for kcat were not determined.
| Protease | Substrate | Bond cleaved | kcat | Km | Catalytic efficiency |
|---|---|---|---|---|---|
| min−1 | μm | μm−1 min−1 | |||
| fXIa-WT | fIX | Arg145 | 10 ± 1 | 0.27 ± 0.07 | 40 ± 10 |
| fXIa-WT | fIXα | Arg180 | 7.1 ± 0.3 | 0.09 ± 0.01 | 80 ± 10 |
| fXIa-WT no Ca2+ | fIX | Arg145 | 4.6 ± 0.4 | 1.8 ± 0.3 | 3 ± 1 |
| fXIa-CD | fIX | Arg145 | 2.4 ± 0.4 | 3 ± 1 | 0.8 ± 0.3 |
| fXIa-CD | fIXα | Arg180 | NDa | >4 | 0.018 ± 0.001 |
| fXIa/PKA3 | fIX | Arg145 | 6 ± 1 | 3.9 ± 0.9 | 1.6 ± 0.4 |
| fXIa/PKA3 | fIXα | Arg180 | ND | >4 | 0.029 ± 0.003 |
| fXIa-CD/PK-HC | fIX | Arg145 | 5 ± 1 | 3 ± 1 | 2 ± 1 |
| fXIa-CD/PK-HC | fIXα | Arg180 | ND | >4 | 0.02 ± 0.01 |
a ND, not determined.
Cleavage of FIX and FIXα by FXIa Species Lacking the A2 or A3 Domains
Although the fXIa heavy chain clearly contains a substrate-binding exosite (18–20), different studies indicate the site is on the A2 (21) or A3 (22, 23) domain. A second fIX-binding exosite may reside on the catalytic domain (17). We used recombinant fXIa variants to identify the region of the molecule involved in high-affinity binding to fIX and fIXα.
FXIa in which the A3 domain is replaced with the PK A3 domain (fXIa/PKA3), and the isolated fXIa catalytic domain (fXIa-CD) were tested for their ability to cleave fIX and fIXα (Fig. 3 and Tables 1 and 2). The cleavage rates of the Arg145–Ala146 and Arg180–Val181 bonds were markedly reduced compared with fXIa-WT, with pronounced fIXα accumulation. Km values for fIX cleavage after Arg145 by fXIa/PKA3 and fXIa-CD were increased ∼25-fold compared with fXIa-WT, with 60- and 90-fold decreases in catalytic efficiency, respectively. The catalytic efficiency of cleavage after Arg180 was impaired to an even greater degree, but only kcat/Km could be estimated accurately because saturation was not achieved for the reactions (Fig. 3E). The values for Km in Tables 1 and 2 should, therefore, be considered lower limits for the actual Km. These data indicate that the A3 domain is important for binding of fIX and fIXα to fXIa, and that loss of the A3 binding site has a deleterious effect on both cleavages. FIXα accumulation is the result of an 11–14-fold higher catalytic efficiency for Arg145–Ala146 cleavage compared with Arg180–Val181 cleavage. Progress curve simulations for cleavage of isolated fIXα by fXIa/PKA3 and fXIa-CD suggested that Arg180–Val181 is cleaved with similar, low catalytic efficiency when fIX or fIXα is the starting substrate, indicating that the exosite is important for efficient binding of both fIX and fIXα as substrates. Cleavage of fIX or fIXα by fXIa in which the A2 domain was replaced with PK A2 (fXIa/PKA2) was similar to cleavage by fXIa-WT (Table 1) indicating that the A2 domain does not contain a high affinity fIX or fIXα binding site.
FIGURE 3.

FIX and fIXα cleavage by fXIa/PKA3. A, nonreducing 17% polyacrylamide-SDS gels of 1000 nm fIX (top) or fIXα (bottom) in assay buffer with Ca2+ incubated at room temperature with fXIaPKA3 (60 nm and 240 nm active sites for fIX and fIXα, respectively). Positions of standards for fIX, fIXα, and fIXaβ are indicated at the right of each panel. B, progress curves of fIX disappearance (●), and fIXα (○), and fIXaβ (▴) generation from panel A (top). Lines represent least-squares fits to the data. C, initial velocities of cleavage after Arg145 (conversion of fIX to fIXα) by 1 μm fXIa/PKA3 active sites as a function of fIX concentration. The initial rates were obtained from the slopes of the linear portions of progress curves documenting disappearance of fIX with time. D, progress curves of fIXα cleavage by fXIa/PKA3 determined from panel A (bottom). E, initial velocities of cleavage after Arg180 (conversion of fIXα to fIXaβ) by 1 μm fXIa/PKA3 active sites as a function of fIXα concentration.
The fXIa catalytic domain is attached to the heavy chain by the Cys362–Cys482 disulfide bond (11, 12). The absence of the heavy chain or the substitution of Cys482 with serine may have caused changes to fXIa-CD that reduced its ability to activate fIX, independent of the loss of the A3 exosite. FXIa-CD/PK-HC consists of the fXIa catalytic domain attached to the heavy chain of PK. Like the fXI heavy chain, the PK heavy chain contains four apple domains, and it is connected to the catalytic domain through a single disulfide bond. Activation of fIX and fIXα by fXIa-CD/PK-HC (Tables 1 and 2) was similar to activation by fXIa/PKA3 and fXIa-CD. Taken as a whole, the data indicate that a major exosite for binding of fIX and fIXα is located on the fXIa A3 domain. Loss of this exosite, either in chimeras (fXIa/PKA3 or fXIa-CD/PK-HC) or through absence of the heavy chain (fXIa-CD) results in loss of the high affinity site and a marked increase in Km for fIX activation.
The anti-human fXI monoclonal IgG O1A6 binds to the A3 domain (24). In the presence of O1A6, fIX activation by fXIa-WT appears similar to activation by fXIa/PKA3, with decreased rates of cleavage for both bonds and fIXα accumulation (compare Figs. 4, A and B, to 3B). An IgG that binds to the A2 domain (14E11; Ref. 24) had no effect on fIX activation by fXIa (not shown).
FIGURE 4.

FIX cleavage by fXIa-WT in the presence of monoclonal antibodies, and in the absence of Ca2+ ions. Panels A–C, effects of antibodies. Shown are progress curves of fIX disappearance (●), and fIXα (○) and fIXaβ (▴) generation. FIX (100 nm) was activated by 3 nm fXIa-WT (active sites) in the presence of (A) vehicle, (B) 50 nm IgG OA16, or (C) 1000 nm IgG SB 249417. Panels D and E, importance of Ca2+. Shown are progress curves of fIX disappearance (●), and fIXα (○) and fIXaβ (▴) generation. FIX (1000 nm) was activated in assay buffer at room temperature by (D) fXIa-WT (5 nm active sites) in the presence of Ca2+, or (E) fXIa-WT (40 nm active sites) in the presence of 25 mm EDTA.
Cleavage of FIX and FIXα in the Absence of Calcium Ions
Although most reactions catalyzed by fXIa do not require Ca2+, fIX activation by fXIa is Ca2+-dependent. The fIX Gla domain is involved in the interaction with fXIa (25), and Ca2+ is likely required for proper Gla domain conformation. In the absence of Ca2+, fIX bond cleavage is slow, with accumulation of fIXα (compare Fig. 4, D and E). Km values for fIX activation in the absence of Ca2+ are similar to those with fXIa/PKA3 in the presence of Ca2+ (Tables 1 and 2). Absence of Ca2+ had little effect on the overall rate of fIX cleavage by fXIa/PKA3 or fXIa-CD (not shown). The data support the hypothesis that the Ca2+-dependent interaction between the fXIa A3 domain and fIX requires the fIX Gla domain. Consistent with this, fIX activation by fXIa in the presence of IgG SB 249417 (25), which binds to the fIX Gla domain, is characterized by significant accumulation of fIXα and slow generation of fIXaβ (Fig. 4C).
Binding of FXIa to FIX and FIXα
We studied binding of active site-blocked fXIa to immobilized fIX, fIXα, and fIXaβ using surface plasmon resonance. Zymogen fXI-WT did not bind to any species in the presence of Ca2+, nor did fXIa-WT in the presence of 10 mm EDTA (data not shown). In the presence of Ca2+, fXIa-WT bound to fIX, fIXα, and fIXaβ. Binding at equilibrium was plotted as a function of the fXIa-WT concentration (Figs. 5, A–C). Kd for binding to fIX (130 ± 20 nm), fIXα (140 ± 20 nm), and fIXaβ (60 ± 10 nm) were comparable (Table 3). Isolated fXIa heavy chain also bound to these species in a Ca2+-dependent manner (Fig. 5, D and E) with Kd values of 90 ± 20, 70 ± 10, and 70 ± 10 nm, respectively (Table 3). We were unable to demonstrate binding of fXIa-CD, fXIa/PKA3, or fXIa-CD/PK-HC (Fig. 5F and Table 3) to fIX, fIXα, or fIXaβ at analyte concentrations up to 5 μm. These results indicate that most of the binding energy for the fIX-fXIa interaction requires the fXIa A3 domain. The Western blots in Fig. 5G show that FXIa-HC inhibits fIX conversion to fIXaβ by fXIa, indicating the A3 exosite engages fIX in the absence of the catalytic domain, and accumulation of FIXα in the presence of fXIa-HC supports the premise that fIXα is released from fXIa.
FIGURE 5.

FXIa binding to fIX, fIXα, and fIXaβ. Surface plasmon resonance was used to assess binding of fXIa perfused over immobilized fIX, fIXα, or fIXaβ in the presence of Ca2+ ions at 10 μl/min for 6 min. Dissociation was monitored for 10 min. Panels A–C, fXIa-WT binding. FXIa-WT concentrations tested were: 1, 5, 10, 25, 37.5, 75, 150, 300, and 500 nm. Affinity and kinetic parameters were determined after subtraction of nonspecific binding from the control surface. Nonlinear regression fitting of the steady state equilibrium binding of fXIa-WT to (A) fIX, (B) fIXα, and (C) fIXaβ was performed using a bivalent model. Panels D–F, surface plasmon resonance data for Ca2+-dependent binding of fXIa-WT, fXIa-HC, and fXIa-CD binding to fIX. D, shown are curves for fXIa-WT binding to immobilized fIX at analyte concentrations listed above. E, binding curves for fXIa-HC at the same concentrations used for fXIa-WT. F, binding curve for fXIa-CD at a single analyte concentration (5000 nm). Panel G, effect of fXIa-HC on fIX activation by fXIa-WT. Shown are Western blots of time courses of fIX (100 nm) activated by fXIa-WT (2 nm active sites) in the presence of vehicle control (C) or 1 μm fXIa-HC (HC). Samples collected at various times (shown at bottom) into nonreducing SDS sample buffer were size fractionated by SDS-PAGE. Detection was with a polyclonal anti-human fIX antibody and chemiluminescence. Positions of standards for fIX, fIXα, and fIXaβ are shown between the images.
TABLE 3.
Affinity for fXIa binding to fIX, fIXα, and fIXaβ
Using surface plasmon resonance, fXIa species (1 to 5000 nm) in Ca2+-containing HBS-P buffer were perfused across sensor chips coated with fIX, fIXα, or fIXaβ. HBS-P buffer without fXIa was then perfused for 10 min to follow dissociation. Data were corrected for nonspecific binding by subtracting signals obtained with analytes infused through a flow cell without coupled protein. Binding was analyzed using a bivalent binding model for all species except fXIa-CD/PK-HC and fXIa-CD, which were evaluated with a 1:1 binding model. Kd values were calculated from the quotient of the derived dissociation (kd) and association (ka) rate constants.
| Analyte |
Kd for binding of analyte to ligand |
||
|---|---|---|---|
| Factor IX | Factor IXα | Factor IXaβ | |
| nm | |||
| fXIa-WT | 130 ± 20 | 140 ± 30 | 60 ± 10 |
| fXIa-HC | 90 ± 20 | 70 ± 10 | 70 ± 10 |
| fXIa-CD | >5000 | >5000 | >5000 |
| fXIa/PKA3 | >3000 | >3000 | >3000 |
| fXIa-CD/PK-HC | >5000 | >5000 | >5000 |
| fXIa/PKA2-Ser140 | 40 ± 10 | 40 ± 10 | NDa |
a ND, not done.
DISCUSSION
The current study was undertaken to establish the mechanism by which fXIa converts fIX to fIXaβ. The data need to be interpreted in light of published work in the field, some of which is in disagreement with the results presented here. It is clear that fIX activation by fVIIa or fXIa involves sequential proteolysis of the Arg145–Ala146 and Arg180–Val181 peptide bonds (2, 5). The observation that fIX containing an R145A substitution is cleaved poorly by fXIa (14) supports the premise that the Arg145–Ala146 cleavage site is presented to the fXIa active site after binding of fIX, and that conformational changes resulting from cleavage after Arg145, and perhaps repositioning of fIXα on the protease, enhances cleavage after Arg180.
Although fIXα forms during fIX activation by either fVIIa or fXIa, it does not accumulate during the latter reaction (13, 14). The fact that fXIa is a dimer initially suggested that the two subunits may cleave the two fIX bonds prior to releasing fIXaβ; however, subsequent work demonstrated that the ability to cleave fIX without fIXα accumulation is intrinsic to each fXIa subunit (14, 15). Wolberg et al. (13) observed that cleavage rates of Arg145–Ala146 and Arg180–Val181 in the intermediates fIXaα (fIX cleaved after Arg180) and fIXα (cleaved after Arg145) are similar and comparable with the overall rate for conversion of fIX to fIXaβ. They suggested that any intermediate formed may not be released prior to conversion to fIXaβ. However, significant fIXα accumulation was subsequently observed in competitive reactions in which fIX is activated by fXIa in the presence of active site-inhibited fXIa (14). Some portion of fIXα must be released from fXIa, arguing against a mechanism based strictly on the intermediate remaining bound to fXIa. There are parallels between fIX activation and prothrombin activation, in this regard. During ordered bond cleavage of prothrombin by fXa, the 150-fold higher catalytic efficiency of the second cleavage relative to the first results in formation of an intermediate (meizothrombin) that does not accumulate, but that can be captured with a tripeptide chloromethylketone (28).
The results reported here support an exosite- and Ca2+-mediated release-rebind mechanism for fIX activation by fXIa (summarized in Fig. 6), in which the efficiency of the second cleavage is enhanced by conformational changes resulting from the first cleavage. We propose that binding of fIX to the fXIa A3 domain is followed by docking with the active site and cleavage after Arg145 to form fIXα. This is followed by a second docking step with the active site after rebinding of fIXα to A3, and cleavage after Arg180. The ratio of the catalytic efficiencies of the two cleavages determine whether fIXα accumulates. If the A3 domain exosite is available, catalytic efficiency for cleavage after Arg180 is 7-fold greater than for cleavage after Arg145, and fIXα does not accumulate. In the absence of a functional exosite, the rates of both bond cleavages are markedly decreased, but the predominance of the catalytic efficiency of the second cleavage is lost, leading to fIXα accumulation.
FIGURE 6.

Model for the mechanism of fIX activation by fXIa. In the schematic images representing fXIa, apple domains are shown as four clustered gray circles with the exosite on A3 indicated in black (E). The fXIa catalytic domain is a white ellipse with the active site indicated by a black circle (A). For fIX, the catalytic domain (dark gray ellipse) and light chain (light gray ellipse) are connected by a line representing a disulfide bond. The fIX activation peptide is the white oval between the heavy and light chains. Bi-directional arrows represent reversible binding, and uni-directional arrows represent proteolytic cleavage. FIX is activated by a fXIa subunit by sequential cleavage after Arg145 and Arg180, with the intermediate fIXα released and then rebound to the fXIa A3 domain. Details of the model are described in the text. The dashed lines indicate the possibility that some fraction of fIXα may be converted to fIXaβ without release from the fXIa A3 domain.
The observations that fIXα is captured by active site-inhibited fXIa (14) or fXIa-HC in competition assays, and that a small amount of fIXα is transiently observed on gels during fIX cleavage by fXIa-WT, support a mechanism involving fIXα release. However, the data do not exclude the possibility that release is partial, with a fraction of fIXα repositioned for cleavage after Arg180, while still bound to fXIa (a transition from fIXα·XIa in Equation 1 directly to fIXα*·fXIa in Equation 2). Such a mechanism has been termed “processive” in prior studies (13). Using our data, we simulated a model that includes conformational repositioning of fIXα·fXIa to the productive complex fIXα*·fXIa without release (represented by dashed arrows in Fig. 6), in parallel with release rebinding. Because the true ratio of fIXα·fXIa to fIXα*·fXIa is unknown, we examined a range of values for the ratio (0.05–1), and found that the model fits data sets for cleavage after Arg180 for both nascent and purified fIXα by fXIa-WT reasonably well at a ratio of 0.5, a Kd of ∼100 nm for binding of fIXα to fXIa in a productive complex, and a kcat of ∼20 min−1. Independently determined parameters for cleavage of the first bond, obtained by Michaelis-Menten analysis, and Ki values for surface plasmon resonance binding of fIX, fIXα, and IXaβ to active site-blocked fXIa-WT were kept constant. The results suggest a possible upper limit of ∼40% (the difference between kcat of 35 min−1 and 20 min−1) of fIXα proceeding to fIXaβ through repositioning without release-rebinding. If the mechanism only involved repositioning of fIXα without release, no transient intermediate should be observed in time courses with fXIa-WT, no fIXα would competitively bind to active site-blocked fXIa, and kcat for the second cleavage would have to be exceedingly large (up to 60 to 150 min−1). Our data indicate this latter scenario is not likely.
The 3-fold greater efficiency of cleavage of fIXα generated in situ compared with purified fIXα recruited from the aqueous phase in the release-rebind model deserves comment. Although it must be recognized that this result may simply reflect structural perturbations in fIXα acquired during purification, the difference in catalytic efficiency disappears in reactions with exosite-impaired proteases, perhaps weakening this argument somewhat. The difference in catalytic efficiency may actually reflect some degree of fIXα repositioning without release, or perhaps induced fit related to conformational changes in fXIa, fIXα, or both triggered by fIX cleavage after Arg145.
Coagulation proteases are trypsin-like enzymes, but with more restricted substrate specificity than trypsin. Binding interactions at exosites on the proteases distinct from the active site are key to the mechanisms of action of these enzymes, and have been shown to be primary determinants of substrate affinity and specificity during activation of prothrombin (29–33) and factor X (34). Binding of the substrate at the exosite precedes substrate docking with the active site and catalysis (31, 33). In the current study, high affinity binding between fIX or fIXα and fXIa was largely, if not completely, due to a Ca2+-dependent interaction with the A3 domain. FXIa/PKA3 and the isolated fXIa protease domain (fXIa-CD) did not engage fIX or fIXα with high affinity, resulting in >25-fold increases in Km for cleavage after both Arg145 and Arg180. These results are not consistent with a model based on the fXIa catalytic domain containing a high affinity fIX binding exosite. Sinha et al. (17) reported that the slow rate of fIX conversion to fIXaβ by an isolated fXIa protease domain was due to reduced kcat, with Km for the overall reaction similar to that for fXIa-WT. It was concluded that fIX, but not fIXα, bound to an exosite on the catalytic domain in a Ca2+-independent manner. A mechanism was proposed that required fIX to bind to both the catalytic domain and heavy chain exosites, although Km data imply that the putative catalytic domain site would dominate the binding interaction. Our results do not support this model. Although we noted that fXIa-CD activates fIX similarly in the presence or absence of Ca2+, the kinetic and binding data fail to support the presence of a fIX binding site on fXIa-CD. Furthermore, our results for fIX activation by fXIa-WT and direct binding studies do not indicate that fIX binds with high affinity to fXIa-WT in the absence of Ca2+. The reason for the differences between our results and those of Sinha et al. (17) are not clear; however, their study used a chromogenic assay to examine fIX activation, which would not facilitate examination of the initial cleavage converting fIX to fIXα with the detail we were able to achieve with our densitometry-based approach.
The same group posited that catalytic domain amino acids Glu458 and Lys550 (Glu98 and Lys192 in chymotrypsin numbering) may be part of a fIX binding exosite (35). However, substitutions for these residues resulted in >100-fold reductions in kcat for fIX conversion to fIXaβ, more consistent with a catalytic defect. Preliminary work from our group indicated that fXIa with a Lys550 substitution activates fIX with normal Km, with the reduction in kcat likely due to disruption of the interaction between the Lys550 side chain and the substrate P3′ residue (36). Recently, Marcinkiewicz et al. (37) reported that cross-talk between the fXIa catalytic domain and heavy chain is required for expression of the fIX binding site on the heavy chain, based on observing that isolated fXIa heavy chain (fXIa-HC) does not bind to fIX. In the current study, in contrast, fXIa-HC and fXIa-WT bound to fIX and fIXα with similar affinity in the presence of Ca2+. Furthermore, fXIa-HC inhibited fIX activation by fXIa. These findings are most consistent with a mechanism in which fIX binds initially to the fXIa heavy chain, with binding being independent of a contribution from the catalytic domain.
Although our data do not support the presence of a fIX-binding exosite on the fXIa catalytic domain that is required for initial substrate recognition, they do not rule out the possibility that such a binding site is expressed as the result of initial binding of fIX to the A3 exosite (although such a process would not explain the differences between our results and published work). Previously, we noted that binding of fIX to fXIa results in a mixed-type inhibition of cleavage of a tripeptide substrate by fXIa, consistent with fIX binding to A3 causing changes in the protease domain, in addition to competing with the tripeptide at the active site (18).
The Gla domains of the coagulation proteases fVIIa, fIXa, and factor Xa contain γ-carboxylated glutamic acid residues that bind Ca2+ and facilitate binding to phosphatidylserine-rich membranes. Km for reactions involving these proteases are strongly influenced by the Gla domain-phospholipid interaction. FXIa lacks a Gla domain (10–12), and fXIa activation of fIX is not influenced appreciably by phospholipid. However, the fIX Gla domain is required for binding to fXIa (25), and may be the structure that interacts with the A3 domain. Supporting this is the observation that the kinetic parameters of fIX activation by fXIa-WT in the absence of Ca2+ are similar to those for activation by fXIa/PKA3 (Table 1), and that an antibody to the fIX Gla domain results in accumulation of fIXα (Fig. 4C). It is possible, therefore, that during fIX activation by fXIa, the A3 domain performs the Gla domain binding role that phospholipid performs during fIX activation by fVIIa.
FXI is a relatively new component of the blood coagulation mechanism, making its appearance during mammalian evolution as the result of a duplication of the PK gene (38). Human PK and fXI share a high degree of structural homology, and are 58% identical in amino acid sequence (10). However, the sequence around the putative fIX binding site in the fXI A3 domain, which is highly conserved in mammals, is distinctly different from the corresponding sequence in PK (38), supporting the conclusion that changes to the A3 domain were critical to the ability of fXIa to engage fIX as a substrate.
This work was supported, in whole or in part, by National Institutes of Health Grants HL58837 and HL81326 (to D. G.), HL080018 (to I. M. V.), and HL36365 (to S. P. B.) from the NHLBI.
- fIX
- factor IX
- fIXα
- factor IXα
- fIXaβ
- factor IXaβ
- fXI or fXIa
- factor XI or XIa
- fVII or fVIIa
- factor VII or VIIa
- fX or fXIa
- factor X or Xa
- PK
- prekallikrein
- A1
- A2, A3, and A4, apple domains 1, 2, 3, and 4
- fXIIa
- factor XIIa.
REFERENCES
- 1. Roth D. A., Freeman S. J., Furie B. (2009) in Hematology: Basic Principles and Practice (Hoffman R., Benz E. J., Shattil S. J., Furie B., Silberstein L. E., McGlave P., Heslop H., eds) 5th Ed., pp. 1899–1910, Churchill Livingstone-Elsevier, Philadelphia, PA [Google Scholar]
- 2. Bajaj S. P., Thompson A. R. (2006) in Hemostasis and Thrombosis: Basic Principles and Clinical Practice (Colman R. W., Marder V. J., Clowes A. W., George J. N., Goldhaber S. Z., eds) 5th Ed., pp. 131–150, Lippincott, Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 3. Furie B., Furie B. C. (2009) Hematology: Basic Principles and Practice (Hoffman R., Benz E. J., Shattil S. J., Furie B., Silberstein L. E., McGlave P., Heslop H., eds) 5th Ed., pp. 1819–1836, Churchill Livingstone-Elsevier, Philadelphia, PA [Google Scholar]
- 4. Lindquist P. A., Fujikawa K., Davie E. W. (1978) Activation of bovine factor IX (Christmas factor) by factor XIa (activated plasma thromboplastin antecedent) and a protease from Russell's viper venom. J. Biol. Chem. 253, 1902–1909 [PubMed] [Google Scholar]
- 5. Smith S. B., Gailani D. (2008) Update on the physiology and pathology of factor IX activation by factor XIa. Expert Rev. Hematol. 1, 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zur M., Nemerson Y. (1980) Kinetics of factor IX activation via the extrinsic pathway. Dependence of Km on tissue factor. J. Biol. Chem. 255, 5703–5707 [PubMed] [Google Scholar]
- 7. Nemerson Y. (1992) The tissue factor pathway of blood coagulation. Semin. Hematol. 29, 170–176 [PubMed] [Google Scholar]
- 8. Lawson J. H., Mann K. G. (1991) Cooperative activation of human factor IX by the human extrinsic pathway of blood coagulation. J. Biol. Chem. 266, 11317–11327 [PubMed] [Google Scholar]
- 9. Bouma B. N., Griffin J. H. (1977) Human blood coagulation factor XI. Purification, properties, and mechanisms of activated factor XII. J. Biol. Chem. 252, 6432–6437 [PubMed] [Google Scholar]
- 10. Fujikawa K., Chung D. W., Hendrickson L. E., Davie E. W. (1986) Amino acid sequence of human factor XI, a blood coagulation factor with four tandem repeats that are highly homologous with plasma prekallikrein. Biochemistry 25, 2417–2424 [DOI] [PubMed] [Google Scholar]
- 11. McMullen B. A., Fujikawa K., Davie E. W. (1991) Location of the disulfide bonds in human coagulation factor XI. The presence of tandem apple domains. Biochemistry 30, 2056–2060 [DOI] [PubMed] [Google Scholar]
- 12. Papagrigoriou E., McEwan P. A., Walsh P. N., Emsley J. (2006) Crystal structure of the factor XI zymogen reveals a pathway for transactivation. Nat. Struct. Mol. Biol. 13, 557–558 [DOI] [PubMed] [Google Scholar]
- 13. Wolberg A. S., Morris D. P., Stafford D. W. (1997) Factor IX activation by factor XIa proceeds without release of a free intermediate. Biochemistry 36, 4074–4079 [DOI] [PubMed] [Google Scholar]
- 14. Smith S. B., Verhamme I. M., Sun M. F., Bock P. E., Gailani D. (2008) Characterization of novel forms of coagulation factor XIa. Independence of factor XIa subunits in factor IX activation. J. Biol. Chem. 283, 6696–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu W., Sinha D., Shikov S., Yip C. K., Walz T., Billings P. C., Lear J. D., Walsh P. N. (2008) Factor XI homodimer structure is essential for normal proteolytic activation by factor XIIa, thrombin, and factor XIa. J. Biol. Chem. 283, 18655–18664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Orcutt S. J., Krishnaswamy S. (2004) Binding of substrate in two conformations to human prothrombinase drives consecutive cleavage at two sites in prothrombin. J. Biol. Chem. 279, 54927–54936 [DOI] [PubMed] [Google Scholar]
- 17. Sinha D., Marcinkiewicz M., Navaneetham D., Walsh P. N. (2007) Macromolecular substrate-binding exosites on both the heavy and light chains of factor XIa mediate the formation of the Michaelis complex required for factor IX activation. Biochemistry 46, 9830–9839 [DOI] [PubMed] [Google Scholar]
- 18. Ogawa T., Verhamme I. M., Sun M. F., Bock P. E., Gailani D. (2005) Exosite-mediated substrate recognition of factor IX by factor XIa. The factor XIa heavy chain is required for initial recognition of factor IX. J. Biol. Chem. 280, 23523–23530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sinha D., Seaman F. S., Walsh P. N. (1987) Role of calcium ions and the heavy chain of factor XIa in the activation of human coagulation factor IX. Biochemistry 26, 3768–3775 [DOI] [PubMed] [Google Scholar]
- 20. Baglia F. A., Sinha D., Walsh P. N. (1989) Functional domains in the heavy-chain region of factor XI. A high molecular weight kininogen-binding site and a substrate-binding site for factor IX. Blood 74, 244–251 [PubMed] [Google Scholar]
- 21. Baglia F. A., Jameson B. A., Walsh P. N. (1991) Identification and chemical synthesis of a substrate-binding site for factor IX on coagulation factor XIa. J. Biol. Chem. 266, 24190–24197 [PubMed] [Google Scholar]
- 22. Sun Y., Gailani D. (1996) Identification of a factor IX binding site on the third apple domain of activated factor XI. J. Biol. Chem. 271, 29023–29028 [DOI] [PubMed] [Google Scholar]
- 23. Sun M. F., Zhao M., Gailani D. (1999) Identification of amino acids in the factor XI apple 3 domain required for activation of factor IX. J. Biol. Chem. 274, 36373–36378 [DOI] [PubMed] [Google Scholar]
- 24. Kravtsov D. V., Matafonov A., Tucker E. I., Sun M. F., Walsh P. N., Gruber A., Gailani D. (2009) Factor XI contributes to thrombin generation in the absence of factor XII. Blood 114, 452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aktimur A., Gabriel M. A., Gailani D., Toomey J. R. (2003) The factor IX γ-carboxyglutamic acid (Gla) domain is involved in interactions between factor IX and factor XIa. J. Biol. Chem. 278, 7981–7987 [DOI] [PubMed] [Google Scholar]
- 26. Cheng Q., Sun M. F., Kravtsov D. V., Aktimur A., Gailani D. (2003) Factor XI apple domains and protein dimerization. J. Thromb. Haemost. 1, 2340–2347 [DOI] [PubMed] [Google Scholar]
- 27. Johnson K. A., Simpson Z. B., Blom T. (2009) Global kinetic explorer. A new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 [DOI] [PubMed] [Google Scholar]
- 28. Carlisle T. L., Bock P. E., Jackson C. M. (1990) Kinetic intermediates in prothrombin activation. Bovine prethrombin 1 conversion to thrombin by factor X. J. Biol. Chem. 265, 22044–22055 [PubMed] [Google Scholar]
- 29. Betz A., Krishnaswamy S. (1998) Regions remote from the site of cleavage determine macromolecular substrate recognition by the prothrombinase complex. J. Biol. Chem. 273, 10709–10718 [DOI] [PubMed] [Google Scholar]
- 30. Orcutt S. J., Pietropaolo C., Krishnaswamy S. (2002) Extended interactions with prothrombinase enforce affinity and specificity for its macromolecular substrate. J. Biol. Chem. 277, 46191–46196 [DOI] [PubMed] [Google Scholar]
- 31. Krishnaswamy S. (2005) Exosite-driven substrate specificity and function in coagulation. J. Thromb. Haemost. 3, 54–67 [DOI] [PubMed] [Google Scholar]
- 32. Page M. J., Macgillivray R. T., Di Cera E. (2005) Determinants of specificity in coagulation proteases. J. Thromb. Haemost. 3, 2401–2408 [DOI] [PubMed] [Google Scholar]
- 33. Bock P. E., Panizzi P., Verhamme I. M. (2007) Exosites in the substrate specificity of blood coagulation reactions. J. Thromb. Haemost. 5, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baugh R. J., Dickinson C. D., Ruf W., Krishnaswamy S. (2000) Exosite interactions determine the affinity of factor X for the extrinsic Xase complex. J. Biol. Chem. 275, 28826–28833 [DOI] [PubMed] [Google Scholar]
- 35. Su Y. C., Miller T. N., Navaneetham D., Schoonmaker R. T., Sinha D., Walsh P. N. (2011) The role of factor XIa (FXIa) catalytic domain exosite residues in substrate catalysis and inhibition by the Kunitz protease inhibitor domain of protease nexin 2. J. Biol. Chem. 286, 31904–31914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmidt A. E., Agah S., Sun M. F., Padmanabhan K., Cascio D., Gailani D., Bajaj S. P. (2008) Structural and functional significance of amino acid lysine 192 (chymotrypsin numbering) in factor XIa and factor VIIa. Blood 112, 708a (abstract 2011)18502829 [Google Scholar]
- 37. Marcinkiewicz M. M., Sinha D., Walsh P. N. (2012) Productive recognition of factor IX by factor XIa exosites requires disulfide linkage between the heavy and light chains of factor XIa. J. Biol. Chem. 287, 6187–6195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ponczek M. B., Gailani D., Doolittle R. F. (2008) Evolution of the contact phase of vertebrate blood coagulation. J. Thromb. Haemost. 6, 1876–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]