

Abstract
Although T cells can mediate potent antitumor responses, immune tolerance mechanisms often result in the deletion or inactivation of T cells that express T-cell receptors (TCRs) against potentially effective target epitopes. Various approaches have been devised to circumvent this problem. In one approach, the gene encoding an antibody against a cancer-associated antigen is linked, in the form of a single-chain variable fragment (scFv), to genes that encode transmembrane and signaling domains. This chimeric antigen receptor (CAR) is then introduced into T cells for adoptive T-cell therapy. In another approach, the anti-cancer scFv is fused to a scFv that binds to the CD3ε subunit of the TCR/CD3 complex. This fusion protein serves as a soluble, injectable product that has recently been termed bispecific T-cell engager (BiTE). Both strategies have now been tested in clinical trials with promising results, but the comparative efficacies are not known. Here, we performed a direct comparison of the in vitro sensitivity of each strategy, using the same anti-cancer scFv fragments, directed against a tumor-specific glycopeptide epitope on the sialomucin-like transmembrane glycoprotein OTS8, which results form a cancer-specific mutation of Cosmc. While both approaches showed specific responses to the epitope as revealed by T cell-mediated cytokine release and target cell lysis, CAR-targeted T cells were more sensitive than BiTE-targeted T cells to low numbers of antigens per cell. The sensitivity scale described here provides a guide to the potential use of these two different approaches.
Keywords: bispecific T-cell engager (BiTE), chimeric antigen receptors (CARs), gene-modified adoptive T-cell transfer, T-cell tumor therapy, tumor-specific epitope
Introduction
Tumor cells express various proteins and epitopes on their surface that differentiate them from healthy cells, either by levels of expression or by revealing novel epitopes not seen in normal “self.” Hence, it is possible for the adaptive immune system to target these cells (reviewed in Refs. 1–3). Antibodies against tumor-associated epitopes, which are limited to antigens presented on the cell surface of tumors, have been identified and exploited against multiple types of cancers using passive immunization.4 Notable examples include rituximab (anti-CD20 for B-cell lymphomas5) and trastuzumab (anti-HER-2/neu for certain breast cancers6). Therapeutic antibodies have had success against tumors, eliciting both complement-mediated responses and antibody-dependent cellular cytotoxicity (ADCC). However, administration of an anti-cancer antibody as a monotherapy is rare, and these are often combined with more traditional chemotherapy (reviewed in ref. 4).
It is known that T cells are capable of inducing anti-tumor responses that are quite potent. However, those T cells that would most efficiently respond to peptide-MHC epitopes on the surface of tumors are often subjected to clonal tolerance or deletion, as many of these epitopes are very similar or identical to self epitopes. T-cell epitopes, recognized by clonotypic T-cell receptors (TCRs), are also sometimes compromised due to downregulation of class I MHC, dysfunction of antigen processing in the tumor,7-9 poor binding of the antigenic peptide to the MHC,10 and anergy or tolerance of T cells which recognize the complex.11 The issue of anergy or tolerance can in part be addressed by removal of tumor-infiltrating lymphocytes (TILs) and conditioning in vitro before re-introduction into a patient, giving objective responses in some cases.12,13 Further efforts with adoptive T-cell therapy has involved genetic modification of T cells in vitro by introduction of TCRs against tumor-associated T-cell epitopes.14-16 This strategy has shown promise, but various challenges surrounding T-cell epitopes in general, as well as potential mispairing of introduced TCR with endogenous TCR, remain (reviewed in ref. 17). The latter problem results in a reduction in the expression level of the introduced TCR,18 and may drive potentially harmful off-target reactions.19
To most efficiently harness the power of T cells in the fight against tumors, several methods have been designed that allow T cells to respond to traditional antibody epitopes. Chimeric antigen receptors (CARs), consisting of extracellular antibody fragments directed against a tumor epitope fused to intracellular T-cell signaling domains, have been transduced into T cells, endowing them with a novel specificity toward a non-MHC-restricted epitope.20 The most common CAR formats currently being evaluated include a scFv targeting domain linked to a spacer, transmembrane domain, and intracellular domains from the T-cell receptor CD3ζ subunit and co-stimulatory domains, such as CD28, OX40, or 4–1BB.21 CAR-based strategies continue to be pursued against a number of tumor-associated epitopes, including CD19.22 Results from recent clinical trials demonstrate the effectiveness of CAR-transduced T cells targeted against the B cell epitope CD19 in achieving long-term remission from refractory chronic lymphocytic leukemia (CLL) when transferred as a monotherapy following lymphodepleting chemotherapy.23,24
Another strategy to target T cells to precise antibody epitopes takes advantage of a long-studied type of molecule called “bispecific antibody,”25,26 which links an anti-cancer antibody with an antibody recognizing CD3 subunits. These have recently been termed BiTEs (bispecific T-cell engagers27). A single-chain variable fragment (scFv) that binds a tumor epitope is linked to a second scFv that binds an invariant portion of the T-cell receptor complex, resulting in activation and targeting of effector T cells against the tumor epitope, regardless of the TCR-mediated specificity of the T cells. Evidence shows that these reagents are considerably more potent than antibodies against tumor cells alone.28 BiTEs have been constructed targeting more than ten tumor-associated epitopes, including blinatumomab against CD19 (for B cell leukemias),29 and MT-110 against EpCAM (for various adenocarcinomas and cancer stem cells),30 both being currently evaluated in clinical trials. High response rates for relapse-free survival and elimination of minimal residual disease were found in refractory acute lymphoblastic leukemia (ALL) patients receiving blinatumomab in clinical trials.31
While both of these strategies have shown promising results, it is not yet clear under what conditions the CAR approach vs. the BiTE approach might be preferred. In order to begin to explore this important question, here we have compared both systems using the same scFv (which hence targets the same epitope). We have constructed a CAR and a BiTE, both with the specificity of the tumor-specific antibody 237, a tumor-specific glycopeptide epitope on a murine fibrosarcoma that spontaneously arose in an old mouse.32 The epitope includes an altered glycosylation on Thr77 of the sialomucin-like transmembrane protein OTS833 resulting from a mutation in the gene coding for the Core 1 β3Gal-T–specific molecular chaperone (Cosmc). Overexpression of the wild-type Cosmc leads to complete disappearance of the epitope. The structure of this IgG2a antibody has been solved in complex with the glycopeptide that constitutes the minimal epitope, and its affinity against this glycopeptide has been measured as 140 nM.34
Our side-by-side comparisons of the BiTE and CAR strategies revealed the sensitivity of each approach for cytokine or cytotoxic responses by CD4+ and CD8+ T cells to cells expressing different levels of the 237 epitope, based on the number of 237 antibodies detected by staining at high concentration. While specific, sensitive responses were seen for both approaches, in all cases, CAR-expressing T cells were sensitive to lower numbers of 237 epitopes per cell than T cells targeted by BiTEs. For example, CAR-directed CTLs were capable of lysing Jurkat T cells that had the lowest detectable levels of antigen. The results provide a guide to the appropriate use of CARs or BiTEs toward recruiting an effective T-cell response, taking into consideration the expression profile and densities of a given tumor epitope in cancerous compared with normal cells.
Results
Characterization of target cells expressing the 237 epitope
The monoclonal antibody 237 was identified as a cancer-specific antibody after immunization of mice with the spontaneous murine fibrosarcoma cell line Ag104A.32 To compare the relative sensitivities of T cells expressing CARs or targeted by BiTEs, we took advantage of three transformed target cell lines, each of which exist in a 237high and a 237low or 237low/int state, exposing a range of target epitopes per cell (Fig. 1). Ag104A cells express both the OTS8 transmembrane protein and a mutated Cosmc chaperone, resulting in naturally high levels of expression of the 237 epitope.32,33 A Cosmc rescue mutant of this line (ACosmc) expresses nearly undetectable levels of this epitope, even when stained at high levels of antibody (Fig. 1A and B). The Neuro2A line is derived from a murine neuroblastoma that has a truncation mutation in its Cosmc chaperone, and expresses the Tn antigen, but it does not express the OTS8 membrane protein.33,35 The 237 epitope is not detected on Neuro2A cells at saturating 237 mAb levels; however, transduction of Neuro2A with the OTS8 gene (Neuro2A/OTS8) resulted in high levels of the 237 epitope expression (Fig. 1A and B).

Figure 1. Characterization of 237 antibody binding to target cells. (A) 5-fold dilutions of 237 antibody starting from 1 mg/mL were used to stain Ag104A (top panel), ACosmc (second panel), Neuro2A (third panel), Neuro2A+OTS8 (fourth panel), Jurkat (fifth panel), and JOE (bottom panel) cells. The shaded peak in each panel represents staining by the secondary detecting antibody only. (B) Plot of the median fluorescence values (top panel), and the fold-increase over staining with secondary only (bottom panel) for Ag104A (closed diamonds), ACosmc (open diamonds), Neuro2A (open circles), Neuro2A+OTS8 (closed circles), JOE (closed triangles), and Jurkat (open triangles) cells. (C) Quantification of the number of 237 antibodies bound per target cell when stained at 1 mg/mL.
The third cell type that we examined was the human lymphoblastoid T-cell line Jurkat (reviewed in ref. 36). Jurkat cells have a truncation mutation in the Cosmc chaperone and express Tn antigens,37 but do not express murine OTS8. Despite this, there was low level of binding to these cells at the highest antibody concentrations (Fig. 1A and B). The detection only at the highest antibody concentrations suggests that the affinity of the 237:epitope interaction is considerably lower, perhaps because the structure of the neoantigen differs from that of the glycopeptides found on Ag104A cells.34 Consistent with this notion, when Jurkat cells were transduced with the murine OTS8 protein (Jurkat with OTS8 Epitope, or JOE), the maximal binding and binding curves were very similar to those obtained with murine Ag104A and Neuro2A/OTS8 cells.
To obtain a more quantitative view of the levels of 237 antibody binding on these cells, we compared staining levels at 1mg/mL 237 antibody to calibrated IgG2a-binding beads, both detected with the same secondary antibody. This allowed for the quantification of the number of epitopes seen on the surface of Ag104A cells as roughly 14,000 (Fig. S1). By comparison to Ag104A and the standards, approximate 237 epitope detection levels were established for each line (Fig. 1C). Collectively, the these target cell lines provided a spectrum of antigen densities to associate with effector T-cell responses elicited using targeting strategies with BiTEs or CARs.
Design and characterization of 237 BiTE
The bispecific antibody fusion consisted of the 145–2C11 scFv (VH-linker-VL, that binds to the CD3 ε chain in the TCR complex),38 here called 2C11, linked to the 237 scFv (VL-linker-VH). The gene encoding these two scFvs, joined by a flexible, soluble linker, was expressed as a single polypeptide in stably transfected human embryonic kidney 293F cells (Fig. 2A). A C-terminal His6 tag was included to facilitate purification by binding to a nickel-NTA resin. The resulting protein (referred to as the 237 BiTE) was further purified by size exclusion chromatography, yielding a single band on SDS-PAGE (Fig. 2B).

Figure 2. Design and characterization of 237 BiTE. (A) Schematic diagram of the mature 237 BiTE, expressed in HEK-293F cells. (B) SDS-PAGE gel of purified 237 BiTE protein. (C) Stimulation of interleukin-2 (IL-2) release from a TCR+ 58−/− T-cell hybridoma when stimulated by immobilized 2C11 antibody or immobilized 237 BiTE. (D) Stimulation of target cell lysis by activated 2C T cells with various concentrations of 237 BiTE for Ag104A (closed diamonds) and ACosmc (open diamonds). Error bars represent the standard deviation of triplicate measurements .
To examine the functional capacity of the anti-CD3 fragment of the purified 237 BiTE, the fusion was immobilized on tissue culture plates. The full-length 145–2C11 antibody was immobilized for comparison, and TCR+ T-cell hybridoma cells were added to examine stimulation as monitored by interleukine 2 (IL-2) release. Immobilized 237 BiTE induced a similar maximum level of activation as the 2C11 antibody (Fig. 2C), indicating that the 2C11 scFv fragment of the BiTE was properly folded and functional.
To further test the full functionality of the 237 BiTE, ex vivo-stimulated 2C effector T cells were co-cultured with 51Cr-labeled Ag104A or ACosmc target cells, and the BiTE was titrated in at various levels. Specific lysis of Ag104A, but not ACosmc, cells was seen for BiTE concentrations as low as 1nM (Fig. 2D). Thus, the 237 BiTE was able to specifically redirect T cells against targets that expressed the 237 epitope.
Design and characterization of 237 CAR
A CAR containing the 237 scFv (here called 237 CAR) was also constructed to evaluate its ability to redirect the specificity and activity of T cells against targets that expressed the 237 epitope. The 237 CAR consisted of the 237 scFv (VL-linker-VH) linked in frame to the CD8 hinge domain, CD28 transmembrane and intracellular signaling domains, and CD3 ζ intracellular signaling domain (Fig. 3A). This construct was cloned into the pMP71 retroviral vector for transduction into activated T cells.
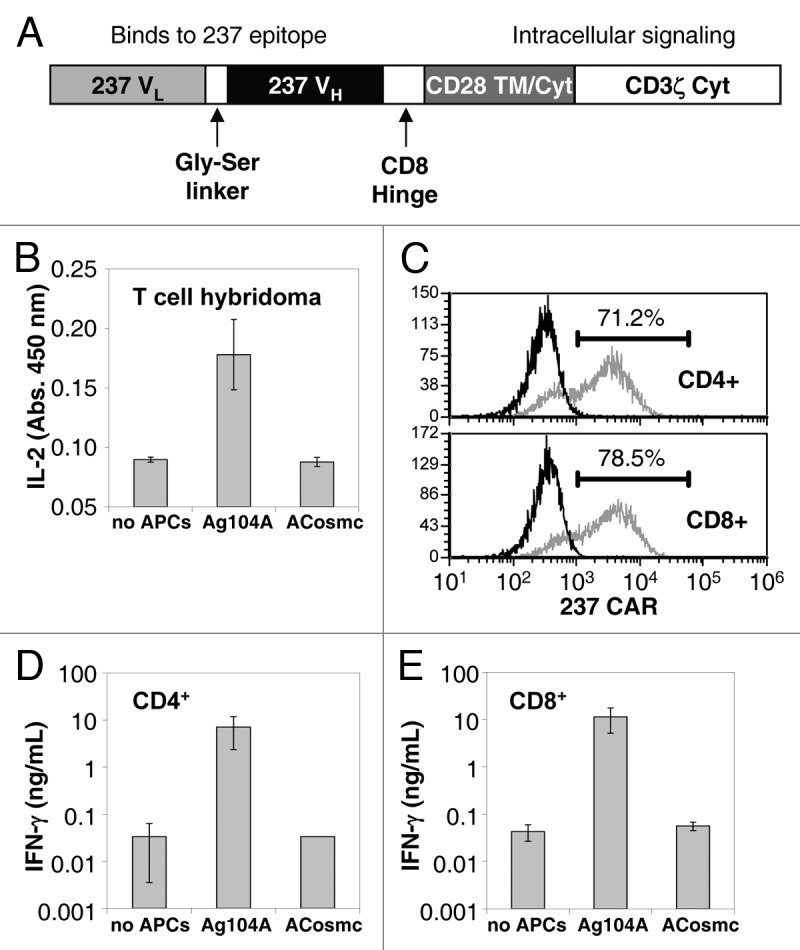
Figure 3. Design and characterization of 237 CAR. (A) Schematic diagram of the mature 237 CAR, expressed on the surface of T cells or T-cell hybridomas. (B) Stimulation of interleukin-2 (IL-2) release by 58−/− T-cell hybridomas (co-receptor negative) transduced with 237 CAR and co-cultured with Ag104A or ACosmc cells. (C) Transduction efficiencies of activated polyclonal T cells that were purified for CD4+ (top panel) or CD8+ (bottom panel). Efficiencies ranged from 40 to 80%. (D-E) Stimulation of polyclonal effector T cells transduced with 237 CAR and co-cultured with Ag104A or ACosmc cells. Interferon γ (IFNγ) release was detected for CD4+ (D) and CD8+ (E) T cells. Error bars represent the standard deviation of triplicate measurements.
To evaluate the expression and function of the 237 CAR, the construct was packaged and transduced into the αβ TCR-negative T-cell hybridoma line 58−/−. Expression was verified by detection with polyclonal goat anti-mouse antibodies that cross-react with the 237 variable regions. The CAR-positive population was enriched by fluorescence-activated cell sorting (data not shown). To examine their function and specificity, the CAR-positive T-cell hybridomas were incubated with Ag104A or ACosmc overnight and IL-2 secretion was measured (Fig. 3B). Only the Ag104A cell line was capable of inducing IL-2 secretion by CAR-transduced T-cell hybridomas.
To determine if normal, activated CD4+ and CD8+ T cells could be transduced with the 237 CAR for redirecting activity against Ag104A, splenocytes from C57 mice were activated in vitro with anti-CD3/anti-CD28 beads. A day later, CD4+ and CD8+ cells were isolated and transduced with the 237 CAR. The efficiency of transduction, as measured with the anti-mouse Ig reagent, ranged from 40 to 80% (Fig. 3C). CAR-transduced and mock-transduced CD4+ and CD8+ populations were incubated with the Ag104A or ACosmc target cells, and the concentration of interferon γ (IFNγ) in supernatants was measured one day later (Fig. 3D, E). As with the T-cell hybridoma line, only the Ag104A line stimulated release of IFNγ, and target cells were capable of stimulating cells in a co-receptor independent process since both the CD4+ and CD8+ populations were positive for IFNγ release.
Cytokine release stimulated by target cells with various levels of antigen
To further explore the specificity of the two approaches, ex vivo activated CD4+ and CD8+ T cells were used either as CAR-transduced effector cells, or in combination with the 237 BiTE (tested at 100 nM), together with the characterized antigen-presenting cell lines (Fig. 1). T cells were co-cultured with antigen-presenting cells at a 1:1 ratio for 24 h, and supernatants were analyzed for IFNγ levels (Fig. 4). In line with the the results reported above, both 237 CAR-transduced T cells (Fig. 4A) and the 237 BiTE-targeted T cells (Fig. 4D) were able to specifically recognize Ag104A (about 14,000 epitopes/cell) but not ACosmc (< 300 epitopes/cell) cells. This was true for both CD8+ and CD4+ T cells.

Figure 4. Cytokine release stimulated by 237 targeting strategies. Interferon γ (IFNγ) was detected in the supernatants of 237 CAR- or Mock-transduced T cells (A-C), or T cells treated or not with 100 nM 2C11:237 BiTE (D-F); each co-cultured with murine fibrosarcoma Ag104A (237high, open bars) or ACosmc (237low, shaded bars) cells (A,D); murine neuroblastoma Neuro2A+OTS8 (237high, open bars) or Neuro2A (237low, shaded bars) cells (B,E); human JOE (Jurkat+OTS8, 237high, open bars) or Jurkat (237low-int, shaded bars) cells (C,F). In each panel, CD8+ T-cell responses are shown on the left and CD4+ T-cell responses of are shown on the right. Samples were set up in triplicate with error bars indicating standard deviation. Data shows representative results from at least 3 independent experiments. Unpaired, two-tailed Student’s t-test was used to calculate p values.
Responses to the mouse Neuro2A (< 300 epitopes/cell) and Neuro2A/OTS8 (~30,000 epitopes/cell) cell lines were consistent with the mouse Ag104A system. Thus, both the CAR and BiTE approaches showed specificity in that only the antigen-positive Neuro2A/OTS8 cells stimulated IFNγ release (Fig. 4B and E). CAR-transduced cells showed comparable release of IFNγ by both the CD8+ and CD4+ T cells, BiTE-transduced signals were more efficient in CD8+ than in CD4+ T cells. The reduced stimulation of BiTE-targeted CD4+ T cells by Neuro2A/OTS8 cells was somewhat unexpected, compared with the Ag104A result (Fig. 4B) as the Neuro2A/OTS8 cell line expresses more epitopes (~30,000/cell) that Ag104A (~14,000/cell). As other factors such as the LFA-1/ICAM binding can influence the T-cells response, it is possible that lower levels of ICAM in Neuro2A/OTS8 cells, as compared with Ag104 cells, may account for this difference.
To compare the CAR and BiTE systems further, we used human Jurkat (~770 epitopes/cell) and JOE (~9,900 epitopes/cell) cells as antigen-presenting cells (Fig. 4C and F). In this setting, the JOE cell line was able to specifically stimulate IFNγ release from both CAR-expressing CD8+ and CD4+ T cells. In contrast, the 237 BiTE was unable to stimulate both CD8+ and CD4+ T cells upon exposure to JOE or Jurkat cells. The reduced stimulation seen with the human target cell lines might also be related to the LFA-1/ICAM interaction, as it is known that human ICAM does not work as efficiently with mouse LFA-1 as does mouse ICAM.39
Target cell lysis by 237 CAR-expressing and BiTE-targeted T cells
Effector T cells targeted by either the 237 CAR or the 237 BiTE were also evaluated for their ability to lyse target cells in a 51Cr-release assay, as cytotoxicity has been reported to be among the most sensitive activities of T cells. Indeed, in the case of CAR-transduced CD8+ and CD4+ T cells, a 5:1 ratio of effector:target (E:T) yielded significant lysis of not only JOE target cells but also Jurkat cells that expressed less than 1,000 epitopes per cell (Fig. 5A). While it appears surprising that there is no discrimination of 237 CAR-expressing T cells to target cells with a very large difference in epitope levels (over 12-fold), these results are consistent with previous results showing saturation of CAR-targeted lysis across a wide range of E:T ratios,40 and a diminished effect of varying epitope levels per target above a threshold, even at low E:T ratios.41 The 237 BiTE was capable of mediating the lysis of JOE target cells by both CD8+ and CD4+ T cells at a 5:1 E:T ratio (Fig. 5B). However, BiTE-targeted cells mediated considerably lower levels of Jurkat cell lysis than CAR-expressing cells, consistent with the generally lower sensitivity of the BiTE approach.

Figure 5. Specific lysis of human target cells stimulated by 237 targeting strategies. Lysis of human JOE (Jurkat+OTS8, 237high, open bars), or Jurkat (237low-int, shaded bars)cells, by T cells that were transduced with the 237 CAR or subjected to mock transduction (A), or incubated with or without 100 nM 237 BiTE (B). In each panel, CD8+ T-cell responses are shown on the left and CD4+ T-cell responses of are shown on the right. Lysis was measured by 51Cr release into the culture medium. Triplicate samples were measured, and error bars represent standard deviation. Results are typical for at least three independent experiments. Unpaired, two-tailed Student’s t-test was used to calculate p values.
Specific lysis of Ag104A and Neuro2A cells was also observed (Fig. S2), although the level of lysis was reproducibly low for both these target cell lines. The lower level of lysis could be overcome by using activated 2C TCR transgenic T cells that had been stimulated in vitro by specific peptide and IL-2 (Fig. 2D). While the reduced lysis appears to be in part due to the polyclonal effector cells, it is unclear what accounts for their ability to lyse JOE and Jurkat cells efficiently. It is worth noting that Jurkat cells are non-adherent and thus do not require trypsinization, whereas both Ag104A and Neuro2A cell lines require trypsinization. It is therefore possible that this affects an adhesion interaction that is important for the polyclonal T cells. The low level of lysis seems unlikely to be mediated by PD-1/PD-L1 interaction, since the polyclonal effector T cells are negative for PD-1 by flow cytometry (data not shown). As a matter of fact, significant levels of PD-1 can be seen on the 2C transgenic effector cells (data not shown), which lyse the Ag104A cells efficiently (Fig. 2D).
Discussion
Based on the results of the activation assays, combined with 237 epitope quantification on the various target cells tested, a sensitivity scale for the two approachs (CAR and BiTE) was constructed. The scale indicates the ranges of epitopes/cell over which particular responses might be expected from targeting by these T-cell-directed strategies (Fig. 6). The x-axis shows the number of epitopes per cell, from high levels on the left (predicted to give maximal responses) to low levels on the right. For each activation response (T-cell type, and targeting strategy), a bar stretches from high numbers of epitopes per cell to the approximate level where that response is no longer observed, at the lower epitope densities.
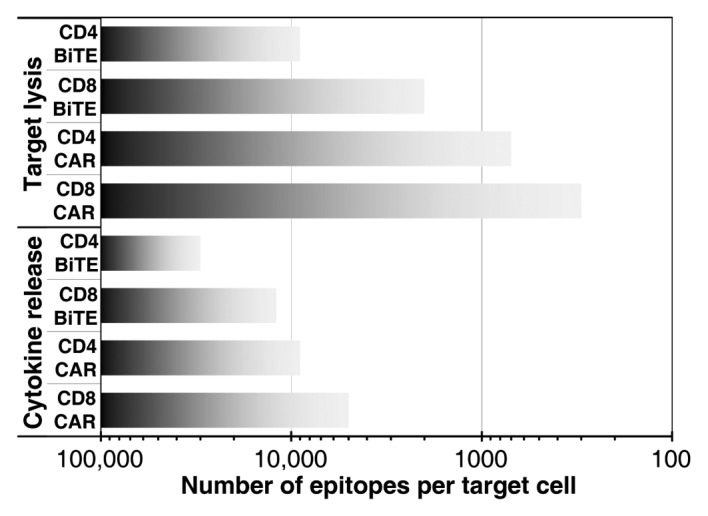
Figure 6. Sensitivity scale for T-cell targeting strategies by number of epitopes per target cell. The approximate range over which each response is expected for CD8+ or CD4+ T cells expressing CARs or upon the use of BiTEs is shown with shaded bars.
As can be seen in Figure 6, target cell lysis occurs for all targeted T cells at lower levels of epitope than IFNγ secretion. Likewise, CD8+ T cells are more sensitive than CD4+ T cells for the same targeting strategy and activation response. Finally, it can be seen that cells expressing the 237 CAR are more sensitive to lower levels of antigen than cells treated with the 237 BiTE. Of course, the specific antigen levels described relate to this particular epitope/scFv pair, the interaction of which exhibits an affinity for the glycopeptide of 140 nM.34 It is unknown if the affinity of the 237 antibody might be higher for the native epitope. Nevertheless, two previous studies examined the impact of affinity by comparing CARs constructed with scFv fragments that had a wide range of affinities (over 1000-fold).42,43 Interestingly, the impact of affinity on T cell targeting efficiency was minimal, whereas the effects of antigen density and CAR density were critical. Additional experimental results and mathematical models of CAR/antigen interactions with various affinities support this relationship between antigen levels on target cells and expression levels of CARs both influencing the response to targets.41,44,45
There are a few reports on the effects on BiTE efficiency, as related to tumor target affinity. In the case of clinical targeting of CD19, both the CAR and BiTE strategies appear to use scFv fragments with affinities well below 10 nM (0.4 nM for the FMC63 antibody for the CAR,24,46 and 0.7 nM for the HD37 antibody for the MT-103 BiTE blinatumomab).28,29 The relative sensitivity trends exhibited here would be expected to be maintained for most epitopes, especially for scFv fragments with KD values > 10 nM. Nevertheless, it is also clear that the targeting sensitivity of the 237 BiTE is likely to be considerably greater than for effector functions associated with conventional, unarmed antibody (e.g., see epitope density discussion, below). Furthermore, it is also quite possible that the in vivo sensitivity and efficacy of a BiTE might benefit from the pharmacokinetic properties that drive the removal of unbound forms of the BiTE. For example, the ability of a T cell to actively carry the monovalent-bound BiTE to a tumor may be critically important for activity. In the in vitro assays, such monovalent-bound forms could actually act as inhibitors of the active form which requires simultaneous binding of both cancer antigen and T cell. These issues remain to be examined in the 237 system.
Quantitative measurements of epitopes per cancer cell are rarely performed. However, for the well-studied epitopes CD19 and CD20, which are expressed by both normal and malignant C cells, several measurements exist. In normal B cell, CD19, which is clinically targeted by both CAR-expressing and BiTE-treated T cells,24,47 has been reported to be at levels of ~20,000–30,000 epitopes per cell, while CD20 has been reported to be at ~40,000–160,000 epitopes per cell.48-51 Interestingly, in B-cell neoplasms, the levels of both epitopes are often lower: CD19 has been reported at ~5,400–13,300/cell and CD20 at ~10,500–30,500/cell.51 Downregulation of these epitopes on tumor cells makes selective targeting of transformed vs. normal B cells difficult. In B cell malignancies, maximal reactivity against the tumor cells is desired and depletion of normal B cells along with malignant cells can be tolerated. For the epidermal growth factor receptor 1 (EGFR, or ERBB1), one study found the average number of epitopes per cell to be downregulated from ~30,700 on normal epithelial cells to ~14,700 on cervical squamous cell and adenocarcinomas. This situation was further complicated as 63% of tumors had downregulated the epitope while 10% had upregulated it.52
Another tumor-associated epitope, the epidermal growth factor receptor 2 (HER2, or ERBB2), is normally expressed by multiple tissues, including respiratory, gastrointestinal, urogenital, and cardiac cancers. However, tumors of various types can experience up to a 40-fold increase in expression, from ~50,000 epitopes per normal cell to ~2,000,000 epitopes per transformed cell (reviewed in ref. 53). Overexpression occurs in 15–30% of breast cancers, where enhanced expression is correlated with worse prognosis. Successful treatments with the anti-HER2 antibody trastuzumab have been developed, although eventually resistance occurs. There have been some efforts to direct T cells to HER2 in the clinic, including the trifunctional antibody ertumaxomab, which binds HER2 and CD3, and contains a mouse-rat hybrid Fcγ domain, shown to promote targeting to both T cells and FcγR-expressing cells.54 Promising results from a Phase I clinical trial showed that also patients with low levels of HER2 expression responded to the drug, rather than only those with very high levels (which respond to trastuzumab).55,56 Thus, the therapeutic window accessible by soluble T-cell targeting molecules was expanded beyond single monoclonal antibodies for this epitope by taking advantage of the enhanced sensitivity of T cells. However, the apparently enhanced sensitivity to this epitope deriving from the CAR approach resulted in a serious adverse effect, leading to death of a patient associated with recognition of normal levels of HER2 in the lung, presumably activating ultra-sensitive T cells.57
While the levels of epitopes on normal and tumor tissues should be an important factor in the decision to use monoclonal antibodies, BiTEs, or CARs, there are other factors to be considered. For example, BiTE therapy seeks to redirect the activity of resident T cells in a patient, some of which may be tolerized or exhausted under the conditions of the tumor microenvironment.58,59 Conversely, adoptively transferred CAR-expressing T cells are conditioned in vitro, away from tolerizing influences, and could be further transformed to deliver immunostimulatory factors upon activation to counteract tumor-mediated immunosuppression.60 Methods to achieve optimal persistence of CAR-expressing T cells remain to be determined (reviewed in ref. 61), while safety checks such as the inclusion of suicide genes62 are also being explored to control potential harmful effects. BiTEs provide a conventional drug approach in terms of storage, delivery, and dosage; however, due to generally short half-lives in vivo (2–3 h for blinatumomab47) compared with full antibodies, they must be delivered frequently or by continuous infusion pumps.
In summary, CAR-expressing T cells can provide greater sensitively than BiTE-treated T cells for a given epitope (Fig. 6). Accordingly, CARs might be considered when epitope densities are low, or when downregulation of targeted epitopes is known to limit effectiveness. The approach might be particularly effective for truly tumor-specific epitopes, with an expression that is exquisitely restricted to transformed cells, as it is thought to be the case for 237,33 or when the destruction of normal cells expressing the antigen can be tolerated, as it is the case for CD19. Conversely, BiTE-based approaches may be best used in scenarios where a particular therapeutic window is available to select between normal and malignant cells. For example, BiTEs may be beneficial in tumors in which a particular epitope is overexpressed compared with normal tissue, or when there may be cross-reactivity to similar lower density epitopes on normal tissues.
Materials and Methods
Antibodies
The monoclonal antibody 237 is an IgG2a secreted by a hybridoma derived from a C3H/HeN mouse that had been immunized with irradiated Ag104A tumor cells,32,33 and was purified from mouse ascites fluid. The monoclonal antibody 145–2C11 was purified from hybridoma culture supernatant by ammonium sulfate precipitation and protein A affinity chromatography. Anti-TCR Cβ:phycoerythrin (Cat. # 561081), anti-CD8α:Alexa 647 (Cat. # 557682), and anti-CD4:Alexa 647 (Cat. # 557681) antibodies were purchased from BD PharMingen, and Alexa 647 goat F(ab’)2 anti-mouse IgG (H+L) was purchased from Invitrogen (Cat. # A21237).
Cell lines and culture conditions
Ag104A, a spontaneous fibrosarcoma isolated from an aging mouse (C3H/HeN),32 its wild-type Cosmc chaperone mutant ACosmc,33 Neuro2A, a murine neuroblastoma,35 and its OTS8-trandsuced line Neuro2A+OTS833 were maintained in complete DMEM media (supplemented with 10% fetal calf serum, L-glutamine, penicillin, and streptomycin, and β mercaptoethanol) and passaged by trypsinizing from tissue culture plastic; T-cell hybridoma 58−/−,63 Jurkat E6–1, a human T-cell leukemia line,36 and the OTS8-transduced line JOE were maintained in suspension in complete RPMI 1640 media. Plat-E64 (Cell Biolabs, Inc., Cat. # RV-101) retroviral packaging cell line was maintained in complete DMEM media supplemented with 1 μg/mL puromycin and 10μg/mL blasticidin (InvivoGen, Cat. # ant-pr-1 and ant-bl-1, respectively), and were passaged by trypsinizing from tissue culture plastic. HEK-293F cells were grown in serum-free Freestyle 293 medium (Life Technologies, Cat. #12338–018). All cell lines were maintained at 37°C, 5% CO2.
Quantitation of cell surface epitopes by flow cytometry
Ag104A cells were stained at 4◦C with saturating amounts of purified 237 for 2 h, side by side with calibrated mouse IgG-binding beads (Dako QIFIKIT, Dako North America, Inc., Cat. # K0078). The cells and beads were then washed for 8 min at 4◦C in a large excess of phosphate-buffered saline (PBS, pH 7.4) + 1% bovine serum albumin (PBS-BSA), and stained for one hour on ice with a fluorescein-labeled goat anti-mouse antibody (Dako North America, Inc.). The cells and beads were washed again in a large excess of PBS-BSA, and resuspended immediately prior to analysis by flow cytometry (BD Accuri C6 flow cytometer, BD Accuri Cytometers, Inc.). Quantification of bound 237 antibodies per cell was accomplished by comparing the increase in fluorescence of Ag104A when stained with 237 vs. secondary goat anti-mouse only, and a calibration curve determined by the increase in fluorescence for the IgG-binding beads related to the number of IgG molecules bound per peak (Fig. S1). Ag104A measurement is based on three independent measurements on different days. For other target cells (ACosmc, Jurkat, JOE, Neuro2A, and Neuro2A+OTS8), staining was performed similarly side-by-side with Ag104A, but detection was performed with goat anti-mouse IgG labeled with Alexa 647, to avoid interference with green fluorescent protein (GFP) included in several of the lines. Quantification of bound 237 antibodies per cell were determined by comparison to Ag104A in at least two independent experiments. Labeling of Dako QIFIKIT beads with Alexa 647-labeled antibody was linear in the range of the target cell stains, but was nonlinear outside that range (data not shown).
Design of 237 BiTE construct
The gene encoding the 237 bispecific T cell engager (BiTE) was constructed from the sequences of the 145–2C11 antibody (anti-murine CD3ε38) and the 237 antibody.32 Single-chain Fv fragments for each were arranged as follows from 5′ to 3′: 145–2C11 VH – (GGGGS)3 – 145–2C11 VL – Gly-Ser linker (GGGSGGGGSGSGGGSGGGGSGGG) – 237 VL – (GGGGS)3 – 237 VH, making the 145–2C11 scFv N-terminal in the soluble molecule for improved function (personal communication with Roman Kischel, Micromet/Amgen). A C-terminal His6 tag was included for ease of purification. The gene was flanked with BglII and NotI restriction sites on the 5′ and 3′ ends, respectively, and was synthesized and codon-optimized (Genscript). The gene was cloned in-frame into the pDisplay vector for mammalian expression (Life Technologies, Cat. # V66020).
Expression and purification of 237 BiTE
The 237 BiTE (2C11:237) in the pDisplay plasmid was transfected into HEK-293F cells using 293fectin according to manufacturer’s instructions (Life Technologies Cat. # 12347–019). Transfected cells were grown and expanded in Freestyle 293 media supplemented with 250μg/mL G418 (Mediatech, Inc. Cat. # 61–234). Expression-scale cultures were grown in shake flasks for 8–10 d at 120 rpm, 37°C, 5% CO2, and then the cultures were spun to remove cells, and the supernatants were incubated with Ni-NTA agarose beads (Qiagen Cat. # 30230) overnight at 4°C, stirring gently. The beads were then collected on a scintered glass funnel, washed in phosphate-buffered saline (PBS, pH 7.4), and protein was eluted by incubation of the beads in PBS containing 0.5M imidazole. The beads were removed by 0.2 μm filtration, and protein was purified by size exclusion chromatography over a Superdex 200 column (GE Healthcare Life Sciences Cat. # 17–5175–01) on a Perseptive Biosystems BioCAD Sprint HPLC (Applied Biosystems).
Construction of 237 CAR
The plasmid containing the 237 scFv was provided by Hans Schreiber. A XhoI restriction site within the 237 coding sequence was removed by introducing a silent mutation into the protein sequence by QuikChange II (Agilent Technologies Cat. # 200523) to allow for incorporation of the 237 scFv into a larger CAR construct via a 3′ XhoI site, introduced by overlap extension PCR. The Human IgKappa antibody leader sequence (MDFQVQIFSFLLISASVIMSRG65), including a NotI restriction site before the gene, was fused by overlap extension to the 5′ end of the 237 scFv. Separately, for generation of the CAR, a gene fusion consisting of the murine CD8α hinge,66 transmembrane and cytoplasmic CD28, and cytoplasmic CD3ζ was designed based on sequences from the NIH database (GenBank accession #AAS07035.1), and the resulting gene was synthesized and codon optimized (Genscript), including a 3′ EcoRI restriction site. The two genes (NotI-leader-237 scFv-XhoI; and XhoI-CD8α hinge-CD28-CD3ζ-EcoRI) were ligated into the pMP71 retroviral vector via the NotI and EcoRI sites.
Transduction of 237 CAR into T cells
PlatE retroviral packaging cells were plated at 3x106 cells per 10cm poly-L-Lysine coated tissue culture plate and allowed to grow overnight at 37°C, 5% CO2. The cells were then transfected with 40μg 237 CAR in pMP71 plasmid or Mock (no vector DNA) and 60μL Lipofectamine 2000 reagent (Life Technologies Cat. # 11668030) in OptiMEM medium (Life Technologies Cat. # 11058–021) according to manufacturer’s instructions. After transfection, the cells were washed once using complete IMDM medium, and fresh IMDM medium was added. The transfected cells were cultured for two days at 37°C, 5% CO2, and then culture supernatants were harvested, filtered through a 0.45μm filter, and an additional 50μL Lipofectamine 2000 was added to each retroviral supernatant.
Splenocytes were isolated from C57BL/6 mice and treated with ACK lysis buffer to remove red blood cells. Subsequently, CD4+ or CD8+ T cells were purified using magnetic beads (MACS, Miltenyi Biotec, Cat. # 130–095–248 and 130–095–236, respectively). One x 106 purified cells per well of a 24-well plate were stimulated with anti-CD3/anti-CD28 Dynabeads (Life Technologies Cat. # 11452D) per manufacturer’s instructions, and 30 U/mL of IL-2 (Roche Applied Science, Cat. # 11271164001) in complete IMDM medium.
One day after activation, the primary T cells (CD4+ or CD8+) were resuspended and separated from magnetic beads, but maintained in their activation medium. Wells of a 24-well plate were coated with 15 μg/mL RetroNectin (Takara Bio Inc., Cat. # T100A) for 2 h at room temperature, and then blocked with sterile 1% bovine serum albumen in PBS for 30 min at room temperature. To each well, 1mL of activated T cells, 1mL of retroviral or mock supernatant, and 60 units of IL-2 were added. Cells were centrifuged at 2000 × g, 30°C for 1 h (spinfection), then placed at 37°C, 5% CO2. The cells were split 1:2 into complete IMDM medium on day 3 post-activation, and activation or CTL assays were performed on day 4 post-activation. T cells were checked for purity by flow cytometry using anti-TCR Cβ and anti-CD8 or anti-CD4 antibodies, and for transduction with 237 CAR using goat anti-mouse Alexa 647 F(ab’)2.
Transduction and spinfection of 58−/− T-cell hybridomas with 237 CAR or Mock PlatE retroviral supernatants was performed similarly to above, but without requirement for stimulation or inclusion of IL-2 as previously described.67,68
Target cell lysis by 51Cr-Release
Target cells were incubated with 200 μCi of 51Cr in saline (MP Biomedicals) in phosphate-buffered saline (PBS, pH 7.4) with 15% fetal bovine serum for 2 h at 37°C, 5% CO2, with periodic agitation. After incubation, target cells were washed 3x into fresh complete IMDM medium. Labeled targets and effector T cells (40,000 and 200,000, respectively) on day four post activation were added together in the wells of 96-well round-bottom plates, with or without transduction of 237 CAR or inclusion of 237 BiTE at 100nM, and briefly spun down at 200xg. The plate was incubated for four hours at 37◦C, 5% C02. After incubation, supernatants were harvested, and released 51Cr was measured by a Packard Cobra II Series gamma counter, model 5002 (Perkin-Elmer). Percent max release was calculated based on spontaneous release of unstimulated, labeled target cells (minimum) and release of fully lysed target cells in 1.3% triton (maximum). Samples were measured in triplicate, and error bars represent standard deviation. Results are typical for at least three independent experiments. For the level of statistical significance of differences between CD4+ and CD8+ cells, targeted by the same method toward the same target cells, a student’s unpaired, two-tailed t test was performed, and p values are listed.
Measurement of cytokine release by ELISA
50,000 effector T cells, either mock transduced, transduced with 237 CAR, or untransduced, were cultured with and without various stimuli (such as 50,000 target cells, with or without 100nM 237 BiTE) for 20 h at 37°C, 5% CO2. Samples were set up in triplicate with error bars indicating standard deviation, and data shows representative results from at least 3 independent experiments. After the incubation period, supernatants were harvested from each condition, and the supernatant was analyzed for released IFN-γ by sandwich ELISA, using the Ready-Set-Go kit (eBioscience Cat. # 88–7314). Briefly, the wells of a 96-well high-binding ELISA plate were coated with IFNγ capture antibody in phosphate-buffered saline (PBS, pH 7.4) overnight at 4°C, and then blocked overnight with 1% bovine serum albumin in PBS (PBS-BSA) at 4°C. The supernatants and/or standard dilutions of IFNγ in dilution solution (0.1% bovine serum albumin and 0.03% Tween-20 in PBS) were added to the assay wells and incubated overnight at 4°C. The wells were then washed three times with PBS containing 0.1% Tween-20 (PBST), and a solution of biotinylated anti-IFNγ detection antibody in dilution solution was added to each well. The plate was incubated for 2 h at room temperature, then washed three times with PBST, and a solution of avidin-horseradish peroxidase in dilution solution was added to each well. The plate was incubated for 45 min at room temperature, then washed three times with PBST, and 3,3′,5,5′-tetramethylbenzidine (TMB, KPL Cat. # 50–76–00) solution was added to each well and allowed to develop. When adequate color had developed to distinguish wells, the reaction was stopped by adding an equal volume of 0.1N sulfuric acid to the TMB in each well. The absorbance at 450 nm in each well was measured using an ELx800 universal microplate reader (Bio-Tek Instruments). Concentration of IFNγ for each sample was calculated by comparison with a standard curve of IFNγ in the same assay. For the statistical significance of differences between CD4+ and CD8+ cells, targeted by the same method toward the same target cells, a student’s unpaired, two-tailed t test was performed, and p values are listed.
For IL-2 release from 58−/− T-cell hybridomas transduced with 237 CAR, 50,000 hybridomas were incubated alone or with 50,000 target cells (Ag104A or ACosmc) at 37°C, 5% CO2 for 24 h. IL-2 in the supernatant was analyzed for each well by ELISA as described.69 Briefly, 96-well plates (Immulon 2HB, VWR Cat. # 62402–972) were coated with 2.5 μg/ml anti-murine IL-2 (BD PharMingen Cat. # 554424) in PBS for 2 h at room temperature, then blocked with 1% BSA in PBS for 12 h at 4°C. To each well, 50 μl of cell supernatant was added, and incubated for 12 h at 4°C. Wells were washed three times with PBST, and then 6.7 μg/ml biotinylated anti-murine IL-2 (BD PharMingen Cat. # 554426) in dilution solution was added for 1 h at room temperature. Wells were washed three times with PBST, and then incubated with a 1/10,000 dilution of streptavidin-HRP (BD PharMingen Cat. # 554066) for 30 min at room temperature. Plates were washed three times with PBST, and developed with 50 μl TMB substrate. The reaction was stopped with 50 μl of 1N sulfuric acid, and absorbance at 450 nm was measured in each well using an ELx800 universal microplate reader.
Supplementary Material
Disclosure of Potential Conflicts of Interest
The authors have no conflicts or financial interests to disclose.
Acknowledgments
We would like to thank Roman Kischel (Amgen) for helpful discussion and advice, and Samanthi Narayanan (Department of Biochemistry, University of Illinois at Urbana-Champaign) for experimental assistance. This work was supported by NIH grants CA097296 (to DMK and HS) CA22677 (to HS and DMK) and CA37156 (to HS and DMK). JDS was supported by the Samuel and Ruth Engelberg/Irvington Institute Fellowship of the Cancer Research Institute.
Supplemental Materials
Supplemental materials can be found at:
www.landesbioscience.com/journals/oncoimmunology/article/20592
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/20592
References
- 1.Schreiber H. Tumor-specific immune responses. Semin Immunol. 2008;20:265–6. doi: 10.1016/j.smim.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schreiber H, Rowley JD, Rowley DA. Targeting mutations predictably. Blood. 2011;118:830–1. doi: 10.1182/blood-2011-06-357541. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber H. Cancer Immunology. Philadelphia, PA: Lippincott-Williams & Wilkins, 2012. [Google Scholar]
- 4.Karyampudi L, Knutson KL. Antibodies in cancer immunotherapy. Cancer Biomark. 2010;6:291–305. doi: 10.3233/CBM-2009-0141. [DOI] [PubMed] [Google Scholar]
- 5.Grillo-López AJ, White CA, Varns C, Shen D, Wei A, McClure A, et al. Overview of the clinical development of rituximab: first monoclonal antibody approved for the treatment of lymphoma. Semin Oncol. 1999;26(Suppl 14):66–73. [PubMed] [Google Scholar]
- 6.Goldenberg MM. Trastuzumab, a recombinant DNA-derived humanized monoclonal antibody, a novel agent for the treatment of metastatic breast cancer. Clin Ther. 1999;21:309–18. doi: 10.1016/S0149-2918(00)88288-0. [DOI] [PubMed] [Google Scholar]
- 7.Seliger B, Cabrera T, Garrido F, Ferrone S. HLA class I antigen abnormalities and immune escape by malignant cells. Semin Cancer Biol. 2002;12:3–13. doi: 10.1006/scbi.2001.0404. [DOI] [PubMed] [Google Scholar]
- 8.Garrido F, Cabrera T, Concha A, Glew S, Ruiz-Cabello F, Stern PL. Natural history of HLA expression during tumour development. Immunol Today. 1993;14:491–9. doi: 10.1016/0167-5699(93)90264-L. [DOI] [PubMed] [Google Scholar]
- 9.Meidenbauer N, Zippelius A, Pittet MJ, Laumer M, Vogl S, Heymann J, et al. High frequency of functionally active Melan-a-specific T cells in a patient with progressive immunoproteasome-deficient melanoma. Cancer Res. 2004;64:6319–26. doi: 10.1158/0008-5472.CAN-04-1341. [DOI] [PubMed] [Google Scholar]
- 10.Yu Z, Theoret MR, Touloukian CE, Surman DR, Garman SC, Feigenbaum L, et al. Poor immunogenicity of a self/tumor antigen derives from peptide-MHC-I instability and is independent of tolerance. J Clin Invest. 2004;114:551–9. doi: 10.1172/JCI21695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cloosen S, Arnold J, Thio M, Bos GM, Kyewski B, Germeraad WT. Expression of tumor-associated differentiation antigens, MUC1 glycoforms and CEA, in human thymic epithelial cells: implications for self-tolerance and tumor therapy. Cancer Res. 2007;67:3919–26. doi: 10.1158/0008-5472.CAN-06-2112. [DOI] [PubMed] [Google Scholar]
- 12.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol. 1999;163:507–13. [PubMed] [Google Scholar]
- 16.Kessels HW, Wolkers MC, van den Boom MD, van der Valk MA, Schumacher TN. Immunotherapy through TCR gene transfer. Nat Immunol. 2001;2:957–61. doi: 10.1038/ni1001-957. [DOI] [PubMed] [Google Scholar]
- 17.Schmitt TM, Ragnarsson GB, Greenberg PD. T cell receptor gene therapy for cancer. Hum Gene Ther. 2009;20:1240–8. doi: 10.1089/hum.2009.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, et al. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 2007;109:2331–8. doi: 10.1182/blood-2006-05-023069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med 2010; 16:565-70, 1p following 70. [DOI] [PubMed]
- 20.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720–4. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bridgeman JS, Hawkins RE, Hombach AA, Abken H, Gilham DE. Building better chimeric antigen receptors for adoptive T cell therapy. Curr Gene Ther. 2010;10:77–90. doi: 10.2174/156652310791111001. [DOI] [PubMed] [Google Scholar]
- 22.Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101:1637–44. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 23.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mack M, Riethmüller G, Kufer P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci U S A. 1995;92:7021–5. doi: 10.1073/pnas.92.15.7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Staerz UD, Kanagawa O, Bevan MJ. Hybrid antibodies can target sites for attack by T cells. Nature. 1985;314:628–31. doi: 10.1038/314628a0. [DOI] [PubMed] [Google Scholar]
- 27.Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009;69:4941–4. doi: 10.1158/0008-5472.CAN-09-0547. [DOI] [PubMed] [Google Scholar]
- 28.Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–7. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- 29.Löffler A, Kufer P, Lutterbüse R, Zettl F, Daniel PT, Schwenkenbecher JM, et al. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood. 2000;95:2098–103. [PubMed] [Google Scholar]
- 30.Brischwein K, Schlereth B, Guller B, Steiger C, Wolf A, Lutterbuese R, et al. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol. 2006;43:1129–43. doi: 10.1016/j.molimm.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 31.Topp MS, Kufer P, Gökbuget N, Goebeler M, Klinger M, Neumann S, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29:2493–8. doi: 10.1200/JCO.2010.32.7270. [DOI] [PubMed] [Google Scholar]
- 32.Ward PL, Koeppen H, Hurteau T, Schreiber H. Tumor antigens defined by cloned immunological probes are highly polymorphic and are not detected on autologous normal cells. J Exp Med. 1989;170:217–32. doi: 10.1084/jem.170.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schietinger A, Philip M, Yoshida BA, Azadi P, Liu H, Meredith SC, et al. A mutant chaperone converts a wild-type protein into a tumor-specific antigen. Science. 2006;314:304–8. doi: 10.1126/science.1129200. [DOI] [PubMed] [Google Scholar]
- 34.Brooks CL, Schietinger A, Borisova SN, Kufer P, Okon M, Hirama T, et al. Antibody recognition of a unique tumor-specific glycopeptide antigen. Proc Natl Acad Sci U S A. 2010;107:10056–61. doi: 10.1073/pnas.0915176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olmsted JB, Carlson K, Klebe R, Ruddle F, Rosenbaum J. Isolation of microtubule protein from cultured mouse neuroblastoma cells. Proc Natl Acad Sci U S A. 1970;65:129–36. doi: 10.1073/pnas.65.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abraham RT, Weiss A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat Rev Immunol. 2004;4:301–8. doi: 10.1038/nri1330. [DOI] [PubMed] [Google Scholar]
- 37.Ju T, Cummings RD. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc Natl Acad Sci U S A. 2002;99:16613–8. doi: 10.1073/pnas.262438199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leo O, Foo M, Sachs DH, Samelson LE, Bluestone JA. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc Natl Acad Sci U S A. 1987;84:1374–8. doi: 10.1073/pnas.84.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnston SC, Dustin ML, Hibbs ML, Springer TA. On the species specificity of the interaction of LFA-1 with intercellular adhesion molecules. J Immunol. 1990;145:1181–7. [PubMed] [Google Scholar]
- 40.James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, et al. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol. 2008;180:7028–38. doi: 10.4049/jimmunol.180.10.7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weijtens ME, Hart EH, Bolhuis RL. Functional balance between T cell chimeric receptor density and tumor associated antigen density: CTL mediated cytolysis and lymphokine production. Gene Ther. 2000;7:35–42. doi: 10.1038/sj.gt.3301051. [DOI] [PubMed] [Google Scholar]
- 42.Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol. 2004;173:7647–53. doi: 10.4049/jimmunol.173.12.7647. [DOI] [PubMed] [Google Scholar]
- 43.Turatti F, Figini M, Balladore E, Alberti P, Casalini P, Marks JD, et al. Redirected activity of human antitumor chimeric immune receptors is governed by antigen and receptor expression levels and affinity of interaction. J Immunother. 2007;30:684–93. doi: 10.1097/CJI.0b013e3180de5d90. [DOI] [PubMed] [Google Scholar]
- 44.Alvarez-Vallina L, Russell SJ. Efficient discrimination between different densities of target antigen by tetracycline-regulatable T bodies. Hum Gene Ther. 1999;10:559–63. doi: 10.1089/10430349950018634. [DOI] [PubMed] [Google Scholar]
- 45.James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Budde LE, et al. Mathematical modeling of chimeric TCR triggering predicts the magnitude of target lysis and its impairment by TCR downmodulation. J Immunol. 2010;184:4284–94. doi: 10.4049/jimmunol.0903701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nicholson IC, Lenton KA, Little DJ, Decorso T, Lee FT, Scott AM, et al. Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol Immunol. 1997;34:1157–65. doi: 10.1016/S0161-5890(97)00144-2. [DOI] [PubMed] [Google Scholar]
- 47.Nagorsen D, Baeuerle PA. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp Cell Res. 2011;317:1255–60. doi: 10.1016/j.yexcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 48.Bikoue A, George F, Poncelet P, Mutin M, Janossy G, Sampol J. Quantitative analysis of leukocyte membrane antigen expression: normal adult values. Cytometry. 1996;26:137–47. doi: 10.1002/(SICI)1097-0320(19960615)26:2<137::AID-CYTO7>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 49.Gratama JW, D’hautcourt JL, Mandy F, Rothe G, Barnett D, Janossy G, et al. European Working Group on Clinical Cell Analysis Flow cytometric quantitation of immunofluorescence intensity: problems and perspectives. Cytometry. 1998;33:166–78. doi: 10.1002/(SICI)1097-0320(19981001)33:2<166::AID-CYTO11>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 50.Lenkei R, Gratama JW, Rothe G, Schmitz G, D’hautcourt JL, Arekrans A, et al. Performance of calibration standards for antigen quantitation with flow cytometry. Cytometry. 1998;33:188–96. doi: 10.1002/(SICI)1097-0320(19981001)33:2<188::AID-CYTO13>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 51.Olejniczak SH, Stewart CC, Donohue K, Czuczman MS. A quantitative exploration of surface antigen expression in common B-cell malignancies using flow cytometry. Immunol Invest. 2006;35:93–114. doi: 10.1080/08820130500496878. [DOI] [PubMed] [Google Scholar]
- 52.Kimmig R, Pfeiffer D, Landsmann H, Hepp H. Quantitative determination of the epidermal growth factor receptor in cervical cancer and normal cervical epithelium by 2-color flow cytometry: evidence for down-regulation in cervical cancer. Int J Cancer. 1997;74:365–73. doi: 10.1002/(SICI)1097-0215(19970822)74:4<365::AID-IJC1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 53.Brand FX, Ravanel N, Gauchez AS, Pasquier D, Payan R, Fagret D, et al. Prospect for anti-HER2 receptor therapy in breast cancer. Anticancer Res. 2006;26(1B):463–70. [PubMed] [Google Scholar]
- 54.Jones KL, Buzdar AU. Evolving novel anti-HER2 strategies. Lancet Oncol. 2009;10:1179–87. doi: 10.1016/S1470-2045(09)70315-8. [DOI] [PubMed] [Google Scholar]
- 55.Kiewe P, Hasmüller S, Kahlert S, Heinrigs M, Rack B, Marmé A, et al. Phase I trial of the trifunctional anti-HER2 x anti-CD3 antibody ertumaxomab in metastatic breast cancer. Clin Cancer Res. 2006;12:3085–91. doi: 10.1158/1078-0432.CCR-05-2436. [DOI] [PubMed] [Google Scholar]
- 56.Nielsen DL, Andersson M, Kamby C. HER2-targeted therapy in breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. Cancer Treat Rev. 2009;35:121–36. doi: 10.1016/j.ctrv.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 57.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Du C, Wang Y. The immunoregulatory mechanisms of carcinoma for its survival and development. J Exp Clin Cancer Res. 2011;30:12. doi: 10.1186/1756-9966-30-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim PS, Ahmed R. Features of responding T cells in cancer and chronic infection. Curr Opin Immunol. 2010;22:223–30. doi: 10.1016/j.coi.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chmielewski M, Abken H. CAR T cells transform to trucks: chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol Immunother. 2012 doi: 10.1007/s00262-012-1202-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future direction. J Gene Med. 2012 doi: 10.1002/jgm.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Straathof KC, Pulè MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247–54. doi: 10.1182/blood-2004-11-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Letourneur F, Malissen B. Derivation of a T cell hybridoma variant deprived of functional T cell receptor alpha and beta chain transcripts reveals a nonfunctional alpha-mRNA of BW5147 origin. Eur J Immunol. 1989;19:2269–74. doi: 10.1002/eji.1830191214. [DOI] [PubMed] [Google Scholar]
- 64.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–6. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 65.Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol. 2009;183:5563–74. doi: 10.4049/jimmunol.0900447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haynes NM, Trapani JA, Teng MW, Jackson JT, Cerruti L, Jane SM, et al. Single-chain antigen recognition receptors that costimulate potent rejection of established experimental tumors. Blood. 2002;100:3155–63. doi: 10.1182/blood-2002-04-1041. [DOI] [PubMed] [Google Scholar]
- 67.Chervin AS, Aggen DH, Raseman JM, Kranz DM. Engineering higher affinity T cell receptors using a T cell display system. J Immunol Methods. 2008;339:175–84. doi: 10.1016/j.jim.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stone JD, Chervin AS, Aggen DH, Kranz DM. T cell receptor engineering. Methods Enzymol. 2012;503:189–222. doi: 10.1016/B978-0-12-396962-0.00008-2. [DOI] [PubMed] [Google Scholar]
- 69.Chervin AS, Stone JD, Holler PD, Bai A, Chen J, Eisen HN, et al. The impact of TCR-binding properties and antigen presentation format on T cell responsiveness. J Immunol. 2009;183:1166–78. doi: 10.4049/jimmunol.0900054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.