


Abstract
To elucidate how microRNA (miRNA)-regulated networks contribute to the uncontrolled growth of hepatoma cells (HCCs), we identified several proliferation-related miRNAs by comparing miRNA expression patterns in clinical HCC samples and growth-arrested HepG2 cells. To explore the molecular functions targeted by these miRNAs, we classified genes differentially expressed in clinical HCC samples into six functional clusters based on their functional similarity. Using target enrichment analysis, we discovered that targets of three proliferation-related miRNAs—miR-101, miR-199a-3p and miR-139-5p—were significantly enriched in the ‘transcription regulation’ functional cluster. An interactome network consisting of these three miRNAs and genes in the ‘transcriptional control’ cluster revealed that all three miRNAs were highly connected hubs in the network. All three miRNA-centered subnetworks displayed characteristics of a two-layer regulatory architecture, with transcription factors and epigenetic modulators as the first neighbors and genes involved in cell-cycle progression as second neighbors. The overexpression of miR-101 in HepG2 cells reduced the expression of transcription regulators and genes in cell-cycle progression and suppressed the proliferation and colony formation of HepG2 cells. This study not only provides direct experimental data to support the ‘miRNA-centered two-layer regulatory network’ model, but our results also suggest that such a combinatorial network model may be widely used by miRNAs to regulate critical biological processes.
INTRODUCTION
MicroRNAs (miRNAs) are a family of small RNA molecules that negatively regulate the expression levels of protein-coding genes. Mature miRNAs are incorporated into the RNA-induced silencing complex (RISC) and guide the RISC to interact with messenger RNAs based on partial sequence complementarity, leading to the degradation or translational repression of target mRNA. There is accumulating evidence that indicates miRNAs play critical roles in diverse cellular processes, including cell growth, survival, differentiation and maintenance of cellular homeostasis, while dysregulation of miRNAs may be responsible for various disorders, including cancers (1–3). A genome-wide analysis revealed that >50% of human miRNAs are located in the chromosomal fragile sites that are strongly associated with chromosomal alterations in human malignancy (2,4). Indeed, numerous profiling studies have revealed that the expression patterns of miRNAs are significantly different in cancer tissues. In addition the results of those studies indicate that the expression levels of certain miRNAs are frequently altered in tumor tissues. Functional studies further demonstrate that these dysregulated miRNAs can function either as oncogenes or tumor suppressors. Experimental perturbation of these miRNAs is associated with profound changes in all aspects of tumor phenotype both in vitro and in vivo.
The mechanism by which miRNA exerts its functional impact is a topic of great interest in cancer biology. Several computational algorithms based on sequence complementarity have been developed to predict miRNA targets. This computational approach estimates that each miRNA may have hundreds to thousands of mRNA targets (5,6). Similarly, individual protein-coding genes may be targeted by tens to hundreds of miRNAs (6). Recent large-scale transcriptomics and proteomics studies have confirmed that individual miRNAs may alter the transcript and protein levels of hundreds to thousands of genes. However, despite the profound functional consequences observed, the effect of miRNAs on the expression levels of most target genes is very subtle. Two potential mechanisms have been proposed to explain the apparent discrepancy between the biochemical and biological effects. The first mechanism hypothesizes that miRNAs may achieve a profound biological impact by targeting multiple genes in the same signaling pathway. This mechanism was initially proposed following analysis of the pathway and enrichment of computationally predicted miRNA targets and subsequently verified using in vitro studies. For example, the miR-16 family has been shown to trigger G0/G1 arrest by silencing multiple cell-cycle genes simultaneously (7). Similarly, miR-17-5p has been shown to regulate cell-cycle progression by suppressing, in a coordinated manner, more than 20 genes involved in the G1/S transition (8). The second mechanism suggests that miRNAs may effectively regulate a biological function by selectively targeting critical hubs such as transcription factors in the signaling network. These transcription factors can amplify the subtle effect of miRNAs throughout the network to produce profound biological consequences. By analyzing the molecular functions of the predicted targets of miRNAs, Cui et al. found that transcription factors are the most frequently targeted protein-coding genes (9). In addition, by analyzing the interaction network of miRNAs, Tu et al. observed a miRNA-centered two-layer regulatory cascade in which transcription factors function as key mediators of miRNA-initiated regulatory effects (10). These complex interactions between miRNAs and their targets fine tune the expression levels of critical genes to maintain a stable homeostasis of biological processes such as cell growth and development. The alteration of miRNA levels disrupts the network and can lead to severe consequences.
While early studies suggested that miRNAs mainly function through translational suppression, recent studies (11–13) indicate that in mammalian cells miRNAs predominantly exert their effects by decreasing the levels of target mRNAs. This observation provides the rationale to integrate a microarray approach and miRNA target prediction for investigating miRNA-regulated networks and functional consequences. Indeed, through the experimental manipulation of single miRNAs in cultured cells, several groups have successfully constructed miRNA-regulated networks of biological functions by examining the inverse expression of miRNAs and their targets with microarrays (14,15). However, inferring miRNAs-regulated networks and function from clinical tissue microarray data remains highly challenging. Unlike the in vitro experimental models that allow dramatic alterations of individual miRNA expression levels, the magnitude of changes in miRNA levels is much smaller in clinical samples, thus making the recognition of miRNA targets in microarrays more difficult. In addition, the simultaneous alteration of multiple miRNAs is commonly observed in clinical samples, therefore making the target assignment more complicated.
In this study, we describe a stepwise approach to investigating how miRNA-regulated networks contribute to the uncontrolled growth of hepatoma cells (HCCs) (Figure 1). Several tactics were employed to resolve the issues mentioned above. First, although many miRNAs were significantly altered in HCC samples, we incorporated an in vitro cell growth arrest model to narrow down the proliferation-related miRNA candidates. Second, a computational algorithm for miRNA target prediction typically yielded targets with varying degrees of efficacy. We used a stringent cutoff threshold to eliminate low-efficacy targets. Third, we classified differentially expressed genes (DE genes) based on their biological functions and used an R package, GOSim, developed by Frohlich et al.(16), to resolve the redundancy issues in GO annotation. Fourth, we calculated the enrichment score between DE genes and predicted miRNA targets to identify functional cluster regulated by proliferation-related miRNAs. A shortest-path algorithm was then used to build the miRNA-regulated network. A topological analysis was used to identify critical hubs in miRNA-regulated networks. The biological function of miRNA-regulated networks was deduced by analyzing the pathways embedded in the network. Through this approach, we discovered a network regulated by three proliferation-related miRNAs, including miR-101, miR-139-5p and miR-199a-3p, in HCC cells. All three subnetworks regulated by miRNAs exhibited a two-layer regulatory cascade with transcriptional regulators, including both transcription factors and epigenetic modulators, acting as key mediators to regulate the expression of multiple proliferation-related genes. We carried out in vitro experiments to confirm the biological functions of proliferation-related miRNAs and the interactions between miRNAs and their neighbors in the network. Our observations are consistent with the ‘miRNA-centered two-layer regulatory network’ hypothesized by Tu et al. (10) and illustrate the complex and intricate relationship between miRNAs and transcriptional regulators.
Figure 1.

Overview of analysis procedure. MiRNAs involved in the growth control of HCC cells were identified by combining profiling data from HCC tissue samples and growth-arrested cultured HCC cells. The targets for each proliferation-related miRNA were predicted using TargetScan. In parallel, protein-coding genes DE in HCC samples were identified from microarray data. The DE genes were clustered based on their functional similarity in GO biological process using GOSim. Enrichment of individual functional clusters with targets of proliferation-related miRNAs was assessed by Fisher’s exact test and compared with targets of randomly selected miRNAs. The gene set from the functional cluster specifically enriched with proliferation-related miRNAs was uploaded into GeneGO MetaCore to build a network based on the shortest-path algorithm. Network topology was visualized and analyzed with Cytoscape to identify critical hubs and nodes in the miRNA-regulated subnet. Finally, an in vitro study using cultured HepG2 was carried out to validate the predicted function and targets of the miRNA-regulated network.
MATERIALS AND METHODS
Cell culture
The human hepatocellular carcinoma cell line HepG2 was maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM l-glutamine and 5% CO2 at 37°C.
Overexpression of miR-101 in cultured HepG2 cells
Chemically modified double-stranded RNA mimicking endogenous mature miR-101 and scrambled control were purchased from Ambion (Austin, TX, USA). Scrambled control and miR-101 mimetic (15 nM) were transfected into cultured HepG2 cells using RNAiMAX reagent following the manufacture’s protocol (Invitrogen, Carlsbad, CA, USA). The expression level of mature miR-101 was analyzed using stem-loop qRT–PCR (Supplementary Figure S1).
Proliferation and colony formation assay
Cultured HepG2 cells were plated at 1000 cells/well in 96-well plate. Cells were transfected with scrambled control or miR-101, respectively. After 2 and 4 days, cells were fixed and stained with DAPI. Cell numbers in each well were quantified by counting the DAPI-stained nuclei using the IN Cell 1000 image analyzer (GE Healthcare Lifesciences, Piscataway, NJ, USA). For colony formation assay, HepG2 cells were plated at 2000 cells/well in a six-well plate. Cells were transfected with miR-101 mimetic and scramble control, respectively. After 14 days, cells were stained with crystal violet. The colony number and size was determined by Image J (NIH, Bethesda, MD, USA).
Luciferase reporter assay for FOS and TGFB1 3′-UTR
To evaluate the direct effect of miR-101 on FOS and TGFB1, the full-length 3′-UTR of FOS which contains one miR-101-binding site (Supplementary Figure S2A) and TGFB1 was cloned into the pMIR-REPORT vector (Invitrogen). Adherent HepG2 cells in 24-well plate at 80% confluency were transfected with 15 nM of miR-101 mimetic or scrambled control, respectively. After 24 h, cells were cotransfected with 0.33 µg/well of control pMIR-REPORT or fos-pMIR-REPORT and 0.033 µg/well of control Relina luciferase expression vector using Lipofectamine 2000 (Invitrogen). Cells were lysed 24 h later and luciferase activity was analyzed using the dual luciferase reporter system (Promega). The firefly luciferase activity was normalized to the Relina luciferase control.
TGFB1 promoter activity assay
To evaluate the effect of miR-101 on TGFB1 promoter activity, the TGFB1 promoter region with two AP1-binding sites (Supplementary Figure S2B) was cloned into the pGL3-Basic Reporter Vector (Promega). HepG2 cells were grown in 24-well plate to 80% confluence and transfected with 15 nM of scrambled control or miR-101 mimetic for 24 h. Cells were then cotransfected with 0.33 µg/well of control pGL3-Basic reporter or TGFB1-pGL3-basic reporter and 0.033 µg/well of control Relina luciferase expression vector using Lipofectamine 2000 (Invitrogen). Eighteen hours after transfection, cells were treated with TPA (100 nM) to activate AP1 activity for 6 h. Cells were lyzed and luciferase activity was determined using the dual luciferase reporter system (Promega). The firefly luciferase activity was normalized to the control Relina luciferase activity.
RNA preparation
HCC tissue samples and adjacent normal liver tissues were obtained from patients undergoing surgery and were frozen immediately after surgical resection. Tissue collection was performed in accordance with regulations of the Institutional Regulation Board of Chang Gung Memorial Hospital, Taiwan. A total of 23 paired normal and hepatoma tissues were used in this study, twenty paired tissues were used in miRNA quantification while the remaining three were used for microarray study to identify DE genes. Total RNA was extracted from biopsied samples and cultured HepG2 cells using the TRIzol reagent following the manufacturer’s protocol (Invitrogen). RNA concentration was determined using a NanoDrop spectrophotometer. RNA integrity was evaluated using an Agilent 2100 BioAnalyzer (Agilent Technologies, Palo Alto, CA, USA). Only samples with an RNA integrity number >7.5 were used for miRNA and mRNA quantification.
Microarray analysis
The RNA was processed, labeled and hybridized to Affymetrix U133 Plus2 chips according to the manufacturer’s protocol. The raw Affymetrix HG-U133 Plus2 data were MAS5 normalized using the R package. DE genes were identified using Partek Genomics Suite (version 6.4) with the following settings: the gene was confidently detected in at least 50% of the samples, the fold-change was >2 and the paired t-test (two-tailed) P-value was < 0.05. The expression values of the DE genes were subjected to hierarchical clustering using Pearson’s dissimilarity distance and Ward’s method.
miRNA expression analysis
The expression levels of 270 miRNAs were determined by stem-loop RT–PCR as previously described (17). Briefly, 1 µg of total RNA from tissues or cultured cells was added to a reverse transcription (RT) reaction along with pooled miRNA-specific stem-loop RT primers. The RT products were diluted and quantified with a miRNA-specific forward primer and a universal reverse primer using an ABI Prism 7900 Fast Real-Time PCR system (Foster City, CA, USA). The expression levels of individual miRNAs were expressed as 39 substrate the value of threshold cycle (Ct), defined as the cycle number at which the change of SYBR green intensity crosses the threshold of 0.2, after global median normalization. A Student’s t-test was performed to identify significantly and DE miRNAs.
mRNA quantification with qRT–PCR
For quantitating mRNA expression, the total RNA was reverse transcribed using oligo-dT primers. The reverse transcribed products were diluted and quantified using gene-specific PCR primers (Supplementary Table S1) designed by LightCycler and quantified using an ABI Prism 7900 Fast Real-Time PCR system (Foster City) as previously described (17). The average Ct of two internal controls, EEF1A1 was used for data normalization.
Target prediction
To perform target prediction, the 3′-UTR sequence of 17 330 human protein-coding genes was retrieved from the UCSC genome database, and the sequences of all mature miRNAs were downloaded from miRBase (http://www.mirbase.org/). Target prediction and context score analyses were performed using the TargetScan_50.pl and TargetScan_Context_Scores.pl scripts downloaded from the TargetScan website (http://www.targetscan.org/vert_50/). A sum context score < −0.2 was used as a filter to select high efficacy targets (5).
Functional classification of DE genes
DE genes were separated into different functional groups based on their Gene Ontology (GO) Annotation database biological process annotations. Their functional similarity was calculated using the R package GOSim (16). The package measures the semantic similarity of genes based on their distances to the closest common ancestor term and/or the annotation statistics of their common ancestor terms. We used the Jiang and Conrath’s method to evaluate the GO term similarity because of the strong correlation between GO and family similarity (18). The similarity scores calculated using this method reflects the similarity between genes based on their complete GO annotation. The maximum similarity between any pair of genes was used to generate the distance matrix for functional classification. Functionally similar genes were partitioned using the K-means clustering algorithm, with a k-range of 3 to 20. A Davies–Bouldin (DB) score (19) was calculated using 200 iterations to estimate the ratio of within-cluster scatter to between-cluster separation. The clustering results were exported to spreadsheet and visualized by scatter plot.
Statistical analysis
We used Fisher’s exact test, calculated using the R software package, to examine the specific enrichment of proliferation-related miRNA targets in a functional cluster. The 2D contingency matrix consisted of predicted targets of a specific proliferation-related miRNA and DE genes in a specific functional cluster. The Fisher’s exact test P-value was converted into a −log scale and reported as enrichment score.
Network modeling and topological analysis
To construct the miRNA-regulated network, we uploaded the DE genes within each cluster and putative miRNA to the target relationship as customized interactions in a MetaCore database (GeneGo, St Joseph, MI, USA). For each cluster, the uploaded dataset was used to construct a separate network consisting of the shortest paths (i.e. having the smallest possible number of directed one-step interactions) between pairs of initial objects in each direction based on standard Dijkstra’s shortest paths algorithm. In addition to the protein–protein interactions, the networks also contained interactions based on transcription regulations, including transcription factors and miRNAs to their targets. The resulting networks were exported to Cytoscape (http://cytoscape.org/index.php) for visualization and network topology analysis. For the network topological analysis, we defined the network as a directed graph, G, let G = (V, E). The CentiScaPe 1.0 pug-in was used to calculate network centralities, including node degrees and Betweenness, for all genes and miRNAs in the network (20). The degree of connections, deg(v), reflects the number of nodes directly connect to a given node v. A node assessed to have a high degree of connection is likely to be a regulatory hub. The SP (shortest path) Betweenness values were determined to analyze the importance of a given node in maintaining the functionally coherent network. The SP betweenness was scored by counting the number of shortest paths linking s and t through a node v and divided by the total shortest paths between s and t:
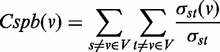 |
where 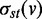 indicates the number of shortest paths linking s and t through a node v and
indicates the number of shortest paths linking s and t through a node v and 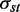 indicates the total number of shortest paths between s and t. A node with a higher Betweenness score is more crucial to maintaining network stability. Finally, a functional enrichment analysis was performed using the Database for Annotation, Visualization and Integrated Discovery 2008 Tool (21,22).
indicates the total number of shortest paths between s and t. A node with a higher Betweenness score is more crucial to maintaining network stability. Finally, a functional enrichment analysis was performed using the Database for Annotation, Visualization and Integrated Discovery 2008 Tool (21,22).
RESULTS
Identification of miRNAs involved in the growth control of HCC cells
Recent studies clearly demonstrate that miRNAs are involved in multiple aspects of cancer initiation and progression, including the regulation of cell growth, survival, angiogenesis and metastasis (2,23). To identify miRNAs that are differentially expressed in HCC cells, we profiled the expression levels of 270 miRNAs in 20 pairs of HCC and adjacent normal liver tissue samples using a stem-loop RT–qPCR method previously established in our laboratory (17). DE miRNAs were identified using paired t-tests after global median normalization. We identified 12 upregulated miRNAs and 26 downregulated miRNAs in the HCC samples (paired t-test, P-value ≤ 0.05, fold-change ≥ 2; Supplementary Table S2). These DE miRNAs include several well-characterized oncogenic miRNAs, such as miR-10b, miR-18a, miR-93, miR-221, miR-222 and tumor suppressive miRNAs, such as miR-22, miR-101, miR-122, miR-125b, miR-139-5p, miR-199a-3p, miR-199a-5p and miR-451. The overall miRNA expression patterns observed in our HCC samples were similar to the miRNA expression profiles reported by two previous studies (24–26). Unsupervised hierarchical clustering using these 38 DE miRNAs clearly separated normal and HCC samples into two different groups (Figure 2A).
Figure 2.

miRNAs inversely modulated in HCC tissues and TPA-treated cultured HCC cells. (A) Hierarchical clustering of 20 normal liver tissues samples and 20 HCC samples based on the expression levels of 38 DE miRNAs. Hierarchical clustering was generated using Pearson’s dissimilarity distance and the average linkage method. (B) Expression levels of 270 miRNAs in cultured HepG2 cells after being treated with DMSO or 100 nM TPA for 3 days. The dotted red lines indicate the 3-fold change boundary. (C) A Venn diagram of miRNAs DE in HCC samples and miRNA altered upon TPA treatment. Shown in the intersection are the miRNAs inversely expressed in the HCC samples and TPA-treated samples.
To further identify miRNAs involved in the proliferation of HCC cells, we took advantage of a well-characterized TPA (12-O-tetradecanoylphorbol 13-acetate)-induced growth arrest model of cultured HepG2 cells (27). Cultured HepG2 cells were treated with 100 nM of TPA for 3 days to induce growth arrest, and the expression patterns of 270 miRNAs in the TPA-treated HepG2 cells were compared against dimethyl sulfoxide (DMSO)-treated cells (Figure 2B). Using a fold-change ≥3 as the cutoff, we identified 15 upregulated and nice downmodulated miRNAs in TPA-arrested HepG2 cells (Supplementary Table S3).
We reasoned that a proliferation-related miRNA would display an inverse expression pattern in uncontrolled growing HCC tissues and growth-arrested HepG2 cells. Therefore, we examined the miRNA expression patterns to locate miRNAs whose expression was inversely altered in the HCC samples and in the TPA-treated HepG2 cells (Figure 2C). We identified five miRNAs, including miR-22, miR-100, miR-101, miR-139-5p and miR-199a-3p, whose expression levels were upmodulated by TPA treatment and downmodulated in HCC samples. In addition, we also identified miR-10b, whose expression was upmodulated in HCC samples but downmodulated in HepG2 cells upon TPA-induced growth arrest. These six inversely expressed miRNAs were selected as proliferation-related miRNA candidates for subsequent studies (Table 1).
Table 1.
MicroRNAs inversely altered in HCC tissues and TPA-treated HepG2 cells
| microRNA name | Chromosomal location | Tissue miRNA levelsa |
Fold-change (T versus N) | Paired t-test P-value | HepG2 miRNA levelsa |
Fold change (TPA/DMSO) | Predicted targets | ||
|---|---|---|---|---|---|---|---|---|---|
| Normal | Tumor | DMSO | TPA | ||||||
| miR-139-5p | 11q13.4 | 12.73 ± 0.47 | 10.32 ± 1.27 | −5.35 | 1.54 × 10−7 | 6.72 | 9.25 | 5.79 | 617 |
| miR-199a-3p | 1q24.3, 19p13.2 | 10.83 ± 1.22 | 7.63 ± 2.29 | −9.16 | 9.02 × 10−6 | 4.65 | 7.02 | 5.17 | 531 |
| miR-101 | 1p31.3, 9p24.1 | 6.07 ± 0.84 | 4.98 ± 0.90 | −2.13 | 1.48 × 10−5 | 5.54 | 7.28 | 3.35 | 1004 |
| miR-10b | 2q31.1 | 5.21 ± 0.73 | 7.20 ± 1.53 | 3.98 | 5.57 × 10−5 | 11.10 | 9.13 | −3.92 | 329 |
| miR-100 | 11q24.1 | 12.47 ± 0.83 | 10.39 ± 2.90 | −4.22 | 5.94 × 10−3 | 3.90 | 7.74 | 14.36 | 39 |
| miR-22 | 17p13.3 | 12.45 ± 1.53 | 11.41 ± 1.92 | −2.06 | 7.08 × 10−3 | 6.50 | 8.09 | 3.00 | 459 |
amiRNA levels expressed as 39-Ct, Tissue miRNA levels expressed as mean ± SD.
Functional classification of DE protein-coding genes in HCC
To identify candidate genes regulated by proliferation-related miRNAs, we analyzed the expression patterns of protein-coding genes in three additional pairs of normal and HCC tissues using microarrays and identified a total of 1648 DE genes, including 1136 upmodulated and 512 downmodulated genes in the HCC samples (Supplementary Table S4). Unsupervised hierarchical clustering analysis using these DE genes clearly separated the HCC tissues from the normal liver tissue samples (Figure 3A). To explore how these DE genes reflect the underlying biological processes differentiating HCC from normal hepatocytes, we used GO terms to annotate the biological processes associated with these DE genes and then calculated their functional similarity based on the semantic similarity of their GO terms using the GOSim package (16). These annotated DE genes were then separated into multiple functional classes based on their functional similarity using the k-means clustering method. A total of 1104 DE genes were annotated with GO biological processes, and a matrix of similarity distance was generated for k-means clustering. To determine the optimal number for a functional cluster, the k-means clustering was performed for cluster numbers from 3 to 20, and the DB value was calculated for each cluster number. The lowest DB value was detected when the cluster number was set at 6 (Figure 3B). These results suggest that the annotated DE genes in HCC cells can be grouped into six major functional clusters. A Principle Component Analysis using similarity distance confirmed that the annotated DE genes within each cluster were closely associated with each other, and all six clusters were separated with little or no overlap (Figure 3C). These six functional clusters comprised genes coding for protein transporters (c1), immune response (c2), cell-cycle regulation (c3), signal transduction (c4), protein translation (c5) and regulation of transcription (c6). For each functional cluster, the cluster size, enrichment statistics and the most informative GO terms are summarized in Table 2.
Figure 3.

Functional clustering of genes DE in HCC tissues. (A) Unsupervised hierarchical clustering of 1648 genes DE in normal liver tissue samples and HCC samples. (B) Distribution of DB values of the k-means clustering of the functional similarity of DE genes. (C) Principal component analysis of the functional clusters generated by k-means clustering method based on the similarity distance calculated by GOSim.
Table 2.
Functional classification of differentially expressed genes in HCC
| Cluster name | No. of genes | No. of enriched GO termsa | Enrichment P-valuesb | Biological function |
|---|---|---|---|---|
| C1 | 98 | 37 (109) | 1.29 × 10−4 | GO:0015031∼protein transport |
| GO:0045184∼establishment of protein localization | ||||
| GO:0055085∼transmembrane transport | ||||
| GO:0046907∼intracellular transport | ||||
| GO:0016192∼vesicle-mediated transport | ||||
| C2 | 166 | 36 (127) | 1.45 × 10−2 | GO:0002526∼acute inflammatory response |
| GO:0042158∼lipoprotein biosynthetic process | ||||
| GO:0006954∼inflammatory response | ||||
| GO:0042157∼lipoprotein metabolic process | ||||
| GO:0007596∼blood coagulation | ||||
| C3 | 81 | 97 (138) | 2.63 × 10−9 | GO:0000087∼M phase of mitotic cell cycle |
| GO:0000280∼nuclear division | ||||
| GO:0007067∼mitosis | ||||
| GO:0051726∼regulation of cell cycle | ||||
| GO:0000075∼cell cycle checkpoint | ||||
| C4 | 242 | 284 (459) | 1.07 × 10−3 | GO:0007165∼signal transduction |
| GO:0007242∼intracellular signaling cascade | ||||
| GO:0007243∼protein kinase cascade | ||||
| GO:0006915∼apoptosis | ||||
| GO:0042981∼regulation of apoptosis | ||||
| C5 | 208 | 84 (135) | 5.89 × 10−4 | GO:0006412∼translation |
| GO:0006397∼mRNA processing | ||||
| GO:0008380∼RNA splicing | ||||
| GO:0019752∼carboxylic acid metabolic process | ||||
| GO:0010608∼posttranscriptional regulation of gene expression | ||||
| C6 | 306 | 68 (119) | 2.04 × 10−12 | GO:0045449∼regulation of transcription |
| GO:0010468∼regulation of gene expression | ||||
| GO:0031323∼regulation of cellular metabolic process | ||||
| GO:0006325∼chromatin organization | ||||
| GO:0016568∼chromatin modification |
aTerms with enrichment P-value < 0.05; total number of GO terms are shown in parenthesis.
bEnrichment P-value calculated using DAVID.
Functional clusters significantly and specifically enriched by targets of proliferation-related miRNAs
To understand the relationship between proliferation-related miRNAs and the DE functional cluster in HCC cells, we predicted the target spectrum of these proliferation-related miRNAs and assessed which functional cluster was enriched with their putative targets. The targets for proliferation-related miRNAs were predicted using the TargetScan algorithm, and high-efficacy targets were selected by context score filtering (≤−0.2) (5). A total of 2954 unique targets were predicted for the six proliferation-related miRNAs. The extent of miRNA target enrichment in each functional cluster was analyzed using Fisher’s exact test (Table 3). A total of 248 predicted targets for all six proliferation-related miRNAs were mapped to the HCC DE genes, with a highly significant enrichment P-value (P-value = 1.85 × 10−8). The targets of three proliferation-related miRNAs, including miR-101, miR-139-5p and miR-199a-3p, were found to be significantly enriched in the regulation of a transcription cluster (c6) with P-values of 0.001, 0.006 and 0.015, respectively. In addition, targets of miR-139-5p were found enriched in immune response cluster (c2, P-value = 0.029) and targets of miR-199a-3p were found enriched in the signal transduction cluster (c4, P-value = 0.018). To our surprise, genes in the cell-cycle control cluster (c3) were not significantly enriched by targets of any proliferation-related miRNA, implying that genes involved in cell-cycle progression are not the primary targets for proliferation-related miRNAs.
Table 3.
Enrichment of miRNA targets in functional clusters
| miRNA name | Predicted target | Enrichment of miRNA targets in clustera |
||||||
|---|---|---|---|---|---|---|---|---|
| C1 (98) | C2 (166) | C3 (81) | C4 (242) | C5 (208) | C6 (306) | C1–C6 (1104) | ||
| miR-100 | 39 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| miR-101 | 1004 | 6 | 14 | 3 | 18 | 10 | 31*** | 82*** |
| miR-10b | 329 | 2 | 2 | 2 | 7 | 4 | 4 | 21 |
| miR-139-5p | 617 | 6 | 11* | 4 | 13 | 8 | 20** | 62*** |
| miR-22 | 531 | 3 | 6 | 3 | 7 | 4 | 11 | 34 |
| miR-199a-3p | 459 | 0 | 5 | 4 | 14* | 8 | 17* | 48*** |
| all 6 miRs | 2954 | 17 | 38* | 16 | 59** | 34 | 84*** | 248*** |
aP-value calculated by Fisher’s exact test.
*P-value < 0.05; **P-value < 0.01; ***P-value < 0.005.
The size of these six functional clusters ranged from 81 to 306 genes, with c6 being the largest cluster. It is possible that the difference in cluster size may affect the results of the enrichment analysis. To rule out the potential biases caused by cluster size, we calculated the baseline enrichment score for each functional cluster by randomly selecting six miRNAs whose expression level was not significantly altered in HCC cells and conducted target prediction and target enrichment analysis for these six functional clusters. Baseline enrichment fraction and enrichment score were estimated from 10 such random sets to avoid potential bias generated by miRNA selection. The details on the selected miRNAs, predicted targets and the enrichment score in each functional cluster for these 10 random sets used to derive the baseline estimate are provided in Supplementary Table S5. Both the enrichment fraction and enrichment score of the proliferation-related miRNA targets were significantly higher in the c6 cluster (one sample t-test P-values < 0.0001, Figure 4A and B) compared with the baseline level, implying that the proliferation-related miRNAs exert their biological function by targeting genes in the transcription regulation (c6) cluster. These results are consistent with a previous report that indicates that miRNAs preferentially target nuclear proteins, most of which are transcription factors (9).
Figure 4.

Enrichment of individual functional clusters with targets of proliferation-related miRNAs and randomly selected miRNAs. (A) Number of proliferation-related miRNAs (GR-miRs) and numbers of randomly selected miRNAs (Random-miRs) with targets significantly enriched in individual functional clusters. Target enrichment analysis was performed using a one-sided Fisher’s exact test, and the threshold for significantly enriched miRNA was P < 0.05. Data for random miRs are presented as the average ± SD from ten sets of randomly selected miRNAs. (B) Aggregate enrichment score in individual functional cluster by proliferation-related miRNAs (GR-miRs) and randomly selected miRNAs (Random-miRs). Enrichment scores for individual miRNA are calculated as −log(P-value) where the P-value was calculated by Fisher’s exact test. Data for random miRs are presented as the average ± SD from ten sets of randomly selected miRNAs. For both 4A and B, the comparisons between GR-miRs and Random-miRs were performed using one-sample t-tests (two-tailed). *P < 0.05; ***P < 0.0001.
Building a miRNA-regulated network based on interactome and identifying critical nodes and hubs
To understand how these three proliferation-related miRNAs, miR-101, miR-139-5p and miR-199a-3p, regulate genes in the cluster c6, we generated an interactome network to connect these three miRNAs and all the genes in the c6 cluster. The network was modeled using the shortest path algorithm in GeneGO, with a maximum allowance of two intermediate nodes. The resulting network contained 811 nodes connected by 2975 links (Figure 5A). To assess the contribution of individual nodes to the stability of the modeled network, topological properties, including the degree of connection and Betweenness, were calculated and ranked for each node. Nodes with a high (top 5%) degree of connection were considered hubs, and nodes with high (top 5%) Betweenness were defined as bottlenecks (Table 4). Among the 31 ‘hub-bottleneck’ nodes, >50% were transcription factors, for example, FOS, EGR1, HNF4A, MYC, SP1, NFYA, LEF1 and TP53. Interestingly, the highest ranked node (both of Betweenness and degree of connection), FOS, is targeted by all three miRNAs (Table 4). The remaining ‘hub-bottleneck’ nodes included signaling molecules (CDKN2A, ERK1/2, MAP3K1 and MTOR) and genes involved in epigenetic regulation (EZH2, SUZ12). In addition to the protein-coding genes, the three miRNAs, miR-101, miR-139-5p and miR-199a-3p, also exhibited a high-ranking ‘hub-bottleneck’ property in the network. All three miRNAs were ranked within the top 3% in Betweenness and degree of connection among the 811 nodes in the network, suggesting that these miRNAs play critical roles in stabilizing the c6 cluster network connections (Table 4). Among these three miRNAs, miR-101 is the node with the highest ranking in both Betweenness and degree of connection.
Figure 5.

microRNA-centered interactome network from cluster 6. Nodes with a top 5% Betweenness value from cluster 6 interactome and miR-101, miR-139-5p and miR-199a-3p are highlighted. The distribution of nodes was based on the Betweenness values rather than the regulatory level of the interactions. The higher the rank percentile, the more centered the node. The entire regulatory network (811 nodes and 2975 edges) is shown in (A). (B–D) depicts the miR-101-, miR-139-5p- and miR-199a-3p-centered subnetworks, respectively. The subnetworks for each specific miRNA were constructed by a two-step shortest path of regulatory interactions. There are 382 nodes and 552 edges in the miR-101-centered subnetwork (B), 287 nodes and 354 edges in the miR-139-5p-centered subnetwork (C) and 142 nodes and 166 edges in miR-199a-3p-centered subnetwork (D). Genes are represented by nodes and functional associations by edges.
Table 4.
Topological properties of the top 5% objects in cluster C6 network
| GeneGO object name | Mappedgene symbol | Molecular function | DEGs | Predicted targetsa | miRNA subnetworkb | Betweenness |
No. of interactions (degree) |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Value | Rank* (%) | Total | In | Out | Rankc (%) | ||||||
| AP-1 | FOS | TF | 101, 139-5p, 199-3p | 101, 139-5p, 199a-3p | 249208.61 | 0.14 | 291 | 112 | 179 | 0.14 | |
| EGR1 | EGR1 | TF | Yes | 101, 139-5p, 199a-3p | 72295.33 | 0.27 | 125 | 32 | 93 | 0.28 | |
| miR-101 | miR-101 | 56924.00 | 0.41 | 77 | 4 | 73 | 0.42 | ||||
| HNF4-alpha | HNF4A | TF | 101, 139-5p | 44757.16 | 0.54 | 69 | 8 | 61 | 0.56 | ||
| miR-139-5p | miR-139-5p | 30350.00 | 0.68 | 50 | 1 | 49 | 0.99 | ||||
| c-Myc | MYC | TF | 101, 139-5p, 199a-3p | 26864.92 | 0.82 | 63 | 25 | 38 | 1.41 | ||
| SP1/SP3 complex | SP1 | TF | 199-3p | 101, 139-5p, 199a-3p | 26196.43 | 0.95 | 70 | 14 | 56 | 0.70 | |
| NF-Y | NFYA | 199a-3p | 25680.85 | 1.09 | 63 | 12 | 51 | 0.85 | |||
| Tcf(Lef) | LEF1 | TF | 101, 139-5p, 199a-3p | 22192.08 | 1.22 | 59 | 24 | 35 | 1.55 | ||
| ESR1 (nuclear) | ESR1 | TF | 101, 139-5p, 199a-3p | 21792.08 | 1.36 | 54 | 22 | 32 | 1.83 | ||
| p53 | TP53 | TF | 101, 139-5p, 199a-3p | 21048.43 | 1.50 | 57 | 28 | 29 | 2.11 | ||
| IGF-1 | IGF-1 | 101, 139-5p, 199a-3p | 20782.87 | 1.63 | 46 | 36 | 10 | 7.46 | |||
| KLF4 | KLF4 | TF | Yes | 101, 139-5p | 20556.33 | 1.77 | 55 | 25 | 30 | 1.97 | |
| C/EBPdelta | CEBPD | TF | Yes | 101, 139-5p, 199a-3p | 19309.54 | 1.90 | 61 | 36 | 25 | 2.54 | |
| miR-199a-3p | miR-199a-3p | 101 | 18887.15 | 2.04 | 55 | 13 | 42 | 1.13 | |||
| NF-kB | RELA | TF | 101, 139-5p | 17556.05 | 2.18 | 41 | 15 | 26 | 2.39 | ||
| ERK1/2 | ERK1/2 | 16240.25 | 2.31 | 27 | 13 | 14 | 4.93 | ||||
| EZH2 | EZH2 | EPIGENES | Yes | 101 | 101, 139-5p, 199a-3p | 15792.27 | 2.45 | 53 | 13 | 40 | 1.27 |
| Androgen receptor | AR | TF | 101, 139-5p | 13974.19 | 2.59 | 40 | 20 | 20 | 3.24 | ||
| JAK2 | JAK2 | 101 | 101, 139-5p | 13876.60 | 2.72 | 26 | 11 | 15 | 4.65 | ||
| c-Src | SRC | 101, 139-5p | 13570.23 | 2.86 | 22 | 13 | 9 | 9.01 | |||
| MEKK1(MAP3K1) | MAP3K1 | 12517.24 | 2.99 | 10 | 7 | 3 | 24.08 | ||||
| N-CoR | NCOR1 | TF | Yes | 101, 139-5p | 12444.68 | 3.13 | 47 | 14 | 33 | 1.69 | |
| STAT3 | STAT3 | TF | 101, 139-5p | 12201.45 | 3.27 | 28 | 12 | 16 | 4.08 | ||
| p16INK4 | CDKN2A | Yes | 101, 139-5p, 199a-3p | 11640.78 | 3.40 | 45 | 41 | 4 | 18.17 | ||
| mTOR | MTOR | Yes | 101, 139-5p | 9691.05 | 3.54 | 30 | 20 | 10 | 7.46 | ||
| SUZ12 | SUZ12 | EPIGENES | Yes | 199a-3p | 9046.93 | 3.67 | 40 | 12 | 28 | 2.25 | |
| Caspase-3 | CASP3 | 101, 139-5p | 101, 139-5p | 8983.41 | 3.81 | 12 | 3 | 9 | 9.01 | ||
| IRF8 | IRF8 | TF | Yes | 8469.37 | 3.95 | 35 | 12 | 23 | 2.82 | ||
| Ubiquitin | UBB | 101, 139-5p, 199a-3p | 8159.22 | 4.08 | 31 | 9 | 22 | 2.96 | |||
| LRH1 | NR5A2 | TF | Yes | 139-5p | 101, 139-5p | 7736.99 | 4.22 | 38 | 27 | 11 | 6.34 |
| ATF-3 | ATF3 | TF | Yes | 101, 139-5p, 199a-3p | 7376.23 | 4.35 | 39 | 22 | 17 | 3.94 | |
| Cathepsin L | Cathepsin L | 101, 139-5p, 199a-3p | 7358.72 | 4.49 | 26 | 22 | 4 | 18.17 | |||
| ZNF143 | ZNF143 | TF | Yes | 101 | 101 | 7337.00 | 4.63 | 15 | 5 | 10 | 7.46 |
aPredicted by TargetScan and context score <−0.2.
bThe components in specific miRNA-subnetworks.
cRank percentile.
The miRNA-regulated subnetwork exhibits a two-layer regulatory architecture
To further understand the regulatory architecture and the biological function of these proliferation-related miRNAs, we extracted the subnetwork regulated by individual miRNAs and evaluated their topological properties (Supplementary Tables S6–S9). Notably, when the miRNA-regulated subnets were plotted based on the topological ranking of individual nodes, all three miRNA-regulated subnets exhibited a two-layer regulatory architecture, with multiple transcriptional regulators clustered in the inner layer, while genes with other molecular functions were scattered in the outer layer. The subnetwork regulated by miR-101 consisted of 381 nodes connected by 552 links (Figure 5B). The high-ranking nodes, which are with higher Betweenness value, in the miR-101 subnet included multiple transcription factors (FOS, SP1, LEF1, MYC, EGR1, TP53, STAT3, NFYA, RELA, ZNF143 and CEBPD), epigenetic modulators (EZH2, SUZ12 and NCoR) and signaling molecules (MTOR, JAK2, ERK1/2, CDKN2A and IGF1). Some of these top-ranking nodes, including FOS, ZNF143 and EZH2, were predicted targets of miR-101. These high-ranking nodes were clustered in the inner layer of the miR-101 subnet. Notably, miR-199a-3p was also a high-ranking node within the miR-101 subnet. miR-199a-3p itself also regulated a subnet with 142 nodes connected by 166 links with multiple high-ranking nodes including transcription factors (FOS, SP1, MYC, EGR1, LEF1, NFYA, TP53, CEBPD and ESR1), epigenetic modulators (EZH2, SUZ12) and signaling molecules (IGF1 ad CDKN2A) (Figure 5C). Similarly, the miR-139-5p subnet contained 287 nodes connected by 354 links (Figure 5D). The top-ranking nodes of the subnet included several transcription factors (FOS, LEF1, SP1, MYC, HNF4A, EGR1, CEBPD, RELA, ESR1, ATF3, STAT3 and TP53), epigenetic modulator (EZH2) and signaling molecules (MTOR, SRC, JAK2, CDKN2A and IGF1).
We chose miR-101 among the three candidate miRNAs for further characterization based on the following considerations. First, miR-101 was the highest ranking miRNA in the c6 interactome network (Table 4), and the targets of miR-101 were found to be enriched only in C6 (Table 3), suggesting that miR-101 may preferentially regulate genes in C6 cluster. Second, in our interaction network, miR-101 also regulates the expression of another highly ranked miRNA, miR-199a-3p, through the epigenetic modulation (28). Third, while miR-101 has been found to promote serum starvation-induced apoptosis in HCC cells by targeting Mcl-1 (29), a miR-101-regulated growth control network has not been investigated.
To confirm the validity of our miR-101 regulatory network, we manually examined the interactions between the highest ranked transcriptional factor (FOS) and epigenetic modulator (EZH2) and their down-stream targets listed in MetaCore database. In the miR-101 regulatory subnetwork, FOS contains 56 out-going interactions which are experimentally supported in 174 different publications (Supplementary Table S10). Similarly, the 51 out-going interactions from EZH2 within the miR-101 subnetwork were also experimentally supported by 32 unique studies (Supplementary Table S11). These data suggest that miR-101 may exert a broad effect on gene expression by targeting these critical transcriptional modulators.
To further confirm the two-layer architecture of miRNA regulatory network, we selected the miR-101-FOS-TGFB1 circuit (Figure 6A) for experimental validation. TGF-β-signaling pathway exhibited the highest fold-enrichment score in the pathway enrichment analysis of miR-101 second neighbors in both KEGG and BioCarta pathway databases (Supplementary Table S12). Indeed, overexpression of miR-101 in HepG2 cells suppressed the FOS mRNA level up to 75% (Figure 6B) and reduced the mRNA level of TGF-β1 up to 70% (Figure 6C), confirming the regulatory effect of miR-101 on FOS and TGF-β 1. Overexpressing miR-101 in HepG2 cells caused a 50% reduction in the FOS 3′-UTR reporter activity but showed no effect on TGFB1 3′-UTR reporter activity (Figure 6D), demonstrating that FOS is a direct target of miR-101 whereas TGFB1 is a second neighbor. These results agree with the TargetScan prediction which found a high efficacy miR-101-binding site on the 3′-UTR of FOS (Supplementary Figure S2A) but no miR-101-binding site on TGFB1 3′-UTR. To further confirm the two-layer regulatory circuit, we constructed a reporter containing the TGFB1 promter and analyzed the reporter activity in HepG2 cells upon miR-101 overexpression. The TGFB1 promoter contains two AP1-binding sites and can be activated by TPA treatment in HepG2 cells (Supplementary Figure S2C). As shown in Figure 6E, overexpressing miR-101 reduced the promoter activity of TGFB1 by 36%. These data support the two-layer regulatory mechanism in the miR-101-FOS-TGFB1 circuit.
Figure 6.

miR-101 regulates expression level of TGFB1 through AP-1. (A) The proposed miR-101-to-FOS-to-TGFB1 regulatory circuit. (B) Effect of miR-101 on FOS mRNA level. Cultured HepG2 were transfected with 15 nM of miR-101 or scrambled control for 48 h. FOS expression level was determined using qRT-PCR. ***P < 0.001. (C) Effect of miR-101 on TGFB1 expression level. Cultured HepG2 were transfected with 15 nM of miR-101 or scrambled control for 48 h. TGFB1 expression level was determined using qRT–PCR. ***P < 0.001. (D) Effect of miR-101 on FOS 3′-UTR and TGFB1 3′-UTR reporter activity. Cultured HepG2 were transfected with 15 nM of miR-101 or scrambled control for 24 h. Cells were cotransfected with plasmids containing firefly luciferase reporter carrying FOS 3′-UTR or TGFB1 3′-UTR and control Relina luciferase and incubated for additional 24 h. Luciferase activity was determined as described in materials and methods. The firefly reporter activity was normalized to the Relina luciferase activity. Data are means ± SEM of three independent experiments each analyzed in duplicates. *P < 0.05. (E) Effect of miR-101 on TGFB1 promoter activity. Cultured HepG2 were transfected with 15 nM of miR-101 or scrambled control for 24 h. Cells were cotransfected with plasmids containing firefly luciferase fused to TGFB1 promoter and control Relina luciferase and incubated for additional 24 h. Cells were treated with DMSO or TPA (100 nM) for 6 h in a fresh culture medium before the luciferase activity assay. TGFB1 promoter activity was normalized to the Relina luciferase activity. Data are means ± SEM of three independent experiments each analyzed in duplicates. *P < 0.05.
miR-101-regulated multiple cell-cycle-related genes in HCC cells
To understand the molecular mechanism of the miR-101-regulated growth control, we analyzed 297 nodes in the second layer of the miR-101 subnet for pathway enrichment using the KEGG and BioCarta databases along with the online bioinformatics service tool DAVID. A total of 162 nodes were mapped in the KEGG pathway database, and 144 nodes were mapped in the BioCarta pathway database (Supplementary Table S10). The most significantly regulated pathway in the KEGG database was the cell-cycle pathway (P-value = 3.28 × 10−12, fold-enrichment = 6.03), followed by the TGF-β-signaling pathway (p-value = 1.01 × 10−9, fold-enrichment = 6.49), MAPK-signaling pathway, multiple growth factor-mediated signaling pathways, cell adhesion pathways, immune response pathways and apoptosis pathways. Similarly, the most significantly regulated pathway in the BioCarta database was the G1/S check point in cell cycle (P-value = 1.06 × 10−7, fold-enrichment = 5.16), followed by the p38 MAPK-signaling pathway and the TGF-β-signaling pathway in addition to multiple growth and differentiation-related pathways. These data revealed that the secondary nodes regulated by miR-101 are highly enriched with genes involved in cell growth and proliferation, particularly effector genes involved in cell-cycle progression (Figure 7A).
Figure 7.

miR-101 regulates multiple cell-cycle-related genes and controls the growth of HCC cells. (A) Distribution of objects from the miR-101-regulated sub-network in the KEGG Cell-cycle pathway. The genes labeled with grey color indicate the nodes in miR-101-regulated subnetwork. (B–C) Expression levels of genes that are the first-layered regulators (B) and second-layered effectors, which are cell-cycle-related genes (C) of the cell cycle in HepG2 cells transfected with negative control and miR-101. Total RNA was obtained 48 h after transfection, and mRNA measurements were performed in duplicate. Data are presented as the means ± SEM of three independent experiments. (D) Effect of miR-101 on growth of HepG2 cells. HepG2 cells were transfected with control or miR-101 and maintained for 2 and 4 days, respectively. The cells were fixed and stained with DAPI. The cell number was determined using an InCell 1000 imaging system. Data are the average of nine fields. (E) Effects of miR-101 on colony-forming activity of cultured HepG2 cells. Cells were transfected with control or miR-101 and plated at 2000 cells/well in six-well plates for 14 days. After staining with crystal violet, the colony number and size were determined using Image J.
miR-101 has been shown to promote apoptosis in serum-starved HepG2 cells by targeting the Mcl-1 gene. However, the effect of miR-101 on cell growth and proliferation has not been explored in HCC cells. To test the effect of miR-101, synthetic miR-101 was transfected into cultured HepG2 cells and cell proliferation was evaluated by cell counting and colony formation assay under regular culture condition. Compared with a scrambled control, HepG2 cells overexpressing miR-101 demonstrated a significant reduction in cell number and colony counts (Figure 7B, C and Supplementary Figure S3). To confirm the miR-101-regulated subnet, we analyzed the expression levels of multiple nodes in the first and second layer in cultured HepG2 cells overexpressing miR-101. As shown in Figure 7D, overexpression of miR-101 significantly suppressed the expression levels of nodes in the first layer, including multiple transcription factors (FOS, EGR1, SP1, CEBPD and RELA) and epigenetic modulators (EZH2 and SUZ12). Similarly, the expression levels of second layer targets, including multiple G1-phase regulators (CCNE1 and CDK4) and S-phase regulators (MCM family), were also reduced in miR-101 expressing HepG2 cells (Figure 7E). In addition, the second layer target, cyclin-dependent kinase inhibitor CDKN1B, was upregulated in HepG2 cells overexpressing miR-101 (Figure 7E). The results indicated that miR-101 is a proliferation-related miRNA that regulates cell growth and proliferation by concordantly modulating the expression of multiple transcription regulators and cell-cycle related genes in HCC.
DISCUSSION
In this study, we combined computational and biochemical approaches to identify proliferation-related miRNAs and the biological networks regulated by these miRNAs in HCC cells. By comparing the miRNA expression patterns in clinical HCC samples and miRNAs differentially expressed in growth-arrested HepG2 cells, we identified six candidates as proliferation-related miRNAs in HCC. As these proliferation-related miRNAs are differentially expressed in HCC tissues, their targets and associated pathways were expected to present corresponding alterations in HCC cells. Through functional clustering using GoSim, we identified six major functional categories, including ‘cell-cycle control’ and ‘transcriptional regulation’ in the protein-coding genes differentially expressed in HCC tissues. To explore the relationship between these proliferation-related miRNAs and the DE functional categories in HCC, we analyzed the enrichment of miRNA targets in each functional cluster. Interestingly, none of the six miRNAs presented significant target enrichment in the ‘cell-cycle control’ functional cluster. Instead, the targets of three miRNAs, miR-101, miR-199a-3p and miR-139-5p, were significantly enriched in the ‘transcription regulation’ functional cluster. These three proliferation-related miRNAs identified in this study have also been found downmodulated in HCC samples when compared with adjacent benign tissues (25).
To understand how these three miRNAs may regulate proliferation of HCC cells through ‘transcription control’, we built an interactome network with the three miRNAs and all genes in the ‘transcriptional control’ cluster using the shortest-path algorithm. Topological analysis revealed that all three miRNAs were highly connected hubs in the network, with the ‘Betweenness’ and ‘degree of connection’ all ranked in the top 3% of the entire network. More importantly, all three miRNA-centered subnetworks displayed characteristics of a two-layer regulatory architecture, with multiple transcription factors and epigenetic modulators as the first neighbors and genes involved in other functions as the second neighbors. Analysis of the miR-101-regulated subnetwork revealed that nodes in the second neighbors are enriched with genes involved in growth control, including multiple genes in cell-cycle progression. Overexpression of miR-101 reduced the expression of multiple genes involved in G1- and S-phase transition and suppressed proliferation and colony formation of HepG2 cells. In addition, the expression levels of multiple transcription regulators were significantly altered upon miR-101 overexpression. These data indicate that miR-101 regulates cell growth by directly targeting transcription regulators rather than targeting genes involved in cell-cycle progression. Our observation implies that miR-101 uses both mechanisms; targeting critical hubs in the network and coregulating multiple genes in the same pathway to achieve growth control in HCC. It is possible that such a combined regulatory mechanism is widely used by miRNAs to regulate critical biological processes.
Previously, most studies tend to attribute the miRNA-mediated biological effect to a single or few selected targets whose levels were dramatically modulated by the miRNA. However, recent large-scale transcriptomics and proteomic studies clearly demonstrate that individual miRNAs may alter the expression of hundreds to thousands of genes at both the transcript and protein levels. Except for a few targets, the impact of miRNAs on the expression levels of most targets is usually very subtle. However, a miRNA may still exert profound biological impacts when its targets are mapped to the same pathways. Therefore, several computational approaches have been developed to integrate target prediction and pathway analysis in order to infer the biological functions of miRNA. The most widely used approach is to predict targets for miRNAs of interest and then deduce biological function by performing pathway enrichment analysis on the predicted targets. This approach is straightforward, but the deduced biological functions may not be biologically relevant if a significant portion of the predicted targets is not expressed by the cells under investigation. The second approach is to identify predicted miRNA targets which show inverse expression patterns with the miRNA of interest in matching samples. Biological functions of miRNA under investigation were then inferred from these inversely expressed targets. Due to the subtle effect of miRNAs on their targets, the number of inversely correlated targets could be limited and thus makes the functional interpretation more difficult. In this study, we built the regulatory network using the interactome data from the GeneGO database to include both physical and regulatory interactions, thus allowing transcriptional regulation to be included in the network. As a result, the interactions between the miRNAs and transcriptional regulators can be thoroughly examined, and the topological weight of direct and indirect neighbors can be simultaneously evaluated. The resulting two-layer regulatory network reveals a concerted interaction of miRNAs and transcription regulators in regulating critical cellular function such as cell-cycle progression. Such intricate interactions would have been obscured using the protein–protein interactome alone.
In our study, several genes involved in proliferation and cell-cycle progression were found differentially expressed in HCC cells, and these genes were grouped into functional cluster 3. Most of these genes were catalytic enzymes required for an orderly progression of cell division. Interestingly, this cluster was not enriched by targets of proliferation-related miRNAs, suggesting that these catalytic molecules are not the main targets of proliferation-related miRNAs. On the other hand, HCC differentially expressed transcription regulators in functional cluster 6 were enriched with targets of proliferation-related miRNAs, suggesting that these differentially expressed transcription regulators are the primary targets of miRNAs. More importantly, downstream effectors of these transcription regulators are enriched with genes involved in cell-cycle control, suggesting that modulating these transcriptional regulators can significantly amplify the effect of a single miRNA through the activity of transcriptional control. Thus, the observed two-layer regulatory network explains how miRNA can induce a dramatic phenotype change by causing a collection of slight but concordant changes in target genes.
Aberrant expression of miRNAs has been observed in various pathological conditions, including in most types of cancer. The three proliferation-related miRNAs identified in this study include miR-101, miR-139-5p and miR-199a. All three miRNAs have been found to be downmodulated in a previous study using HCC samples from a different geographical location (25), suggesting that these miRNAs play important role in regulating the growth of HCC cells regardless of the cancer etiology. It is possible that these miRNAs also regulate proliferation of other cell types through the two-layer regulatory mechanism, and downmodulation of these miRNAs may contribute to uncontrolled proliferation in other cancer types. Indeed, miR-101 have also been found downmodulated in lung cancer (30), glioblastoma (31) and gastric cancer (32). Similarly, miR-199 have been found to be downmodulated in human parathyroid carcinomas (33), oral squamous cell carcinoma (34) gastric cancer (35), prostate cancer (36) and serous ovarian cancer (37), while reduced level of miR-139 have been found in head and neck cancer (38), lung cancer (30), colorectal cancer (39), oral squamous cell carcinoma (40) and pancreatic cancer (41). Previous studies have shown that miR-101, miR-139 and miR-199 directly target c-fos and c-Myc, two well-characterized oncogenic transcription factors involved in various types of malignant disorders (42). The loss of these proliferation-related miRNAs could result in the upregulation of these oncogenes and lead to the uncontrolled growth of tumor cells.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–12 and Supplementary Figures 1–3.
FUNDING
Chang Gung Memorial Hospital [CMRPD-190412 to C.K.C.]; National Science Council of Taiwan, ROC [NSC99-2311-B182-004-MY3 to C.K.C., NSC-99-2320-B182-016-MY3 to S.J.C.]; Ministry of Education, ROC to Chang Gung University. Funding for open access charge: National Science Council of Taiwan, ROC [NSC99-2311-B182-004-MY3 to C.K.C.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Ka-Wai Leong and Yu-Ting Hong for excellent technical assistance on the reporter assays.
REFERENCES
- 1.Bar M, Wyman SK, Fritz BR, Qi J, Garg KS, Parkin RK, Kroh EM, Bendoraite A, Mitchell PS, Nelson AM, et al. MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells. 2008;26:2496–2505. doi: 10.1634/stemcells.2008-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ren J, Jin P, Wang E, Marincola FM, Stroncek DF. MicroRNA and gene expression patterns in the differentiation of human embryonic stem cells. J. Transl. Med. 2009; 7:20. doi: 10.1186/1479-5876-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calin G, Sevignani C, Dumitru C, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl Acad. Sci. USA. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Q, Fu H, Sun F, Zhang H, Tie Y, Zhu J, Xing R, Sun Z, Zheng X. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 2008;36:5391–5404. doi: 10.1093/nar/gkn522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cloonan N, Brown MK, Steptoe AL, Wani S, Chan W, Forrest AR, Kolle G, Gabrielli B, Grimmond SM. The miR-17-5p microRNA is a key regulator of the G1/S phase cell cycle transition. Genome Biol. 2008;9:R127. doi: 10.1186/gb-2008-9-8-r127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui Q, Yu Z, Purisima EO, Wang E. Principles of microRNA regulation of a human cellular signaling network. Mol. Syst. Biol. 2006;2:46. doi: 10.1038/msb4100089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tu K, Yu H, Hua Y-J, Li Y-Y, Liu L, Xie L, Li Y-X. Combinatorial network of primary and secondary microRNA-driven regulatory mechanisms. Nucleic Acids Res. 2009;37:5969–5980. doi: 10.1093/nar/gkp638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hendrickson DG, Hogan DJ, McCullough HL, Myers JW, Herschlag D, Ferrell JE, Brown PO. Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS Biol. 2009;7:e1000238. doi: 10.1371/journal.pbio.1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 15.Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, Wilson M, Wang X, Shelton J, Shingara J, et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007;67:7713–7722. doi: 10.1158/0008-5472.CAN-07-1083. [DOI] [PubMed] [Google Scholar]
- 16.Frohlich H, Speer N, Poustka A, Beissbarth T. GOSim–an R-package for computation of information theoretic GO similarities between terms and gene products. BMC Bioinformatics. 2007;8:166. doi: 10.1186/1471-2105-8-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H-C, Chen G-H, Chen Y-H, Liao W-L, Liu C-Y, Chang K-P, Chang Y-S, Chen S-J. MicroRNA deregulation and pathway alterations in nasopharyngeal carcinoma. Br. J. Cancer. 2009;100:1002–1011. doi: 10.1038/sj.bjc.6604948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang JJ, Conrath DW. In Proceedings of International Conference Research on Computational Linguistics (ROCLING X) 1997. Semantic Similarity Based on Corpus Statistics and Lexical Taxonomy. Taiwan. [Google Scholar]
- 19.Davies DL, Bouldin DW. A cluster separation measure. IEEE Trans. Pattern Anal. Mach. Intell. PAMI-1, 224–227. [PubMed] [Google Scholar]
- 20.Scardoni G, Petterlini M, Laudanna C. Analyzing biological network parameters with CentiScaPe. Bioinformatics. 2009;25:2857–2859. doi: 10.1093/bioinformatics/btp517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 22.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esquela-Kerscher A, Slack F. Oncomirs - microRNAs with a role in cancer. Nat. Rev. Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 24.Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H, Okanoue T, Shimotohno K. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene. 2006;25:2537–2545. doi: 10.1038/sj.onc.1209283. [DOI] [PubMed] [Google Scholar]
- 25.Jiang J, Gusev Y, Aderca I, Mettler TA, Nagorney DM, Brackett DJ, Roberts LR, Schmittgen TD. Association of MicroRNA expression in hepatocellular carcinomas with hepatitis infection, cirrhosis, and patient survival. Clin. Cancer Res. 2008;14:419–427. doi: 10.1158/1078-0432.CCR-07-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X-M. MicroRNA signatures in liver diseases. World J. Gastroenterol. 2009;15:1665–1672. doi: 10.3748/wjg.15.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiang C-W, Huang Y, Leong K-W, Chen L-C, Chen H-C, Chen S-J, Chou C-K. PKCalpha mediated induction of miR-101 in human hepatoma HepG2 cells. J. Biomed. Sci. 2010; 17:35. doi: 10.1186/1423-0127-17-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong Q, Qin L, Wu X, Zheng Y, Yang Y, et al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell. 2011;19:232–243. doi: 10.1016/j.ccr.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Su H, Yang J, Xu T, Huang J, Xu L, Yuan Y, Zhuang S. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res. 2009;69:1135–1142. doi: 10.1158/0008-5472.CAN-08-2886. [DOI] [PubMed] [Google Scholar]
- 30.Mascaux C, Laes JF, Anthoine G, Haller A, Ninane V, Burny A, Sculier JP. Evolution of microRNA expression during human bronchial squamous carcinogenesis. Eur. Respir. J. 2009;33:352–359. doi: 10.1183/09031936.00084108. [DOI] [PubMed] [Google Scholar]
- 31.Smits M, Nilsson J, Mir SE, van der Stoop PM, Hulleman E, Niers JM, de Witt Hamer PC, Marquez VE, Cloos J, Krichevsky AM, et al. miR-101 is down-regulated in glioblastoma resulting in EZH2-induced proliferation, migration, and angiogenesis. Oncotarget. 2010;1:710–720. doi: 10.18632/oncotarget.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H-J, Ruan H-J, He X-J, Ma Y-Y, Jiang X-T, Xia Y-J, Ye Z-Y, Tao H-Q. MicroRNA-101 is down-regulated in gastric cancer and involved in cell migration and invasion. Eur. J. Cancer. 2010;46:2295–2303. doi: 10.1016/j.ejca.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 33.Corbetta S, Vaira V, Guarnieri V, Scillitani A, Eller-Vainicher C, Ferrero S, Vicentini L, Chiodini I, Bisceglia M, Beck-Peccoz P, et al. Differential expression of microRNAs in human parathyroid carcinomas compared with normal parathyroid tissue. Endocr. Rel. Cancer. 2010;17:135–146. doi: 10.1677/ERC-09-0134. [DOI] [PubMed] [Google Scholar]
- 34.Yu T, Wang X-Y, Gong R-G, Li A, Yang S, Cao Y-T, Wen Y-M, Wang C-M, Yi X-Z. The expression profile of microRNAs in a model of 7,12-dimethyl-benz[a]anthrance-induced oral carcinogenesis in Syrian hamster. J. Exp. Clin. Cancer Res. 2009;28:64. doi: 10.1186/1756-9966-28-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song G, Zeng H, Li J, Xiao L, He Y, Tang Y, Li Y. miR-199a regulates the tumor suppressor mitogen-activated protein kinase kinase kinase 11 in gastric cancer. Biol. Pharm. Bull. 2010;33:1822–1827. doi: 10.1248/bpb.33.1822. [DOI] [PubMed] [Google Scholar]
- 36.Porkka KP, Pfeiffer MJ, Waltering KK, Vessella RL, Tammela TLJ, Visakorpi T. MicroRNA expression profiling in prostate cancer. Cancer Res. 2007;67:6130–6135. doi: 10.1158/0008-5472.CAN-07-0533. [DOI] [PubMed] [Google Scholar]
- 37.Nam EJ, Yoon H, Kim SW, Kim H, Kim YT, Kim JH, Kim JW, Kim S. MicroRNA expression profiles in serous ovarian carcinoma. Clin. Cancer Res. 2008;14:2690–2695. doi: 10.1158/1078-0432.CCR-07-1731. [DOI] [PubMed] [Google Scholar]
- 38.Wong TS, Liu XB, Wong BYH, Ng RWM, Yuen APW, Wei WI. Mature miR-184 as potential oncogenic microRNA of squamous cell carcinoma of tongue. Clin. Cancer Res. 2008;14:2588–2592. doi: 10.1158/1078-0432.CCR-07-0666. [DOI] [PubMed] [Google Scholar]
- 39.Arndt GM, Dossey L, Cullen LM, Lai A, Druker R, Eisbacher M, Zhang C, Tran N, Fan H, Retzlaff K, et al. Characterization of global microRNA expression reveals oncogenic potential of miR-145 in metastatic colorectal cancer. BMC Cancer. 2009;9:374. doi: 10.1186/1471-2407-9-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kozaki KI, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68:2094–2105. doi: 10.1158/0008-5472.CAN-07-5194. [DOI] [PubMed] [Google Scholar]
- 41.Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner MR, Frankel WL, Morgan DL, Postier RG, Brackett DJ, Schmittgen TD. Expression profiling identifies microRNA signature in pancreatic cancer. Int. J. Cancer. 2007;120:1046–1054. doi: 10.1002/ijc.22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li S, Fu H, Wang Y, Tie Y, Xing R, Zhu J, Sun Z, Wei L, Zheng X. MicroRNA-101 regulates expression of the v-fos FBJ murine osteosarcoma viral oncogene homolog (FOS) oncogene in human hepatocellular carcinoma. Hepatology. 2009;49:1194–1202. doi: 10.1002/hep.22757. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.