

Abstract
To fully describe gene expression dynamics requires the ability to quantitatively capture expression in individual cells over time. Automated systems for acquiring and analyzing real-time images are needed to obtain unbiased data across many samples and conditions. We developed a microfluidics device, the RootArray, in which 64 Arabidopsis thaliana seedlings can be grown and their roots imaged by confocal microscopy over several days without manual intervention. To achieve high throughput, we decoupled acquisition from analysis. In the acquisition phase, we obtain images at low magnification and segment to identify regions of interest. Coordinates are communicated to the microscope to record the regions of interest at high magnification. In the analysis phase, we reconstruct 3D objects from stitched high magnification images, and extract quantitative measurements from a virtual medial section of the root. We tracked hundreds of roots to capture detailed expression patterns of 12 transgenic reporter lines under different conditions.
Introduction
A major challenge in biology today is mapping genotype to phenotype. A key aspect of the mapping function is gene regulation, and it is increasingly evident that we need to capture the dynamics of gene expression if we are to understand the complexity of its regulation. This is particularly challenging in multicellular organisms in which spatial as well as temporal aspects of gene expression need to be monitored.
One way to address this issue is through time-series expression analyses using microarrays or sequencing. Generally, this has been performed on whole organisms or entire organs, but methods have been developed to isolate specific cell types and/or developmental stages 1, 2. Nevertheless, extracting specific cell populations, and the cost of profiling them at different times, can be prohibitive. An alternative is to use fluorescent or light-emitting reporters and to image over time at the resolution of individual cell types 3, 4.
Previous work has addressed the technical challenges of gene expression monitoring using light microscopy in multicellular organisms. For example, image analysis methods developed for Caenorhabditis elegans can identify individual cell types and map levels of fluorescence to an atlas, retaining a manual component to resolve ambiguities 5-8. Applications in plants have focused on the shoot apical meristem 9, 10, achieving high resolution by fusing of images from different angles 11. While these approaches have used computational methods to extract quantitative features, they were carried out one individual at a time and still required manual intervention during both imaging and analysis.
The growing root of Arabidopsis thaliana is particularly well suited for real-time imaging. The typical diameter (around 100 μm) is above the thickness limit of resolution for a confocal microscope 12. However, the four outer cell types are organized as concentric cylinders around the central vascular tissue (Supplementary Fig. 1). This means that a 2D section through the center of the root provides a representative sample of most tissue types 13. The challenge thus lies in conducting live microscopy on a large number of specimens growing under controlled conditions to assess spatiotemporal expression. Recently, a small number of Arabidopsis roots were grown in channels and transferred to a microfluidics device. Media perfusions were performed and data acquired using non-confocal light microscopy 14, 15. While these approaches were motivated by the same questions, they may not be straightforward to scale up, as they do not presently incorporate automation for image acquisition or analysis. With the goal of quantifying dynamic gene expression in a large number of samples, we developed the RootArray, a microfluidics device in which 64 seedlings can be grown in parallel and their roots imaged repeatedly by confocal microscopy. For efficient and flexible image registration we developed an image analysis platform that includes automated real-time detection and tracking of samples. Off-line algorithms then reconstruct the 3D shape of each root and identify a virtual medial section, which is used to map the fluorescence intensity to specific cell types. We applied this integrated platform to systematically quantify expression patterns of 12 reporter genes in roots growing in four different media conditions over time, resulting in thousands of images of individual roots. Comparisons to gene expression profiles acquired by microarrays showed good correlations, but more importantly, identified several cases of transient or heterogeneous expression.
Results
The RootArray enables live imaging of multiple roots
We designed the “RootArray” microfluidics device (Figure 1a,b) to provide a growth chamber in which the plants germinate and grow and can be subjected to treatments before or during imaging. Its core component is a translucent and biologically inert photopolymer scaffold, manufactured using stereolithography, which contains 64 (16 × 4) wells. Each of the wells is filled with agar in which a single seed is planted. Upon assembly, the complete RootArray is sterilized with a bleach-containing solution and connected to a peristaltic pump, which supplies gas and liquid through ports to the shoot and root chambers, respectively. This setup enables a continuous exchange of growth media and the application of compounds, stains and other treatments. RootArrays can be reused, and we typically used one device for 10 – 20 experiments. Assembled RootArrays are mounted vertically on a rack and exposed to continuous light causing seeds to germinate approximately four days after planting. The roots penetrate the agar plug and emerge into the liquid filled chamber. We then mount the RootArray on the motorized stage of a confocal microscope and image over time (Figure 1c,d).
Figure 1.
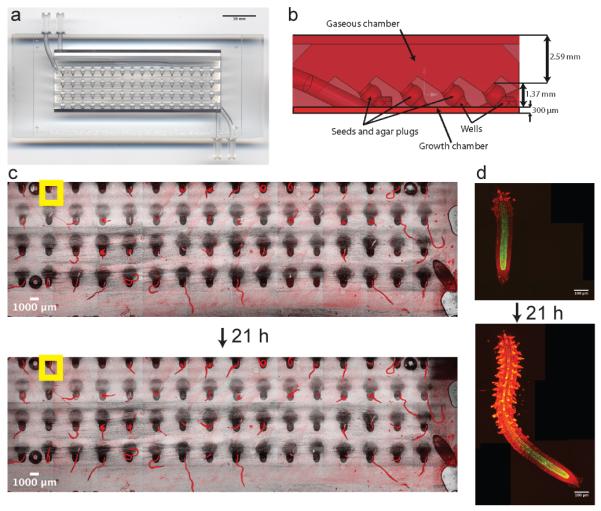
The RootArray. (a) Image shows the RootArray with 64 wells (b) Cross-section with dimensions of the design features. The wells connect both chambers allowing the roots to grow into the liquid chamber and the shoots to emerge into the gaseous chamber. The upper and lower limits indicate the coverslips that are used to seal the array. The growing roots can be imaged over many hours (c,d). An example is given of an overview image at low resolution (c) and the stitched high-resolution images of individual roots (d).
Automated Image Acquisition
Acquiring confocal images of the entire RootArray volume at high resolution would take several days, but the actual roots occupy only a small fraction of the growth chamber volume. To achieve in vivo imaging at high throughput, we developed a robust image processing platform, the RootArray Controller, which performs real-time region-of-interest detection by tracking the roots and acquiring images of only those parts of the chamber in which growing roots are detected. We then perform off-line analyses on the high resolution images (Figure 2). The software can be adapted to work with a wide array of microscope systems.
Figure 2.
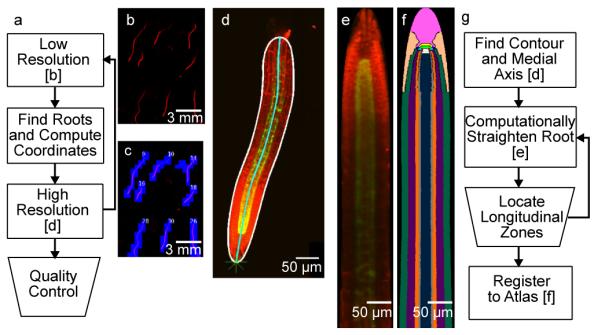
Acquisition and analysis workflow. (a) Summary of the real-time acquisition platform. Rectangles represent automated processes and the trapezoids represent manual steps. (b) A portion of a RootArray imaged at low resolution. The channel shown is fluorescence of the propidium iodide dye. (c) The detected roots from the low resolution image are scheduled for high resolution imaging. The blue squares represent the individual high resolution images required to tile each root. (d) A high resolution image (maximum projection) of an automatically acquired root. The red channel again shows the propidium iodide dye, the green channel depicts expression of a GFP reporter gene. The border of the root is highlighted in white and the medial axis of the root in cyan. (e) The same root after computationally straightening. (f) The straightened root mapped to a tissue atlas. (g) Summary of the offline analysis steps.
In the acquisition phase, imaging rounds alternate between low and high magnification. In the low resolution phase, images cover the complete RootArray (Supplementary Fig. 2). Each image contains a propidium iodide stain channel (indicating cell walls) and a channel capturing the autofluorescence of the RootArray polymer. This provides a high contrast image in which the individual wells of the RootArray are visible.
To identify the physical location of each root (the microscope stage coordinates at which to take images), we needed to determine which pixels in the low resolution image correspond to roots. While a number of simple data-driven segmentation approaches are available and applicable to biological data, RootArray images posed specific challenges including variable background (due to autofluorescence and accumulation of dye over long periods of exposure) and non-root objects visible in the foreground (air bubbles, scratches, etc.), all of which led to considerable variability in input images (Supplementary Fig. 3). Furthermore, plants germinated at different times, leading to different numbers and sizes of objects. To overcome these challenges, we implemented a hierarchical graphical model for image segmentation, which is based on Factor Graphs 16, 17. The model annotates all pixels as roots or non-roots using the maximum projection of the low resolution image (Supplementary Fig. 4, Online Methods). It is trained on a small set of user-labeled images, and effectively learns domain-specific correlations on the pixel intensities, as well as on the object labels at different scales. Compared to many widely used segmentation algorithms, this approach delivered the best tradeoff between robust and efficient performance (Supplementary Fig. 5).
The output of the above segmentation is a labeled connected component image, where each non-zero intensity pixel represents part of an object that needs to be imaged at high resolution, and each intensity labels a group of pixels. This labeled image is provided to a tiling algorithm that calculates the locations of a sufficient number of high resolution images to completely cover all of the labeled components, while taking the direction of growth at the root tips into account. The resulting coordinates are provided to the proprietary microscope software. The user specifies a time interval or number of imaging rounds.
The microscope acquires a high resolution Z stack image for each entry in the coordinate file. It takes several high resolution images to cover an entire root, and images are stitched prior to further analysis 18. In experiments that only require data from the root tip, the throughput is increased by an order of magnitude by excluding the mature part of the root. We implemented a topological tip detection algorithm that identifies the two ends of the root, the meristem and the part emerging from the well plate 19. Well locations were previously identified via the generalized Hough transform on the plate autofluorescence channel 20 (Supplementary Figs. 2b,c and 6).
Registration at high resolution
To quantify spatial features of reporter gene expression, we took advantage of the radially symmetric anatomy of the root (Supplementary Fig. 1). Any 2D surface that intersects the center of the root (a medial section) will be a representative sample in which almost all tissue types can be uniquely identified. Accordingly, the analysis is comprised of the following steps: compute a 2D medial section image, perform manual quality control and identification of developmental zones, and register the medial section image to a root atlas.
The medial section computation faces strict performance requirements due to the large number of images and the size of each stitched image (50 – 500 megavoxels, 300 MB to 2 GB uncompressed file size). First, the 3D shape of the root is identified via edge detection and an efficient active contour algorithm 21. Using this information, we locate the surface of the root and compute its diameter along its length. Surface and diameter are used to derive the medial axis (the curve passing through the middle of the root). We then simultaneously reconstruct the medial section and computationally straighten the root by sampling along lines perpendicular to the medial axis and parallel to the Z axis (Figure 3).
Figure 3.
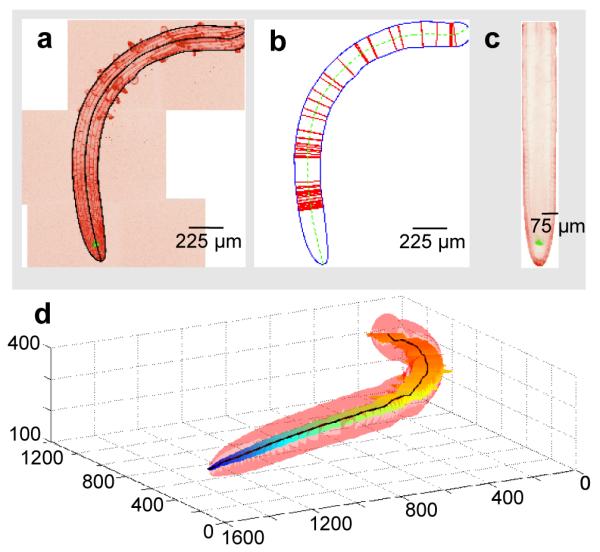
Computation of the medial section of a plant root. (a) A false-colored image shows the maximum projection of a stitched 3D high resolution image. Black lines indicate the computed border and medial axis of the root. (b) Extracted root, with sampling points used in the computational straightening marked as red lines. (c) A computed straightened medial section of the same root. (d) A 3D plot of the identified root (in pink), the medial section (with a heat map indicating depth from the well surface), and the medial axis (black line). The range of depth covered by this root is approximately 160 microns; this root thus spans half of the depth of the RootArray growth chamber.
The active contour algorithm is parametric, and due to the large variance in image contrast it may fail on some images for a given parameter. We therefore made the choice to manually confirm the quality of the medial section computation and mark the developmental zones using the ZoneMarker Fiji plugin (see Online Methods). If the manual review indicates that the medial section is incorrect, we recompute it with a different parameter set (using one of only four different parameter sets led to successful segmentation of most images despite large variation in the input images). The varying image contrast also provides significant challenges for the automatic identification of the developmental zones. Therefore, once an image has passed quality control, an expert user identifies the starting point of each zone (meristem, elongation, maturation).
The final step is the registration of the medial section to the root atlas, which provides labels of tissue types for every pixel in a prototypical root. The relative sizes of each tissue type and the contour of the atlas were estimated from manually annotated training data. We use the developmental zone information and the contour of the medial section to perform a piece-wise, linear registration to the atlas image. The result is a labeled image mask that can be used to extract tissue-specific phenotype information (Figure 4). We distinguish 9 tissue types, with a subset of tissues further divided by developmental zone resulting in a total of 18 tissue regions. Benchmarking against manual region annotation indicated an accuracy of 19–98% depending on the cell type with an average accuracy of 66% (Supplementary Table 1). Low-accuracy tissue regions were generally small relative to the other cell types in the vicinity (pericycle and endodermis in the meristem).
Figure 4.
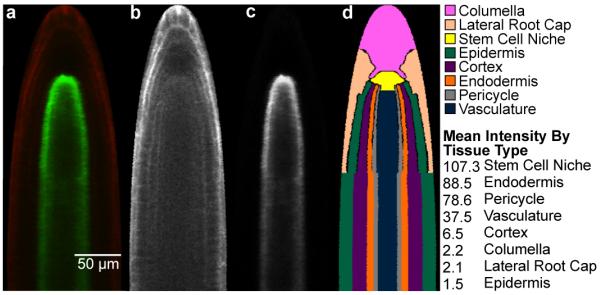
Extraction of expression information. (a) Example image of a computationally straightened root after crosstalk removal, showing the meristem and elongation zones. (b) The corresponding propidium iodide channel (c) The corresponding GFP channel; expression is conferred by the SCR promoter. (d) The atlas mapped onto the root. mean pixel intensity extracted for the individual cell types of the root meristem is indicated.
Experimental design and quality control
For initial testing of the RootArray, we investigated the dynamic changes of gene expression in response to several abiotic stresses. Transgenic lines containing 12 reporter genes, which included important regulators of cellular identity and/or developmental processes (Supplementary Table 2) were grown under four conditions: standard media (MS), low pH (pH), iron deficiency (−Fe), and sulfur deficiency (−S). Each array was set up with 16 individuals of 4 transgenic lines distributed diagonally over the array to reduce positional effects. Additionally, each line and treatment was tested multiple times. Because roots emerging from the lower growth channels touch the borders of the RootArray much sooner than roots growing from the upper rows, seeds of each genotype were distributed equally between rows.
For all experiments, seedlings were initially grown in standard MS media. Prior to imaging, the root growth chamber was perfused with the treatment medium. Most of the images were collected over 48 hours. While image acquisition was fully automated, expert quality control was required to maintain high quality standards during analysis. For each root, we mapped its well position and performed a rigorous multistep quality control. We first excluded roots that were unhealthy, not imaged completely, or of insufficient quality. The second step excluded roots that had not been segmented correctly or in which the position of the root tip was not detected correctly. These steps, as well as marking the borders of the developmental zones were conducted in a single interface (Supplementary Fig. 7) and typically required less than 10 seconds per root image. Thus, quality control and assignment of wells adds up to a maximum of 16 minutes per time point for several hours of automated acquisition (Supplementary Table 3). Our dataset of 73 different RootArrays allowed us to benchmark the performance and the throughput of our platform (Supplementary Fig. 8, Supplementary Table 3). On average, roots were visible for 54% of the seeds planted. From those, 65% could be fully segmented in at least one time point. From tracked roots, 42% passed a quality control step that excluded roots of insufficient quality. Of these, 45% could be registered and quantified correctly. While the remaining roots were only a fraction of all roots, we retained a substantial dataset that included 1393 whole root time course images of 730 distinct roots from 73 different RootArrays (Supplementary Table 4).
We calculated the growth rate, counted cells expressing a cell-cycle marker indicative of division and measured the dimensions of the different developmental zones (Supplementary Fig. 9, Supplementary Table 5). The average growth rate of 75μm/h under MS conditions in the first 24h (Supplementary Fig. 9) of imaging was reduced compared to the reported growth rate of older plants on vertical agar plates 22, 23. Since root growth rate is dependent on seed size, growth conditions and seedling age24, this was not unexpected. We saw no evidence of cell death in slower growing roots that passed our quality control or deviations from normal root morphology indicating that slower growth was not adversely affecting the plants. Moreover, it provided the benefit of allowing to sample more time points before the roots reached the borders of the RootArray. Finally, we only compared roots grown under the same conditions, so any growth-rate specific effects would be similar under different treatments. In fact, while there were significant changes of growth rate and in the size of developmental zones upon certain treatments, the effect size was small and there was no significant change in cell division rate after any treatment (Supplementary Fig. 9).
Quantitative Analysis of Phenotypes
From these data we were able to address several important questions related to gene expression in a complex organ system. For instance, it is often assumed that gene expression patterns are identical among genetically identical individuals under the same conditions. To assess the extent of heterogeneity of gene expression under uniform conditions, we evaluated images from isogenic plants grown in the same RootArray (Supplementary Fig. 10a). For some promoters (S17, S18, S4 and A8) we observed high levels of heterogeneity due to the absence or strong reduction of the fluorescent signal in some individuals (Supplementary Fig. 11). In contrast, the cell identity regulators WER (Supplementary Fig. 10b-g) and SCR (Supplementary Fig. 10h-k) displayed a relatively low level of variation. Misexpression of these cell identity regulators might lead to detrimental effects 25. Consistent with these observations, genes with low heterogeneity showed good agreement with tissue-specific expression levels as measured by microarrays, while lines exhibiting higher levels of heterogeneity had less agreement (Supplementary Fig. 12).
The heterogeneity underlined the importance of using a robust procedure to compare expression patterns of genes between different conditions. We therefore used a method that subtracted background and assigned a rank of average pixel intensity for each tissue in each root. We then conducted a nonparametric Wilcoxon rank sum test on each genotype for each condition as compared to roots grown in MS media for each tissue type. We corrected for multiple testing by calculating a false discovery rate (FDR) for each p-value (Supplementary Table 6, Figure 5). By applying a rigorous statistical test, the high heterogeneity of some reporters within one condition should not affect our results. In fact, the detection of significant changes in spite of the variability underlines the magnitude of the observed effects. Using a cut-off FDR of 5% we identified 10 of the 12 reporters as being affected in at least one tissue for one or more abiotic stresses. The most significant changes of expression were detected in low pH media and sulfur deficient media. Lack of iron was the weakest stressor during the observed timeframe, which could partially be due to the lower sample numbers of the −Fe condition. We visually inspected the images with the highest contribution to the results to confirm that genes were expressed in the tissues in which significant changes were detected (Supplementary Table 6).
Figure 5.

Significance of gene expression changes in different growth conditions (−Fe: Iron deficient medium; pH 4.6: low pH medium; −S: Sulfur deficient medium). Different tissue types are shown along the x-axis, the −log10 FDRs for change of expression compared to expression in MS medium for each color-coded reporter gene are shown along the y axis. The dashed line indicates the significance threshold of 5% FDR.
We also looked for time-dependent changes in expression in response to different treatments, focusing on the WOX5 and UPB1 lines, which had shown the most dramatic expression changes. Progressive changes were observed for UPB1 expression in low pH and in −S (Supplementary Fig. 13). For WOX5, we also observed a progressive change of expression in sulfur deficient and low pH media in the meristem and elongation zones (Figure 6). Under low pH, a few cells started to express GFP ectopically after 6 hours. Expression expanded into other cells between 6 h and 15 h and then retracted to the initial expression pattern at later time points (Figure 6 i,n). In the case of −S, a similar transient expression pattern was observed. The WOX5 expressing cells in the elongation zone are different individuals in each image (see Figure 6 j,o), indicating that the WOX5 promoter undergoes a burst of expression in some cells, producing a standing wave of expression in the elongation zone through which the cells travel on their developmental path. This would suggest the presence of a self-sustaining regulatory network controlling WOX5 expression.
Figure 6.

Progression of expression change in WOX5 reporter lines. MS medium (a–e), Sulfur deficient (−S) medium (f–j), low pH (pH 4.6) medium (k–o). Insets for f–n are magnified portions indicated by the box. e,j,k are low-resolution images of the root at the last time point and the squares indicate the tip position at the time points shown. One time point of the root in o not depicted; k-n correspond to the black, blue, red and cyan squares in o.
Discussion
The RootArray platform allowed us to monitor and compare reporter gene expression in Arabidopsis roots at the level of tissue types in different developmental zones. A practical limitation to achieving cellular resolution is the propidium iodide dye, which accumulates over time and is unevenly incorporated. We expect that transgenic cellular markers will make it possible to register individual cells and to calibrate expression levels. We also noted significant morphological variation, for instance in the location of the quiescent center, making it necessary to manually mark the developmental zones. Additional transgenic markers would allow for a completely automated and more fine-grained, probabilistic approach to the segmentation of tissue types.
While we expect that additional markers can provide for single-cell resolution, the speed of confocal microscopy is not rapid enough to allow for the analysis of all cellular events. For example, imaging of even a significant proportion of the 64 roots takes several hours, and isnot fast enough to track individual cells in a quickly growing root undergoing cell divisions. To achieve a higher temporal and spatial resolution, the RootArray could be used to observe a smaller number of roots limited to the meristematic zone, which would lead to an imaging rate of several images per hour and thus the necessary resolution to track individual cells. Alternatively, other imaging modalities such as light-sheet microscopy could provide faster image acquisition times 22, 26.
The RootArray is a technology still in development, and we expect improvements in several aspects of the workflow, such as ways to increase the yield of automatically analyzed roots and the accuracy of the mapping and quantification of GFP. Nevertheless, we expect a wide application of the RootArray technology since it is not only suitable for studying gene expression, but holds promise for a broad spectrum of applications, including quantitative methods based on fluorescent proteins such as FRET, co-localization studies, mutant screens using fluorescent markers, variation in natural populations, and high throughput determination of growth characteristics using cell wall dyes.
Online Methods
RootArray
RootArrays were manufactured using a WaterShed XC 11122 high-resolution stereolithography build in 0.002” Layers with substrate build style option at Fineline Prototyping, Raleigh, NC, USA. The RootArray (Figure 1) contains 64 wells that are filled with agar (see below). After having inserted the seeds, the assembly of the RootArray is sealed by placing adhesives on both sides and attaching cover slips to the adhesives (see below). The custom cut adhesives (basically a double layer of 3M 9485PC adhesive) are ~300μm thick. The assembled RootArray contains two chambers. The one in which the roots grow has a depth of ~300μm. The volume in which the shoots grow is ~ 2.61mm deep on top of the ridges and 3.99mm at the bottom of the ridges.
Plant Materials and Growth Conditions
Seeds were kept dry at 4°C for stratification. Prior to RootArray assembly, an aliquot was suspended in deionized, autoclaved water. This aliquot was used up to 7 days for RootArray preparation. First, all wells of the RootArray were filled with 3μl liquid agar (3%). Then, a coverslip was attached to the liquid side. Subsequently, the seeds were placed on top of the agar in the wells. To fix the position of the seed, 1μl drops of agar were placed onto the seeds. After this the wells were covered with a thin layer of agar.
Then, the air side of the RootArray was sealed with a coverslip using the adhesive. After a brief period at 4°C, the RootArray was sterilized using a 25% solution of household bleach. After sterilization the array was flushed 5 times with sterile water. A manual illustrating the RootArray set-up protocol is available upon request.
Seeds were germinated and grown in the sterile RootArray environment. MS liquid media was pumped through the RootArray using a Watson-Marlow Bredel pump (323 DU/D Pump 400 RPM US) with 1.6mm silicone tubing (Bio-Rad) at a flow rate of approximately 300μl/min. Plants were imaged 4-7 days after being planted in the RootArray under continuous light at 22-23°C. Before imaging, the RootArray was separated from the pumping system and the root chamber was flushed with 4ml of treatment media. Finally 1ml of a treatment/stain mixture containing 980 μl treatment medium and 20 μl (1mg/ml) propidium iodide was injected in the root chamber. The composition of the different growth/treatment media was as described in. 27 Treatments and treatment dates are given in Supplementary Table 7.
Image Acquisition
The RootArray was mounted on a Zeiss LSM510 meta Axiovert 200M microscope system. There was no ambient illumination during imaging and the ambient temperature was 21-22°C. The microscope was supplied with two proprietary applications (ZEN2009 and Multitime 7.3). ZEN2009 is an interactive graphical user interface that can be used to manually view and acquire images, set various microscope parameters and save them as profiles, as well as the ability to save manually marked reference points on the microscope stage. Multitime is a plug-in for ZEN and can perform several additional functions, including interfacing ZEN with third-party applications via a control file and a coordinate file containing stage coordinates and configuration instructions for image acquisition. Our acquisition software, the RootArray Controller, was used to instruct Multitime to take a low resolution (2.5x objective) fixed grid of the entire growth chamber. We first identified regions of interest via image segmentation (e.g. roots, root meristems). The RootArray Controller then provided Multitime with the coordinates for taking high resolution images (20x objective) of only the regions of interest. The high-resolution images were 3-dimensional z stacks, typically containing 40 slices spaced 7.2μm apart. This sequence of low resolution scan, feature detection, and subsequent high resolution imaging was repeated, thus providing images at consecutive time points for a given RootArray. The number of high resolution images acquired at each imaging round was the limiting time factor (it took ~1.1 minutes for each individual z stack). We implemented a graphical user interface for a manual review of acquired images and for associating each image with its corresponding plant.
Low Resolution Mosaic
A fixed grid of low resolution images covering the growth chamber was acquired. The number of rows and columns in the image grid was variable and depended on the bounding box specified by the user. Each image contained two channels; the first identified fluorescence from the propidium iodide dye that outlined the cells, and the second showed the autofluorescence from the back of the RootArray. This second channel provided an image in which the wells were identifiable.
Hierarchical Model for Root Detection
The low resolution mosaic images of the RootArray provided significant challenges for a segmentation algorithm. There were occasional scratches, inconsistencies in the background, and varying intensity of the roots depending on imaging conditions and age of the array. We designed a model-based classification algorithm to overcome these challenges. Our model is a generalization of typical Markov Random Field models for segmentation, in which the observable pixel intensities are dependent on a single layer of hidden labels 16, to multiple intermediate layers that represent spatial dependencies at different resolutions. Furthermore, we included rotation invariance in the model to account for the varying directions of root growth. A detailed mathematical description of the model can be found in Supplementary Note 1.
Training the Root Model
The hierarchical model contained several parameters that needed to be estimated; namely, we needed the mean and covariance for the multivariate normal distributions, and the probabilities for the events in the multinomial distributions. We trained the model using hand-labeled low resolution images (0 for background, 1 for root). We trained the parameters to optimize the Maximum Likelihood using expectation maximization (EM). Training and test images are available as part of the Supplementary Software which is available via: http://www.genome.duke.edu/labs/ohler/research/image_analysis/rootarray/nature_methods-supp/rootarray-software.tgz
Well Detection
To assign images to phenotypes, we needed to connect the roots to the wells. The locations of the wells were fixed by the RootArray specification. Therefore, we could detect the location of the holes by determining the translation and rotation of a RootArray. The Generalized Hough transform is a way of identifying a translated, scaled, and rotated template image, called the kernel, in an image 20. We constructed a template well image by averaging 12 hand-selected well images. Each well image was selected from the autofluorescence channel of a low resolution mosaic and cropped so that the well was centered. The kernel used in the Generalized Hough transform was created by taking the template well image, masking it by thresholding it for high values of its gradient magnitude, and taking the resulting points and their gradient vectors.
The output of the Generalized Hough transform is a M × N × R × S discrete count matrix called accumulator (where M × N) is the size of the input image, R is the number of rotation angles to sample at, and S is the number of scales to sample at). Its elements are the number of votes for the kernel image at a specific location with a specific rotation and scale. We thresholded the accumulator, and took the elements with the maximum number of votes from proximal elements (elements that are within a thresholded distance from each other). We located the element closest to the upper left corner of the low resolution mosaic, and the element that is closest to the bottom right corner. We determined the translation of the wells by the value of the upper left element, and determined the orientation of the wells by looking at the orientation between the two corner elements.
Detection of Tips and Potential Growth Regions
We used a topological algorithm for tip detection 19. For each foreground pixel in the root mask image, we centered a closed disk of fixed radius. We subtracted the root mask image from this closed disk and counted the number of resulting connected components (i.e., we determined whether the disk has been cut into pieces or simply pierced). For the branch (middle portion) of the roots, the disk was cleaved into two components. For the tips, the disk remained as one component (see Supplementary Fig. 6). For a given root, this algorithm will return two tips: the root meristem and the point at which the root enters the growth chamber. To disambiguate between the two, we considered any tip that overlaps with the well image mask to be a non-meristem tip.
Even though we restricted our high resolution imaging to regions of interest, the duration between the low resolution imaging and the high resolution imaging was sometimes significant. This allowed for the tips of roots to extend beyond the identified region of interest before high resolution imaging. We also observed that in some experiments, the uptake of the propidium iodide dye was weaker near the root meristem. In order to ensure that we fully captured the root meristem during high resolution imaging, we predicted the region of potential growth (including the potentially undetectable portion of the meristem).
We calculated the direction of current root growth, and then imaged a cone of fixed size in that direction allowing for the possibility of curvature. The tip detection algorithm returns the root meristem tips in the low resolution mosaic. These tips are not points, but regions of the root whose size is proportional to the width of the root and the radius of the disk used in the tip detection. To determine the orientation of the root, we detected the orientation of this tip region.
Let Tk = {(x, y) : (x, y) are pixel coordinates in the tip} be the identified tip region of the kth root. The orientation vector of Tk is given by the angle of the eigenvector with the largest eigenvalue of the covariance matrix of Tk. This vector may be pointing into the root, and we therefore found a branch pixel that touches a pixel in Tk. We constructed a vector from the centroid of Tk to this branch pixel, and calculate the angle between this vector and the eigenvector. If this angle was less than 90 degrees, we rotated the orientation angle by 180 degrees. We defined the region of growth as a triangle of fixed size at the centroid of Tkin the calculated orientation.
Tiling
Once regions of interest were defined (whether complete roots or root meristems), we scheduled high resolution images that covered the entirety of each region. In images acquired early in the project, we placed a fixed grid corresponding to the size of the high resolution images over the entire low resolution mosaic using stage coordinates and allowing for overlap. A high resolution tile from the grid was scheduled if it overlapped with a region of interest. This resulted in redundant imaging; if a root grew along the edge between two tiles, both tiles would be scheduled even if one tile could cover the width of the root.
We therefore defined a revised algorithm that we currently use. The vertical span of the region of interest was calculated as the difference between the minimum and maximum y. We divided the vertical span by the height of a high resolution image to determine how many rows our tiling would have. We grouped the points in the region of interest by which row their y coordinate was contained in. We sorted the points in a given row by their x coordinate. If any of the sorted points was not covered by a high resolution image, we scheduled a high resolution image at the y coordinate for the row, and the x coordinate for the point.
Stitching of high resolution images
For a given region of interest, multiple high resolution images were required to cover the entire region. The tiles were scheduled in such a way that a small portion of each high resolution image would overlap with adjacent images. We stitched these high resolution images together using a command-line wrapper around the Fiji Stitching plugin 18. The plugin took as input the stage coordinates of each high resolution image mapped to relative pixel coordinates and an estimate of expected overlap. The output of the plugin was a large image of the entire region of interest.
Well-mapping and quality control
Scratches, air bubbles, and other artifacts may be mistakenly identified as roots. Subsequent failures in the well detection step should not prevent a RootArray experiment from being executed. In a final quality control step, the maximum projection of each stitched high resolution image was manually evaluated in a graphical user interface application. At the same time, each region of interest was mapped to its corresponding well (allowing for recorded genotype data to be mapped to the image) (see Supplementary Fig. 7).
Root Detection Performance
To evaluate the factor graph segmentation performance, we compared it against 17 segmentation algorithms available in Fiji [http://www.fiji.sc]. We evaluated each algorithm on a set of 15 manually labeled, low resolution mosaics obtained at different time points and experimental conditions. Both our algorithm and the Robust Automatic Threshold Segmentation (RATS) required training, carried out on a separate set of 15 images (each image contained one or two hand-labeled roots). The RATS algorithm was trained using the Apache Commons library direct search optimizer.
The performance of segmentation algorithms may be quantified by false positive and false negative rates. Each of these metrics reflects a different view on accuracy, and their associated real world cost may be difficult to infer. The rate-limiting step in acquiring and analyzing RootArray data is the amount of time for the confocal microscope to take images. Therefore, the primary measure of performance for any segmentation algorithm is the number of correctly detected roots that can be imaged per unit of time. This metric combines both the false positive (images needlessly imaged by the microscope) and false negative (missed roots) rates.
Each algorithm takes an input image and outputs a binary classification per pixel. Certain errors are less problematic than others. For example, a false positive pixel on the boundary of the root will most likely not result in an additional image being acquired by the microscope (each high resolution tile covers multiple low resolution pixels), while missing an entire portion of a root renders that data useless. Therefore, we do not score each segmentation algorithm on its direct output; instead, we run each algorithm through the tiling step and quantify its performance on the number of erroneous and missing tiles.
Formally, we defined the metric of roots per minute as , where R is the number of roots acquired and T is the number of tiles scheduled. The constant 1.1 reflects the amount of time (in minutes) it takes for the microscope to acquire a high-resolution tile. A higher value of this constant penalizes false positives more, a lower value similarly weights against false negatives. This ratio highlights the trade-off between sensitivity and specificity of the segmentation algorithms. We considered a root to be successfully acquired if more than 90% of it was identified (i.e. 90% of its pixels were covered by adjacent tiles). We took the output of each algorithm (the binary root mask) and applied the tiling algorithm, thereby calculating the number of tiles that would be acquired. Since the number of tiles required is dependent on the size of each root, we normalized the metric to the optimal throughput. We calculated the optimal throughput per image by applying the tiling algorithm to each hand-labeled image in the evaluation set (see Supplementary Fig. 5).
Software
The RootArray Controller was written in Java 1.6 and installed on the microscope computer. It contains the tip detection, growth region, and tiling code. The root detection software was written in Java with a Java Native Interface (JNI) wrapper around the C++ ITK library 28. It was installed on a computation server and remotely executed by the RootArray Controller via SSH. The microscope was operated with Zen 2009 and Multitime Series Plus 2009-2010-25.
Offline registration and virtual cross-section of high-resolution images
High resolution quasi-3D images were processed for quantification in 4 steps: segmentation of the foreground root from the background, location of the medial axis of the root, computation of a virtual longitudinal medial image, and registration of the resulting image to an atlas. The longitudinal medial image is a representative slice through the middle of the root; details about its estimation are found in Supplementary Note 2.
Registration to Tissue Atlas
Medial longitudinal section images were registered to a representative tissue atlas. The tissue atlas was created from manually annotated images and contains 18 different tissue types: the lateral root cap, the columella, the stem cell niche that includes the quiescent center and the root meristem initial cells, and five remaining tissues (epidermis, endodermis, cortex, vasculature, pericycle) further separated into the three longitudinal zones (meristem, elongation, and maturation zones).
The tissue atlas had five longitudinal landmark points: the tip (i.e. the first point in the root), the QC, the first point (from the tip) at which the root’s width remained constant, the start of the maturation zone, and the end of the root (i.e. a point to crop the medial section image). These five points determine four regions. The tip and point of constant width were automatically detected in the virtually reconstructed medial section images. The user manually selected the QC, elongation start, maturation start, and end of the root during the quality control (see Supplementary Fig. 7). Each landmark is only a longitudinal point in the image, and therefore only one click per landmark was required from the user.
Each medial section image was registered using piecewise linear registration. That is, the computationally and manually defined landmark points separated an image into four sections. Each section’s height was linearly scaled to the corresponding atlas section’s height. Each row in the medial image was then linearly scaled to match the width of the corresponding row in the tissue atlas. The final result was a label of tissue and longitudinal zone for each pixel in the medial section image.
Performance Analysis of Tissue Registration
We evaluated the performance of our tissue registration by examining random samples of our final dataset. Specifically, we randomly chose 50 medial section images from our data. From each image, we randomly selected points from each tissue type. The number of points per tissue type was weighted by the size of the tissue type, and we selected a total of 50 points (over all tissues) per medial image. These points were then overlaid onto the original medial section fluorescence image (containing GFP and propidium iodide channels), and a biologist manually assigned the correct tissue type per point in a blind experiment. We reported a mean accuracy of 66% across all tissue types and longitudinal zones (see Supplementary Table 1).
Analysis methods to quantify gene expression changes
Removal of Cross-Talk and Background
Prior to cross-talk removal, we subtracted the background from the GFP channel by subtracting the mode intensity of the image (and setting resulting negative values to zero). For every step after this, we masked out any pixels that had corresponding propidium iodide channel pixels that were saturated (intensity = 255). We used the same parameters of the algorithm as previously described 29, except that we increased the pixel count threshold for samples from the joint histogram from 100 to 250. An example is shown in Supplementary Fig. 14.
Comparison between the RootArray image data and the RootMap microarray data
The cell-type spatiotemporal enriched microarray dataset (RootMap) contains up to 14 different cell types across 13 tissue sections, with each section encompassing approximately 3 to 5 cells along the longitudinal axis 30. The expression of each region was profiled using the Affymetrix ATH1 microarray containing 22,246 probe sets. In contrast, each RootArray image identifies a total of 18 tissue types. We manually identified which RootMap entries corresponded to a given RootArray tissue region; we used this one-to-one correspondence in calculating the mean over the microarray values and estimating Pearson correlations. All data and code developed for this study are available at http://labs.genome.duke.edu/ohler/research/image_analysis/rootarray
Assessing significance of expression changes
Background subtracted ranks of average pixel intensity for each tissue in each individual root were used as the basis for the analysis. A nonparametric Wilcoxon rank sum test was conducted on each genotype for each condition as compared to roots grown in MS media for each tissue type. We corrected for multiple testing by calculating a false discovery rate (FDR; 31) for each p-value (Supplementary Table 6, Fig 5). We used a cut-off at 5% FDR. This analysis was done in R 32.
Analysis of growth rate and cell division
For growth rate analysis in an unbiased manner, we took into consideration experiments with more than 4 time points and marked the position of the root tips on the low resolution images of several early time points. In total, 57 low resolution images with 3636 well/root positions were taken into account (Supplementary Table 8). Each position of each root was marked with the Fiji/ImageJ multipoint tool at the first time point of analysis. In the subsequent rounds the marks were shifted to the new positions of the root tips to allow for accurate measurements. When no root had emerged from the well, the mark was made at the lowest point of the well. After measurement of all positions in all images, the Euclidean distance between the coordinates of the root tip positions at each time point was calculated. This pixel value was converted to μm using the scale from the low resolution images in which each pixel corresponds to 39.7748 μm. This growth rate was divided by the hours between acquisitions of the images. To stratify the results, we took into account only data of the first 24h of treatment. The box plots were made using R.
For easier visual evaluation, for each image of all maximum projections of all CyclinB1;1 images contained in the data set, the green signal was converted to the fire colormap lookup table (LUT) in Fiji. On these images, all signal spots that were confined to cells on the images were counted.
Data Deposition
We deposited the image data of the high resolution stitched data, as well as the corresponding low resolution data on a local installation of the BISQUE image data server 33. This allows for online viewing of the data, as well as querying and downloading of the data. The interface is available under the URL: http://rootarray.biology.duke.edu/experiments-wb-2010
Supplementary Material
Acknowledgments
We are grateful for bioinformatic assistance from Mikhail Kovtun and Vladimir Popov and for technical assistance from Niel Lebeck, Yufuko Nishimura, Alexis Woods, Kimberly Lewis, Bonnie Wohlrab and Santosh Satbhai. We thank Hironaka Tsukagoshi, Ross Sozzani, Christopher Topp and Jaimie Van Norman for valuable discussions and critical reading of the manuscript. We also acknowledge John Harer and Yiping Wang for their assistance in algorithm design and software development, and Zbigniew Iwinski of Carl Zeiss MicroImaging Inc. for developing an interface for the Multitime application. We are grateful for seed donations from Nico Geldner, University of Lausanne (pCASP1::GFP, pCASP2::GFP, pCASP4::GFP) and Peter Doerner, University of Edinburgh (pCycB1;1::CycB1;1-GFP). This work was supported by US National Science Foundation award DBI-0953184 to U.O. and National Institutes of Health award P50-GM081883 and NSF award IOS-1021619 to P.N.B. and U.O. Some of the authors have a financial interest in and/or were/are consultants for GrassRoots Biotechnology which owns the rights to the RootArray technology.
Footnotes
AUTHOR CONTRIBUTIONS
W.B. designed and conducted the RootArray experiments, B.Ma., B.Mo., D.L.M., U.O. conceived and implemented the computational methods, B.Ma., B.Mo., I.P. and W.B. analyzed the data, P.N.B, R.W.T., J.J., S.J.K.,G.K.F.,R.L.C. conceived and developed the RootArray, U.O. and P.N.B. participated in experimental designs, W.B., B.Mo., B.Ma., U.O. and P.N.B wrote the paper.
The RootArray Controller software can be assessed via: http://www.genome.duke.edu/labs/ohler/research/image_analysis/rootarray/nature_methods-supp/rootarray-software.tgz
References
- 1.Birnbaum K, et al. Cell type-specific expression profiling in plants via cell sorting of protoplasts from fluorescent reporter lines. Nat Methods. 2005;2:615–619. doi: 10.1038/nmeth0805-615. [DOI] [PubMed] [Google Scholar]
- 2.Salmand P-A, Iche-Torres M, Perrin L. Tissue-specific cell sorting from Drosophila embryos: application to gene expression analysis. Fly (Austin) 2011;5:261–265. doi: 10.4161/fly.5.3.16509. [DOI] [PubMed] [Google Scholar]
- 3.Lecuyer E, Tomancak P. Mapping the gene expression universe. Curr Opin Genet Dev. 2008;18:506–512. doi: 10.1016/j.gde.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Held M, et al. CellCognition: time-resolved phenotype annotation in high-throughput live cell imaging. Nat Methods. 2010;7:747–754. doi: 10.1038/nmeth.1486. [DOI] [PubMed] [Google Scholar]
- 5.Bao Z, et al. Automated cell lineage tracing in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2006;103:2707–2712. doi: 10.1073/pnas.0511111103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aydin Z, Murray JI, Waterston RH, Noble WS. Using machine learning to speed up manual image annotation: application to a 3D imaging protocol for measuring single cell gene expression in the developing C. elegans embryo. BMC Bioinformatics. 2010;11:84–84. doi: 10.1186/1471-2105-11-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X, et al. Analysis of cell fate from single-cell gene expression profiles in C. elegans. Cell. 2009;139:623–633. doi: 10.1016/j.cell.2009.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Long F, Peng H, Liu X, Kim SK, Myers E. A 3D digital atlas of C. elegans and its application to single-cell analyses. Nat Methods. 2009;6:667–672. doi: 10.1038/nmeth.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu M, et al. Adaptive cell segmentation and tracking for volumetric confocal microscopy images of a developing plant meristem. Mol Plant. 2011;4:922–931. doi: 10.1093/mp/ssr071. [DOI] [PubMed] [Google Scholar]
- 10.Jonsson H, Heisler MG, Shapiro BE, Meyerowitz EM, Mjolsness E. An auxin-driven polarized transport model for phyllotaxis. Proc Natl Acad Sci U S A. 2006;103:1633–1638. doi: 10.1073/pnas.0509839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernandez R, et al. Imaging plant growth in 4D: robust tissue reconstruction and lineaging at cell resolution. Nat Methods. 2010;7:547–553. doi: 10.1038/nmeth.1472. [DOI] [PubMed] [Google Scholar]
- 12.Ntziachristos V. Going deeper than microscopy: the optical imaging frontier in biology. Nat Methods. 2010;7:603–614. doi: 10.1038/nmeth.1483. [DOI] [PubMed] [Google Scholar]
- 13.Mace DL, et al. Quantification of transcription factor expression from Arabidopsis images. Bioinformatics. 2006;22:e323–331. doi: 10.1093/bioinformatics/btl228. [DOI] [PubMed] [Google Scholar]
- 14.Grossmann G, et al. The RootChip: an integrated microfluidic chip for plant science. Plant Cell. 2011;23:4234–4240. doi: 10.1105/tpc.111.092577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parashar A, Pandey S. Plant-in-chip: Microfluidic system for studying root growth and pathogenic interactions in Arabidopsis. Appl Phys Lett. 2011;98:263703–2637033. [Google Scholar]
- 16.Frey BJ, Jojic N. A comparison of algorithms for inference and learning in probabilistic graphical models. IEEE Trans Pattern Anal Mach Intell. 2005;27:1392–1416. doi: 10.1109/TPAMI.2005.169. [DOI] [PubMed] [Google Scholar]
- 17.Kschischang FR, Frey BJ, Loeliger HA. Factor graphs and the sum-product algorithm. Ieee T Inform Theory. 2001;47:498–519. [Google Scholar]
- 18.Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 2009;25:1463–1465. doi: 10.1093/bioinformatics/btp184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edelsbrunner H, Harer J. Computational Topology, An Introduction. American Mathematical Society; 2010. [Google Scholar]
- 20.Ballard DH. Generalizing the Hough Transform to Detect Arbitrary Shapes. Pattern Recogn. 1981;13:111–122. [Google Scholar]
- 21.Xu CY, Prince JL. Gradient vector flow: A new external force for snakes. Proc Cvpr Ieee. 1997:66–71. [Google Scholar]
- 22.Maizel A, von Wangenheim D, Federici F, Haseloff J, Stelzer EHK. High-resolution live imaging of plant growth in near physiological bright conditions using light sheet fluorescence microscopy. Plant J. 2011;68:377–385. doi: 10.1111/j.1365-313X.2011.04692.x. [DOI] [PubMed] [Google Scholar]
- 23.Beemster GT, Baskin TI. Analysis of cell division and elongation underlying the developmental acceleration of root growth in Arabidopsis thaliana. Plant Physiol. 1998;116:1515–1526. doi: 10.1104/pp.116.4.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks TL, Miller ND, Spalding EP. Plasticity of Arabidopsis root gravitropism throughout a multidimensional condition space quantified by automated image analysis. Plant Physiol. 2010;152:206–216. doi: 10.1104/pp.109.145292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee MM, Schiefelbein J. WEREWOLF, a MYB-related protein in Arabidopsis, is a position-dependent regulator of epidermal cell patterning. Cell. 1999;99:473–483. doi: 10.1016/s0092-8674(00)81536-6. [DOI] [PubMed] [Google Scholar]
- 26.Sena G, Frentz Z, Birnbaum KD, Leibler S. Quantitation of cellular dynamics in growing Arabidopsis roots with light sheet microscopy. PLoS One. 2011;6:e21303. doi: 10.1371/journal.pone.0021303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iyer-Pascuzzi AS, et al. Cell Identity Regulators Link Development and Stress Responses in the Arabidopsis Root. Developmental Cell. 2011;21:770–782. doi: 10.1016/j.devcel.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoo TS, et al. Engineering and algorithm design for an image processing Api: a technical report on ITK--the Insight Toolkit. Stud Health Technol Inform. 2002;85:586–592. [PubMed] [Google Scholar]
- 29.Luengo Hendriks CL, Keranen SV, Biggin MD, Knowles DW. Automatic channel unmixing for high-throughput quantitative analysis of fluorescence images. Opt Express. 2007;15:12306–12317. doi: 10.1364/oe.15.012306. [DOI] [PubMed] [Google Scholar]
- 30.Brady SM, et al. A high-resolution root spatiotemporal map reveals dominant expression patterns. Science. 2007;318:801–806. doi: 10.1126/science.1146265. [DOI] [PubMed] [Google Scholar]
- 31.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995;57:289–300. [Google Scholar]
- 32.R-Development_Core_Team . R Foundation for Statistical Computing; Vienna, Austria: 2005. [Google Scholar]
- 33.Kvilekval K, Fedorov D, Obara B, Singh A, Manjunath BS. Bisque: a platform for bioimage analysis and management. Bioinformatics. 2010;26:544–552. doi: 10.1093/bioinformatics/btp699. RootArray. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.