

Abstract
The aryl hydrocarbon receptor (AHR) is a transcription factor that responds to diverse ligands and plays a critical role in toxicology, immune function, and cardiovascular physiology. The structural basis of the AHR for ligand promiscuity and preferences is critical for understanding AHR function. Based on the structure of a closely related protein HIF2α, we modeled the AHR ligand binding domain (LBD) bound to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and benzo(a)pyrene (BaP) and identified residues that control ligand preferences by shape and H-bond potential. Mutations to these residues, particularly Q377 and G298, resulted in robust and opposite changes in the potency of TCDD and BaP and up to a 20-fold change in the ratio of TCDD/BaP efficacy. The model also revealed a flexible “belt” structure; molecular dynamic (MD) simulation suggested that the “belt” and several other structural elements in the AHR-LBD are more flexible than HIF2α and likely contribute to ligand promiscuity. Molecular docking of TCDD congeners to a model of human AHR-LBD ranks their binding affinity similar to experimental ranking of their toxicity. Our study reveals key structural basis for prediction of toxicity and understanding the AHR signaling through diverse ligands.
Key Words: dioxin, Ah receptor, structural modeling, structural determinants, ligand preferences, ligand tolerance.
The aryl hydrocarbon receptor (AHR) is a PAS (PER, ARNT, SIM) family, ligand-activated transcription factor, and responds to numerous xenobiotic and endogenous ligands for regulating toxicology, immune function, and cardiovascular physiology. Enormous efforts have been devoted to identification of AHR ligands over the past four decades since the striking observation that the AHR mediates the toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (Poland et al., 1976). Thousands of environmental toxins have been suggested to be the AHR ligands and mediate broad physiological responses (Nguyen and Bradfield, 2008; Schmidt and Bradfield, 1996). Some AHR ligands induce the expression of xenobiotic metabolizing enzymes that mediates toxin metabolism and adaptive response (Hayes et al., 2007; Whitlock, 1999). Halogenated aromatic hydrocarbons induce the expression of a battery of proteins that lead to severe toxic endpoints such as chloracne, thymic involution, cleft palate, and cancer (Bock and Köhle, 2006; McMillan and Bradfield, 2007). The AHR is also found to be essential for the normal function of the vascular and immune systems (Esser et al., 2009; Korashy and El-Kadi, 2006; Savouret et al., 2003; Stevens et al., 2009), presumably through activation by unknown endogenous ligands. Interestingly, aberrant accumulation of a tryptophan metabolite, Kynurenine, was recently shown to affect immune function and induce cancer progression through induction of the AHR signaling (Mezrich et al., 2010; Opitz et al., 2011). A natural AHR ligand, indole-3-carbinol, was shown to improve the maintaining of innate immunity for intestinal homeostasis and resistance to infection via the AHR signaling (Li et al., 2011). Thus, deregulation of the AHR by environmental toxicants, natural, or endogenous ligands has important implications for human diseases.
The physiological and toxicological consequences of AHR ligands are mediated through the AHR signaling pathway. The unliganded AHR is located in cytosol and associated with protein chaperones (i.e., Hsp90, p23, and AHR-interacting protein 1) (Carver et al., 1998; Kazlauskas et al., 1999; Meyer and Perdew, 1999; Meyer et al., 1998; Perdew, 1988). Upon ligand binding, the AHR is translocated to the nucleus (Ikuta et al., 1998; Petrulis et al., 2003), where the liganded AHR forms a heterodimeric complex with the AHR transcription partner, AHR nuclear translocator (ARNT) (Schmidt and Bradfield, 1996). The AHR-ARNT heterodimer then binds to cognate dioxin-responsive enhancers (DREs) and induces the expression of a variety of target genes that mediate broad physiological consequences from xenobiotic metabolism and cytotoxicity to normal immune and vascular function (Chan et al., 1994; Reyes et al., 1992).
The minimal ligand binding domain (LBD) of the AHR has been mapped to a hydrophobic region encompassing PAS-B and the sequence immediately following PAS-B (Coumailleau et al., 1995; Dolwick et al., 1993). The structure of the AHR-LBD had been modeled by different groups (Bisson et al., 2009; Pandini et al., 2009; Procopio et al., 2002). Little is known, however, about the structural basis of the AHR ligand preferences and ligand promiscuity. Analysis of three-dimensional quantitative structure-activity relationship (3D-QSAR) of the AHR ligands had been performed based on the experimental data on one or a few families of AHR ligands, such as TCDD congeners, to determine how different shape and chemical properties acquired through various modifications affect the toxicity and efficacy of the ligands (Arulmozhiraja and Morita, 2004). These analyses support a general preference of the AHR toward chemicals with a planar shape and with less extension at the medial positions and larger extensions at the lateral positions. Although such information is useful, the structural determinants of the AHR that control ligand preferences remain unclear.
Here we built a model of the mouse AHR-LBD bound to TCDD and benzo(a)pyrene (BaP), two model ligands that exhibit different size, shape, and chemical properties. The model reveals an elongated, planar ligand binding pocket that makes close contacts to the medial positions and loose contacts to the lateral positions of the ligands. Structural and mutational analysis identified AHR residues that control ligand preferences and revealed a flexible “belt” structure, which, together with several other flexible structural elements in the AHR-LBD, likely contributes to ligand tolerance. In silico docking of TCDD congeners to a model of human AHR-LBD gave a ranking of their binding affinities similar to experimental ranking. Our results identified key AHR structural elements that control ligand preference and tolerance and can be used to extrapolate structural signatures of ligands that favor AHR binding. This will aid identification of unknown AHR ligands and prediction of ligand toxicity.
MATERIALS AND METHODS
Modeling AHR-LBD bound to TCDD. The sequence of AHR-LBD was threaded stepwise through the structure of human HIF2α (PDB code: 3H7W) in Sybyl (Tripos). Residues lining the cavity of the HIF2α structure were first changed to AHR sequence and energy minimized. After TCDD was docked to the AHR ligand binding cavity using program Autodock (Goodsell et al., 1996), the remaining α-helices, β-sheets, and loop regions with the same length in sequence were gradually changed to AHR sequence in Sybyl. The backbone conformation and rotamer usage were examined and improved in Coot, followed by energy minimization in Sybyl. Loop regions that differ from HIF2α in length were built manually in combination with loop conformation search to roughly resemble the overall fold in HIF2α. Iterative model building, ligand docking, energy minimization, and optimization of backbone conformation and rotamer usage were performed until satisfying results were obtained.
Automatic docking of AHR ligands to the model of AHR-LBD. AHR ligands were docked individually or as a group to the ligand binding cavity of the model of AHR-LBD using Autodock (Goodsell et al., 1996). The structure of AHR ligands was built in Sybyl and energy minimized. The receptor was either set in a fixed conformation or, with a selected group of residues lining the ligand binding pocket, set as flexible residues as described. The latter allows automatic search of residue conformations that would give optimized ligand binding. Top ten ligand binding modes were selected based on ligand binding energy and visually examined before they are used for in silico ranking of TCDD congeners.
MD simulation. Explicit solvent simulations are carried out using GROMACS v4 (Frenkel and Smit, 1996; Hess et al., 2008). The systems are first minimized and then equilibrated for 5 ns before production runs at 300 K. TIP3P, the generic 3-point water, is used as the solvent (Jorgensen et al., 1983); 0.015M KCl is included to be near physiological conditions. Periodic boundary conditions are applied. Electrostatic interactions are treated using Particle Mesh Ewald (PME) with a PME order of 6 and a real-space cut-off of 14 Å. For van der Waals interactions, the switch scheme is used, in which the Lennard-Jones potential is normal up to 10 Å and switched off to reach zero at 12 Å beyond 10 Å. The temperature is controlled by the Berendsen thermostat at 300 K. Bonds are constrained via the LINCS algorithm. Pressure is controlled by an isotropic Berendsen coupling and held at 1 bar with a compressibility factor of 4.5 × 10−5. The 200 ns of production trajectories are collected for both the AHR-LBD and HIF2α.
Cloning and expression of recombinant mAHR. The wild-type (WT) and mutant mAHR were cloned into the XhoI/SalI cloning sites of pTARGET (Promega, Madison, WI) using routine PCR methods and molecular cloning procedures using the pSport-mAHR plasmid (PL65) as template (Dolwick et al., 1993). For recombinant expression of mAHR, COS-1 cells were cultured in 6-cm dishes and transfected with 2 μg of WT or mutant mAHR expression vector. Twenty-four hours after transfection, cells were collected, and the whole cell extracts were prepared by celLytic M reagent (Sigma Aldrich, St Louis, MO). Twenty microgram of whole cell extract was analyzed by Western blot using antibodies that specifically recognize the mAHR (bear-2) and β-actin (Sigma).
Luciferase reporter gene assay. COS-1 cells were cultured in 24-well plates and transiently transfected with pTarget vector containing WT or mutant mAHR, or empty vector (20 ng), together with pGudLu6.1 DREs-driven luciferase reporter vector (100ng) (Han et al., 2004) and TK-renilla luciferase vector (20 ng) (Invitrogen, Carlsbad, CA). Six hours after transfection, cells were treated with TCDD, BaP, or vehicle alone (0.1% dimethyl sulfoxide) for 4h and assayed with dual luciferase reporter assay system (Promega). The expressed luciferase activity was measured using Microlumat Plus luminometer (Berthold Technologies, Hertfordshire, U.K.). The transcriptional activity of all mAHR mutants were tested using a fixed concentration of TCDD (0.2 nM) and BaP (0.4 nM), and their response curves were measured as described. Data analysis and simulation were performed using GraphPad Prism 4 (GraphPad software Inc., La Jolla, CA). The experiments were performed in triplicate and repeated at least twice. Representative results are shown in mean ± SEM.
RESULTS
Modeling of the AHR-LBD Bound to TCDD
The AHR harbors an internal PAS domain with two degenerate repeats known as PAS-A and PAS-B (Coumailleau et al., 1995; Dolwick et al., 1993). The minimal LBD of mouse AHR has been mapped to a hydrophobic region encompassing PAS-B and the sequence immediately following PAS-B (residues 270–384) (Coumailleau et al., 1995; Dolwick et al., 1993). Similar to previous modeling of the structure of AHR-LBD, we utilized the crystal structure of HIF2α PAS-B motif (Scheuermann et al., 2009) as an initial model, which shares almost 30% sequence similarity to AHR-LBD (Supplementary figure S1). A stepwise manual model building and iterative ligand docking was utilized to build the structure of AHR-LBD bound to TCDD as described in the Materials and Methods section, until 99% of residues fell into the “most favored” or “allowed” regions of the Ramachandran plot. Despite the relatively low sequence similarity, all the residues in the AHR ligand binding core were readily registered to the structure of HIF2α, except a long loop that we named the “belt” (Supplementary figure S1; Fig. 1A). Another loop (Hβ-Iβ) that failed to be registered connects β-strands Hβ and Iβ and extends away from the ligand binding cavity (Supplementary figure S1; Fig. 1A).
FIG. 1.

Overall structure of the AHR-LBD. (A) Alignment of the model of mouse AHR-LBD (magenta) and the crystal structure of HIF2α (PDB code: 3H7W, blue) in perpendicular view. The “belt” in HIF2α and the AHR are shown in cyan and coral, respectively. The “belt” sequences of HIF2α and the AHR and their alignment are shown (inset). (B) A slice of surface illustrating the internal surface of the ligand binding pocket bound to TCDD. Green arrows, lateral positions; red arrows, medial positions. (C) The electrostatic potential of the AHR ligand binding pocket shows that the ligand binding pocket is largely hydrophobic. Two polar AHR residues near the medial positions of TCDD are shown (ball-and-stick, colored magenta).
The model of the AHR-LBD aligns very well to the crystal structure of HIF2α bound to an artificial ligand (PDB code: 3H7W) (Fig. 1A). Similar to other PAS domain proteins, the AHR-LBD adopts a glove-shaped protein fold with a central β-sheet flanked by two α-helices and long connecting loops (Fig. 1A). The model revealed an elongated, planar ligand binding pocket formed predominately by the central β-sheet, together with the Fα helix and “belt” (Fig. 1A). The “belt” is the longest loop in the AHR (residues 307–322) that spans half the equatorial circumference of the ligand binding cavity. The long edges of the ligand binding pocket, formed by the central β-sheet and “belt”, make close contacts to the medial positions of TCDD, and the short edges of the pocket, formed by the Fα helix and Aβ/Bβ strands, make loose contacts to the lateral positions of TCDD (Fig. 1B). This mode of ligand binding is consistent with the previous observation that the medial extension of the AHR ligand reduces ligand binding affinity (Ashek et al., 2006) and that addition of bulky groups to the lateral positions of the ligand favors AHR binding (Arulmozhiraja and Morita, 2004). For convenience, we refer to the residues near the medial and lateral positions of the ligands as medial and lateral AHR residues, respectively.
Similar to that predicted previously, the ligand binding pocket of the AHR is highly hydrophobic (Fig. 1C) and is formed predominantly by hydrophobic residues, F281, F289, P291, L302, Y304, F318, I319, C327, M334, M337, F345, L347, V357, and A375 (Supplementary figure S2A) (Pandini et al., 2009). Several residues lining the ligand binding pocket were suggested to be important for ligand binding by previous mutational analysis (Pandini et al., 2009), consistent with their close contact or proximity to the ligand (Supplementary figure S2B). Interestingly, a few hydrophilic residues at the central β-sheet were found near the ligand medial positions (Fig. 1C), which might affect ligand preferences by size and H-bond potential.
Identification of AHR Medial Residues That Affect Ligand Preferences
Although the AHR ligand binding pocket is primarily formed by hydrophobic residues, our models of the AHR-LBD bound to TCDD and BaP show that several polar residues in the central β-sheet are located near the medial positions of the ligands (Fig. 2A). These residues form two H-bond networks: one is formed by the sidechains of Thr283 and His285 in the Aβ strand and a backbone carbonyl group in the loop following Aβ and the other is formed between the sidechains of Gln377 and Ser359 (Fig. 2A). When TCDD is bound, Gln377 also forms an H-bond to the medial oxygen atom of TCDD (Fig. 2A). The former H-bond network is in a linear arrangement near the planar hydrophobic surface of the ligand (Fig. 2A). The hydrophobic contacts would strengthen this H-bond network, which in turn energetically favor ligand binding. Ala375, a residue whose genetic variations have a strong effect on the AHR activity (Chang et al., 1993; Ema et al., 1994; Poland et al., 1994), is located between the two H-bond networks and in a close proximity to the ligand medial positions (Fig. 2A).
FIG. 2.

The AHR medial residues in ligand binding. (A) The interactions of the AHR medial residues to TCDD (dark grey) and BaP (light grey). TCDD and BaP are in cylinder. The AHR is in ribbon. AHR residues are in ball-and-stick, except A375, whose Cα/Cβ atoms are shown in grey spheres. The H-bond interactions are shown in dashed lines. The schematic structures of TCDD and BaP are shown (right). (B) The expression level of WT mAHR and the mAHR medial-residue mutants in COS-1 cells. (C) Induction of the transcriptional activity of the WT and mAHR mutants bearing medial-residue mutations by TCDD and BaP. COS-1 cells expressing WT or mutant mAHR were treated by TCDD (0.2 nM) or BaP (0.4 nM) followed by determination of the induced luciferase activity. The level of induction was normalized to that of the WT mAHR. Experiments were performed in triplicate and repeated twice. Mean ± SEM were calculated and shown. (D) The response curves of the WT and mutant mAHR to TCDD. COS-1 cells expressing WT or mutant mAHR were treated by increasing concentrations of TCDD for 4h followed by determination of the induced luciferase activity. The graph represents normalized data from 0 to 100% response. Experiments were performed in triplicates and repeated twice; representative results are shown. (E) The response curves of the WT and mutant mAHR to BaP, which were determined similar to that for TCDD (D).
Genetic variations at residue 375 distinguish the B- and D-allele of mAHR. The D-allele harbors a valine instead of alanine (Supplementary figure S3) and exhibits a reduced binding affinity between the AHR and TCDD (Chang et al., 1993; Ema et al., 1994; Poland et al., 1994). Based on our model (Fig. 2A), a bulkier residue at this position creates repulsive contacts to the ligand and alters the adjacent H-bond networks, which explain the weakened ligand binding of the D-allele (Chang et al., 1993; Ema et al., 1994; Poland et al., 1994). A nonresponsive Ahr allele identified in zebrafish (drAHR1a) (Andreasen et al., 2002) also harbors a genetic variation for this residue, A375T (Supplementary figure S3), which would weaken ligand binding similar to A375V. In addition, drAHR1a harbors a genetic variation at residue 285, H285Y, which is expected to disrupt the geometry required for the H-bond interactions and exert a steric effect on ligand binding (Fig. 2A). Combination of A375T and H285Y would completely abolish ligand binding, underlying why the drAHR1a allele is not responsive and unable to bind ligands (Andreasen et al., 2002).
The close contact of the AHR medial residues to the ligands and the distinct H-bond interactions led us to determine how the size and H-bond potential of these residues control ligand preferences. BaP and TCDD are ideal model ligands because BaP is bulkier at the medial axis and shorter at the lateral axis than TCDD. In addition, TCDD possesses medial oxygen atoms that are absent in BaP and form a unique H-bond interaction with the sidechain of Gln377 (Fig. 2A). The recombinant WT or mutant mAHRs (Q337S, Q377A, S359A/Q377A, T283A, H285S, H285A, T283A/H285L, A375V) were expressed in COS-1 cells that stably express a luciferase reporter driven by a DRE (dioxin response element)-containing segment of the mouse CYP1A promoter (DRE-Luc). The recombinant AHR proteins showed similar expression levels and same protein size (Fig. 2B). The COS-1 cells expressing the WT and mutant mAHRs were treated with TCDD (0.2nM) and BaP (0.4nM) (which correspond to the EC25 doses of TCDD and BaP in COS-1 cells expressing the WT mAHR) for 4h, and the induction level of the AHR activity was measured and normalized to that of WT mAHR (Fig. 2C). The cells expressing mAHR Q377S, which has a smaller residue that maintains the H-bond potential at 377, displayed a 3-fold higher AHR activity induced by BaP compared with the cells expressing WT mAHR but exhibited no change in the AHR activity induced by TCDD. The cells expressing Q377A mutant, however, resulted in a decrease of AHR activity induced by either TCDD or BaP. This is likely because Ser359 becomes nonengaged in H-bond interaction and its polarity perturbs the local conformation of the ligand binding pocket. Indeed, the mAHR S359A/Q377A displayed a 2.7-fold higher AHR activity induced by BaP and a 0.6-fold AHR activity induced by TCDD. These results show that a smaller residue at 377 selectively enhances BaP binding and a loss of the H-bond potential specifically affects TCDD binding. The H285S, T283A, H285A, and T283A/H285L mutant proteins lost their basal AHR activity and ligand binding activity to both TCDD and BaP. The A375V mutation remarkably reduced the induction level of AHR activity by either TCDD or BaP.
To estimate the change of the AHR ligand preferences by mutations at residue 377, we further determined the response curves of TCDD and BaP and calculated the EC50 values (Figs. 2D and E). In COS-1 cells expressing Q377A and S359A/Q377A, the EC50s of TCDD were 2.8- and 2.3-fold higher than that in the WT mAHR expressing cells, whereas the Q377S mutant barely affected the EC50 of TCDD (Fig. 2D; Table 1). In contrast, the EC50s of BaP for Q377S and S359A/Q377A mutations were 14- and 10-fold lower than that for WT mAHR (Fig. 2E; Table 1). These results further demonstrate how the size and H-bond potential of residue 377 differentially affect BaP and TCDD binding. Notably, S359A/Q377A alters the relative efficacy of TCDD and BaP by more than 20-fold (Table 1).
Table 1.
Demographic and clinical characteristics of BCC case subjects and control subjects, the Childhood Cancer Survivor Study*
| Characteristics | Case subjects (n = 199), N (%) | Control subjects (n = 597), N (%) | OR† (95% CI) | OR‡ (95% CI) | ||||
|---|---|---|---|---|---|---|---|---|
| Sex | ||||||||
| Male | 106 (53.3) | 285 (47.7) | 1.0 (referent) | – | ||||
| Female | 93 (46.7) | 312 (52.3) | 0.9 (0.6 to 1.5) | – | ||||
| Race | ||||||||
| White | 182 (91.5) | 533 (89.3) | 1.8 (0.8 to 4.3) | – | ||||
| Other | 17 (8.0) | 64 (10.7) | 1.0 (referent) | – | ||||
| First primary cancer diagnosis | ||||||||
| Wilms tumor, soft tissue sarcoma, bone cancer, or neuroblastoma | 20 (10.1) | 191 (32.0) | – | 1.0 (referent) | ||||
| Central nervous system cancer | 17 (8.5) | 59 (9.9) | – | 2.3 (0.9 to 5.9) | ||||
| Hodgkin lymphoma | 100 (50.3) | 144 (24.1) | – | 2.2 (1.0 to 4.9) | ||||
| Leukemia | 48 (24.1) | 151 (25.3) | – | 1.5 (0.7 to 3.2) | ||||
| Non–Hodgkin lymphoma | 14 (7.0) | 52 (8.7) | – | 1.8 (0.7 to 4.8) | ||||
| Year of first primary cancer diagnosis | ||||||||
| 1970–1973 | 59 (29.6) | 196 (32.8) | 0.4 (0.2 to 0.9)§ | – | ||||
| 1974–1977 | 65 (32.7) | 186 (31.2) | 0.6 (0.3 to 1.2) | – | ||||
| 1978–1981 | 44 (22.1) | 145 (24.3) | 0.6 (0.3 to 1.3) | – | ||||
| 1982–1986 | 31 (15.6) | 70 (11.7) | 1.0 (referent) | – | ||||
| Age at first primary cancer diagnosis||, y | ||||||||
| 0–4 | 32 (16.1) | 96 (16.1) | – | – | ||||
| 5–9 | 28 (14.1) | 84 (14.1) | – | – | ||||
| 10–14 | 47 (23.6) | 141 (23.6) | – | – | ||||
| 15–20 | 92 (46.2) | 276 (46.2) | – | – | ||||
| Age at BCC diagnosis, y | ||||||||
| 10–19 | 13 (6.5) | 48 (8.0) | – | – | ||||
| 20–29 | 61 (30.7) | 197 (33.0) | – | – | ||||
| 30–39 | 105 (52.8) | 297 (49.7) | – | – | ||||
| 40–50 | 20 (10.1) | 55 (9.2) | – | – | ||||
| Time from first primary cancer diagnosis to BCC diagnosis||, y | ||||||||
| 5–14 | 54 (27.1) | – | – | – | ||||
| 15–19 | 76 (38.2) | – | – | – | ||||
| 20–24 | 44 (22.1) | – | – | – | ||||
| 25–29 | 25 (12.6) | – | – | – | ||||
| Location of BCC | ||||||||
| Abdomen | 9 (4.5) | – | – | – | ||||
| Arm | 2 (1.0) | – | – | – | ||||
| Back | 41 (20.6) | – | – | – | ||||
| Chest | 42 (21.1) | – | – | – | ||||
| Head or face | 69 (34.7) | – | – | – | ||||
| Leg | 3 (1.5) | – | – | – | ||||
| Neck | 29 (14.6) | – | – | – | ||||
| Pelvis | 3 (1.5) | – | – | – | ||||
| Unknown | 1 (0.5) | – | – | – | ||||
| Skin color¶ | ||||||||
| Pale or milky white | 91 (48.4) | 200 (41.7) | 1.8 (0.9 to 3.7) | – | ||||
| Very light brown, sometimes freckles | 67 (35.6) | 148 (30.8) | 2.7 (1.3 to 5.6) | – | ||||
| Light tan, brown, olive, or dark brown | 30 (16.0) | 126 (26.3) | 1.0 (referent) | – | ||||
| Unknown | 0 (0) | 6 (1.3) | – | – | ||||
| Hair color¶ | ||||||||
| Light blond or blond | 19 (10.1) | 50 (10.5) | 2.6 (1.0 to 6.9) | – | ||||
| Strawberry blond, red, or red-brown | 16 (8.5) | 42 (8.7) | 1.6 (0.6 to 4.6) | – | ||||
| Light brown or medium brown | 121 (64.4) | 265 (55.2) | 3.1 (1.5 to 6.4) | – | ||||
| Dark brown or black | 31 (16.5) | 116 (24.2) | 1.0 (referent) | – | ||||
| Unknown | 1 (0.5) | 7 (1.5) | – | – | ||||
*OR = odds ratio; CI = confidence interval; BCC = basal cell carcinoma; – = not applicable.
†Multivariable analysis; model adjusted for sex, race, year of diagnosis, radiation dose, and Hodgkin lymphoma diagnosis.
‡Multivariable analysis; model adjusted for sex, race, year of diagnosis, and radiation dose.
§P trend = .20 (two-sided).
||Indicates matching variable.
¶Sun sensitivity descriptors as listed in the 2003 follow-up survey. Number of respondents to 2003 survey: 188 case subjects, 480 control subjects. Each sun sensitivity variable was assessed in a separate risk model.
Identification of AHR Residues That Favor Ligand Lateral Extension
Similar to our studies on the AHR medial residues, we expect that structure-based alterations of the AHR lateral residues would also differentially affect the AHR binding preference for TCDD and BaP. Our models of AHR-LBD bound to BaP and TCDD show that the lateral positions of the AHR ligand binding pocket have extra space between ligands and the AHR (Figs. 1C and 3A). Gly298 in helix Fα is located near the lateral positions of the ligands and is farther away from BaP than from TCDD (Fig. 3A), consistent with the shorter lateral dimension of BaP.
FIG. 3.
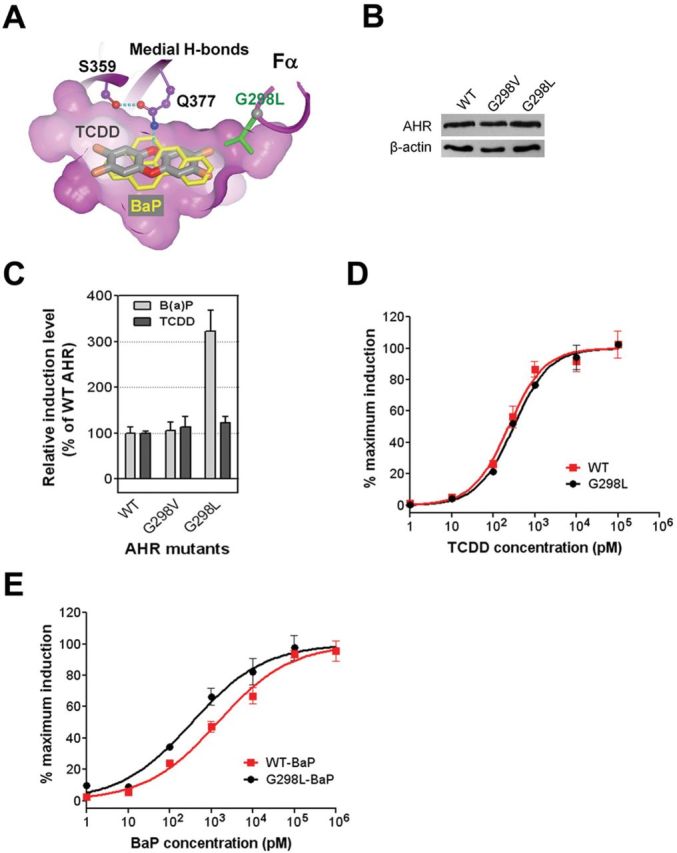
The AHR lateral residues in ligand binding. (A) The interactions of the AHR lateral residue to TCDD (dark) and BaP (light). A slice of surface of the ligand binding pocket shows the extra space at lateral positions, highlighting G298. The Cα atom of G298 is in grey sphere. G298L places the extended sidechain (cylinder) into the extra lateral space. (B) The expression level of the WT mAHR and mAHR mutants bearing mutations to G298. (C) Induction of the transcriptional activity of the WT mAHR and mAHR mutants bearing mutations to G298 by TCDD and BaP as in Figure 2C. (D) The response curves of the WT mAHR and mAHR G298L to TCDD determined as in Figure 2D. (E) The response curves of the WT mAHR and mAHR G298L to BaP determined as in Figure 2E.
We replaced Gly298 of the mAHR to valine and leucine to mimic ligand lateral extension by the length of two-carbon and three-carbon chains, respectively. Using the same methodology shown in Figs. 2C and E, we investigated the effect of these mutations on the AHR ligand preferences. The replacements of Gly298 (G298V, G298L) did not impact the expression level and the size of the protein (Fig. 3B). The G298L mutation led to increase in the induction level of AHR activity by the EC25 concentration of BaP (3.2-fold), whereas the TCDD-induced AHR activity was slightly increased (1.2-fold) (Fig. 3C). In contrast to G298L, the G298V mutation barely influenced the AHR ligand preferences (Fig. 3C). The response curves showed that the G298L mutant barely affected the EC50 of TCDD but reduced the EC50 of BaP by 5-fold (Figs. 3D and E, Table 1). This result revealed that residue Gly298 is an important structural determinant that controls ligand preferences by favoring ligand lateral extension.
The “Belt” and Other Flexible Structural Elements in AHR Ligand Binding and Tolerance
The close contacts of the “belt” to the medium positions of the ligand (Figs. 1B and 4A) suggest that the AHR “belt” is critical for ligand binding. Two hydrophobic residues within the “belt”, F318 and I319, are predicted to directly contact the ligand (Supplementary figure S2A), among which F318 was previously shown to be important for ligand binding (Pandini et al., 2007; Pandini et al., 2009). The extended length of the “belt” along the long axis of the ligand binding cavity suggests that the “belt” might be flexible and contribute to the ability of the AHR to tolerate diverse ligands. The “belts” in the AHR and HIF2α are very similar and differ in length by one residue (Fig. 1A). Alignment of several crystal structures of HIF2α bound to ligands and its NMR structures in the absence of ligands showed that the “belt” is in distinctly different positions in the ligand-bound form versus the unbound form (Supplementary figure S4). Although the AHR “belt” contains only one extra glycine residue compared with that of HIF2α (Fig. 1A), it defines a much larger ligand binding pocket (Fig. 1A). This suggests that the AHR “belt” can undergo larger and more diverse changes upon binding to ligands, underlying why the AHR, but not HIF2α, can bind to diverse ligands.
To test this hypothesis, we created mAHR mutants harboring deletion of G313 or G315 to reduce the flexibility of the “belt” to a level similar to that of HIF2α (Fig. 4A). Neither mutation affected the expression level and size of the AHR protein (Fig. 4B). Deletion of G313 (∆G313) exhibited a stronger effect on the AHR signaling in response to BaP at the EC25 concentration than the signal induced by TCDD (Fig. 4C). Deletion of G315 (∆G315) abolished the AHR signaling in response to both ligands (Fig. 4C). Next, we measured the response curves of mAHR∆G313 to TCDD and BaP. The EC50 of TCDD was increased 2-fold by deletion of G313 (Fig. 4D; Table 1), and that for BaP was increased 2.7-fold (Fig. 4E; Table 1). These observations suggest that the “belt” contributes to the ability of the AHR to tolerate diverse ligands. Gly315 in the “belt” is critical for ligand binding, which is due to its direct contacts to the ligand and its contribution to the flexibility of the “belt” (Fig. 4A).
FIG. 4.
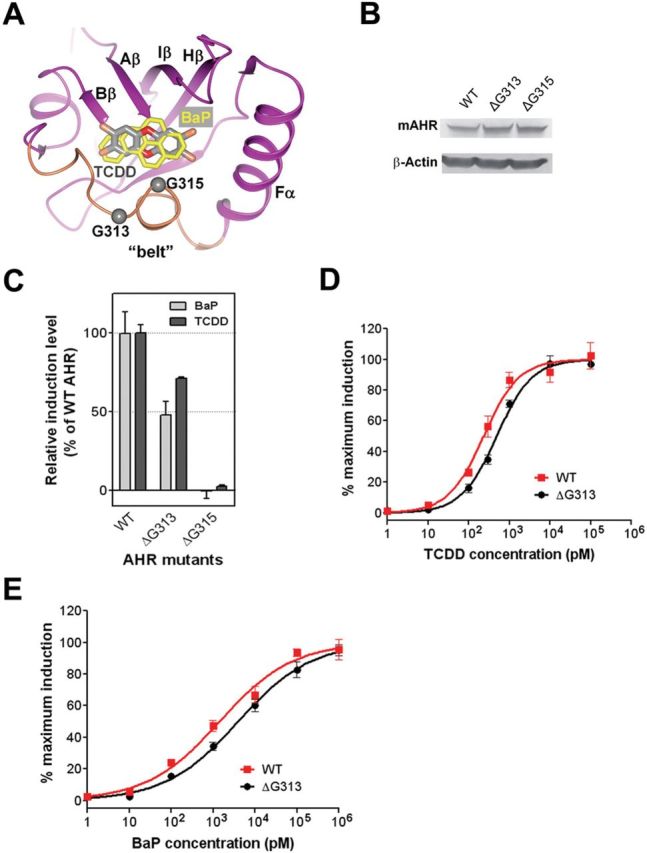
The AHR “belt” in ligand binding. (A) Overlay of the TCDD (dark grey) and BaP (light grey) in the ligand binding pocket of the AHR. The model is shown similar to Figure 2A. The Aβ-Bβ loop is not shown for visualization of the ligands. The Cα atoms of G313 and G315 are shown in grey spheres. (B) The expression level of recombinant WT mAHR and the mAHR mutants bearing glycine residue deletions in COS-1 cells. Deletion of G313 or G315 did not affect the protein level of recombinant mAHR. (C) Induction of the transcriptional activity of WT or mutant mAHR by TCDD and BaP measured as in Figure 2C. (D) The response curves of the WT mAHR and mAHR ∆G313 to TCDD determined as in Figure 2D. (E) The response curves of the WT mAHR and mAHR ∆G313 to BaP determined as in Figure 2E.
To identify other structural elements in the AHR-LBD that might contribute to ligand promiscuity, we performed MD simulation of the AHR-LBD and HIF2α. Several structural elements in the AHR-LBD appear to have a higher degree of flexibility than those in HIF2α (Fig. 5). Besides the “belt”, the Aβ-Bβ loop, Hβ/Iβ strands, and Fα helix are significantly more flexible in the AHR-LBD than in HIF2α, particularly the Aβ-Bβ loop and the Hβ strand (Fig. 5). Although these structural elements are not the most flexible regions in the structure and each may adopt slightly different conformations, collectively they might make important contributions to the ability of the AHR to tolerate different ligands.
FIG. 5.

Molecular dynamics of the human AHR-LBD and HIF2α. The structures are illustrated in worm and colored based on B factors. Blue indicates the most rigid regions and red the most flexible regions of the structures. Among the structural elements lining the ligand binding pocket, the AHR “belt”, Aβ-Bβ loop, and Hβ strand are significantly more flexible than those in HIF2α, and the AHR Fα helix and Iβ strand have a slightly higher flexibility than those in HIF2α.
In Silico Docking and Ranking of Binding Affinity of TCDD Congeners
The toxicity of the AHR ligands has been estimated by Toxic Equivalency Factors, in which the potencies of individual compounds to activate the AHR signal have been normalized to that of TCDD (Van den Berg et al., 2006). An important application of the model of the AHR-LBD is the in silico estimation of ligand binding affinity to the AHR and predictions of their relative toxicity. If such an approach can be shown to be successful, it will provide a rapid and unbiased method to determine the potential threat of environmental toxicants on human health (Hartung et al., 2009). To test our model for such use, we first generated 10 models of human AHR in Sybyl with slightly different conformations in the “belt” and the Aβ-Bβ loop based on the MD simulation result and screened for a model that most accurately reflects the known rank order of the binding affinities of dioxin congeners. We hypothesize that these structural elements adopt different conformations to accommodate different families of the AHR ligands (i.e., dioxins vs. PAHs). After energy minimization of each AHR model, TCDD was docked with the sidechain of residues lining the ligand binding pocket set in a flexible mode. This allows optimization of the model to obtain a maximum binding affinity for TCDD. Each optimized model was then used for systematic docking of seven TCDD congeners (Figs. 6A and B) in a fixed mode to obtain the docking energy and binding affinity.
The binding energy of TCDD to the 10 models varied in between −7.17 kcal/mol and −7.84 kcal/mol, consistent with the subtle modifications made to each model. The calculated binding affinity is still three orders of magnitude smaller than the experimental binding affinity, reflecting the systematic errors in the model and the algorithm for calculating docking energy. Nonetheless, the systematic errors of modeling and molecular docking might not affect the accuracy of in silico ranking because the predicted binding affinity can be normalized to that of TCDD. To determine which of the models above were useful for ranking halogenated dioxin congeners, the in silico binding affinity of all six test congeners to each model was normalized to that of TCDD and then compared to the experimentally determined affinity or toxicity that was also normalized to TCDD. Experimental data on the binding affinity or toxicity of TCDD congeners were summarized from published data sets (Ashek et al., 2006; Safe, 1986). Based on the correlation between the in silico ranking and the experimental ranking, the model that gave the most accurate ranking was identified, which correlated well with the experimental ranking (Fig. 6C). The correlation coefficient is calculated to be 0.98±0.07.
FIG. 6.

Correlation of the computational and experimental ranking of the binding affinity of TCDD congeners to the AHR. (A) Numbering of replacement positions in dibenzo-p-dioxin. (B) A list of TCDD congeners with known measured binding affinity. (C) The relative binding affinity based on the binding energy of computational docking versus the relative binding affinity measured in comparison to TCDD. The calculated correlation coefficient is 0.98±0.07.
DISCUSSION
The structure of the AHR-LBD had been modeled by different groups (Bisson et al., 2009; Jogalekar et al., 2010; Pandini et al., 2007; Pandini et al., 2009; Procopio et al., 2002), and residues that line the ligand binding cavity with a critical role in ligand binding had been identified by mutational analysis (Pandini et al., 2007; Pandini et al., 2009). Identification of these residues, however, provides little information on how the AHR recognizes diverse ligands with distinctly different shapes and chemical properties and what structural features of the AHR affect ligand preferences.
Built on our models of the AHR-LBD bound to TCDD and BaP, our structural and mutational analysis revealed novel structural elements that play a key role in controlling ligand preferences. We showed that the AHR medial residues control ligand preferences by both shape and H-bond potential. Gln377, a polar AHR medial residue, makes a unique H-bond to TCDD oxygen atom (Fig. 2A). Disruption of this H-bonding interaction by single mutation Q377A or double mutation Q377A/S359A reduced the AHR signaling to TCDD (Fig. 2D). In contrast, Q377S and Q377A/S359A increased the efficacy of BaP (Fig. 2E), presumably due to the reduced residue size that relieves the repulsive contacts to the medial positions of BaP. By modulating the size and H-bond potential of AHR residues, we showed vividly that the Q377A/S359A mutation decreased the ratio of TCDD/BaP efficacy by more than 20-fold, and BaP becomes 3- to 7-fold more potent than TCDD (Table 1). Similarly, we identified an AHR lateral residue Gly298, which affects the ligand preferences by residue size. Extension of this residue by the size of a two-carbon chain (G298V) barely affected the AHR signaling, whereas extension by a three-carbon chain (G298L) exhibited a significant effect on the AHR signaling to BaP but exhibited little effect on that to TCDD (Figs. 3C and E). As a consequence, G298L reduced the ratio of TCDD/BaP efficacy by about 5-fold (Table 1).
Our studies also provide insights into the structural basis of the AHR to tolerate diverse ligands. MD simulation suggests that a long, flexible “belt” structure and several other structural elements lining the AHR ligand binding pocket exhibit a higher flexibility than those in HIF2α (Fig. 5). These structural elements likely undergo conformational changes and collectively contribute to AHR ligand promiscuity. Most importantly, Gly315 in the AHR “belt”, which is missing in HIF2α, is critical for the AHR signaling induced by either TCDD or BaP, likely by contributing to a greater flexibility of the AHR “belt” and by directly lining the ligand binding pocket of the AHR (Fig. 4). Understanding the conformational flexibility of the AHR-LBD is important for modeling such that proper models of the AHR-LBD can be made to facilitate in silico ranking of the binding affinities of AHR ligands (Fig. 6).
Elucidation of the structural determinants of the AHR that control ligand binding and ligand preferences not only provide clear explanations for previous observations from genetic, biochemical, and 3D-QSAR studies but more importantly also provide detailed structural knowledge for extrapolating the structural features of the ligands that favor or disfavor ligand binding with a greater accuracy and confidence (Fig. 7). Using BaP as a model ligand, which is bulky at medial positions and short at lateral positions, our results suggest that reducing the size of the medial residue 377 by the length of a two-carbon chain without disrupting the H-bond potential (Gln → Ser) or increasing the size of the lateral residue 298 by the length of a three-carbon chain (Gly → Leu) (Fig. 7) significantly increased the efficacy of BaP (Figs. 2 and 3). These results allow us to predict that an AHR ligand with the shape similar to BaP would have an increased efficacy by shortening the medial positions of the ligand by the size of a two-carbon chain and extending the lateral positions of the ligand by the size of a three-carbon chain (Fig. 7). Furthermore, the novel H-bond interaction between the AHR medial residue 377 and TCDD suggest that the AHR ligands can be categorized by medial H-bond potential. More studies along this line for defining AHR structural determinants that control ligand preferences would allow us to define the range of chemical shape and pattern of H-bond potential suitable for binding to the AHR. This would greatly facilitate prediction of the efficacy of AHR ligands and identification of unknown ligands.
FIG. 7.

A model illustrating extrapolation of the structural features of the AHR ligands that would favor receptor binding. Q377S and G298L enhance the AHR response to BaP through decrease of the medial residue by the size of a two-carbon chain and increase of the lateral residue by the size of a three-carbon chain, respectively. Thus, the AHR ligands that are shorter at medial positions than BaP by the distance of a two-carbon chain and longer at lateral positions than BaP by the size of a three-carbon chain are expected to have a significantly higher efficacy in stimulating the AHR signaling than BaP.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
University of Wisconsin at Madison startup fund (Y.X.); National Institutes of Health (P30CA014520 to C.A.B.); American Cancer Society Research Scholar Grant (RSG-10–153-01-DMC to Y.X.); National Institutes of Health (2R56AI063325-06A1 to Q.C.).
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Dr Norman Drinkwater for suggestions on data statistics.
REFERENCES
- Andreasen E. A., Hahn M. E., Heideman W., Peterson R. E., Tanguay R. L. (2002).. The zebrafish (Danio rerio) aryl hydrocarbon receptor type 1 is a novel vertebrate receptor. Mol. Pharmacol. 62, 234––249 [DOI] [PubMed] [Google Scholar]
- Arulmozhiraja S., Morita M. (2004).. Structure-activity relationships for the toxicity of polychlorinated dibenzofurans: Approach through density functional theory-based descriptors. Chem. Res. Toxicol. 17, 348––356 [DOI] [PubMed] [Google Scholar]
- Ashek A., Lee C., Park H., Cho S. J. (2006).. 3D QSAR studies of dioxins and dioxin-like compounds using CoMFA and CoMSIA. Chemosphere 65, 521––529 [DOI] [PubMed] [Google Scholar]
- Bisson W. H., Koch D. C., O’Donnell E. F., Khalil S. M., Kerkvliet N. I., Tanguay R. L., Abagyan R., Kolluri S. K. (2009).. Modeling of the aryl hydrocarbon receptor (AhR) ligand binding domain and its utility in virtual ligand screening to predict new AhR ligands. J. Med. Chem. 52, 5635––5641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock K. W., Köhle C. (2006).. Ah receptor: Dioxin-mediated toxic responses as hints to deregulated physiologic functions. Biochem. Pharmacol. 72, 393––404 [DOI] [PubMed] [Google Scholar]
- Carver L. A., LaPres J. J., Jain S., Dunham E. E., Bradfield C. A. (1998).. Characterization of the Ah receptor-associated protein, ARA9. J. Biol. Chem. 273, 33580––33587 [DOI] [PubMed] [Google Scholar]
- Chan W. K., Chu R., Jain S., Reddy J. K., Bradfield C. A. (1994).. Baculovirus expression of the Ah receptor and Ah receptor nuclear translocater. Evidence for additional dioxin responsive element-binding species and factors required for signaling. J. Biol. Chem. 269, 26464––26471 [PubMed] [Google Scholar]
- Chang C., Smith D. R., Prasad V. S., Sidman C. L., Nebert D. W., Puga A. (1993).. Ten nucleotide differences, five of which cause amino acid changes, are associated with the Ah receptor locus polymorphism of C57BL/6 and DBA/2 mice. Pharmacogenetics 3, 312––321 [DOI] [PubMed] [Google Scholar]
- Coumailleau P., Poellinger L., Gustafsson J. A., Whitelaw M. L. (1995).. Definition of a minimal domain of the dioxin receptor that is associated with Hsp90 and maintains wild type ligand binding affinity and specificity. J. Biol. Chem. 270, 25291––25300 [DOI] [PubMed] [Google Scholar]
- Dolwick K. M., Swanson H. I., Bradfield C. A. (1993).. In vitro analysis of Ah receptor domains involved in ligand-activated DNA recognition. Proc. Natl. Acad. Sci. U.S.A. 90, 8566––8570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ema M., Ohe N., Suzuki M., Mimura J., Sogawa K., Ikawa S., Fujii-Kuriyama Y. (1994).. Dioxin binding activities of polymorphic forms of mouse and human aryl hydrocarbon receptors. J. Biol. Chem. 269, 27337––27343 [PubMed] [Google Scholar]
- Esser C., Rannug A., Stockinger B. (2009).. The aryl hydrocarbon receptor in immunity. Trends Immunol. 30, 447––454 [DOI] [PubMed] [Google Scholar]
- Frenkel D., Smit B. (1996).. Understanding Molecular Simulations: From Algorithms to Applications. Academic Press, San Diego, CA [Google Scholar]
- Goodsell D. S., Morris G. M., Olson A. J. (1996).. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 9, 1––5 [DOI] [PubMed] [Google Scholar]
- Han D., Nagy S. R., Denison M. S. (2004).. Comparison of recombinant cell bioassays for the detection of Ah receptor agonists. Biofactors 20, 11––22 [DOI] [PubMed] [Google Scholar]
- Hartung T., Blaauboer B., Leist M. (2009).. Food for thought… on education in alternative methods in toxicology. ALTEX. 26, 255––263 [DOI] [PubMed] [Google Scholar]
- Hayes K. R., Zastrow G. M., Nukaya M., Pande K., Glover E., Maufort J. P., Liss A. L., Liu Y., Moran S. M., Vollrath A. L., Bradfield C. A. (2007).. Hepatic transcriptional networks induced by exposure to 2,3,7,8- tetrachlorodibenzo-p-dioxin. Chem. Res. Toxicol. 20, 1573––1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008).. Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435––447 [DOI] [PubMed] [Google Scholar]
- Ikuta T., Eguchi H., Tachibana T., Yoneda Y., Kawajiri K. (1998).. Nuclear localization and export signals of the human aryl hydrocarbon receptor. J. Biol. Chem. 273, 2895––2904 [DOI] [PubMed] [Google Scholar]
- Jogalekar A. S., Reiling S., Vaz R. J. (2010).. Identification of optimum computational protocols for modeling the aryl hydrocarbon receptor (AHR) and its interaction with ligands. Bioorg. Med. Chem. Lett. 20, 6616––6619 [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983).. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926––935 [Google Scholar]
- Kazlauskas A., Poellinger L., Pongratz I. (1999).. Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin (aryl hydrocarbon) receptor. J. Biol. Chem. 274, 13519––13524 [DOI] [PubMed] [Google Scholar]
- Korashy H. M., El-Kadi A. O. (2006).. The role of aryl hydrocarbon receptor in the pathogenesis of cardiovascular diseases. Drug Metab. Rev. 38, 411––450 [DOI] [PubMed] [Google Scholar]
- Li Y., Innocentin S., Withers D. R., Roberts N. A., Gallagher A. R., Grigorieva E. F., Wilhelm C., Veldhoen M. (2011).. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 147, 629––640 [DOI] [PubMed] [Google Scholar]
- McMillan B. J., Bradfield C. A. (2007).. The aryl hydrocarbon receptor sans xenobiotics: Endogenous function in genetic model systems. Mol. Pharmacol. 72, 487––498 [DOI] [PubMed] [Google Scholar]
- Meyer B. K., Perdew G. H. (1999).. Characterization of the AhR-hsp90-XAP2 core complex and the role of the immunophilin-related protein XAP2 in AhR stabilization. Biochemistry 38, 8907––8917 [DOI] [PubMed] [Google Scholar]
- Meyer B. K., Pray-Grant M. G., Vanden Heuvel J. P., Perdew G. H. (1998).. Hepatitis B virus X-associated protein 2 is a subunit of the unliganded aryl hydrocarbon receptor core complex and exhibits transcriptional enhancer activity. Mol. Cell. Biol. 18, 978––988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezrich J. D., Fechner J. H., Zhang X., Johnson B. P., Burlingham W. J., Bradfield C. A. (2010).. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 185, 3190––3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L. P., Bradfield C. A. (2008).. The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 21, 102––116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz C. A., Litzenburger U. M., Sahm F., Ott M., Tritschler I., Trump S., Schumacher T., Jestaedt L., Schrenk D., Weller M., et al. (2011).. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197––203 [DOI] [PubMed] [Google Scholar]
- Pandini A., Denison M. S., Song Y., Soshilov A. A., Bonati L. (2007).. Structural and functional characterization of the aryl hydrocarbon receptor ligand binding domain by homology modeling and mutational analysis. Biochemistry 46, 696––708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandini A., Soshilov A. A., Song Y., Zhao J., Bonati L., Denison M. S. (2009).. Detection of the TCDD binding-fingerprint within the Ah receptor ligand binding domain by structurally driven mutagenesis and functional analysis. Biochemistry 48, 5972––5983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdew G. H. (1988).. Association of the Ah receptor with the 90-kDa heat shock protein. J. Biol. Chem. 263, 13802––13805 [PubMed] [Google Scholar]
- Petrulis J. R., Kusnadi A., Ramadoss P., Hollingshead B., Perdew G. H. (2003).. The hsp90 Co-chaperone XAP2 alters importin beta recognition of the bipartite nuclear localization signal of the Ah receptor and represses transcriptional activity. J. Biol. Chem. 278, 2677––2685 [DOI] [PubMed] [Google Scholar]
- Poland A., Glover E., Kende A. S. (1976).. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J. Biol. Chem. 251, 4936––4946 [PubMed] [Google Scholar]
- Poland A., Palen D., Glover E. (1994).. Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol. Pharmacol. 46, 915––921 [PubMed] [Google Scholar]
- Procopio M., Lahm A., Tramontano A., Bonati L., Pitea D. (2002).. A model for recognition of polychlorinated dibenzo-p-dioxins by the aryl hydrocarbon receptor. Eur. J. Biochem. 269, 13––18 [DOI] [PubMed] [Google Scholar]
- Reyes H., Reisz-Porszasz S., Hankinson O. (1992).. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 256, 1193––1195 [DOI] [PubMed] [Google Scholar]
- Safe S. H. (1986).. Comparative toxicology and mechanism of action of polychlorinated dibenzo-p-dioxins and dibenzofurans. Annu. Rev. Pharmacol. Toxicol. 26, 371––399 [DOI] [PubMed] [Google Scholar]
- Savouret J. F., Berdeaux A., Casper R. F. (2003).. The aryl hydrocarbon receptor and its xenobiotic ligands: A fundamental trigger for cardiovascular diseases. Nutr. Metab. Cardiovasc. Dis. 13, 104––113 [DOI] [PubMed] [Google Scholar]
- Scheuermann T. H., Tomchick D. R., Machius M., Guo Y., Bruick R. K., Gardner K. H. (2009).. Artificial ligand binding within the HIF2alpha PAS-B domain of the HIF2 transcription factor. Proc. Natl. Acad. Sci. U.S.A. 106, 450––455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J. V., Bradfield C. A. (1996).. Ah receptor signaling pathways. Annu. Rev. Cell Dev. Biol. 12, 55––89 [DOI] [PubMed] [Google Scholar]
- Stevens E. A., Mezrich J. D., Bradfield C. A. (2009).. The aryl hydrocarbon receptor: A perspective on potential roles in the immune system. Immunology 127, 299––311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berg M., Birnbaum L. S., Denison M., De Vito M., Farland W., Feeley M., Fiedler H., Hakansson H., Hanberg A., Haws L., et al. (2006).. The 2005 World Health Organization reevaluation of human and Mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol. Sci. 93, 223––241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock J. P., Jr. (1999).. Induction of cytochrome P4501A1. Annu. Rev. Pharmacol. Toxicol. 39, 103––125 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.