

Abstract
Huntington's disease (HD) is an incurable neurological disorder caused by an abnormal glutamine repeat expansion in the huntingtin (Htt) protein. In the present studies, we investigated the role of Transducers of Regulated cAMP response element-binding (CREB) protein activity (TORCs) in HD, since TORCs play an important role in the expression of the transcriptional co-regulator peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α), whose expression is impaired in HD. We found significantly decreased TORC1 expression levels in STHdhQ111 cells expressing mutant Htt, in the striatum of NLS-N171-82Q, R6/2 and HdhQ111 HD transgenic mice and in postmortem striatal tissue from HD patients. TORC1 overexpression in wild-type (WT) and Htt striatal cells increased CREB mRNA and protein levels, PGC-1α promoter activity, mRNA expression of the PGC-1α, NRF-1, Tfam and CytC genes, mitochondrial DNA content, mitochondrial activity and mitochondrial membrane potential. TORC1 overexpression also increased the resistance of striatal cells to 3-nitropropionic (3-NP) acid-mediated toxicity. In cultured WT and mutant Htt striatal cells, small hairpin RNA-mediated TORC1 knockdown resulted in decreased PGC-1α expression and increased susceptibility to 3-NP-induced toxicity. Overexpression of PGC-1α partially prevented TORC1 knockdown-mediated increased susceptibility of Htt striatal cells to 3-NP. Specific knockdown of TORC1 in the striatum of NLS-N171-82Q HD transgenic mice induced neurodegeneration. Lastly, knockdown of Htt prevents transcriptional repression of TORC1 and CREB in Htt striatal cells. These findings show that impaired expression and function of TORC1, which results in a reduction in PGC-1α, plays an important role in mitochondrial dysfunction in HD.
INTRODUCTION
Huntington's disease (HD) is an incurable and fatal autosomal-dominant neurodegenerative disease and characterized by neuronal degeneration in the striatum, and other regions of the brain, with progressive behavioral and cognitive deficits and involuntary choreiform movements. HD is caused by an abnormal CAG repeat expansion in exon 1 of the HD gene, resulting in formation of an increased polyglutamine region in the mutant huntingtin (Htt) protein. How the mutant Htt protein elicits its toxic effects remains elusive, but several mechanisms have been postulated including transcriptional dysregulation, abnormalities in mitochondrial energy metabolism, protein aggregation, protein disposal and oxidative damage (1).
Mitochondrial dysfunction and bioenergetic defects in HD occur by several different mechanisms (1–5). First, mutant Htt may interact directly with mitochondria. Studies in an HD mouse model and cultured HD striatal cells (STHdhQ111) show that mutant Htt associates directly with the outer mitochondrial membrane (6). Electron microscopic studies in neurons show the localization of N-terminal mutant Htt on mitochondria (7). Second, mutant Htt impairs in vitro and in vivo trafficking of mitochondria in neurons, leading to loss of mitochondrial motility and eventually to mitochondrial dysfunction (8–10). Third, mutant Htt impairs mitochondrial function by altering gene transcription (11,12). Mutant Htt directly interacts with and down-regulates the activity of several transcription factors including p53, cAMP response element-binding (CREB) protein, TAFII130 and SP1 (13–16). CREB is reduced in HD and mutant Htt represses the expression of CREB by a direct interaction with the CREB-binding protein (CBP) (13,14,17–20). Overexpression of CBP rescues polyglutamine-induced neuronal toxicity (14).
Recently, an interaction of mutant Htt with peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) was shown to play a role in HD pathogenesis (21). PGC-1α is a coactivator of several transcription factors and a key regulator of mitochondrial biogenesis, energy homeostasis, adaptive thermogenesis and glucose metabolism. PGC-1α activates a diverse set of metabolic programs in different tissues by forming complexes with several transcription factors, including nuclear respiratory factors (NRF-1 and NRF-2) and nuclear hormone receptors (PPARα, PPARγ, ERRα and thyroid receptor) (22,23). PGC-1α expression and activity are impaired in cell models and transgenic mouse models of HD and in the brain and muscle tissues from HD patients (19,21,24,25). Mutant Htt protein impairs the ability of PGC-1α to activate downstream target genes involved in adaptive thermogenesis, resulting in hypothermia in HD transgenic mice (24). Mutant Htt also binds to the CREB/TAF4 complex which impairs the activation of the PGC-1α promoter and the transcription of PGC-1α leading to the down-regulation of its mitochondrial target genes, such as the mitochondrial transcription factor A (Tfam) and cytochrome c, which leads to abnormalities in mitochondrial function and energy metabolism (21,25). Overexpression of PGC-1α in muscle and brain tissues reduces mitochondrial dysfunction and enhances mitochondrial biogenesis in transgenic HD mice (19,21). On the other hand, the selective ablation of PGC-1α leads to increased striatal neuron degeneration and increased susceptibility to the mitochondrial toxin 3-NP in HD transgenic mice (21). We showed that there are reductions in PGC-1α in HD postmortem brain tissue which correlate with reductions in numbers of mitochondria (26).
Transducers of Regulated CREB Activity (TORCs) are coactivators of CREB, which enhance CREB-dependent gene transcription (27). The TORC family members TORC1, TORC2 and TORC3 not only induce CREB activity, but also strongly regulate PGC-1α promoter activity, transcription and mitochondrial biogenesis (28). TORC1 is highly expressed in the brain, while TORC2 and TORC3 are abundantly expressed in the lung and muscle (28). TORC1 plays a critical role in dendritic growth of developing cortical neurons, through the positive regulation of activity-dependent CREB target gene transcription (29). In Drosophila, TORC promotes stress resistance and maintains energy balance under starvation conditions, by induction of CREB target genes in the brain (30,31). TORCs also enhance neuronal survival after ischemia by the activation of CREB target genes (32).
TORCs are known to be critical regulators of both CREB and PGC-1α expression, whose transcription and activity are decreased in HD. Furthermore, both CREB and PGC-1α knockout mice exhibit the specific degeneration of hippocampal and striatal neurons and phenotypes reminiscent of HD (33–35). We, therefore, investigated TORC activity and function in HD striatal cells, HD transgenic mice and postmortem brain tissue from HD patients.
RESULTS
Impaired TORC1 mRNA transcription and protein levels in the striatum of HD transgenic mice lines and Htt striatal cells
Decreased PGC-1α and CREB transcription appear to play an important role in HD pathogenesis. TORC1 is a strong inducer of CREB transcription, which increases PGC-1α promoter activity, transcription and mitochondrial biogenesis (28). We, therefore, investigated whether TORC dysfunction contributes to the impairment of PGC-1α and CREB expression in HD. We analyzed TORC1, TORC2 and TORC3 mRNA expression, and protein levels, in HD striatal cell lines, in the striatum and cerebral cortex of three different HD transgenic mouse models and in HD postmortem brain tissue. We utilized NLS-N171-82Q, R6/2 and Hdh-Q111 knockin HD transgenic mice and striatal STHdhQ111 cells, an immortalized cell line made from the Hdh-Q111 mice (36–39).
We found significantly decreased TORC1 and TORC2 mRNA expression in the striatum of NLS-N171-82Q HD mice when compared with wild-type (WT) mice (Fig. 1A). TORC3 mRNA expression was not significantly altered in these mice. In the striatum of the R6/2 and Hdh-Q111 HD mice, TORC1 mRNA expression was significantly reduced by 30 and 52%, respectively, when compared with WT; however, there were no significant alterations in TORC2 and TORC3 mRNA expression (Fig. 1B and C). The TORC1 mRNA expression was significantly decreased in STHdhQ111/Q111 striatal cells (expressing endogenous mutant Htt with 111-CAG repeats) when compared with WT STHdhQ7/Q7 (expressing endogenous normal Htt) (Fig. 1D), but there were no significant changes in TORC2 and TORC3 mRNA expression (Fig. 1D). We observed a significant decrease in TORC1 protein levels in the NLS-N171-82Q, R6/2 and Hdh-Q111 HD transgenic mice, when compared with their respective WT mice (Fig. 1E and F). These results show that there are both decreased TORC1 mRNA expression and protein levels in the HD transgenic mice and immortalized striatal cells expressing mutant Htt.
Figure 1.

Decreased TORC1 mRNA expression and protein levels in the HD transgenic mice and HD striatal cells. Relative TORC isoforms (TORC1, TORC2 and TORC3) mRNA expression in the striatum of WT and HD transgenic mice such as NLS-N171-82Q (A), R6/2 (B) and STHdhQ111 (C). TORC1 mRNA expression is significantly reduced, and TORC3 is unchanged in the striatum of all three HD transgenic lines when compared with WT mice. TORC2 mRNA expression is significantly reduced in NLS-N171-82Q HD mice only. TORC mRNA expression was normalized to β-actin. Data are expressed as the mean ± standard error of the mean (SEM). *P < 0.05, versus WT (n = 6 mice in each group). (D) Relative TORC1, TORC2 and TORC3 mRNA expressions in the WT STHdhQ7 and mutant STHdhQ111 striatal cell lines. TORC1 mRNA expression is significantly decreased, while TORC2 and TORC3 are unchanged in the HD STHdhQ111 cell lines when compared with STHdhQ7. Values were normalized to β-actin. Graph represents the mean ± SEM of three independent experiments. *P < 0.05, versus STHdhQ7. (E) Western blot analysis of TORC1 protein levels in the striatum of WT and HD transgenic mice NLS-N171-82Q, R6/2 and HdhQ111. Values were normalized to β-actin, used as a loading control (n = 6 mice in each group). (F) Relative TORC1 protein density expressed after normalization with β-actin. Representative blots showing two samples from each group. A significant decrease in TORC1 protein levels is observed in all three HD transgenic mice lines when compared with WT mice. Mean ± SEM, *P < 0.05, versus WT (n = 6 mice in each group).
Decreased TORC1 transcription and protein levels in the striatum of HD patients
We examined whether the decreased transcriptional expression of TORC1 observed in the HD transgenic mice and Htt striatal cells also occurs in HD patients. We carried out quantitative real-time polymerase chain reaction (qRT-PCR) analysis of the mRNA expression of the TORC isoforms in striatal brain tissue from age-matched control subjects and symptomatic HD patients. We found a significant decrease (52%) of TORC1 mRNA expression in the striatum of HD patients when compared with control subjects (Fig. 2A). We also analyzed the mRNA expression of TORC2 and TORC3. TORC2 mRNA expression was unchanged in the HD patients (Fig. 2A) but TORC3 mRNA was significantly increased in the HD patients when compared with normal controls (Fig. 2A). We examined TORC1 protein levels in the striatum of HD patients and control subjects using western blots. There were significantly decreased TORC1 protein levels in the striatum of HD patients (Fig. 2B and C). These results show that both TORC1 transcription and protein levels are reduced in the striatum of HD patients.
Figure 2.

Altered mRNA expression of TORC isoforms (TORC1, TORC2 and TORC3) and decreased TORC1 protein levels in HD. (A) Total RNA was isolated from putamen of age-matched control subjects and HD patients. qRT-PCR analysis was performed for the relative mRNA expression of TORC1, TORC2 and TORC3 and normalized to β-actin. TORC1 mRNA expression is significantly decreased, TORC2 is unchanged and TORC3 is significantly increased in HD patients when compared with control subjects. Data are expressed as the mean ± SEM. *P < 0.05, versus control subjects (n = 6 subjects in each group). (B) Analysis of TORC1 protein levels by western blot in the putamen of control subjects and HD patients. Western blotting was performed using 60 µg of total striatal protein extracts. β-Actin was used as a loading control. (C) Relative TORC1 protein density expressed after normalization with β-actin. Representative blot showing three samples from each group. Significant decreases in TORC1 protein levels are observed in HD patients when compared with control subjects. Mean ± SEM, *P < 0.05, versus control subjects.
TORC1 overexpression induces TORC1, CREB and PGC-1α transcription and PGC-1α promoter activity in the Htt striatal cells
In order to assess whether TORC1 overexpression can induce CREB and PGC-1α transcription and activity, we overexpressed TORC1 in WT STHdhQ7 (Q7/7) and HD STHdhQ111 (Q111/111) striatal cells using a TORC1 plasmid. We found significantly decreased TORC1 mRNA expression in the STHdhQ111 cells, when compared with STHdhQ7 cells (Fig. 3A). TORC1 overexpression resulted in a significant up-regulation of TORC1 mRNA expression in the STHdhQ111 and STHdhQ7 cells (Fig. 3A). TORC1 overexpression also caused a significant increase in TORC1 protein levels in both cell types (Fig. 3B). TORC1 is a nuclear protein that functions as a transcriptional coactivator and activates CREB transcription through CRE sites (27). TORC1 also regulates activity-dependent CREB target gene expression and maintains the dendrite growth of cortical neurons and long-term potentiation in the hippocampus (29,40).
Figure 3.

Overexpression of TORC1 induces CREB and PGC-1α expression and PGC-1α promoter activity in WT and HD striatal cells. (A) WT STHdhQ7 (Q7/7) and mutant STHdhQ111 (Q111/111) striatal cell lines were transiently transfected with TORC1 plasmids or empty vector. Overexpression of TORC1 significantly increased mRNA expression of TORC1 in STHdhQ7 and STHdhQ111 striatal cells. TORC mRNA expression was normalized to β-actin. Graph represents the mean ± SEM of three independent experiments. *P < 0.05. (B) Relative TORC1 protein levels in STHdhQ7 and STHdhQ111 striatal cell lines after TORC1 overexpression. A significant decrease in TORC1 protein levels is observed in STHdhQ111 cells, which is up-regulated after TORC1 overexpression. Values were normalized to β-actin, used as a loading control. (C) qRT-PCR analysis of relative mRNA expression of CREB after the overexpression of TORC1 in STHdhQ7 and STHdhQ111 striatal cells. (D) Relative CREB and pCREB protein levels in STHdhQ7 and STHdhQ111 striatal cell lines after TORC1 overexpression. Significant decrease in CREB and pCREB protein levels is observed in STHdhQ111 cells, which is up-regulated after TORC1 overexpression. Values were normalized to β-actin, used as a loading control. (E) Relative mRNA expression of PGC-1α after the overexpression of TORC1 in STHdhQ7 and STHdhQ111 striatal cells. PGC-1α mRNA expression was normalized to β-actin. Graph represents the mean ± SEM of three independent experiments. (F) STHdhQ7 and STHdhQ111 striatal cells were transfected with a PGC-1α promoter (2 kb) construct linked to a luciferase reporter gene in pGL3 vector (pGL3-PGC1α-Luc) alone or co-transfected with pGL3-PGC1α-Luc and a TORC1 plasmid. The luciferase activity is normalized to the firefly luminescence over renilla luminescence. TORC1 overexpression caused significantly increased PGC-1α promoter activity in STHdhQ7 and STHdhQ111 cells. Mean ± SEM of three experiments.
We next investigated whether TORC1 overexpression could affect CREB mRNA expression and protein levels. As shown in Figure 3C, basal CREB mRNA expression was significantly decreased in the STHdhQ111 cells when compared with STHdhQ7 cells, transfected with vector only. TORC1 overexpression caused significantly increased CREB mRNA expression in STHdhQ111 and STHdhQ7 cells (Fig. 3C). Phosphorylation of CREB leads to activation of CREB-mediated gene transcription (41); therefore, we assessed the effects of TORC1 overexpression on CREB phosphorylation in HD striatal cells. We observed significantly decreased basal and phosphorylated CREB protein levels in empty vector transfected STHdhQ111 cells when compared with STHdhQ7 cells (Fig. 3D). The levels of both basal and phosphorylated CREB proteins were markedly up-regulated by TORC1 overexpression in both STHdhQ111 and STHdhQ7 cells (Fig. 3D). Furthermore, we examined the effects of TORC1 overexpression on PGC-1α transcription. PGC-1α transcript expression was significantly increased in the STHdhQ111 and STHdhQ7 cells transfected with a TORC1 plasmid when compared with the empty vector control (Fig. 3E). We next investigated whether TORC1 overexpression affects PGC-1α promoter activity. We transiently transfected the STHdhQ111 and STHdhQ7 striatal cells with a pGL3-PGC-1α promoter–reporter construct containing the PGC-1α promoter encompassing −2533 to +78 of the mouse PGC-1α gene, fused to luciferase termed pGL3-PGC-1α-Luc. We observed a significant inhibition of the luciferase reporter activity in the mutant STHdhQ111 compared with the WT STHdhQ7 cells, showing decreased PGC-1α promoter activity as a consequence of mutant Htt (Fig. 3F). We next did transient co-transfection of pGL3-PGC-1α-Luc with TORC1 plasmids. Overexpression of TORC1 resulted in significantly increased PGC-1α promoter activity in both cell types (Fig. 3F). These results show that TORC1 significantly increases CREB and PGC-1α expression and activity in WT and mutant Htt striatal cells.
Overexpression of TORC1 increases the expression of mitochondrial biogenesis genes and mitochondrial DNA content
Increased PGC-1α promoter activity observed following TORC1 overexpression prompted us to ask whether TORC1 also affects PGC-1α downstream transcription targets including the mitochondrial biogenesis genes and mitochondrial DNA content in the STHdhQ111 and STHdhQ7 cells. PGC-1α plays a prominent role in regulating mitochondrial biogenesis and cellular respiration by modulating the expression of transcription factors such as the mitochondrial transcription factor Tfam, a nuclear-encoded mitochondrial transcription factor and a target of NRF-1, which after translocation to mitochondria increases the transcription and replication of the mitochondrial genome which leads to mitochondrial biogenesis (42–44). PGC-1α, which is a transcriptional coactivator, acts in concert with CREB and NRF-1 to enhance CytC expression.
We, therefore, assessed the effects of TORC1 overexpression on the expression of the PGC-1α downstream target genes, NRF-1, Tfam and CytC in the STHdhQ111 and STHdhQ7 striatal cells (Fig. 4A). As shown in Figure 4A, the mRNA expression of NRF-1, Tfam and CytC was significantly reduced in STHdhQ111 cells when compared with STHdhQ7 cells, following transient transfection with empty vector only. The mRNA expression of NRF-1, Tfam and CytC was significantly up-regulated in both types of cells after TORC1 transient overexpression (Fig. 4A). We next examined the effects of TORC1 expression on numbers of mitochondria, and mitochondrial DNA content, in the STHdhQ7 and STHdhQ111 striatal cells. We examined the ratio of mRNA for COX-II, a mitochondrial DNA-encoded gene, to the mRNA for 18s rRNA, a nuclear encoded transcript, which is a reliable induce of mitochondrial DNA content (45). There was a significant reduction in the COX II/18s rRNA ratio in the STHdhQ111 cells at baseline, when compared with STHdhQ7 cells (Fig. 4B). Following TORC1 overexpression, there was a significant increase in the COXII/18s rRNA ratio in both types of cells, consistent with increased mitochondrial DNA content (Fig. 4B). These results show that TORC1 overexpression significantly increases the expression of PGC-1α downstream target genes and mitochondrial DNA content.
Figure 4.

TORC1 overexpression induces the expression of PGC-1α target genes (mitochondrial biogenesis genes) and increases mitochondrial DNA content in WT and HD striatal cells. (A) WT STHdhQ7 (Q7/7) and mutant STHdhQ111 (Q111/111) striatal cell lines were transiently transfected with a TORC1 plasmid and empty vector. Overexpression of TORC1 increased the mRNA expression of PGC-1α target genes involved in mitochondrial biogenesis including NRF-1, Tfam and CytC in STHdhQ7 and STHdhQ111 striatal cells. mRNA expression was normalized to β-actin. The graph represents the mean ± SEM of three independent experiments. Mean ± SEM of three experiments, *versus vector transfected STHdhQ7 and #versus vector transfected STHdhQ111. (B) Mitochondrial DNA content was assessed by analyzing the COX-II/18s RNA ratio by qRT-PCR. TORC1 overexpression resulted in increased mitochondrial DNA content in STHdhQ7 and STHdhQ111 striatal cells. Mean ± SEM of three experiments. *P < 0.05, versus vector transfected STHdhQ7 and #versus vector transfected STHdhQ111.
TORC1 expression protects against mitochondrial dysfunction and toxicity induced by the mitochondrial toxin 3-NP in both WT and mutant Htt striatal cells
In order to assess the effects of TORC1 expression on mitochondrial dysfunction and mitochondrial energy metabolism in HD, we transiently transfected mutant Htt striatal cells with TORC1 plasmids, followed by treatment with the mitochondrial toxin 3-NP, which inhibits complex II of the respiratory chain. STHdhQ111 striatal cells display low mitochondrial ATP production and show increased vulnerability to 3-NP (46). We studied the effects of TORC1 on mitochondrial membrane potential (MMP) by analyzing fluorescent staining of Mitotracker dye in the STHdhQ111 striatal cells. Mitotracker is a fluorescent dye which after diffusing through the plasma membrane accumulates in mitochondria and fluoresces, and the resulting fluorescence is directly proportional to MMP. We found that treatment of STHdhQ111 striatal cells with 3-NP resulted in significantly decreased red fluorescence staining of Mitotracker dye, when compared with vehicle-treated STHdhQ111 cells (Fig. 5A and B). TORC1 overexpression significantly increased the red Mitotracker fluorescence and MMP (Fig. 5A and B). TORC1 overexpression significantly protected against the 3-NP-mediated decreases in MMP in striatal cells (Fig. 5A and B). Figure 5B is the quantification of the Mitotracker fluorescence observed in Figure 5A in striatal cells after TORC1 and 3-NP treatment. We next studied mitochondrial activity and cell viability using the MTT assay in the STHdhQ111 striatal cells following TORC1 overexpression and 3-NP treatment. The MTT assay relies on the reduction in yellow soluble tetrazolium salt to insoluble purple formazan by the mitochondrial succinate dehydrogenase enzyme, which is an index of mitochondrial activity. Treatment with 3-NP significantly decreased the MTT activity in the STHdhQ111 striatal cells, when compared with untreated cells (Fig. 5C). Transient transfection of STHdhQ111 cells with TORC1 significantly increased the MTT activity and protected striatal cells from the 3-NP-mediated decreases in MTT (Fig. 5C). These results therefore show that TORC1 overexpression increases mitochondrial activity and protects STHdhQ111 cells against 3-NP-induced toxicity.
Figure 5.

Overexpression of TORC1 increases MMP and protects STHdhQ7 and mutant STHdhQ111 striatal cells against the mitochondrial toxin 3-NP acid-induced toxicity. (A) Mutant STHdhQ111 (Q111/111) striatal cell lines were transiently transfected with TORC1 plasmid or empty vector, followed by treatment with the mitochondrial toxin 3-NP. MMPs were measured by assessing Mitotracker staining. Increased Mitotracker staining reflects an increase in MMP. 3-NP treatment in HD striatal cells resulted in decreased Mitotracker staining and MMP, which was protected in TORC1 overexpressing cells. TORC1 overexpression in striatal cells caused an increased MMP. Scale bar = 100 µm. (B) Quantification of Mitotracker staining in term of relative fluorescence. Quantification analysis was performed in ten microscopic fields for each group. Graph represents the mean ± SEM of three independent experiments. *P < 0.05. (C) Mitochondrial metabolic activity was measured by the MTT assay. Mitochondrial activity was decreased by 3-NP in STHdhQ111 striatal cells, TORC1 overexpression resulted in increased mitochondrial activity in 3-NP-treated HD striatal cells. Graph represents the mean ± SEM of three independent experiments. *P < 0.05.
TORC1 knockdown reduces PGC-1α mRNA expression and enhances mitochondrial dysfunction and susceptibility of Htt striatal cells to 3-NP
The neuroprotection provided by TORC1 to the Htt striatal cells against 3-NP-induced toxicity, prompted us to analyze whether knockdown of TORC would make striatal cells more susceptible to mitochondrial dysfunction. We examined the effects of transient knockdown of TORC1 in STHdhQ111 striatal cells using two TORC1 small hairpin RNA (shRNA) sequences. We observed significantly decreased TORC1 mRNA expression following TORC1 shRNA transfection in striatal cells (Fig. 6A), and both the TORC1 shRNAs had almost equal knockdown efficiency, while scrambled sequence had no effect on TORC1 mRNA expression (Fig. 6A). The knockdown of TORC1 did not change the baseline mRNA expression of TORC2 in striatal cells, showing that the shRNA produced a specific knockdown of TORC1 only, and not other TORC isoforms (Fig. 6B). We then investigated whether TORC1-specific knockdown would affect PGC-1α mRNA expression. Basal PGC-1α mRNA expression was significantly decreased in the STHdhQ111 cells when compared with STHdhQ7 cells and was unaffected in scrambled sequence-transfected cells (Fig. 6C). TORC1 knockdown caused significantly decreased PGC-1α mRNA expression in both the STHdhQ111 and STHdhQ7 cells (Fig. 6). The scrambled sequence had no effect on Mitotracker staining and mitochondrial activity as measured by the MTT assay, in STHdhQ111 striatal cells (Fig. 6D and E). TORC1 knockdown significantly decreased the Mitotracker red fluorescence and mitochondrial activity, as did 3-NP treatment, and the fluorescence was further significantly decreased after TORC1 knockdown in combination with 3-NP treatment (Fig. 6D and E). These observations show that TORC1 knockdown in the STHdhQ111 striatal cells significantly worsened 3-NP-mediated reductions in MMP and mitochondrial activity.
Figure 6.
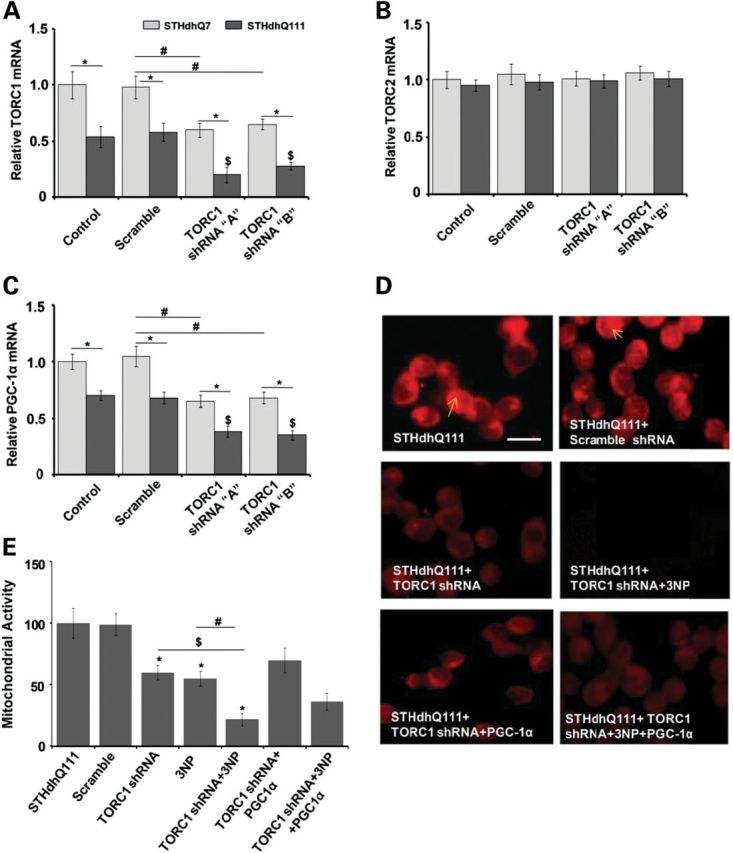
Selective ablation of TORC1 reduces TORC1 and PGC-1α mRNA expression, decreases MMP and makes STHdhQ7 and mutant STHdhQ111 striatal cells more susceptible to 3-NP-induced toxicity. Overexpression of PGC-1α partially prevents TORC1 knockdown-mediated susceptibility of Htt striatal cells to 3-NP. (A–C) STHdhQ7 (Q7/7) and mutant STHdhQ111 (Q111/111) striatal cells were transiently transfected with plasmid vector containing scrambled or two shRNA target sequences 1 and 2 against TORC1. qRT-PCR analysis shows that the knockdown of TORC1 caused a significant decrease in TORC1 (A) and PGC-1α mRNA expression (C), while mRNA expression of TORC2 was unchanged (B). Both of the shRNA target sequences caused a comparable knockdown of TORC1. Values were normalized to β-actin. Data are expressed as the mean ± SEM of three experiments. *P < 0.05, versus STHdhQ7, #versus scramble sequence transfected STHdhQ7 and $versus scramble sequence transfected STHdhQ111. (D) TORC1 knockdown was produced by transient transfection of STHdhQ111 striatal cells with TORC1 specific shRNA sequence, 2followed by treatment with the mitochondrial toxin 3-NP. MMP was measured by assessing Mitotracker staining. TORC1 knockdown in HD striatal cells resulted in decreased Mitotracker staining, which was further decreased by 3-NP treatment. Overexpression of PGC-1α partially increased MMP in TORC1 knockdown and 3-NP-treated HD striatal cells. Scramble sequence had no effect on MMP and Mitotracker staining. Scale bar = 100 µm. (E) Mitochondrial metabolic activity was measured with an MTT assay. Mitochondrial activity was decreased by 3-NP in STHdhQ111 striatal cells. TORC1 knockdown resulted in significantly decreased mitochondrial activity which was further decreased following 3-NP treatment of HD striatal cells. Overexpression of PGC-1α partially, but not significantly, increased MMP in TORC1 knockdown and 3-NP-treated HD striatal cells. Graph represents the mean ± SEM of three independent experiments. *P < 0.05, versus STHdhQ111, $versus STHdhQ111 + TORC1 shRNA and #versus STHdhQ111 + 3-NP.
Overexpression of PGC-1α partially prevents TORC1 knockdown-mediated susceptibility of Htt striatal cells to 3-NP
We next examined whether the overexpression of PGC-1α prevents deleterious effects of TORC1 down-regulation in Htt striatal cells. We transiently co-transfected STHdhQ111 and STHdhQ7 cells with PGC-1α overexpressing plasmids and TORC1 shRNA. TORC1 knockdown significantly decreased the Mitotracker red fluorescence and mitochondrial activity, which was partially but not significantly prevented by PGC-1α overexpression (Fig. 6D and E). TORC1 knockdown in combination with 3-NP treatment further significantly decreased MMP and mitochondrial activity, which was slightly up-regulated by PGC-1α overexpression (Fig. 6D and E). These observations show that PGC-1 overexpression partially prevents TORC1 knockdown-mediated increased susceptibility to 3-NP and reductions in MMP in the STHdhQ111 striatal cells.
TORC1 knockdown enhances neurodegeneration in the striatum of NLS-N171-82Q HD transgenic mice
We next examined whether a knockdown of TORC1 in the striatum of NLS-N171-82Q HD transgenic mice makes them more susceptible to mitochondrial dysfunction and neurodegeneration. We stereotaxically injected TORC1 siRNA or scrambled sequences directly into the striatum of NLS-N171-82Q HD mice. We observed ∼70% decreases in TORC1 mRNA expression in the striatum following TORC1 siRNA but no effect of scrambled sequence (data not shown). We investigated the effects of TORC1 knockdown on neuron viability by observing Fluoro Jade-B staining in the striatum (Fig. 7A and B). Fluoro Jade-B is a fluorescein derivative which specifically binds to neurons undergoing neurodegeneration. Intra-striatal injection of scrambled sequence siRNA did not produce Fluoro Jade-B positive neurons. We observed a significantly increased number of Fluoro Jade-B stained degenerating neurons in the striatum of NLS-N171-82Q HD mice following 3-NP injection (Fig. 7A and B). TORC1 knockdown also resulted in increased numbers of degenerating neurons in the striatum (Fig. 7A and B). TORC1 knockdown made the NLS-N171-82Q HD mice more susceptible to 3-NP toxicity, as shown by the presence of a significantly increased number of degenerating neurons in the TORC1 siRNA with the 3-NP group (Fig. 7A and B). We next investigated nuclear chromatin condensation by Hoechst staining in the striatum of the NLS-N171-82Q mice after TORC1 knockdown. There was significantly increased chromatic condensation, and nuclear fragmentation in the NLS-N171-82Q mice striatum following TORC1 siRNA treatment, when compared with scrambled sequence (Fig. 7C). These results show that a knockdown of TORC1 produces cell death and striatal neurodegeneration in HD mice.
Figure 7.
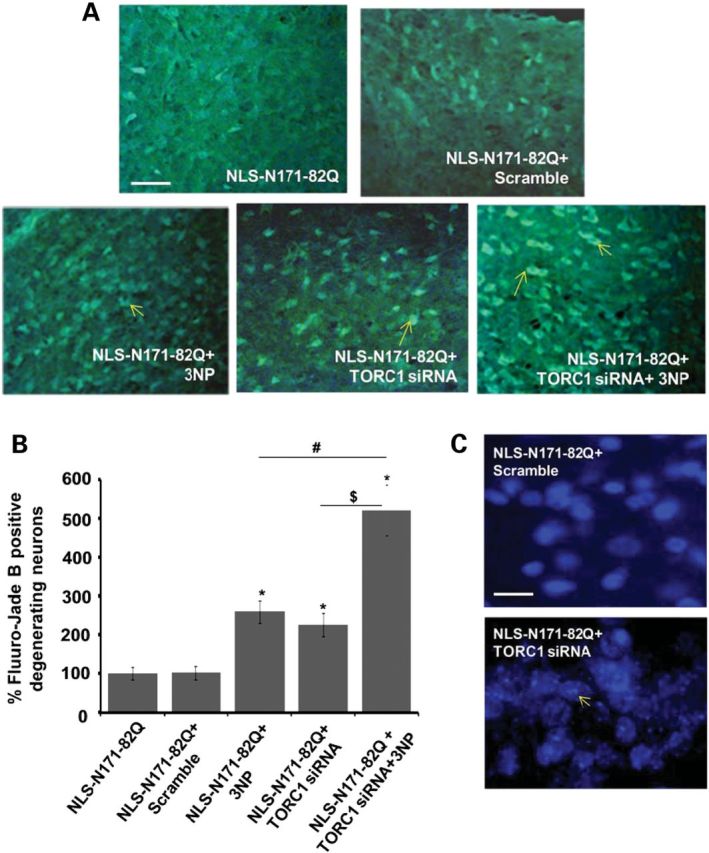
TORC1 ablation enhances neurodegeneration in the striatum of NLS-N171-82Q HD transgenic mice. (A and B) TORC1 knockdown was done by the stereotaxic injection of TORC1 siRNA and scrambled sequences directly into the striatum of NLS-N171-82Q HD mice. Fluoro Jade-B stained degenerating neurons in the striatum of NLS-N171-82Q HD mice are significantly increased by 3-NP injection and TORC1 knockdown. TORC1 knockdown made NLS-N171-82Q HD mice more susceptible to 3-NP toxicity, shown by a significantly increased number of degenerating neurons in the TORC1 + 3-NP-injected group. *P < 0.05, n = 5 mice, *versus NLS-N171-82Q, #versus NLS-N171-82Q + 3-NP and $versus NLS-N171-82Q + TORC1 siRNA. Scale bar = 200 µm. (C) Nuclear chromatin condensation and nuclear fragmentation analysis by Hoechst staining in the striatum of NLS mice after TORC1 knockdown. Increased chromatin condensation, and nuclear fragmentation in the NLS mice striatum, is observed following TORC1 knockdown when compared with scrambled sequence. Scale bar = 50 µm.
Htt knockdown prevents transcriptional repression of TORC1, CREB and PGC-1α in Htt striatal cells
In order to see whether mutant Htt affects TORC1, CREB and PGC-1α transcriptional activity, we knocked down mutant Htt expression in Htt striatal cells using a specific target shRNA sequence against mutant Htt (Fig. 8). At baseline, TORC1, CREB and PGC-1α mRNA expression were significantly decreased by 48, 47 and 42%, respectively, in scrambled shRNA-transfected HD STHdhQ111 cells when compared with control STHdhQ7 cells (Fig. 8). We found that the knockdown of mutant Htt with shRNA resulted in a significant increase in TORC1, CREB and PGC-1α mRNA expression in STHdhQ111 cells when compared with control STHdhQ7 cells (Fig. 8).
Figure 8.

Knockdown of mutant Htt prevents the transcriptional repression of TORC1, CREB and PGC-1α in Htt striatal cells. (A) Increased TORC1, CREB and PGC-1α mRNA expressions in STHdhQ111 HD striatal cells by the knockdown of mutant Htt. Control and HD striatal cells were transiently transfected with plasmid vector containing scramble and shRNA target sequences against mutant Htt. Data are expressed as the mean ± SEM of two experiments. *versus control striatal cells, #versus scramble shRNA. P < 0.05.
DISCUSSION
There is substantial evidence that mitochondrial dysfunction, transcriptional dysregulation and bioenergetic defects play an important role in the pathogenesis of HD (1,11). Mutant Htt causes aberrant transcriptional regulation by binding to several transcriptional regulators and interfering with their function (13–16). In particular, an interaction of mutant Htt with the expression of both CREB and PGC-1α has been implicated in HD pathogenesis (13,17,21,25). Furthermore, mutant Htt interferes with the formation of the CREB/TAF4 complex that regulates transcription and the expression of the gene encoding PGC-1α (21).
Impaired transcription and function of PGC-1α contributes to the mitochondrial dysfunction and plays an important role in HD pathogenesis (19–21,24). An involvement of PGC-1α in HD pathogenesis was initially suggested by the observation that PGC-1α knockout mice show spongiform lesions in the striatum, the brain region most affected in HD (34,35). The initial reports showed reduced PGC-1α expression in both striatal cell lines expressing mutant Htt, as well as in the striatum of HD transgenic mice, and in striatal tissue from the postmortem HD patient brains (21,24). We observed a significant reduction in PGC-1α mRNA expression and activity in the striata and muscle of HD transgenic mice, muscle biopsies from HD patients and cultured myoblasts from HD patients (19–21,24). Furthermore, the expression of 24 of 26 genes coactivated by PGC-1α was significantly reduced in the postmortem brain tissue of HD patients (24). The expression of PGC-1α's downstream target genes such as Tfam, NRF-1, NRF-2 and CytC as well as mitochondrial biogenesis are also impaired in HD (19,20). There is marked hypothermia at baseline and following cold exposure in several HD mouse models (24). Following cold exposure, the expression of UCP-1, a PGC-1α target gene involved in adaptive thermogenesis, does not increase in brown adipose tissue from HD mice relative to WT animals, resulting in hypothermia (24). There is also the impairment of PGC-1α activation and mitochondrial biogenesis in response to energy deprivation conditions in HD mice and myoblasts from HD patients (19,20).
The expression of CREB- and CRE-dependent transcription is also reduced in HD transgenic mice and HD cell models (11,13,14,17–20,46). CREB knockout mice show the apoptosis of striatal neurons and a phenotype similar to HD (33). CREB-deficient mice are more sensitive to 3-NP, and when crossed to YAC128 HD transgenic mice, the motor impairment is significantly accelerated (47). A partial loss of CBP reduces the life expectancy of N171-82Q mice (48).
TORCs are coactivators of CREB which enhance CREB-dependent gene transcription, PGC-1α promoter activity, PGC-1α gene transcription and mitochondrial biogenesis (27,28). A screen for transcriptional activators of PGC-1α expression was carried out with 10 000 full-length human cDNAs, examining the induction of the PGC-1α promoter (28). TORC1 was the most potent activator of PGC-1α expression, but TORC2 and TORC3 were also highly active (28). Forced expression of TORCs in primary muscle cells induced the endogenous mRNA of PGC-1α and its downstream genes and increased numbers of mitochondria. TORC1 enhances the dendritic growth of developing cortical neurons and enhances neuronal survival after ischemia through the positive regulation of CREB-target gene transcription (29,32). TORC1 is required for long-term potentiation in hippocampal neurons (40,49). TORCs also promote stress resistance and maintain energy balance by the induction of CREB target genes in the brains of Drosophila under starvation conditions (30,31). There is strong evidence showing that mutant Htt interferes with CREB and PGC-1α transcription in HD and that this may play an important role in HD pathogenesis. Since TORCs regulate the transcription and function of PGC-1α and CREB, we examined whether the impairment of TORC expression contributes to HD pathogenesis.
We found significantly decreased TORC1 mRNA expression in the striatum of NLS-N171-82Q, R6/2 and Hdh-Q111 HD transgenic mice when compared with their respective WT animals. TORC2 mRNA expression was significantly decreased in NLS-N171-82Q mice, while TORC3 was unchanged in all three HD transgenic mice lines. We also found a significant decrease in TORC1 protein levels in the striatum of all three HD transgenic mouse lines. We observed significantly decreased expression of TORC1 mRNA in HD STHdhQ111 striatal cells. These results show that the down-regulation of TORC1 mRNA and protein levels occurs in both HD transgenic mice and striatal cells, while TORC2 and TORC3 were unchanged or in one instance decreased. TORC1 mRNA and protein levels were also significantly decreased in the striatum of HD patients when compared with age-matched control subjects. We found a significant increase in TORC3 in the striatum of HD patients, which may be compensatory, but is of uncertain significance since TORC3 levels are relatively low in the nervous system.
We investigated the effects of TORC1 overexpression on CREB and PGC-1α expression in HD STHdhQ111 striatal cells. TORC1 overexpression produced a significant increase in TORC1 mRNA and protein levels in the Htt striatal cells. TORC1 overexpression also resulted in a significant up-regulation of CREB transcript expression and protein levels. Several studies have shown that there is reduced CREB and CBP expression in Htt-transfected cell lines and transgenic mice, which can be reversed by increasing cAMP which activates CREB (50,51). The phosphodiesterase type IV inhibitor rolipram increases CREB phosphorylation and improves behavior and neuropathologic abnormalities in the R6/2 transgenic mouse model of HD (51). Rolipram inhibition of phosphodiesterase IV also prevents the sequestration of CBP and protects striatal parvalbumin interneurons in the R6/2 mouse model of HD (50). Inhibition of the striatal-specific phosphodiesterase PDE10A increases phosphorylated CREB and brain-derived neurotrophic factor (BDNF) and ameliorates striatal and cortical pathology, and motor impairments in the R6/2 mice (52). Depletion of CBP has been linked to cell death mediated by mutant Htt in vitro (14,53,54).
Consistent with CREB up-regulation, the overexpression of TORC1 also caused a significant up-regulation of PGC-1α mRNA expression and its promoter activity. These observations are consistent with a previous study, suggesting that TORC-mediated increases in PGC-1α transcription and function are CREB-dependent and were inhibited in the absence of CREB (28). CREB plays an important role in the regulation of PGC-1α and its ability to induce gluconeogenesis and to increase in response to H2O2 (55,56). The regulation of PGC-1α by CREB/TAF4-dependent transcription was shown to be impaired in HD (21). TORC2 promotes fasting induced hepatic gluconeogenesis by inducing the expression of PGC-1α (57,58). Drosophila-deficient in TORC show impaired metabolism and they are strongly sensitive to starvation and oxidative stress (30). Our results show that TORC1 overexpression enhances both CREB and PGC-1α mRNA and protein levels in HD.
Recent studies showed a decrease in the expression of PGC-1α target genes and mitochondrial biogenesis in HD transgenic mice and HD patients (19,20,25). PGC-1α in concert with CREB regulates mitochondrial biogenesis and cellular respiration by modulating the expression of transcription factors such as NRF-1, Tfam and CytC (42–44). Therefore, increased CREB mRNA expression and PGC-1α promoter activity observed following TORC1 overexpression prompted us to study, whether increased TORC1 expression also affects PGC-1α downstream target genes and mitochondrial DNA content in the Htt striatal cells. We found significantly increased mRNA expression of PGC-1α target genes involved in mitochondrial biogenesis (NRF-1, Tfam and CytC) and mitochondrial DNA content following TORC1 overexpression. These results are consistent with an earlier study which showed that the expression of TORCs induce PGC-1α and CREB transcription and mitochondrial biogenesis in muscle cells (28).
Overexpression of TORC1 increased mitochondrial respiration and MMP in the Htt striatal cells. Moreover, TORC1 overexpression provided significant protection against 3-NP-mediated decreases in mitochondrial respiration and MMP. Furthermore, specific knockdown of TORC1 in striatal cells decreased PGC-1α expression and MMP. The TORC1 knockdown made striatal cells more susceptible to 3-NP-mediated decreases in MMP and mitochondrial respiration. We also found that a specific knockdown of TORC1 in the striatum of NLS-N171-82Q HD transgenic mice resulted in increased neurodegeneration and chromatin condensation. The severity of neurodegeneration was exacerbated in the striatum of TORC1-deficient HD mice treated with 3-NP.
These observations are consistent with TORC1 being an activator of PGC-1α, which is a potent suppressor of reactive oxygen species and which exerts neuroprotective effects (56). PGC-1α knockout mice show marked increases in vulnerability to the neurotoxins 1-methyl-4-phenyl-1,2,3,6-tetrahydrodropyridine (a complex I inhibitor) and kainic acid (an excitotoxin) (56). Previous studies showed that PGC-1α overexpression made Htt striatal cells more resistant to 3-NP-induced neurotoxicity, and increased mitochondrial respiration, consistent with our observations (24). Adenoviral vector-mediated overexpression of PGC-1α in the striatum of R6/2 HD mice resulted in increased mean neuronal volume and decreased neuronal atrophy, whereas down-regulation of PGC-1α worsened the behavioral and neuropathological abnormalities in a PGC-1α knockout/HD knockin mouse model. Our results are consistent with observations that TORCs by activating CREB target genes enhance neuronal survival after ischemia (32) and promote stress resistance in Drosophila (30).
Taken together, these findings provide strong evidence of an impairment of TORC1 activity and function in striatal tissue from HD transgenic mice, striatal tissue of HD patients and in HD striatal cells. The present results therefore show that mutant Htt reduces the expression of TORC1, which contributes to the deficiency of PGC-1α and its downstream genes which play an important role in mitochondrial biogenesis and neuronal survival. An impairment of TORC1 expression and the transcriptional activation of CREB and PGC-1α, therefore, may play an important role in the pathogenesis of HD. These findings suggest that the modulation of other pathways regulating TORC1 may be important in neuronal survival in HD. Several kinases that phosphorylate TORC1 including MEKK1, potently activate TORC1 (59). Degradation of salt-inducible kinase 2 leads to the activation of CREB-dependent transcription, by increasing dephosphorylation and nuclear import of TORC1 resulting in powerful neuroprotection from ischemic cell death (60). Another pathway which modulates TORC1 is Sirt1 (61). Sirt1 overexpression improves survival, neuropathology and expression of BDNF in HD transgenic mice. The neuroprotective effect required the presence of TORC1. Sirt1 deacetylates and activates TORC1, by promoting its dephosphorylation and interaction with CREB. Mutant htt interferes with the TORC1–CREB interaction to repress BDNF. Therefore, there are a number of potential therapeutic avenues which can be manipulated to increase TORC1 expression and exert beneficial therapeutic effects in HD. One potential means would be to increase nicotinamide adenine dinucleotide, which activates Sirt1. A pharmacologic activator of TORC1 is tanshinone IIA, which protects against focal ischemic brain injury by inducing the nuclear translocation of TORC1, and up-regulation of expression of TORC1, pCREB and BDNF (62). Our findings therefore suggest that agents which increase TORC1 expression and activity may be an attractive approach for the treatment of HD.
MATERIAL AND METHODS
Transgenic animals
The NLS-N171-82Q mice with a C3H/C57Bl6 background were kind gift from Dr David Borchelt. R6/2 and HdhQ111 mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA). NLS-N171-82Q mice have the human prion promoter driving the expression of an 82-CAG repeat in the first 171 amino acids of the Htt gene. R6/2 mice express exon 1 of the human HD gene, containing 150-CAG repeats, and develop neurological symptoms including impaired motor coordination and cognitive performance (36). Hdh-Q111 mice are CAG knockin into the mouse Htt gene and show neuropathological changes, including the formation of insoluble mutant N-terminal fragments (39). The HD mice were genotyped by a PCR assay of DNA obtained from tail tissue. The animals were housed at Weill Medical College of Cornell University animal house and were kept on a 12-h light/dark cycle, with food and water continuously available. Experiments were carried out using procedures that minimized pain and discomfort. All experiments were conducted within National Institutes of Health guidelines for animal research and were approved by the Institutional Animal Care and Use Committee.
Gene Expression Analysis by qRT-PCR
Total RNA was isolated from the striatum of WT and HD mice and HD patients and striatal cells using the TriZol Reagent. Genomic DNA was removed using RNase free DNase (Ambion, USA). RNA pellets were resuspended in DEPC-treated water (Ambion). Equal amounts of RNA were revere transcribed using the Superscript first-strand synthesis system with Oligo-dT (Invitrogen, USA) and diluted in nuclease-free water (Ambion) to a final concentration of 10 ng/µl. RT-PCR using SYBR green (PE Applied Biosystems, Foster City, CA, USA) was used to detect changes in the mRNA expression of various genes using specific primer pairs (sequences are in Supplementary material). Expression of the cellular house-keeping gene β-actin served as a control to normalize values. All qRT-PCR plating was performed on ice. Targets were detected and quantified in real-time using the ABI Prism 7700 Sequence Detector System (PE Applied Biosystems). Relative expression was calculated using the ΔΔCt method.
Western blot
The striatum of WT and HD mice and striatal cells were homogenized in cell extraction buffer containing 50 mm Tris–HCl, pH 7.4, 150 mm NaCl, 2 mm ethylenediaminetetraacetic acid, 1% sodium dodecyl sulfate, 0.5% nonyl phenoxypolyethoxylethanol, 0.5% deoxycholate supplemented with protease and phosphatase inhibitors (Sigma, USA). Samples were homogenized, sonicated for 5 s and centrifuged at 4000g for 30 min. Protein concentrations of the supernatant were determined using the BCA protein assay, as per the manufacturer's recommended protocol. Equal amounts of protein (45 μg) were loaded on to a 4–20% Tris-glycine gel (Invitrogen). Membranes were then blocked for 1 h at room temperature in Tris-buffered saline/Tween-20 (TBST) (50 mm Tris–HCl, 150 mm NaCl, pH 7.4, 1% Tween-20) containing 5% non-fat dried milk. The membranes were incubated overnight at 4°C for TORC1 (1:1000, Cell Signaling, USA), CREB (1:1000, Cell Signaling), pCREB (1:500, Cell Signaling) and β-actin (1:10 000, Chemicon, USA). Membranes were then washed three times with TBST and incubated for 1 h with HRP-conjugated secondary antibody and the immunoreactive proteins detected using a chemiluminescent substrate (Pierce, USA) according to the manufacturer's instructions. Films were scanned by film processor (Konica Minolta & Graphic Inc., SRX-101A, USA). Protein expression was quantified using Scion Image for Windows (NIH, USA).
Mitochondrial DNA content
Total DNA was isolated by a standard phenol-chloroform extraction procedure. Fragments of the mitochondrial COX-II gene and the nuclear 18S gene were amplified in duplicate by quantitative real-time PCR (45). The absolute content of mitochondrial DNA (mtDNA) was expressed as the mtDNA-to-nuclear-DNA ratio (COX-II mtDNA/18S nuclear DNA).
Striatal cell culture
WT STHdhQ7/Q7 (expressing endogenous normal Htt) and STHdhQ111/Q111 striatal cells (expressing endogenous mutant Htt; 111-CAG repeats) were cultured. Both types of cells were cultured in Dulbecco's modified Eagle medium containing 1 mm l-glutamine, 25 mm d-glucose, 1% G418, 400 mg/ml geneticin and 10% fetal bovine serum at 33°C with 5% CO2.
PGC-1α luciferase reporter assays
STHdhQ7 and STHdhQ111 cells were transiently transfected with the 2 kb PGC-1α promoter luciferase reporter having the sequences of mouse PGC-1α promoter (pGL3-PGC1α-Luc) between +78 and −2533 (Addgene, USA) using Lipofectamine reagent (Invitrogen). Cells were also co-transfected with pGL3-PGC1α-Luc and TORC1 plasmid (kind gift from H. Takemori, Japan). Luciferase activity was determined using a dual luciferase assay as per the manufacturer's instructions (Promega, USA).
TORC1 overexpression and knockdown
TORC1 overexpression in STHdhQ7 and STHdhQ111 cells was carried out by transient transfection with TORC1 plasmid using Lipofectamine. TORC knockdown was carried out by the transfection of cells with shRNA constructs with the target sequence TORC1 (5′-GAACAATCCGCGGAAATTTA-3′) and scramble sequence (5′-AATTCTCCGAACGTGTCACGT-3′), followed by gene expression and protein-level analysis.
Htt knockdown
Htt knockdown in STHdhQ7 and STHdhQ111 cells was carried out by transient transfection with Htt shRNA plasmid which is a pool of three target-specific lentiviral vector plasmids each encoding 19–25 nt plus hairpin shRNAs (Santa Cruz Biotechnology Inc., USA). For transient transfection STHdhQ7 and STHdhQ111 cells were maintained in Dulbecco's modified Eagle medium with 10% FBS in a 6-well plate. Transient transfection was performed with 1 μg Htt shRNA plasmid and scramble plasmid per well, at a confluence of ∼65–70% using Lipofectamine™ 2000 as per the manufacturer's protocol.
PGC-1α and TORC1 co-transfection
STHdhQ7 and STHdhQ111 cells were co-transfected with 1 µg of pcDNA-PGC1α plasmid (Addgene) and TORC1 shRNA and scramble plasmid constructs.
3-NP treatment
After 36 h of TORC1 overexpression and knockdown, cells were treated with 100 µm 3-NP for 2 h, followed by MMP and mitochondrial activity analysis.
Mitotracker staining
MMP in STHdhQ7 and STHdhQ111 cells after TORC1 overexpression or knockdown and 3-NP treatment was measured by analyzing florescence of Mitotracker dye. Cells were stained with MitoTracker Red FM (Invitrogen) following the manufacturer's protocol, and fluorescence was monitored in an inverted fluorescent microscope.
Fluoro-jade B and Hoechst labeling
Fluoro-jade B and Hoechst labeling was performed to monitor degenerating neurons and chromatin condensation, respectively, in the striatum region of NLS-N171-82Q HD transgenic mice after TORC1 knockdown. In brief, 20 µm-thick coronal sections across striatum were cut using a cryostat, mounted on slides, stained with Hoechst nuclear stain (Invitrogen) and Fluoro-jade B (Millipore, USA) as per the manufacturer's instruction.
Statistical analysis
Statistical analysis was performed using GraphPad InStat statistical analysis software version 3.05 for Windows (San Diego, CA, USA). The mean significant difference in the experimental groups was determined using one-way analysis of variance followed by the Tukey–Kramer post hoc multiple comparisons test. P-values of <0.05 were considered to be statistically significant.
Supplementary Material
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENTS
This work was supported by the NS 39258 and Huntington's disease Society of America Coalition for the Cure to M.F.B and CSIR Supra Institutional Project-08 to R.K.C. We are grateful to Dr K.C. Gupta, Director, CSIR-IITR, Lucknow for his support during the study. D.M. and S.A. are recipients of Junior Research Fellowship from CSIR, New Delhi, India. S.K.T. is recipient of Junior Research Fellowship from UGC, New Delhi, India. Dr David Borchett is thanked for providing us with NLS-N171-82Q transgenic HD mice. We are grateful to Dr H. Takemori, Japan for providing us a TORC1 plasmid vector. We are also thankful to Laragen Inc., Los Angels, CA, USA, for mice PCR genotyping. Secretarial assistance of Greta Strong is gratefully acknowledged (IITR manuscript number-3034).
Conflict of Interest statement. None declared.
REFERENCES
- 1.Browne S.E., Beal M.F. The energetics of Huntington's disease. Neurochem. Res. 2004;29:531–546. doi: 10.1023/b:nere.0000014824.04728.dd. doi:10.1023/B:NERE.0000014824.04728.dd. [DOI] [PubMed] [Google Scholar]
- 2.Milakovic T., Johnson G.V. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J. Biol. Chem. 2005;280:30773–30782. doi: 10.1074/jbc.M504749200. doi:10.1074/jbc.M504749200. [DOI] [PubMed] [Google Scholar]
- 3.Oliveira J.M., Jekabsons M.B., Chen S., Lin A., Rego A.C., Goncalves J., Ellerby L.M., Nicholls D.G. Mitochondrial dysfunction in Huntington's disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J. Neurochem. 2007;101:241–249. doi: 10.1111/j.1471-4159.2006.04361.x. doi:10.1111/j.1471-4159.2006.04361.x. [DOI] [PubMed] [Google Scholar]
- 4.Chaturvedi R.K., Beal M.F. Mitochondrial approaches for neuroprotection. Ann. N. Y. Acad. Sci. 2008;1147:395–412. doi: 10.1196/annals.1427.027. doi:10.1196/annals.1427.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mochel F., Haller R.G. Energy deficit in Huntington disease: why it matters. J. Clin. Invest. 2011;121:493–499. doi: 10.1172/JCI45691. doi:10.1172/JCI45691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choo Y.S., Johnson G.V., MacDonald M., Detloff P.J., Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. doi:10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 7.Panov A.V., Gutekunst C.A., Leavitt B.R., Hayden M.R., Burke J.R., Strittmatter W.J., Greenamyre J.T. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat. Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 8.Orr A.L., Li S., Wang C.E., Li H., Wang J., Rong J., Xu X., Mastroberardino P.G., Greenamyre J.T., Li X.J. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 2008;28:2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008. doi:10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trushina E., Dyer R.B., Badger J.D., 2nd, Ure D., Eide L., Tran D.D., Vrieze B.T., Legendre-Guillemin V., McPherson P.S., Mandavilli B.S., et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. doi:10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang D.T., Rintoul G.L., Pandipati S., Reynolds I.J. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol. Dis. 2006;22:388–400. doi: 10.1016/j.nbd.2005.12.007. doi:10.1016/j.nbd.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Sugars K.L., Rubinsztein D.C. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. doi:10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- 12.Cha J.H. Transcriptional signatures in Huntington's disease. Prog. Neurobiol. 2007;83:228–248. doi: 10.1016/j.pneurobio.2007.03.004. doi:10.1016/j.pneurobio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steffan J.S., Kazantsev A., Spasic-Boskovic O., Greenwald M., Zhu Y.Z., Gohler H., Wanker E.E., Bates G.P., Housman D.E., Thompson L.M. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl Acad. Sci. USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. doi:10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nucifora F.C., Jr, Sasaki M., Peters M.F., Huang H., Cooper J.K., Yamada M., Takahashi H., Tsuji S., Troncoso J., Dawson V.L., et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. doi:10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 15.Chen-Plotkin A.S., Sadri-Vakili G., Yohrling G.J., Braveman M.W., Benn C.L., Glajch K.E., DiRocco D.P., Farrell L.A., Krainc D., Gines S., et al. Decreased association of the transcription factor Sp1 with genes downregulated in Huntington's disease. Neurobiol. Dis. 2006;22:233–241. doi: 10.1016/j.nbd.2005.11.001. doi:10.1016/j.nbd.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Dunah A.W., Jeong H., Griffin A., Kim Y.M., Standaert D.G., Hersch S.M., Mouradian M.M., Young A.B., Tanese N., Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. doi:10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 17.Cong S.Y., Pepers B.A., Evert B.O., Rubinsztein D.C., Roos R.A., van Ommen G.J., Dorsman J.C. Mutant huntingtin represses CBP, but not p300, by binding and protein degradation. Mol. Cell. Neurosci. 2005;30:12–23. doi: 10.1016/j.mcn.2005.05.003. doi:10.1016/j.mcn.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Sugars K.L., Brown R., Cook L.J., Swartz J., Rubinsztein D.C. Decreased cAMP response element-mediated transcription: an early event in exon 1 and full-length cell models of Huntington's disease that contributes to polyglutamine pathogenesis. J. Biol. Chem. 2004;279:4988–4999. doi: 10.1074/jbc.M310226200. doi:10.1074/jbc.M310226200. [DOI] [PubMed] [Google Scholar]
- 19.Chaturvedi R.K., Adhihetty P., Shukla S., Hennessy T., Calingasan N., Beal M.F., Yang L., Starkov A., Kiaei M., Cannella M., et al. Impaired PGC-1alpha function in muscle in Huntington's disease. Hum. Mol. Genet. 2009;18:3048–3065. doi: 10.1093/hmg/ddp243. doi:10.1093/hmg/ddp243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaturvedi R.K., Calingasan N.Y., Yang L., Hennessey T., Johri A., Beal M.F. Impairment of PGC-1alpha expression, neuropathology and hepatic steatosis in a transgenic mouse model of Huntington's disease following chronic energy deprivation. Hum. Mol. Genet. 2010;19:3190–3205. doi: 10.1093/hmg/ddq229. doi:10.1093/hmg/ddq229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui L., Jeong H., Borovecki F., Parkhurst C.N., Tanese N., Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. doi:10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 22.Lin J., Handschin C., Spiegelman B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. doi:10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Chaturvedi R.K., Beal M.F. PPAR: a therapeutic target in Parkinson's disease. J. Neurochem. 2008;106:506–518. doi: 10.1111/j.1471-4159.2008.05388.x. doi:10.1111/j.1471-4159.2008.05388.x. [DOI] [PubMed] [Google Scholar]
- 24.Weydt P., Pineda V.V., Torrence A.E., Libby R.T., Satterfield T.F., Lazarowski E.R., Gilbert M.L., Morton G.J., Bammler T.K., Strand A.D., et al. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. doi:10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 25.McGill J.K., Beal M.F. PGC-1alpha, a new therapeutic target in Huntington's disease? Cell. 2006;127:465–468. doi: 10.1016/j.cell.2006.10.023. doi:10.1016/j.cell.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 26.Kim J., Moody J.P., Edgerly C.K., Bordiuk O.L., Cormier K., Smith K., Beal M.F., Ferrante R.J. Mitochondrial loss, dysfunction and altered dynamics in Huntington's disease. Hum. Mol. Genet. 2010;19:3919–3935. doi: 10.1093/hmg/ddq306. doi:10.1093/hmg/ddq306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conkright M.D., Canettieri G., Screaton R., Guzman E., Miraglia L., Hogenesch J.B., Montminy M. TORCs: transducers of regulated CREB activity. Mol. Cell. 2003;12:413–423. doi: 10.1016/j.molcel.2003.08.013. doi:10.1016/j.molcel.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 28.Wu Z., Huang X., Feng Y., Handschin C., Feng Y., Gullicksen P.S., Bare O., Labow M., Spiegelman B., Stevenson S.C. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc. Natl Acad. Sci. USA. 2006;103:14379–14384. doi: 10.1073/pnas.0606714103. doi:10.1073/pnas.0606714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li S., Zhang C., Takemori H., Zhou Y., Xiong Z.Q. TORC1 regulates activity-dependent CREB-target gene transcription and dendritic growth of developing cortical neurons. J. Neurosci. 2009;29:2334–2343. doi: 10.1523/JNEUROSCI.2296-08.2009. doi:10.1523/JNEUROSCI.2296-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang B., Goode J., Best J., Meltzer J., Schilman P.E., Chen J., Garza D., Thomas J.B., Montminy M. The insulin-regulated CREB coactivator TORC promotes stress resistance in Drosophila. Cell Metab. 2008;7:434–444. doi: 10.1016/j.cmet.2008.02.010. doi:10.1016/j.cmet.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hietakangas V., Cohen S.M. TORCing up metabolic control in the brain. Cell Metab. 2008;7:357–358. doi: 10.1016/j.cmet.2008.04.006. doi:10.1016/j.cmet.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Sasaki T., Takemori H., Yagita Y., Terasaki Y., Uebi T., Horike N., Takagi H., Susumu T., Teraoka H., Kusano K., et al. SIK2 is a key regulator for neuronal survival after ischemia via TORC1-CREB. Neuron. 2011;69:106–119. doi: 10.1016/j.neuron.2010.12.004. doi:10.1016/j.neuron.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 33.Mantamadiotis T., Lemberger T., Bleckmann S.C., Kern H., Kretz O., Martin Villalba A., Tronche F., Kellendonk C., Gau D., Kapfhammer J., et al. Disruption of CREB function in brain leads to neurodegeneration. Nat. Genet. 2002;31:47–54. doi: 10.1038/ng882. doi:10.1038/ng882. [DOI] [PubMed] [Google Scholar]
- 34.Lin J., Wu P.H., Tarr P.T., Lindenberg K.S., St-Pierre J., Zhang C.Y., Mootha V.K., Jager S., Vianna C.R., Reznick R.M., et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. doi:10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 35.Leone T.C., Lehman J.J., Finck B.N., Schaeffer P.J., Wende A.R., Boudina S., Courtois M., Wozniak D.F., Sambandam N., Bernal-Mizrachi C., et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. doi:10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S.W., et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. doi:10.1016/S0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 37.Trettel F., Rigamonti D., Hilditch-Maguire P., Wheeler V.C., Sharp A.H., Persichetti F., Cattaneo E., MacDonald M.E. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. doi:10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 38.Schilling G., Savonenko A.V., Klevytska A., Morton J.L., Tucker S.M., Poirier M., Gale A., Chan N., Gonzales V., Slunt H.H., et al. Nuclear-targeting of mutant huntingtin fragments produces Huntington's disease-like phenotypes in transgenic mice. Hum. Mol. Genet. 2004;13:1599–1610. doi: 10.1093/hmg/ddh175. doi:10.1093/hmg/ddh175. [DOI] [PubMed] [Google Scholar]
- 39.Wheeler V.C., Gutekunst C.A., Vrbanac V., Lebel L.A., Schilling G., Hersch S., Friedlander R.M., Gusella J.F., Vonsattel J.P., Borchelt D.R., et al. Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum. Mol. Genet. 2002;11:633–640. doi: 10.1093/hmg/11.6.633. doi:10.1093/hmg/11.6.633. [DOI] [PubMed] [Google Scholar]
- 40.Kovacs K.A., Steullet P., Steinmann M., Do K.Q., Magistretti P.J., Halfon O., Cardinaux J.R. TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc. Natl Acad. Sci. USA. 2007;104:4700–4705. doi: 10.1073/pnas.0607524104. doi:10.1073/pnas.0607524104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayr B.M., Canettieri G., Montminy M.R. Distinct effects of cAMP and mitogenic signals on CREB-binding protein recruitment impart specificity to target gene activation via CREB. Proc. Natl Acad. Sci. USA. 2001;98:10936–10941. doi: 10.1073/pnas.191152098. doi:10.1073/pnas.191152098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larsson N.G., Wang J., Wilhelmsson H., Oldfors A., Rustin P., Lewandoski M., Barsh G.S., Clayton D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. doi:10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 43.Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., Troy A., Cinti S., Lowell B., Scarpulla R.C., et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. doi:10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 44.Kelly D.P., Scarpulla R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. doi:10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 45.D'Aurelio M., Vives-Bauza C., Davidson M.M., Manfredi G. Mitochondrial DNA background modifies the bioenergetics of NARP/MILS ATP6 mutant cells. Hum. Mol. Genet. 2010;19:374–386. doi: 10.1093/hmg/ddp503. doi:10.1093/hmg/ddp503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gines S., Seong I.S., Fossale E., Ivanova E., Trettel F., Gusella J.F., Wheeler V.C., Persichetti F., MacDonald M.E. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington's disease knock-in mice. Hum. Mol. Genet. 2003;12:497–508. doi: 10.1093/hmg/ddg046. doi:10.1093/hmg/ddg046. [DOI] [PubMed] [Google Scholar]
- 47.Choi Y.S., Lee B., Cho H.Y., Reyes I.B., Pu X.A., Saido T.C., Hoyt K.R., Obrietan K. CREB is a key regulator of striatal vulnerability in chemical and genetic models of Huntington's disease. Neurobiol. Dis. 2009;36:259–268. doi: 10.1016/j.nbd.2009.07.014. doi:10.1016/j.nbd.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klevytska A.M., Tebbenkamp A.T., Savonenko A.V., Borchelt D.R. Partial depletion of CREB-binding protein reduces life expectancy in a mouse model of Huntington disease. J. Neuropathol. Exp. Neurol. 2010;69:396–404. doi: 10.1097/NEN.0b013e3181d6c436. doi:10.1097/NEN.0b013e3181d6c436. [DOI] [PubMed] [Google Scholar]
- 49.Zhou Y., Wu H., Li S., Chen Q., Cheng X.W., Zheng J., Takemori H., Xiong Z.Q. Requirement of TORC1 for late-phase long-term potentiation in the hippocampus. PLoS One. 2006;1:e16. doi: 10.1371/journal.pone.0000016. doi:10.1371/journal.pone.0000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giampa C., Middei S., Patassini S., Borreca A., Marullo F., Laurenti D., Bernardi G., Ammassari-Teule M., Fusco F.R. Phosphodiesterase type IV inhibition prevents sequestration of CREB binding protein, protects striatal parvalbumin interneurons and rescues motor deficits in the R6/2 mouse model of Huntington's disease. Eur. J. Neurosci. 2009;29:902–910. doi: 10.1111/j.1460-9568.2009.06649.x. doi:10.1111/j.1460-9568.2009.06649.x. [DOI] [PubMed] [Google Scholar]
- 51.DeMarch Z., Giampa C., Patassini S., Bernardi G., Fusco F.R. Beneficial effects of rolipram in the R6/2 mouse model of Huntington's disease. Neurobiol. Dis. 2008;30:375–387. doi: 10.1016/j.nbd.2008.02.010. doi:10.1016/j.nbd.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 52.Giampa C., Laurenti D., Anzilotti S., Bernardi G., Menniti F.S., Fusco F.R. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington's disease. PLoS One. 2010;5:e13417. doi: 10.1371/journal.pone.0013417. doi:10.1371/journal.pone.0013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang H., Nucifora F.C., Jr, Ross C.A., DeFranco D.B. Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum. Mol. Genet. 2003;12:1–12. doi: 10.1093/hmg/ddg002. doi:10.1093/hmg/ddg002. [DOI] [PubMed] [Google Scholar]
- 54.Jiang H., Poirier M.A., Liang Y., Pei Z., Weiskittel C.E., Smith W.W., DeFranco D.B., Ross C.A. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis. 2006;23:543–551. doi: 10.1016/j.nbd.2006.04.011. doi:10.1016/j.nbd.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 55.Herzig S., Long F., Jhala U.S., Hedrick S., Quinn R., Bauer A., Rudolph D., Schutz G., Yoon C., Puigserver P., et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. doi:10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- 56.St-Pierre J., Drori S., Uldry M., Silvaggi J.M., Rhee J., Jager S., Handschin C., Zheng K., Lin J., Yang W., et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. doi:10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 57.Koo S.H., Flechner L., Qi L., Zhang X., Screaton R.A., Jeffries S., Hedrick S., Xu W., Boussouar F., Brindle P., et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. doi:10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 58.Canettieri G., Koo S.H., Berdeaux R., Heredia J., Hedrick S., Zhang X., Montminy M. Dual role of the coactivator TORC2 in modulating hepatic glucose output and insulin signaling. Cell Metab. 2005;2:331–338. doi: 10.1016/j.cmet.2005.09.008. doi:10.1016/j.cmet.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 59.Siu Y.T., Ching Y.P., Jin D.Y. Activation of TORC1 transcriptional coactivator through MEKK1-induced phosphorylation. Mol. Biol. Cell. 2008;19:4750–4761. doi: 10.1091/mbc.E08-04-0369. doi:10.1091/mbc.E08-04-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sasaki T., Takemori H., Yagita Y., Terasaki Y., Uebi T., Horike N., Takagi H., Susumu T., Teraoka H., Kusano K., et al. SIK2 is a key regulator for neuronal survival after ischemia via TORC1-CREB. Neuron. 2011;69:106–119. doi: 10.1016/j.neuron.2010.12.004. doi:10.1016/j.neuron.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 61.Jeong H., Cohen D.E., Cui L., Supinski A., Savas J.N., Mazzulli J.R., Yates J.R., 3rd, Bordone L., Guarente L., Krainc D. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat. Med. 2011;18:159–165. doi: 10.1038/nm.2559. doi:10.1038/nm.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu L., Zhang X., Wang L., Yang R., Cui L., Li M., Du W., Wang S. The neuroprotective effects of Tanshinone IIA are associated with induced nuclear translocation of TORC1 and upregulated expression of TORC1, pCREB and BDNF in the acute stage of ischemic stroke. Brain Res. Bull. 2010;82:228–233. doi: 10.1016/j.brainresbull.2010.04.005. doi:10.1016/j.brainresbull.2010.04.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.