

Abstract
We describe a novel isoelectric point photoswitching phenomenon in both wild-type Aequorea victoria (av) GFP and the E222G mutant Aequorea coerulescens (ac) GFP. A combination of time-resolved microfluidic isoelectric focusing (IEF) and in situ antibody blotting IEF were employed to monitor dark (non-fluorescent) and bright (fluorescent) GFP populations. Through IEF, each population was observed to exhibit distinct isoelectric points (pI) and, thus, distinct formal electrostatic charges. Experimentally observed interconversion between the dark, higher pI and bright, lower pI GFP populations is tightly controlled by differential UV and blue light exposure. The stoichiometry and kinetics of charge transfer tied to this reversible photobleaching process are deduced. In concert with a reaction-transport model of bistable reversible charge and fluorescence photoswitching, the on-chip measurements of population interconversion rates suggest the potential for both rheostatic and discrete switch-like modulation of the electrostatic charge of GFPs depending on the illumination profile. We estimate that 3–4 formal charges distinguish the bright and dark populations of avGFP, compared to one charge for those of acGFP. Given the proposed role of E222 as a bridge between internal and exit hydrogen bond clusters within the GFP β-barrel, the difference in charge switching magnitude between the two mutants provides intriguing evidence for the proton wire hypothesis of proton transport within the GFP structure, and of proton exchange with the bulk solvent. Our facile dynamic and probed IEF assays should find widespread use in analytical screening and quantitative kinetic analysis of photoswitching and other charge switching processes in response to stimuli including light, temperature, or binding/cleavage events.
INTRODUCTION
Benchtop separations are central to preparative and analytical advances in protein characterization1,2. However, the use of endpoint-measurement gel electrophoresis methods places practical limitations on the observation of protein band dynamics during separations. While capillary electrophoresis technologies have been employed for kinetic characterization of aptamer-ligand3 and enzyme reactions4,5, the methods typically rely on single-point detection that prevents real-time observation of dynamic analyte band interaction, formation, or decay processes6. The planar, compact form factor of microfluidic devices lowers practical barriers to electrophoretic separations operated with wide-field and whole-channel imaging7,8. In this way, microfluidic methods are well suited to facile measurement of dynamic protein reaction processes in real time. Augmenting dynamic readouts, miniaturized electrophoretic separations also offer high analytical performance, owing to reduced migration timescales and high electric fields operation2,9–11. Such quantitative and time-resolved analytical methods may enhance our understanding of dynamic protein processes, including the photophysics of fluorescent proteins.
While of fundamental interest, the diverse photophysical properties of the fluorescent proteins have also driven important practical advances spanning the life and engineering sciences12. Originally extracted from bioluminescent jellyfish and corals, development of protein variants with novel spectral (specifically far-red emitting13 and switchable variants) and physicochemical properties has impacted applications as wide-ranging as cellular transport14 high resolution imaging15 and biological information storage16. To uncover the structural underpinnings of photophysical fluorescent protein behaviors, conventional characterization methods combine spectroscopic tools (including time-resolved fluorescence correlation) with molecular dynamics modeling and crystallography17,18. A challenging and ongoing area of study is the complex proton dynamics of photoswitchable fluorescent proteins, in analogy to well-studied proton shuttling processes (e.g., bacteriorhodopsin, cytochrome c oxidase19,20). Importantly, fluorescent protein chromophore emission properties are tightly coupled to the electrostatic environment of the chromophore pocket21 and to internal and longer-range proton transport influenced by the bulk solvent22,23. Consequently, advanced analytical methods are needed to determine how specific protein mutations impact the coupled electrostatic and photophysical properties of the fluorescent proteins. Such tools will inform rational engineering of extreme and tunable properties across the fluorescent proteins.
In this study we use microfluidic isoelectric focusing (IEF) to study a photoswitching phenomenon observed in both wild-type avGFP and the E222G mutant acGFP (Figure 1). IEF is a powerful electrophoresis technique that separates proteins according to isoelectric point (pI) when an electric field is applied along the axis of a stable pH gradient (formed by polyprotic amphoteric buffers)2. During IEF, a native protein focuses at the channel position where the local pH equals the protein pI. Proteins focus into a stationary zone at this location because the species have a net zero electrostatic charge and thus no net electrophoretic mobility24,25. The IEF pH gradient can be generated in free solution or in a sieving matrix, such as polyacrylamide gel. Protein pI is a physicochemical property determined by amino acid composition, three-dimensional conformation, and modifying chemical groups affecting protein charge. Protein isoforms are common and important. Isoforms are versions of a protein having slight differences in pI. These pI differences are generally attributable to post-translational modifications including enzymatic glycosylation or primary sequence cleavage processes occurring within cells26. IEF can resolve a single electrostatic charge difference between protein isoforms27, making IEF a powerful tool for the study of biophysical phenomena. IEF is particularly suited to study of proton dynamics, which impact pI but have little impact on other measureable protein properties (i.e., molecular weight).
Figure 1.
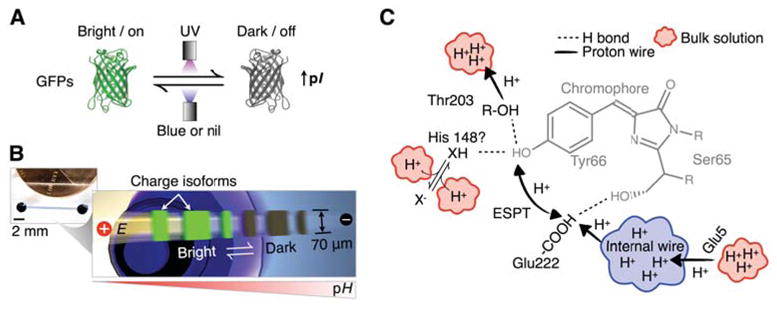
Immunoprobed isoelectric focusing allows dynamic and immunoreactivity-verified monitoring of GFP isoform dynamics during reversible photobleaching. (A) Reversible photobleaching upon UV illumination creates dark GFP isoforms with increased pIs relative to their bright “parents”. (B) Microfluidic chip with three parallel channels between each pair of access wells. Dynamic isoelectric photoswitching processes can be monitored in real time, or isoforms can be captured by the LAVAgel matrix and probed in situ with fluorescently labeled anti-GFP antibody. (C) A sketch of avGFP chromophore and proton wire dynamics29, 40, 41, 56. Hydrogen bond networks allow proton exchange of the chromophore pocket with the external solvent. Glu222 is involved in excited state proton transfer (ESPT) with the chromophore Tyr66. The E222G mutation in acGFP inhibits ESPT and perhaps proton exchange with the internal wire proposed by Agmon et al.41, 55. The dependence of reversible photobleaching magnitude on pH suggests involvement of a titratable residue, X, in the chromophore vicinity that affects chromophore protonation state by hydrogen bonding38 (see text for details).
Using dynamic IEF, we observe and characterize dark (non-fluorescent) and bright (fluorescent) populations of avGFP and acGFP by measuring changes in protein fluorescence and pI. After IEF, we integrate an immunoblotting step to extend our analytical tool to non-fluorescent analytes. Incubation of fluorescently labeled antibodies with IEF-resolved – and immobilized – proteins yields pI and mass distribution for each target, including non-fluorescent forms of GFP. To immobilize proteins, we conduct IEF in a light responsive benzophenone-decorated, polyacrylamide gel (light-activated volume-accessible separation gel or LAVAgel). Brief exposure of the LAVAgel to UV light covalently attaches proteins to the LAVAgel matrix2, allowing subsequent protein probing via introduction of fluorescently labeled antibodies. The ability to blot and probe proteins with near-lithographic spatial control in direct series with controlled light pre-exposure sequences enables quantitation in the absence of an endogenous fluorescence signal from the protein target2. In the case of GFP, immunoblotting corroborates pI photoswitching measurements inferred from intrinsic fluorescence data. The GFP fluorescence signals have complex dependencies on light exposure history28,29 and chemical environment30,31 and, thus, can confound the true protein mass distribution in the microchannel. These dependences make an independent fluorescence probing readout essential.
RESULTS
We first applied the microfluidic IEF assay to analysis of two GFP species of interest, avGFP and acGVP (Figure 1). IEF analysis yielded three predominant isoforms for each variant in the pI 4.8–5.5 range under continuous blue light excitation (isoforms are denoted α, β and γ for avGFP; and δ, ε and ζ for acGFP, see Figure 2). These heterogeneous isoform patterns have been ascribed to differential C-terminal cleavage by non-specific proteases during bacterial expression of the recombinant proteins31. Random terminal cleavage of the tail, which contains two basic (His and Lys, pKa 6.6 ± 1.0 and 10.5 ± 1.1) and two acidic (Asp and Glu, pKa 3.5 ± 1.2 and 4.2 ± 0.9) residues31,32, should produce isoforms differing by roughly one formal charge if the pKas of the differentially cleaved residues are distinct by at least ~1 pH unit from the isoform pIs. High-resolution intact-mass spectrometry of purified recombinant avGFP revealed a 128 Da mass difference between two major peaks (Figure S1). This difference is consistent with cleavage of the C-terminal lysine in the α isoform of avGFP. Given that the cleaved lysine residue contributes a full positive charge, the pI shift attributable to a single electrostatic charge can be estimated at roughly 0.12–0.15 pH units from the relative bright isoform displacements in the pH axis of Figure 2, and from further computational estimation of the expected isoform pIs resulting from differential C-terminal cleavage (data not shown)33. Thus, the magnitudes of isoelectric point photoswitching can be calibrated using a ruler of electrostatic charge to allow direct inference of the stoichiometry of charge transfer events at single-charge resolution34. The effect of charge transfer on the pI of a protein via its titration behavior has complex dependencies on, for example, the pI itself, and the molecular weight and amino acid composition of the protein35,36. Thus, we adopt this “charge ruler” approximation under the assumption that the pI range over which charge shifts are estimated is narrow enough to assume a constant local slope in the charge vs. pH titration curve of the isoforms considered25,37.
Figure 2.
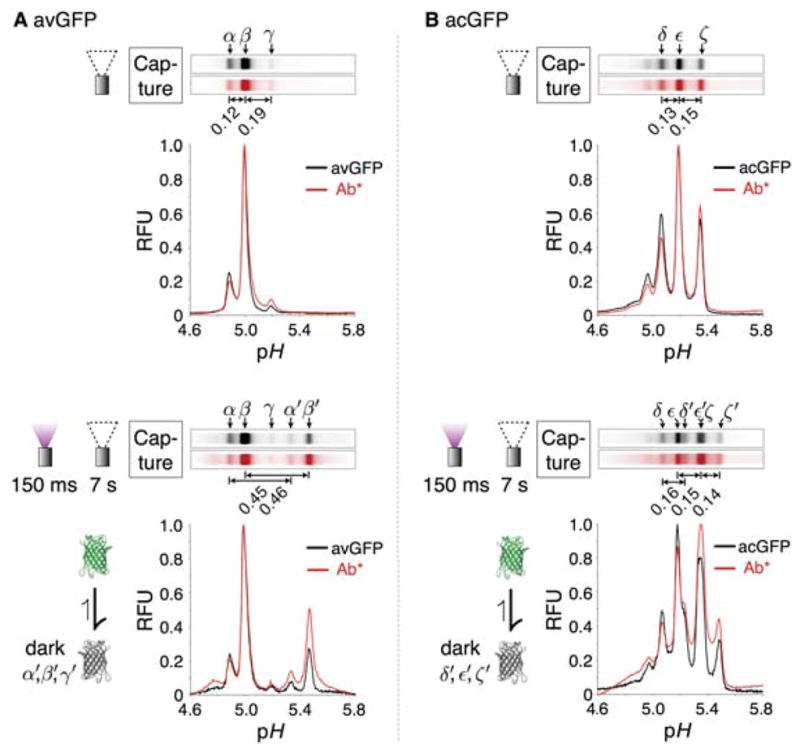
Probed isoelectric focusing of avGFP and acGFP reveals base-shifted reversibly photobleached isoforms. (A) Static LAVAgel fluorescence images and electropherograms of immobilized avGFP isoforms (bright:α, β, γ; dark: α′ and β′; γ′ is below the assay limit of detection) within the microchannel (black; excitation 445–495 nm, emission 508–583 nm) after focusing under - top: nil light exposure conditions, bottom: 150 ms pre-exposure to 100% UV (270 mW cm−2, 300–380 nm). Isoform capture was initiated immediately after the indicated pre-exposure protocol by 15 s UV irradiation of the LAVAgel under non-focusing conditions. Red fluorescence (excitation 525–555 nm, emission >575 nm) gel images and electropherograms are produced following pH gradient washout and LAVAgel probing with 600 nM Texas Red-labeled anti-GFP antibody (Ab*) for immobilized isoforms. N.B. fluorescence of captured dark isoforms (α′ and β′) is switched on during imaging under blue illumination. (B) The corresponding micrographs and electropherograms for immobilized acGFP isoforms (bright:δ, ε, ζ; dark: δ′, ε′ and ζ′) probed with the same anti-GFP antibody.
Next, we sought to investigate the effect of UV and blue light illumination on avGFP and acGFP isoform distributions during dynamic IEF. For both avGFP and acGFP, isoforms exhibited dynamic changes in isoelectric point distributions upon exposure of the focused proteins to sequences of UV and blue light illumination (Figures 3, S2). To summarize the observed photoswitching phenomena: brief exposure of the focused fluorescent isoform bands to UV light induced formation of dark (reversibly bleached) isoform populations with increased pI compared to bright isoforms (Figures 2–4). Following UV exposure, application of blue illumination initiated a dynamic “switch-on” of the fluorescence of the dark isoforms with first-order time constants of 700 ms for avGFP and 720 ms for acGFP (Figures 4A, S3). Concomitantly, we observed migration of the switched-on isoforms to the pIs of their “parent” bright isoforms on the ~5–10 s timescale. More prolonged exposure to UV on the focusing timescale caused a transient increase in the apparent pIs of bright isoforms to values intermediate between the static bright and dark isoform pIs. This apparent bright isoform pI increase reversed when UV illumination was halted (Figure 3B), while dark isoforms assumed their higher pIs until blue light was applied (or until the dark isoforms relaxed back to the bright state during prolonged nil illumination).
Figure 3.
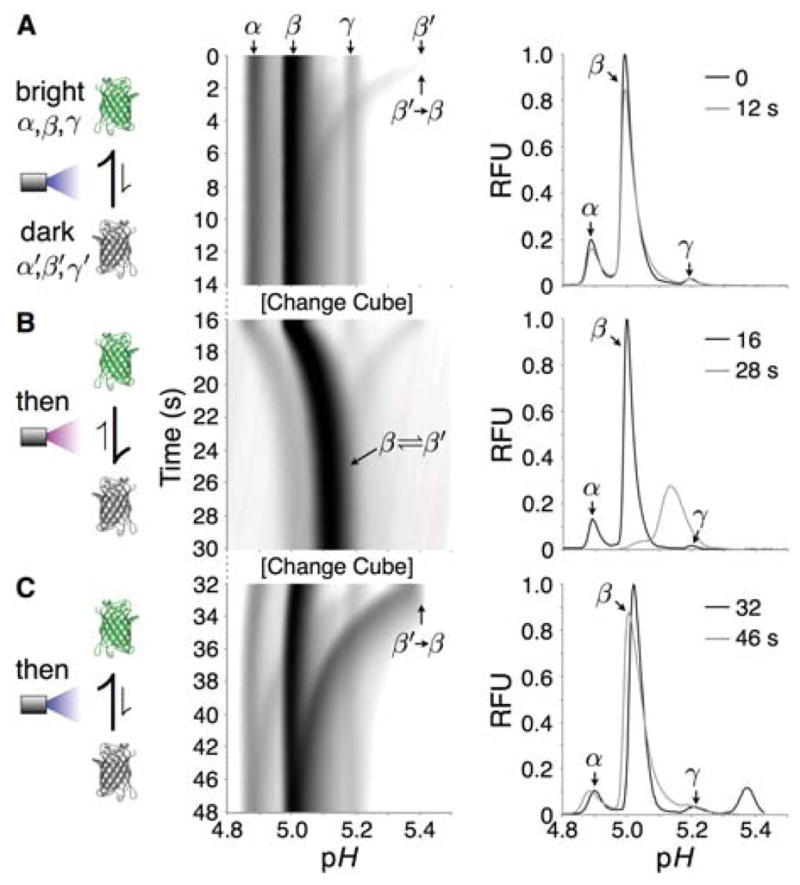
Real-time observation of isoelectric point photoswitching in avGFP. (A–C) Log-transformed fluorescence micrographs corrected for cathodic pH gradient drift showing dynamic isoelectric point evolution of avGFP isoforms during isoelectric focusing over time (100 ms streamed exposures, E = 300 V cm−1). Initial and final electropherograms in linear relative fluorescence units accompany each timelapse micrograph. The three panels show typical behavior upon applying a sequence of microchannel exposure conditions - (A) Nil-to-blue light exposure. (B) 100% UV exposure (excitation 300–380 nm, emission >410 nm). (C) Blue light exposure. Delay between micrographs due to filter cube exchange is ~2 s.
Figure 4.
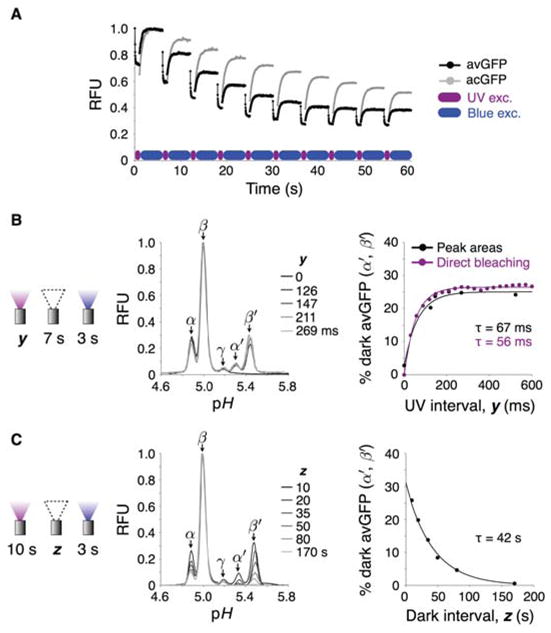
Isoelectric photoswitching kinetics. (A) Fluorescence photoswitching in avGFP and acGFP. Ten consecutive illumination cycles of 100% UV (1 s per cycle) and blue light (5 s per cycle) were conducted via 10x objective and total isoform fluorescence plotted over time for 10 ms frames. (B) Fast reversible bleaching of isoforms under UV illumination. Left: Dark isoforms of avGFP were generated by pre-exposure of bands to 100% UV light for the indicated exposure times y, followed by a focusing equilibration period of 7 s under nil illumination. Focusing field was then halted and dark isoforms converted to the bright state by 3 s blue light exposure and quantified by measuring peak areas. Electropherograms show increase in dark isoform (α′ and β′) representation as a function of UV pre-illumination period. Right: Growth of dark state isoform peak areas in pI 5.25–5.6 range determined from electropherogram data at left (black) and inferred by direct observation of bleaching under 100% UV (purple, from the first exposure cycle of (A)), both fit to RFU = a(1−e−t/τ)+b. (C) Slow isoform fluorescence recovery under nil illumination. Left: Dark isoforms of avGFP were generated by 10 s pre-exposure of bands to 100% UV light via 10x objective, followed by a nil illumination period z of between 10 and 170 s. Focusing field was then halted and remaining dark molecules converted to the bright state by 3 s blue light exposure. Electropherograms show decay in the representation of dark isoform (α′ and β′) peak areas in the pI 5.25–5.6 range as a function of nil illumination period. Right: Dark isoform peak areas fit to a single exponential of the form RFU = RFUmaxe−t/τ (± SD, n = 3, error bar heights smaller than marker size).
Measurement of the dark isoform peak areas as a function of UV pre-exposure time revealed single-exponential switch-off kinetics with a time constant of 67 ms. This kinetic approximately matches the fast bleaching time constant of 56 ms under direct UV exposure of avGFP (Figure 4B). These results suggest reversible bleaching of bright GFP isoforms by UV exposure as the trigger for formation of dark state isoforms with alkaline-shifted pIs. The dark isoform populations, constituting ~25% of the total mass for avGFP after ≳150 ms UV exposure, also decayed back to the bright state under nil illumination conditions (Figure 4C). The relatively short ~5–10 s focusing timescale (compared to the characteristic time of dark population decay) enabled resolution of dark from bright populations, an observation that would be impossible in longer separation-length capillary or slab gel systems. This decay process was again described by single-exponential kinetics, with a time constant of 42.2 s for avGFP, which is similar to those of 58 and 54 s measured by Sinnecker et al. in mammalian cells for ECFP and EYFP respectively (see Discussion)38. Thus, blue photon absorption reduced the fluorescence switch-on time of reversibly bleached dark isoforms by a factor of ~60-fold (from 42 to 0.7 s) under the experimental conditions employed.
We postulate that the distributions of the photoswitched isoforms carry key information about the charge transfer events underlying the changes in isoelectric point. Two mutant-specific differences in these focusing behaviors were observed for avGFP and acGFP. Firstly, prolonged application of UV light caused apparent pI shifts of 0.12 and 0.10 units for all bright avGFP and acGFP isoforms respectively, as well as broadening of the focused zone band widths (4σ) by 2.47- and 1.24-fold respectively (as measured for the major β and ε isoforms, Figures 3B and S2). Secondly, the final pI shifts of each of the dark isoforms (e.g., β′) from their parent bright bands (e.g., β) after halting illumination were ~0.45 and ~0.15 pI units for avGFP and acGFP respectively (~3–4 and ~1 charge units, Figure 2). The fact that these bright-to-dark pI shifts were discrete, rather than spread over a distribution, points to an all-or-nothing conversion process. Thus, these GFPs are hypothesized to exhibit a bistable switch with respect to pI (at least on the fluorescence timescale) and with respect to fluorescence.
We explored the GFP bistable switching hypothesis by modeling the isoelectric photoswitching of the predominant avGFP isoform between bright (β) and dark (β′) states having distinct pIs. GFP molecules interconvert between these states according to an equilibrium reaction of the form β ⇌ β′ with forward and backward rate constants of kβ→β′ and kβ′→β respectively. The rate constants were determined by least squares fitting of the equilibrium bright population distributions obtained from the model to experimentally measured distributions. Focused avGFP isoform distribution fits were made across a range of UV illumination intensities. Governing equations for this model considered focusing and diffusive spreading of peaks (terms 1 and 2 on the right hand side of each equation), as well as first-order interconversion of bright and dark populations (terms 3 and 4):
| (1) |
| (2) |
Where C is concentration, t is time, x is distance along the separation axis, pβ ≈ pβ′ = p is slope in analyte mobility with respect to x, E is applied electric field, and Dβ ≈ Dβ′ = D is the diffusivity of GFP in the separation gel. Non-dimensionalization of the model with respect to the characteristic diffusion time between population peaks (ΔxpI2/D) yielded three parameters 1) a Peclet number Pe=pΔxpI2Ex/D, the ratio of diffusive and convective (focusing) timescales, 2) a Damkohler number κ = kβ→β,ΔxpI2/D, the ratio of diffusive and forward reaction timescales, and 3) an equilibrium constant γ = kβ′→β/kβ→β′, the ratio of backward and forward reaction rates (see SI Text).
The dimensional analysis in Figure 5A captures the diverse behavior of this convection-diffusion-reaction model. The reaction:focusing speed ratio κ/Pe and the equilibrium constant γ divide the (kβ→β′, kβ′→β) parameter space into several behavioral regimes that were directly mapped onto experimental data. At low UV intensities (κ/Pe < 1), the bright and dark populations interchange slowly enough compared to the focusing timescale that distinct bright and dark peaks are formed, producing a wide overall concentration distribution in the pI axis. Notably, as the UV intensity increases, the populations interchanged more rapidly, and the rate constants kβ′→β and kβ→β′ increase at an approximately fixed ratio over the intensity range studied (γ~ 2.9, Figure 5B, C). Rapid interconversion causes the observed bright and dark distributions of the major avGFP isoform to converge along the pI axis at a weighted mean pH of 5.12 at (pIβ′−pIβ)/(1+γ) = 0.12 pH units from pIβ = 5.00, since γ = Cβ/Cβ′ at equilibrium. Simply put, the 5–270 mW cm−2 range in UV intensity we studied produced an intensity-independent equilibrium39 in which each GFP molecule was bright with pI 5.00 around 74% of the time, and dark with pI 5.45 around 26% of the time. Increasing the UV intensity reduced the average time spent by a molecule in each state between switching events. Increases in each rate constant, and thus κ/Pe, explain the transitions in shape (wide to narrow) and position (lower pI to higher pI) of the observed bright isoform distributions, since the ability to resolve the bright and dark populations by focusing is eroded as populations interconvert more and more rapidly.
Figure 5.
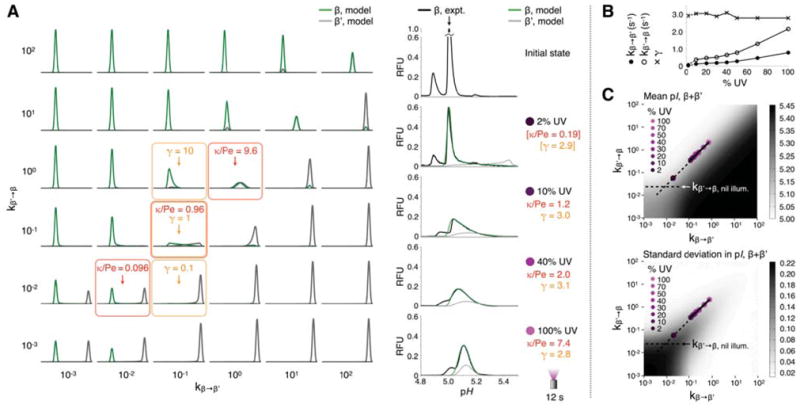
Two-state convection-diffusion-reaction model predicts focusing dynamics during perturbation of avGFP with UV light. (A) At left, sketch of concentration distributions from 1D model of focusing of bright (β) and dark (β′) states of the major avGFP isoform over a range of interconversion rates; x-axes are pI 4.8–5.5, y-axes are arbitrary fluorescence units.κ/Pe compares reaction to focusing speeds, governing the observed morphology of the β peak during focusing. Concentration distributions of the bright and dark states can be distinct (κ/Pe < 1) or overlapping (κ/Pe > 1) depending on the UV intensity, even for the same equilibrium constant γ. Right hand column, least-squares fits of model distributions of β to experiment data for the indicated UV intensities. Fits from 10%–100% UV were global optima, while that at 2% assumed an average γ value from the other fits (square brackets and dashed lines denote this here and in (B) and (C)). (B) Interconversion rates between β and β′ states and their ratio, γ from fits in (A). (C) Intensity plots of pI mean and standard deviation for the sum of β and β′ distributions. Interconversion rates determined from experimental data in (B) are overlayed. Best-fit slope of 0.94 reflects approximately constant γ and mean pI across the UV range.
Dynamic analysis over lower intensity ranges was limited by detection sensitivity, given that the GFP stimulation and imaging conditions were one and the same. Under nil illumination conditions, only the bright isoform populations exist (Figure 2), predicting a transition to the γavGFP ~ 2.9 equilibrium over the 0–5 mW cm−2 UV intensity range and the ability to manipulate the mean avGFP pI from 5.00–5.12 in a rheostatic fashion. Similar fitting of experimental data for acGFP was confounded owing to a low fluorescence SNR caused by the heavily reduced UV absorbance in E222G mutants. However, assuming that the simple two-state model also holds for acGFP, the equilibrium bright distribution pI shift of 0.1 units at 100% UV yields γacGFP ~ 0.5 at most, meaning that at least 67% of the acGFP was in the dark state at equilibrium.
DISCUSSION
The photophysics of the family of green fluorescent proteins derived from Aequorea sp. are characterized by a rich interconnection between spectral properties and short and long-range proton dynamics involving their chromophores29,40–42. The diversity of photophysical phenomena of the fluorescent proteins reflect complex dynamics involving pH-dependent and -independent protonation equilibria22,29,39,42,43, proton exchange with the bulk solvent18,41,44, chromophore and pocket residue conformation21,45–48, electrostatic interactions between the chromophore and surrounding residues in the chromophore pocket23,29, and irreversible chemical reactions at the chromophore or surrounding residues14,49; all of which contribute to the divergent absorption, emission, photoactivation and reversibility aspects of the fluorescence of GFP family members50.
Here we sought to reconcile the unusual UV intensity-dependent IEF focusing behavior of GFP isoforms at the ensemble level by recognizing the interplay between reaction and transport timescales. Specific to the GFP studies conducted here, the wild type Aequorea victoria avGFP, a β-barrel structure containing a buried tripeptide chromophore formed autocatalytically from Ser65, Tyr66 and Gly67, exhibits robust fluorescence with absorption bands at both ~400 and ~475 nm29,40. These absorption bands are comprised of two subpopulations A and B having, respectively, neutral (protonated) and anionic (deprotonated) charge at the hydroxyl group of Tyr66 of the buried chromophore29. While the neutral A form dominates B by 6:1 in wild-type avGFP, S65T (e.g., EGFP) and E222G (e.g., acGFP51) mutants favor the anionic B state in the physiological pH range, thus suppressing the 404 nm absorption peak and simplifying their photophysical behavior29,52. Picosecond spectroscopy studies have detailed an excited-state proton transfer (ESPT) process that occurs upon excitation of the protonated A state, which causes the phenolic proton at Tyr66 to delocalize and transfer to Glu222 via a network of hydrogen bonds due to a drop in the pKa of the tyrosyl phenol in the excited state29,40,43.
As background, the ESPT process is usually reversible, but prolonged, high-intensity exposure of wild type avGFP to UV or blue light can cause nearly permanent (timescale ~ hours) photoisomerization to the anionic B state28,29,43. S65T and E222G mutants favor the anionic chromophore state by suppressing ionization of, and deleting altogether, the glutamate 222 side chain central to the ESPT process29. This conventional picosecond-scale proton transfer phenomenon accounts for short-range migration of the photodissociated proton between Tyr66 and Glu222 within the chromophore cavity. Additionally, nano-to millisecond-scale proton transfer to and from the external solution via a set of “proton wires” consisting of hydrogen bond networks spanning the GFP β-barrel has been hypothesized on the basis of crystallographic, fluorescence autocorrelation, and “pH-jump” spectroscopic evidence41,42,53,54. Proton escape to the bulk solution can occur via at least two proposed exit points, e.g., by rotation of Thr203 via His148 to the protein surface. Additionally, longer-range proton migration along a wire connecting a negatively charged patch of the avGFP surface at Glu5 through Glu222 to its chromophore is expected to allow replenishment of the proton lost by escape to the bulk solvent41, suggesting a proton pumping function55. This hypothesis is to first order compatible with the aforementioned effect of the E222G mutation in acGFP in encouraging chromophore deprotonation, as the proton wire serving as a replenishing proton conduit is likely compromised41. It is important to recognize, however, that in EGFP (in which this conduit is also bisected at Thr6541), chromophore protonation to form a non-fluorescent population proceeds at acidic solution pH with an apparent pKa of 5.842 (similarly in wild-type avGFP with a pKa ~ 531), indicating further redundancy in proton exchange with the bulk solvent, especially at sub-physiological pH56. Indeed, the prevailing view of the effect of mutations targeting E222 in inducing the B chromophore state is that the existence of two negative charges (from E222 and Tyr66 in the excited state) within the spatially confined chromophore pocket is thermodynamically unfavorable, meaning that mutations at E222 that eliminate its charge tend to stabilize the ionized B state chromophore.
While a wide-ranging analysis of all irreversibly (photoactivatable) and reversibly (photochromic) photoswitchable fluorescent proteins is beyond the scope of the present study, several relevant and intriguing phenomena have been recently described and are germane to the present study. Reversible photoswitching processes have been described for green, yellow and cyan avGFP mutants with a range of switching yields38,57,58. Random “blinking” behaviors observed in single molecule studies of fluorescent proteins result from thermodynamic sampling of several chromophore states within individual fluorescent proteins, where at least one is a non-emissive dark state with respect to the excitation wavelength employed42. Several varieties of more directed photoswitching have also been described, in which a given state can be favored through the application of different wavelengths of light. Non-reversible examples necessarily involve permanent biochemical changes to the amino acid scaffold in the vicinity of the chromophore. Examples include UV-induced “photoconversion” of dark wild-type avGFP and PA-GFP to a fluorescent B state by decarboxylation of E22214,59 and “redding” processes caused by as-yet unknown chromophore conversion processes either in the absence of oxygen60 or in the presence of oxidizing agents30.
With these reported phenomena in mind, we hypothesize that the well-known decarboxylation-driven photoconversion process described for wild-type avGFP is likely only a minor contributor to our observations, due to a known irreversibility and to the relatively low illumination intensities applied in this work14,59. Similarly, a contribution from GFP molecules with incorrectly formed chromophores is unlikely, as these species have been observed to remain non-fluorescent even upon successive denaturation and renaturation cycles28. Important here is the immunoprobing functionality; as the probing fluorescence readout is introduced to detect non-fluorescent GFP species (as well as allow similar analysis of other photoactive proteins that lack appreciable fluorescence at their pIs). Thus, immunoblotting allows independent verification of isoelectric points owing to in situ binding of fluorescently labeled antibody to the target of interest.
Examples of both “negative” and “positive” reversible photobleaching have been discovered. Directly relevant to our dynamic IEF observations, Sinnecker et al. characterized a model of positive “reversible photobleaching” in EGFP, EYFP, Citrine, and ECFP. The model included a switch-off transition from a protonated P state to a reversibly bleached form Br upon application of blue 460 nm light (for ECFP) in bulk solution22,38. Decay of the reversibly bleached population to the bright state occurred in the dark on a ~60s timescale. This “kindling” effect was accelerated by application of lower energy green 500 nm light (again, for ECFP). Importantly, reversible bleaching was more efficient under acidic conditions, implicating protonation of a titratable residue in the vicinity of the chromophore in the P state as a prerequisite for transition to the dark state. Likely candidates include Glu222 and His14842, but not the Tyr66 phenol itself, as ECFP (which carries a non-titratable indole ring instead) is also subject to reversible bleaching38. Thus, although the P and Br states likely fall within the canonical A/B/I state framework put forward by Chattoraj et al.43, their properties reflect a more complex involvement of proton dynamics linking the chromophore, its surrounding pocket residues, and the bulk solvent.
We hypothesize that the isoelectric point photoswitching phenomenon in avGFP is consistent with the reversible bleaching observations of Sinnecker et al38. The arrangement of the switching wavelengths (higher energy light shifts equilibrium towards dark state; lower energy light hastens fluorescence recovery), kinetic parameters, and GFP variants studied are most consistent with our observations. Adding to these observations, we observed that the P and Br fluorescence states are characterized by distinct electrostatic charge states at the whole molecule level via direct physicochemical measurement with dynamic IEF. We hypothesize that these distinct electrostatic charge states corroborate proton exchange with the bulk solvent41. The polarity of the observed pI shifts further imply that the transition of GFPs to the dark state involves proton uptake into their structures (causing increased pI), and vice versa, that the return to the bright state involves expulsion of protons (causing decreased pI). More generally, our single-charge resolution pI observations recapitulate the necessity for at least one proton uptake event for the P to Br transition38,42, although the specific residue(s) involved in this process cannot be identified via our whole molecule-level measurements of electrostatic charge. However, the observed difference in protonation stoichiometry between avGFP and acGFP may hold important insights into the structural rearrangements underlying the reversible bleaching process.
Specifically, the all-or-nothing uptake of 3–4 protons by avGFP isoforms during fluorescence switch-off may reflect a cascade of rearrangements in the proposed internal hydrogen bonding network buried within the avGFP structure extending from the chromophore pocket beyond the E222 bridge41,53,61. Indeed, several authors have pointed out the potential for multiple proton storage within the GFP structure at buried hydronium ions and titratable residues41,54. Given that proton travel between the proposed internal and exit hydrogen bond clusters is expected to be restricted or rerouted (if not fully severed) by the E222G mutation in acGFP41, the single-charge shift between bright and dark states observed for the acGFP isoforms may stem from titration dynamics restricted to the chromophore pocket that are facilitated by acid-induced protonation of the unknown pocket residue prescribed by Sinnecker et al.38 (among others39,42) that governs transition between the bright P and dark Br states. While the details of the structural mechanism underlying coupling of photochromism and long-range charge transfer in the GFPs is yet to be elucidated, further studies of key GFP mutants targeted at proposed proton wire pathways is a promising avenue. Specifically, our assay could complement crystallographic and spectroscopic characterization of mutants differing at key hydrogen bonding residues along the proposed internal proton wire that are distant from the chromophore pocket, such as S7241.
Relevant to future study, the coral fluorescent protein Dronpa16 offers a remarkable example of negative reversible photobleaching processes. Dronpa exhibits distinct and stable dark (non-fluorescent, triggered by blue light irradiation) and bright (fluorescent, triggered by violet light) subpopulations18,21. Despite widespread use of Dronpa in high-resolution microscopy, the structural and biophysical mechanisms underpinning photoswitching of Dronpa (and other reversible fluorescent proteins) is still in debate. Cis-trans chromophore isomerization, changes in chromophore protonation state with proton transfer to the bulk solution, and changes in the structural flexibility of the chromophore pocket have all being implicated as playing a role in photoswitching18,21,23,47. Chromophore isomerization and protonation have also been identified in the positive reversible fluorescent protein Padron0.9, in which violet light induces the dark state and blue/green light induces the bright state17 (the opposite of Dronpa, and similar to asFP59547). Still other switching mechanisms have been described, including an unusual chromophore hydration/dehydration process for the Citrine derivative Dreiklang62. The dynamic IEF assay introduced here may prove useful for detailed study of a range of switching mechanisms.
CONCLUSIONS
Microfluidic dynamic IEF brings the advances in separation timescale and resolution necessary for real-time modulation and readout of protein reaction processes, of which photoswitching is an intriguing example explored here. Dynamic IEF analysis generates a rich diversity in GFP focused band morphology from a relatively simple two-state process. Thus, our ability to resolve and track fluorescent protein populations through physicochemical properties rather than by traditional fluorescence or static crystallographic measurements opens the door to a range of dynamic perturbation analyses. Such dynamic analyses should continue to find use in probing the chemical and structural nature of conformational photoswitching processes. We see this analytical capability as relevant to a range of fluorescent and other light-reactive proteins. For example, the ability to characterize GFP constructs genetically encoded with light-switchable charge could benefit a broad range of applications from the study of cellular membrane transport processes to the rational engineering of electrostatic aggregation in signaling molecule or metabolic networks63,64. The fact that reversible fluorescence (and thus isoelectric point) photoswitching is amplified at pH values in the vicinity of the fluorescence pKas of GFPs38 suggests that mutants developed as sensitive pH indicators in the physiologic range56,65 would be attractive candidates for achieving maximum charge switching in biological systems. Further, the more radical photochromism of photoactivatable GFPs (e.g., Dronpa16, Padron0.917 and PA-GFP14, among others12,15) may engender additional benefits in the magnitude and temporal stability of charge conversion, as is currently under study. Beyond studies in cell biology, charge-switchable proteins could be useful in the engineering of biomimetic smart materials with light-actuated transitions in zeta potential, hydrophilicity/wetting behavior, and adhesion properties66–68. The rapid, high resolution dynamic IEF approaches we describe could also be used in high-throughput screening for rational or directed tuning of photoactivatable protein phenomena, from reversible photochromism13 to light-induced protein-protein interactions64.
EXPERIMENTAL PROCEDURES
Reagents and Materials
N-[3-[(4-benzoylphenyl)formamido]propyl] methacrylamide (BPMAC, C21H22N2O3, 350.2 g mol−1) monomer was synthesized via reaction of the succinimidyl ester of 4-benzoylbenzoic acid (323.3 g mol−1; B1577, Invitrogen) with N-(3-aminopropyl)methacrylamide hydrochloride (178.7 g mol−1; 21200, Polysciences, Warrington, PA) in the presence of catalytic triethylamine in dimethylformamide, purified and characterized by 1H NMR and mass spectrometry as previously described2. The monomer was added to BPMAC+ LAVAgel precursor solutions at 4.5 mM (~1 mol % with respect to acrylamide) from a 100 mM stock in dimethylsulfoxide (DMSO) to imbue UV-induced protein photocapture functionality2. BPMAC- precursors were used in dynamic focusing experiments in which protein photocapture was not required, and contained an equivalent volume of DMSO lacking BPMAC.
Purified recombinant wild-type GFP from Aequorea victoria (avGFP) and the Aequorea coerulescens E222G mutant (acGFP) were from Clontech (632373 and 632502, Mountain View, CA). Intact mass analysis of GFP isoforms was performed by high-resolution electrospray-ionization liquid chromatography-mass spectrometry in a Thermo LTQ Orbitrap XL instrument at the QB3 Mass Spectrometry Facility at UC Berkeley. The primary goat polyclonal antibody to GFP was prelabeled with Texas Red by the manufacturer (dye:protein molar ratio = 2.9; ab6660, Abcam, Boston, MA). Equal volumes of a set of fluorescent IEF pI markers with absorption maxima in the near UV (pI 4.0, 4.5, 5.5, 6.2) were mixed in a cocktail and added to GFP samples at 20x dilution (89827 and related products, Sigma).
Microfluidic Chip Fabrication
Microchannels were wet etched in optical white soda lime glass by Caliper Life Sciences (Hopkinton, MA). Each chip contained four straight-channel devices 10.4 mm in length, each consisting of three parallel channels of 10 μm depth and 70 μm width between two 2 mm diameter access wells providing fluidic interfacing via 10 μl press-fit pipet tips. Microchannels were functionalized with acrylate-terminated self-assembled monolayers, as previously described9. Microfluidic LAVAgels were fabricated via introduction of a gel precursor solution by capillary action. The precursor contained 6% total acrylamide (6%T; % concentrations are w/v unless otherwise noted) with 2.6% of the total as the crosslinker bisacrylamide (2.6%C), 15% v/v polybuffer 74 ampholytes (P9652, Sigma, St. Louis, MO), 0.1% v/v Triton X-100 detergent (T8532, Sigma), 4.5 mM BPMAC (see Reagents and Materials). The initiators ammonium persulfate (0.015%, A3678, Sigma), N,N,N′,N′-tetramethylethylenediamine (0.05% v/v, T9281, Sigma) and riboflavin 5′ phosphate (0.0006%, F1392, Sigma) were added just before introduction of degassed precursor to channels. The precursor was polymerized by 6 min flood exposure of chips to 470 nm blue light from a collimated LED source (M470L2, Thorlabs, Newton, NJ) with access wells masked to prevent gel fouling. The blue light intensity at a 470 nm probe setting was ~2.2 mW cm−2 at the chip plane, as measured by a LaserCheck light meter (1098293, Coherent, Santa Clara, CA). Equivalent isoelectric photoswitching behavior was observed in gels chemically polymerized in the absence of riboflavin.
Apparatus and Imaging
Chip imaging was conducted using an Olympus IX71 inverted fluorescence microscope equipped with an EMCCD camera (iXon3 885, Andor, Belfast, Northern Ireland), motorized stage (Applied Scientific Instrumentation, Eugene, OR) and automated filter cube turret controlled through MetaMorph software (Molecular Devices, Sunnyvale, CA). Illumination was provided by a mercury arc lamp mated to an automated shutter and attenuation system (X-Cite Exacte, Lumen Dynamics, Mississauga, ON, Canada). Electric field was applied via a custom high voltage power supply built in-house. LAVAgel photoimmobilization of proteins was conducted via spot UV exposure through a 10x objective (Olympus UPlanFl, NA 0.3) and custom UV-longpass filter cube (excitation 300–380 nm, emission >410 nm; XF1001, XF3097, Omega Optical) at ~269 mW cm−2 as measured via a 365 nm probe (UV513AB meter, General Tools, New York, NY). The same cube was used to observe GFP under UV illumination along with fluorescent pI marker peptides, and channel positions were manually scored (gradient drift between focused-state marker and GFP isoform imaging steps was assumed to be negligible). Green and red fluorescence channels were imaged at 10x using Omega Optical filter cubes optimized for GFP (XF100–3, excitation 445–495 nm at ~89 mW cm−2 for a 470 nm probe setting, emission 508–583 nm) and DsRed2 (XF111–2, excitation 525–555 nm, emission >575 nm). Whole channel imaging at 10x magnification was conducted via stitching of adjacent, overlapping CCD images with 4 × 4 pixel binning in ImageJ (NIH, Bethesda, MD) to produce full gel channel images and electropherograms as previously described9. Imaging scans required ~ 20 s to complete. Real-time single-point imaging of GFP isoform dynamics was conducted in burst acquisition mode.
Transformation of fluorescence data via linear fits to pI markers and associated data processing, including correction for constant cathodic isoform drift velocities in dynamic focusing experiments was performed using MATLAB scripts written in-house (MathWorks, Natick, MA). Least-squares fitting of kinetic data was performed using gnuplot software.
Chip Operation
After precursor gelation, LAVAgel access wells were flushed and replaced with gel buffer consisting of precursor lacking monomers and initiators. Samples were made in gel buffer and introduced at loading wells (~3 μl per well). Sample injection was performed at 200 V cm−1 for 3 minutes. Opposing wells were briefly washed with catholyte (20 mM lysine, 20 mM arginine pH 10.1) and anolyte (70 mM phosphoric acid) and subsequently filled. Focusing was conducted at 200 V cm−1 for 2 min followed by 400 V cm−1 at 1 min (focusing typically completed to equilibrium in 3 minutes or less). Imaging and UV photocapture steps (where applicable) were conducted individually for each device in series. UV photocapture was conducted under stopped electric field for 15 s. Access wells were washed and filled with pH gradient mobilization/probe buffer consisting of 25 mM Tris, 192 mM Glycine pH 8.3, 0.1% v/v Triton X-100 and 3% v/v DMSO. Mobilization and washout of pH gradients to the anodic wells was achieved via a 20 min electrophoretic step. Fluorescently labeled anti-GFP antibody was diluted in probe buffer, loaded, and removed from LAVAgels in 20 min electrophoretic steps. Probe loading and washout were conducted in opposite directions to minimize non-specific signal remaining after washout. Finally, gels were scanned for captured GFP and antibody fluorescence. Removal of the LAVAgel matrix after use was achieved by overnight incubation of the chip in a 2:1 solution of 70% perchloric acid and 30% hydrogen peroxide heated to 75°C, allowing efficient recycling of glass chips, as previously described9.
Supplementary Material
Acknowledgments
This research was made with Government support under and awarded by DoD, Air Force Office of Scientific Research, National Defense Science and Engineering Graduate (NDSEG) Fellowship to A.J.H., 32 CFR 168a. A.J.H. is a 2012 Siebel Scholar. This research was performed under an appointment to the Department of Homeland Security (DHS) Scholarship and Fellowship Program to A.M.T., administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy (DOE) and DHS. ORISE is managed by Oak Ridge Associated Universities (ORAU) under DOE contract number DE-AC05 06OR23100. A.M.T. is also a UC Cancer Research Coordinating Committee (CRCC) pre-doctoral fellow. A.E.H. is an Alfred P. Sloan research fellow in chemistry. This project was supported by New Innovator Award (DP2OD007294 to A.E.H.) from the Office of the Director, National Institutes of Health.
Footnotes
The authors declare no competing financial interest.
Reaction-transport model details and four figures, including further detail on real-time observation of isoelectric point photoswitching in acGFP, kinetics of reversible photoswitching, and total avGFP isoform fluorescence during steady-state focusing under UV illumination.
References
- 1.He M, Herr AE. Nat Protoc. 2010;5:1844. doi: 10.1038/nprot.2010.142. [DOI] [PubMed] [Google Scholar]
- 2.Hughes AJ, Lin RKC, Peehl DM, Herr AE. Proc Natl Acad Sci USA. 2012;109:5972. doi: 10.1073/pnas.1108617109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao J, Krylova SM, Reinstein O, Johnson PE, Krylov SN. Anal Chem. 2011;83:8387. doi: 10.1021/ac2026699. [DOI] [PubMed] [Google Scholar]
- 4.Bao J, Regnier FE. J Chromatogr. 1992;608:217. doi: 10.1016/0021-9673(92)87127-t. [DOI] [PubMed] [Google Scholar]
- 5.Wong E, Okhonin V, Berezovski MV, Nozaki T, Waldmann H, Alexandrov K, Krylov SN. J Am Chem Soc. 2008;130:11862. doi: 10.1021/ja804544x. [DOI] [PubMed] [Google Scholar]
- 6.Petrov A, Okhonin V, Berezovski M, Krylov SN. J Am Chem Soc. 2005;127:17104. doi: 10.1021/ja056232l. [DOI] [PubMed] [Google Scholar]
- 7.Liu Z, Pawliszyn J. J Proteome Res. 2004;3:567. doi: 10.1021/pr034114w. [DOI] [PubMed] [Google Scholar]
- 8.Lemma T, Mandal R, Li XF, Pawliszyn J. J Sep Sci. 2008;31:1803. doi: 10.1002/jssc.200700418. [DOI] [PubMed] [Google Scholar]
- 9.Hughes AJ, Herr AE. Anal Chem. 2010;82:3803. doi: 10.1021/ac100201z. [DOI] [PubMed] [Google Scholar]
- 10.He M, Novak J, Julian BA, Herr AE. J Am Chem Soc. 2011;133:19610. doi: 10.1021/ja207963f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldsmith RH, Moerner WE. Nat Chem. 2010;2:179. doi: 10.1038/nchem.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Physiol Rev. 2010;90:1103. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- 13.Subach FV, Piatkevich KD, Verkhusha VV. Nat Methods. 2011;8:1019. doi: 10.1038/nmeth.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patterson GH, Lippincott-Schwartz J. Science. 2002;297:1873. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- 15.Chang H, Zhang M, Ji W, Chen J, Zhang Y, Liu B, Lu J, Zhang J, Xu P, Xu T. Proc Natl Acad Sci USA. 2012;109:4455. doi: 10.1073/pnas.1113770109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ando R, Mizuno H, Miyawaki A. Science. 2004;306:1370. doi: 10.1126/science.1102506. [DOI] [PubMed] [Google Scholar]
- 17.Brakemann T, Weber G, Andresen M, Groenhof G, Stiel AC, Trowitzsch S, Eggeling C, Grubmuller H, Hell SW, Wahl MC, Jakobs S. JBC. 2010;285:14603. doi: 10.1074/jbc.M109.086314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bizzarri R, Serresi M, Cardarelli F, Abbruzzetti S, Campanini B, Viappiani C, Beltram F. J Am Chem Soc. 2010;132:85. doi: 10.1021/ja9014953. [DOI] [PubMed] [Google Scholar]
- 19.Adelroth P, Brzezinski P. Biochim Biophys Acta. 2004;1655:102. doi: 10.1016/j.bbabio.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 20.Gutman M, Nachliel E. Annu Rev Phys Chem. 1997;48:329. doi: 10.1146/annurev.physchem.48.1.329. [DOI] [PubMed] [Google Scholar]
- 21.Andresen, Stiel AC, Trowitzsch S, Weber G, Eggeling C, Wahl MC, Hell SW, Jakobs S. Proc Natl Acad Sci USA. 2007;104:13005. doi: 10.1073/pnas.0700629104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henderson JN, Ai HW, Campbell RE, Remington SJ. Proc Natl Acad Sci USA. 2007;104:6672. doi: 10.1073/pnas.0700059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizuno H, Mal TK, Walchli M, Kikuchi A, Fukano T, Ando R, Jeyakanthan J, Taka J, Shiro Y, Ikura M, Miyawaki A. Proc Natl Acad Sci USA. 2008;105:9227. doi: 10.1073/pnas.0709599105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Righetti PG. Isoelectric Focusing: Theory, Methodology and Applications. Vol. 11 Elsevier; New York: 1983. [Google Scholar]
- 25.Rilbe H. Ann NY Acad Sci. 1973;209:11. doi: 10.1111/j.1749-6632.1973.tb47520.x. [DOI] [PubMed] [Google Scholar]
- 26.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SMM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Nature. 2011;480:254. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cossu G, Righetti PG. J Chromatogr. 1987;398:211. doi: 10.1016/s0021-9673(01)96506-9. [DOI] [PubMed] [Google Scholar]
- 28.Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW. Biophys J. 1997;73:2782. doi: 10.1016/S0006-3495(97)78307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsien RY. Annu Rev Biochem. 1998;67:509. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 30.Bogdanov AM, Mishin AS, Yampolsky IV, Belousev VV, Chudakov DM, Subach FV, Verkhusha VV, Lukyanov S, Lukyanov KA. Nat Chem Biol. 2009;5:459. doi: 10.1038/nchembio.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward WW. In: Green Fluorescent Protein: Properties, Applications and Protocols. Chalfie M, Kain SR, editors. John Wiley & Sons; Hoboken, NJ: 2006. [Google Scholar]
- 32.Pace CN, Grimsley GR, Scholtz JM. JBC. 2009;284:13285. doi: 10.1074/jbc.R800080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. In: The proteomics protocols handbook. Walker JM, editor. Humana press; Totowa: 2005. [Google Scholar]
- 34.McGrath ME, Vasquez JR, Craik CS, Yang AS, Honig B, Fletterick RJ. Biochemistry. 1992;31:3059. doi: 10.1021/bi00127a005. [DOI] [PubMed] [Google Scholar]
- 35.O’Farrell PH. JBC. 1975;250:4007. [PMC free article] [PubMed] [Google Scholar]
- 36.Bobb D, Hofstee BHJ. Anal Biochem. 1971;40:209. doi: 10.1016/0003-2697(71)90094-7. [DOI] [PubMed] [Google Scholar]
- 37.Weiss GH, Catsimpoolas N, Rodbard D. Arch Biochem Biophys. 1974;163:106. doi: 10.1016/0003-9861(74)90460-3. [DOI] [PubMed] [Google Scholar]
- 38.Sinnecker D, Voigt P, Hellwig N, Schaefer M. Biochemistry. 2005;44:7085. doi: 10.1021/bi047881x. [DOI] [PubMed] [Google Scholar]
- 39.Schwille P, Kummer S, Heikal AA, Moerner WE, Webb WW. Proc Natl Acad Sci USA. 2000;97:151. doi: 10.1073/pnas.97.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brejc K, Sixma TK, Kitts PA, Kain SR, Tsien RY, Ormo M, Remington SJ. Proc Natl Acad Sci USA. 1997;94:2306. doi: 10.1073/pnas.94.6.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shinobu A, Palm GJ, Schierbeek AJ, Agmon N. J Am Chem Soc. 2010;132:11093. doi: 10.1021/ja1010652. [DOI] [PubMed] [Google Scholar]
- 42.Haupts U, Maiti S, Schwille P, Webb WW. Proc Natl Acad Sci USA. 1998;95:13573. doi: 10.1073/pnas.95.23.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chattoraj M, King BA, Bublitz GU, Boxer SG. Proc Natl Acad Sci USA. 1996;93:8362. doi: 10.1073/pnas.93.16.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Q, Byrnes LJ, Shui B, Rohrig UF, Singh A, Chudakov DM, Lukyanov S, Zipfel WR, Kotlikoff MI, Sondermann H. PLoS ONE. 2011;6:e23513. doi: 10.1371/journal.pone.0023513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weber W, Helms V, McCammon JA, Langhoff PW. Proc Natl Acad Sci USA. 1999;96:6177. doi: 10.1073/pnas.96.11.6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baranov MS, Lukyanov KA, Borissova AO, Shamir J, Kosenkov D, Slipchenko LV, Tolbert LM, Yampolsky IV, Solntsev KM. J Am Chem Soc. 2012;134:6025. doi: 10.1021/ja3010144. [DOI] [PubMed] [Google Scholar]
- 47.Andresen M, Wahl MC, Stiel AC, Grater F, Schafer LV, Trowitzsch S, Weber G, Eggeling C, Grubmuller H, Hell SW, Jakobs S. Proc Natl Acad Sci USA. 2005;102:13070. doi: 10.1073/pnas.0502772102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fang C, Frontiera RR, Tran R, Mathies RA. Nature. 2009;462:200. doi: 10.1038/nature08527. [DOI] [PubMed] [Google Scholar]
- 49.Nienhaus K, Nienhaus GU, Wiedenmann J, Nar H. Proc Natl Acad Sci USA. 2005;102:9156. doi: 10.1073/pnas.0501874102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shaner NC, Patterson GH, Davidson MW. J Cell Sci. 2007;120:4247. doi: 10.1242/jcs.005801. [DOI] [PubMed] [Google Scholar]
- 51.Gurskaya NG, Fradkov AF, Pounkova NI, Staroverov DB, Bulina ME, Yanushevich YG, Labas YA, Lukyanov S, Lukyanov KA. Biochem J. 2003;373:403. doi: 10.1042/BJ20021966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ehrig T, O’Kane DJ, Prendergast FG. FEBS Lett. 1995;367:163. doi: 10.1016/0014-5793(95)00557-p. [DOI] [PubMed] [Google Scholar]
- 53.Agmon N. J Phys Chem B. 2007;111:7870. doi: 10.1021/jp071403p. [DOI] [PubMed] [Google Scholar]
- 54.Helms V, Gu W. In: Fluorescent proteins I: From understanding to design. Jung G, editor. Vol. 11 Springer-Verlag; Berlin: 2012. [Google Scholar]
- 55.Agmon N. Biophys J. 2005;88:2452. doi: 10.1529/biophysj.104.055541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bizzarri R. In: Fluorescent proteins I: From understanding to design. Jung G, editor. Vol. 11 Springer-Verlag; Berlin: 2012. [Google Scholar]
- 57.Abbruzzetti S, Bizzarri R, Luin S, Nifosi R, Storti B, Viappiani C, Beltram F. Photochem Photobiol Sci. 2010;9:1307. doi: 10.1039/c0pp00189a. [DOI] [PubMed] [Google Scholar]
- 58.Dickson RM, Cubitt AB, Tsien RY, Moerner WE. Nature. 1997;388:355. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 59.van Thor JJ, Gensch T, Hellingwerf KJ, Johnson LN. Nat Stuct Biol. 2002;9:37. doi: 10.1038/nsb739. [DOI] [PubMed] [Google Scholar]
- 60.Elowitz MB, Surette MG, Wolf PE, Stock J, Leibler S. Curr Biol. 1997;7:809. doi: 10.1016/s0960-9822(06)00342-3. [DOI] [PubMed] [Google Scholar]
- 61.Di Donato M, van Wilderen LJGW, van Stokkum IHM, Stuart TC, Kennis JTM, Hellingwerf KJ, van Grondelle R, Groot ML. Phys Chem Chem Phys. 2011;13:16295. doi: 10.1039/c1cp20387h. [DOI] [PubMed] [Google Scholar]
- 62.Brakemann T, Stiel AC, Weber G, Andresen M, Testa I, Grotjohann T, Leutenegger M, Plessmann U, Urlaub H, Eggeling C, Wahl MC, Hell SW, Jakobs S. Nat Biotechnol. 2011;29:942. doi: 10.1038/nbt.1952. [DOI] [PubMed] [Google Scholar]
- 63.Wang X, Chen X, Yang Y. Nat Methods. 2012;9:266. doi: 10.1038/nmeth.1892. [DOI] [PubMed] [Google Scholar]
- 64.Strickland D, Lin Y, Wagner E, Hope CM, Zayner J, Antoniou C, Sosnick TR, Weiss EL, Glotzer M. Nat Methods. 2012;9:379. doi: 10.1038/nmeth.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bizzarri R, Serresi M, Luin S, Beltram F. Anal Bioanal Chem. 2009;393:1107. doi: 10.1007/s00216-008-2515-9. [DOI] [PubMed] [Google Scholar]
- 66.Burkert S, Bittrich E, Kuntzsch M, Muller M, Eichhorn KJ, Bellmann C, Uhlmann P, Stamm M. Langmuir. 2010;26:1786. doi: 10.1021/la902505q. [DOI] [PubMed] [Google Scholar]
- 67.Oroszi L, Der A, Kirei H, Ormos P. Appl Phys Lett. 2006;89:2635081. [Google Scholar]
- 68.Zhang L, Zhang Z, Wang P. NPG Asia Mater. 2012;4:e8. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.