

Abstract
Until recently, studies of divergence and gene flow among closely-related taxa were generally limited to pairs of sister taxa. However, organisms frequently exchange genes with other non-sister taxa. The “northern oriole” group within genus Icterus exemplifies this problem. This group involves the extensively studied hybrid zone between Baltimore oriole (Icterus galbula) and Bullock's oriole (I. bullockii), an alleged hybrid zone between I. bullockii and black-backed oriole (I. abeillei), and likely mtDNA introgression between I. galbula and I. abeillei. Here, we examine the divergence population genetics of the entire northern oriole group using a multipopulation Isolation-with-Migration (IM) model. In accordance with Haldane's rule, nuclear loci introgress extensively beyond the I. galbula–I. bullockii hybrid zone, while mtDNA does not. We found no evidence of introgression between I. bullockii and I. abeillei or between I. galbula and I. abeillei when all three species were analyzed together in a three-population model. However, traditional pairwise analysis suggested some nuclear introgression from I. abeillei into I. galbula, probably reflecting genetic contributions from I. bullockii unaccounted for in a two-population model. Thus, only by including all members of this group in the analysis was it possible to rigorously estimate the level of gene flow among these three closely related species.
Keywords: Allele sharing, coalescent, Icterus, IMa2, incomplete lineage sorting, introgression, multipopulation, northern orioles
Introduction
The study of phylogeography and population divergence is challenged by the difficulty of distinguishing between shared retained ancestral polymorphism and introgressed alleles (Nielsen and Wakeley 2001). The much larger effective population size of nuclear DNA relative to mtDNA slows the rate of lineage sorting and the speed at which reciprocal monophyly is achieved at the nuclear level (Avise 2004). Slow and stochastic lineage sorting thus causes a pattern of allele sharing among species that can seem similar to patterns caused by gene flow through introgressive hybridization, even between long-divergent lineages (Hudson and Turelli 2003). Multiple unlinked loci and coalescent-based “divergence population genetics” methods are needed to distinguish between these two processes (e.g., Kliman et al. 2000; Machado et al. 2002).
The Isolation-with-Migration (IM) model (Hey and Nielsen 2004, 2007) provides a powerful tool for the study of population divergence and for testing different speciation models and demographic scenarios (Hey 2006; Peters et al. 2007; Kondo et al. 2008; Pinho and Hey 2010). Shared alleles allow multilocus coalescent approaches such as IM to reconstruct the divergence history of closely related species (Nielsen and Wakeley 2001; Hey and Nielsen 2004, 2007). The original IM model was designed for two-population studies between sister taxa that have not exchanged genes with other taxa since they split (Hey and Nielsen 2004, 2007). However, organisms frequently exchange genes with other non-sister taxa (Arnold 2006; Nosil 2008; Petit and Excoffier 2009). Genetic contributions from other source populations not included in the analysis (i.e., “ghost” populations) could thus severely bias or invalidate inference about the history of closely related taxa (Beerli 2004; Slatkin 2005; Strasburg and Rieseberg 2010). Thus, until recently, IM analyses were limited to pairs of taxa that share more recent common ancestry and gene flow with each other. However, this limitation was removed with the release of IMa2, allowing the simultaneous consideration of up to ten populations granted a population tree (i.e., phylogeny) is available for the taxa involved (IMa2: Hey 2010b).
In this study, we used IMa2 to analyze the three species in the “northern oriole” group within the New World orioles (Jacobsen and Omland 2011; Fig. 1). This group includes the well-known hybrid zone between the eastern Baltimore oriole (Icterus galbula) and Bullock's oriole (I. bullockii) (Sutton 1938, 1968; Sibley and Short 1964; Rising 1969, 1970, 1983, 1996; Corbin et al. 1979). I. galbula breeds across the eastern United States south to Louisiana, and across most of Canada from Alberta in the west to Nova Scotia in the east (Fig. 2). The breeding range of I. bullockii extends across western North America from British Columbia, Canada in the north to Durango, Mexico in the south. Both I. galbula and I. bullockii are long-distance migrants and mainly overwinter in southern Mexico and Central America (Rising et al. 1998, 1999). Their winter ranges overlap with the range of the third member of the group, I. abeillei, a Mexican endemic limited to the central Mexican plateau (Jaramillo and Burke 1999) and traditionally considered a close relative and subspecies within I. bullockii due to their overall phenotypic similarity (Jaramillo and Burke 1999).
Figure 1.

Adult males of the three species in the northern oriole group; (a) Baltimore oriole Icterus galbula (photo: Frode Jacobsen), (b) Bullock's oriole I. bullockii (photo: Frode Jacobsen), and (c) black-backed oriole I. abeillei (photo: Jonathan Hiley)
Figure 2.
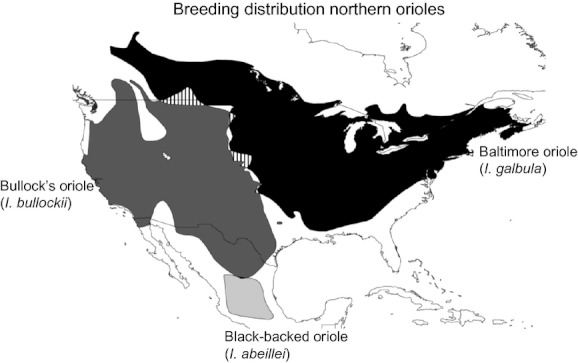
Breeding distributions of the three northern oriole species: Icterus abeillei (light gray), I. bullockii (dark gray), and I. galbula (black). Hatched areas indicate range overlap between I. bullockii and I. galbula. Digital distribution maps for each species were downloaded from NatureServe (Ridgely et al. 2007).
The parapatric breeding ranges of I. galbula and I. bullockii overlap along a long and narrow transect (150 km in Kansas, Allen 2002) that stretches over 1800 km from southern Alberta to central Texas (Rising 1983). The two orioles interbreed extensively in this contact zone, which falls within the “Rocky Mountain-Great Plains” suture zone (Remington et al. 1968), a hotspot of avian hybrid zones between eastern and western species pairs (e.g., Carling and Brumfield 2008; Flockhart and Wiebe 2009; Mettler and Spellman 2009). Documentation of viable hybrid offspring within this contact zone (Sutton 1938; Sibley and Short 1964; Sutton 1968; Rising 1970) led to the lumping of I. galbula and I. bullockii (including I. abeillei) into a single species, the northern oriole (AOU 1973). Later studies found that the I. galbula–I. bullockii hybrid zone was stable (Rohwer and Manning 1990; Rohwer and Johnson 1992; Rising 1996; Allen 2002), and full species status was restored for all three members of the group (AOU 1995). Hybridization has never been firmly documented between I. abeillei and the other two members of the group, but there is some evidence suggesting hybridization with I. bullockii where their ranges abut in northern Durango, Mexico (Miller 1906; but see Rising 1973). We are unaware of any documented instances of hybridization between members of the northern oriole group and other oriole species.
Despite the extensive research on the northern oriole group, we have limited knowledge about the extent of nuclear gene flow within this species complex. Allen (2002) examined clinal variation of phenotypic and molecular markers across the I. galbula–I. bullockii hybrid zone in Kansas, and found that neither male plumage traits nor mtDNA haplotypes introgressed beyond the zone of contact. These findings were recently corroborated by Carling et al. (2011), who estimated the mitochondrial cline to be 328 km wide. In contrast, of 152 neutral AFLP markers examined by Allen (2002), there were no fixed differences between the two species, and only ten AFLP bands differed more than 40% in frequency across the hybrid zone. Allen (2002) interpreted this apparent lack of differentiation in AFLP markers between I. galbula and I. bullockii as evidence of unhampered nuclear introgression since the two species came into secondary contact. However, such extensive allele sharing among closely related species could also result from the slow sorting of retained ancestral polymorphism (Moore 1995; Hudson and Turelli 2003).
The goals of this study were to (1) estimate levels of introgression among all three species in the northern oriole group while accounting for their recent shared ancestry using a three-population IMa2 model and (2) examine the effects of genetic exchange with unsampled taxa in two-population analyses.
Material and Methods
Taxonomic sampling
We sought a broad representation from throughout the ranges of all three species whilst avoiding known areas of sympatry (see Appendix 1 for sampling locations). Our sampling scheme provided a total of 21 to 24 individuals of each of the three species, I. abeillei, I. bullockii and I. galbula. In addition, a single individual of western meadowlark (Sturnella neglecta) was included as an outgroup taxon.
Multilocus nuclear data
This study included sequence data from eight intron-spanning loci that map to six autosomes and the Z sex chromosome in the zebra finch (Taeniopygia guttata) genome (see Table 1for details). The two Z-linked loci are well separated on the zebra finch Z chromosome (>7 Mb apart) and were therefore treated as independent (Backström et al. 2006, 2010). All eight loci were amplified and sequenced following procedures described in previous phylogenetic studies on Icterus (Jacobsen et al. 2010; Jacobsen and Omland 2011). Resulting DNA chromatograms were aligned and edited in SEQUENCHER 4.7 (Gene Codes Corp., Ann Arbor, MI). Edited sequences for all loci were imported into MEGA5 (Tamura et al. 2011) and aligned using MUSCLE (Edgar 2004) with final manual adjustments. The haplotypes of individuals heterozygous at more than one position at a given locus were determined using the software PHASE v.2.1.1 (Stephens et al. 2001; Stephens and Scheet 2005), with the following parameter settings: burn-in = 1000, number of iterations = 10,000, and thinning interval = 1. Each analysis was repeated 10 times using different random starting seed and the run that received the highest log-likelihood was used to infer the haplotypes. To avoid systematic bias in downstream coalescent analyses, all haplotypes resolved at a probability of 0.5 or higher were included in the study (Garrick et al. 2010). An ambiguously aligned single-nucleotide repeat (poly-A) region within locus FGB4 was excluded from the alignment prior to analyses. Linkage analysis of the two Z-linked loci (ALDOB5 and BRM15) using HAPLOVIEW v.4.2 (Barrett et al. 2005) revealed no evidence of LD among polymorphic sites (results not shown). We tested for intralocus recombination using the difference in sums of squares (DSS) test in TOPALi v.2.5 (Milne et al. 2004), with 500 bootstrap replicates to determine statistical significance. A significant DSS peak was detected at locus β-ACT2, and only the largest non-recombinant sequence block (558 bp) was retained for downstream analysis. Accession numbers for sequences obtained from previous studies on this group are listed in Appendix 2. All new sequences collected in this study have been deposited in GenBank under accession numbers JX403068-JX403342 and JX412950-JX412955.
Table 1.
Locus information. Chromosome: intron location determined based on BLAST searches of zebra finch reference genome assembly, with start coordinates for Z-linked loci provided in parentheses; Length: size (in base pairs) of largest non-recombinant alignment segment; N: number of non-recombinant alleles included in the study
| Locus1 | Chromosome | Length | N | Primer source | ||
|---|---|---|---|---|---|---|
| I. abeillei | I. bullockii | I. galbula | ||||
| β-ACT2 | 22 | 558 | 24 | 26 | 24 | (Waltari and Edwards 2002) |
| α-ENO8 | 21 | 252 | 46 | 42 | 46 | (Kondo et al. 2008) |
| FGB4 | 4 | 575 | 24 | 30 | 28 | (Barker 2004) |
| GAPDH11 | 1 | 327 | 36 | 30 | 30 | (Primmer et al. 2002) |
| RDP2 | 12 | 298 | 36 | 28 | 26 | (Waltari and Edwards 2002) |
| TGFβ5 | 3 | 575 | 30 | 32 | 28 | (Bureš et al. 2002) |
| ALDOB5 | Z (400,071) | 253 | 36 | 35 | 24 | (Kondo et al. 2008) |
| BRM15 | Z (7,528,458) | 362 | 40 | 35 | 36 | (Borge et al. 2005) |
Full locus information is as follows: Beta-actin gene, intron 2 (β-ACT2); alpha-enolase gene, intron 8 (α-ENO8); fibrinogen B beta polypeptide gene, intron 4 (FGB4); glyceraldehyde-3-phosphate dehydrogenase gene, intron 11 (GAPDH11); rhodopsin gene, intron 2 (RDP2); and transforming growth factor beta-2, intron 5 (TGFβ5); aldolase-B fructose-biphosphate intron 5 (ALDOB5); Brahma protein gene, intron 15 (BRM15).
Three common measures of nucleotide polymorphism were calculated using ARLEQUIN v.3.11 (Excoffier and Lischer 2010): number of haplotypes (h), haplotype diversity (Hd), and the average number of nucleotide differences per site between two sequences (π). Haplotype networks illustrating the relationships among inferred alleles were constructed for each locus using the median-joining algorithm in NETWORK v.4.5.1.0 (Bandelt et al. 1999, http://www.fluxus-engineering.com). The networks were edited using NETWORK PUBLISHER v.1.1.0.7 (http://www.fluxus-engineering.com).
Tests for selection were conducted using a multilocus HKA test (Hudson et al. 1987) implemented in the HKA program available at http://lifesci.rutgers.edu/∼heylab/HeylabSoftware.htm. We chose western meadowlark (Sturnella neglecta) as outgroup for this test, the most distant relative within the family Icteridae available for comparison across the eight loci examined in this study. Kimura 2P distances between the three northern oriole species and S. neglecta were calculated using DNASP v.5.10.0.1(Librado and Rozas 2009). Statistical significance of the HKA test was determined through 10,000 coalescent simulations based on the number of sampled alleles and observed levels of polymorphism and divergence across loci. These simulations also allowed us to conduct a multilocus test of Tajima's D statistic (Tajima 1989). Fu's (1997) Fs and Ramos-Onsins and Rozas (2002)R2 neutrality statistics were also calculated using DnaSP. Confidence intervals and statistical significance of test values were calculated using 10,000 random permutations and coalescent simulations.
Multilocus coalescent analyses
We examined patterns of gene flow among all three members of the northern oriole group using the Bayesian IM model implemented in the program IMa2 (Hey 2010b), which assumes random mating, constant population sizes, selective neutrality, free recombination among loci and no recombination within loci (Hey and Nielsen 2004, 2007; Hey 2010b).
By default, all model parameters estimated by IMa2 are scaled to the population mutation rate. The parameter estimates were converted into demographic units (i.e., effective population sizes in number of individuals and divergence times in years) using a neutral mutation rate of 1.35*10−9 substitutions per site per year for autosomal loci (Ellegren 2007) and 1.45*10−9 substitutions per site per year for Z-linked loci (Axelsson et al. 2004; Ellegren 2007). To reflect the different modes of inheritance, autosomal (1.0) and Z-linked (0.75) loci were assigned respective inheritance scalars in the IMa2 input file. Population sizes were converted into effective number of individuals using a generation time of 1.7 years, the average age at reproduction documented for multiple passerine lineages (Sæther et al. 2005). The actual generation time in orioles may well be slightly longer, as most oriole species exhibit delayed plumage maturation, which could delay the onset of reproduction (for males in particular) (e.g., Flood 1984; Rohwer and Manning 1990; Butcher 1991; Richardson and Burke 1999). Effective population sizes reported in this study are therefore likely conservative estimates of the actual effective population sizes. The HKY mutation model was specified for all loci.
Three-population IMa2 analysis
First, we estimated levels of gene flow among all three species within the northern oriole group simultaneously in a three-population IMa2 analysis. We conducted separate analyses with two alternative population trees: (1) the nuclear species tree inferred by our recent species tree analysis of Icterus (Jacobsen and Omland 2011): (I. galbula, (I. abeillei, I. bullockii)) and (2) the mtDNA gene tree inferred by Omland et al. (1999): (I. bullockii, (I. abeillei, I. galbula)).
Short preliminary runs were conducted to determine appropriate upper bounds of the demographic parameters. Subsequently, two identical long runs of 3–5*106 MCMC steps with a burnin period of 1*106 steps were started with different starting seeds and run until minimum effective sample sizes (ESS) of 200 were achieved. We assessed convergence by monitoring the mixing properties of the MCMC during each run and by ensuring that similar parameter estimates were obtained from the two independent runs. Acceptable chain mixing (i.e., absence of long-term trends in plots of L[P] and t over the course of each run) and low autocorrelations (<0.1) were achieved by running 100 chains under a geometric heating scheme (with heating parameters set to a = 0.975 and b = 0.75). Population-specific parameter priors were provided in a separate file. We used exponential priors on migration rates (option–j7), and determined appropriate prior distribution means based on the peak posterior parameter estimates from preliminary runs.
Two-population IMa2 analyses
To assess the effects of violating the crucial assumption of no allelic contributions from other source (i.e., “ghost”) populations, we also conducted three separate pairwise IMa2 analyses of the three northern oriole species. Run settings were kept unchanged, except for slight adjustment of parameter priors where posterior density distributions exceeded the upper bounds determined by preliminary runs. Each MCMC (M) mode analysis was replicated using different random seeds to ensure convergence onto similar posterior distributions, using the same criteria for assessing convergence as described above. Finally, we conducted nested model testing by running IMa2 in “Load genealogies” (L) mode to specifically compare the fit of reduced models that do not allow for post divergence gene flow to the full IMa2 model (i.e., including all six parameters Θ1, Θ2, Θa, m1, m2, t). This was performed by sampling 2.5*105 genealogies evenly from the two MCMC runs performed on each population pair and compared the log-likelihood scores of all possible nested models provided in a separate file in the IMa2 distribution package.
Results
Polymorphism and divergence
The three species within the northern oriole group were polymorphic at all loci used in this study, except for I. abeillei that was monomorphic at ALDOB5. Sample sizes and data for individual loci are summarized in Table 1. The three measures of sequence polymorphism were all lower in I. abeillei than in I. bullockii and I. galbula (Table 2), but not significantly so (single factor ANOVA: h: F2,21 = 0.656, P = 0.529; Hd: F2,21 = 0.585, P = 0.566; π: F2,21 = 0.943, P = 0.405; respectively). The differences in mean genetic diversity between I. abeillei and the two other members of the group are mainly driven by the lack of sequence variation at the sex-linked locus ALDOB5 (Table 2).
Table 2.
Locus-specific estimates of number of haplotypes (h), haplotype diversity (Hd), and Jukes & Cantor nucleotide diversity (π)
| I. abeillei | I. bullockii | I. galbula | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Locus | h | Hd | π | h | Hd | π | h | Hd | π |
| β-ACT2 | 5 | 0.656 | 0.00147 | 4 | 0.397 | 0.00094 | 4 | 0.239 | 0.00060 |
| α-ENO8 | 7 | 0.283 | 0.00136 | 9 | 0.490 | 0.00224 | 9 | 0.481 | 0.00234 |
| FGB4 | 10 | 0.786 | 0.00300 | 20 | 0.956 | 0.00741 | 15 | 0.942 | 0.00603 |
| GAPDH11 | 8 | 0.830 | 0.00472 | 8 | 0.823 | 0.00492 | 14 | 0.883 | 0.00590 |
| RDP2 | 7 | 0.711 | 0.00385 | 7 | 0.804 | 0.00438 | 3 | 0.578 | 0.00216 |
| TGFβ5 | 3 | 0.393 | 0.00087 | 6 | 0.585 | 0.00213 | 14 | 0.923 | 0.00430 |
| ALDOB5 | 1 | 0 | 0 | 4 | 0.395 | 0.00217 | 3 | 0.507 | 0.00372 |
| BRM15 | 3 | 0.145 | 0.00068 | 4 | 0.217 | 0.00062 | 2 | 0.413 | 0.00114 |
The haplotype networks revealed extensive allele sharing and few fixed differences among the three northern oriole species (Fig. 3). Some loci were characterized by a distinct star-shaped topology (e.g., α-ENO8, ALDOB5 and BRM15), with a common central (and presumed ancestral) haplotype surrounded by several rare, derived haplotypes one or two mutational steps away from the ancestral haplotype (Fig. 3b,g,h).
Figure 3.
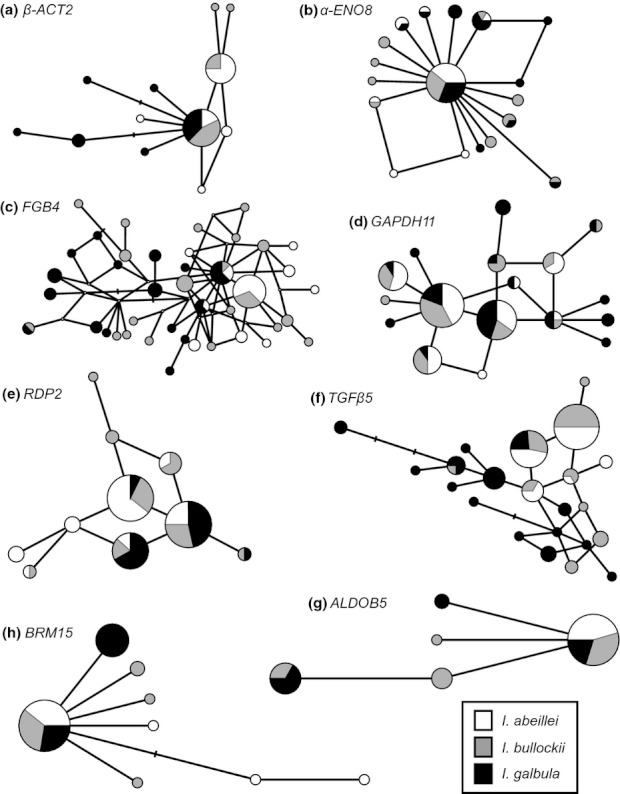
Haplotype networks of eight nuclear intron loci (6 autosomal + 2 Z-linked) revealing extensive haplotype sharing among the three northern orioles: Icterus abeillei (white), I. bullockii (gray), and I. galbula (black). Each circle corresponds to a unique haplotype and the size of each circle reflect the relative total sample size of each haplotype. Small open circles in (c) indicate unsampled/extinct haplotypes, and perpendicular bars along lines connecting haplotypes indicate additional mutations distinguishing neighboring haplotypes.
The three species showed varying degrees of genetic differentiation across loci, but generally exhibited extensive haplotype sharing across loci (Table 3, also see Fig. 3). Strongest differentiation was observed between the highly disjunct I. abeillei and I. galbula, with highly significant FST values at six of the eight loci examined. The two widely hybridizing I. bullockii and I. galbula were strongly differentiated at only half (4/8) of the loci examined. The two sister species, I. abeillei and I. bullockii, were only weakly differentiated and only two loci (FGB4 and ALDO5) remained significant after Bonferroni corrections (Table 3).
Table 3.
Population differentiation among three northern oriole populations, Icterus abeillei, I. bullockii, and I. galbula, estimated based on pairwise genetic distances between populations. Significant FST values are indicated at the 0.05 > P > 0.01 (*) and P < 0.01 (**) level. Bolded values are significant at the Bonferroni-adjusted α-level of 0.00625
| FST | |||
|---|---|---|---|
| Locus | I. abeillei–I. bullockii | I. abeillei–I. galbula | I. bullockii–I. galbula |
| β-ACT2 | 0.151** | 0.372** | 0.096* |
| α-ENO8 | 0.010 | 0.019* | 0.019* |
| FGB4 | 0.102** | 0.383** | 0.173** |
| GAPDH11 | 0 | 0.064* | 0.052* |
| RDP2 | 0.069* | 0.289** | 0.177** |
| TGFβ5 | 0.065* | 0.400** | 0.289** |
| ALDOB5 | 0.131** | 0.241** | 0.017 |
| BRM15 | 0.018 | 0.186** | 0.186** |
The multilocus HKA test indicated no significant departures from neutrality in our dataset. The multilocus Tajima's D test was significant only for I. bullockii, a result that is mainly driven by significantly more negative than expected D values at loci α-ENO8 and BRM15 (Table 4). Fs values were negative at the majority of loci, indicative of recent population growth, but this statistic deviated only significantly from neutral expectations at less than half the loci following Bonferroni corrections (Table 4). The R2 test was only significant at a single locus α-ENO8 (Table 4).
Table 4.
Locus-specific tests for deviations from selective neutrality and constant population sizes in Icterus abeillei (a), I. bullockii (b), and I. galbula (g). Significance of observed values is indicated at the 0.05 > P > 0.01 (*) and P < 0.01 (**) level. Tests were inapplicable for I. abeillei at ALDOB5 due to lack of sequence variation. Bolded values remained significant after Bonferroni corrections for multiple testing (adjusted α-level of 0.00625)
| Tajima's D | Fu's Fs | Ramos-Onsins & Rozas R2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Locus | a | b | g | a | b | g | a | b | g |
| β-ACT2 | 0.048 | −0.821 | −1.884** | −1.406 | −1.227 | −1.398 | 0.14 | 0.108 | 0.118 |
| α-ENO8 | −1.725* | −1.97** | −1.708* | −6.561** | −7.748** | −7.315** | 0.048 | 0.043** | 0.05* |
| FGB4 | −1.412 | −0.24 | 0.711 | −4.64** | −11.431** | −6.177** | 0.085 | 0.118 | 0.152 |
| GAPDH11 | 0.712 | 0.152 | −0.993 | −1.889 | −2.089 | −8.774** | 0.154 | 0.131 | 0.078 |
| RDP2 | −0.136 | 0.019 | 0.474 | −1.978 | −1.962 | 0.464 | 0.114 | 0.126 | 0.16 |
| TGFβ5 | 0.024 | −0.047 | −0.144 | 0.058 | −0.973 | −7.268** | 0.125 | 0.13 | 0.128 |
| ALDOB5 | – | −0.559 | 0.434 | – | −0.88 | 1.258 | – | 0.099 | 0.157 |
| BRM15 | −1.423 | −1.559* | 1.047 | −1.112 | -2.963** | 1.369 | 0.061 | 0.08 | 0.206 |
Three-population IMa2 analysis
The inclusion of all three members of this species complex in a single IMa2 analysis revealed some interesting patterns of gene introgression (Fig. 4). The two competing population trees (nDNA vs. mtDNA) yielded similar parameter estimates (with one important exception discussed below). We therefore only report the results based on the nuclear population tree (I. galbula, (I. abeillei, I. bullockii)).
Figure 4.
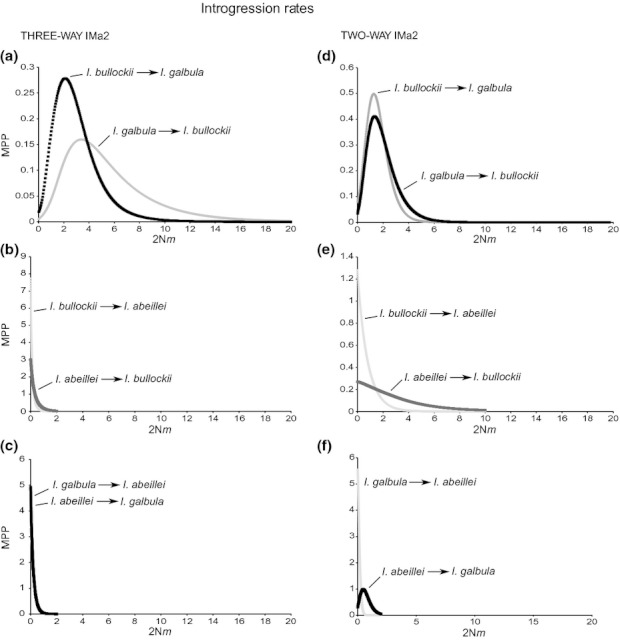
Posterior probability plots of bi-directional migration rates (given in effective number of gene copies - 2Nm) estimated between Icterus bullockii and I. galbula (a,d), between I. abeillei and I. bullockii (b,e), and between I. abeillei and I. galbula (c,f) based on a combined three-population analysis and three separate pairwise analyses in IMa2.
First, we found clear evidence of substantial introgression across the hybrid zone between I. galbula and I. bullockii, (2NmI. galbula > I. bullockii = 3.388, 95% HPD = 0.41–12.30, vs. 2NmI. bullockii > I. galbula = 2.125, 95% HPD = 0.193–6.504, Fig. 4a). Interestingly, the gene flow appears to be slightly skewed in the direction of I. bullockii. In contrast, we did not detect any gene flow between the sister species I. bullockii (Fig. 4b). The gene flow estimates virtually peaked at zero (2Nm = 0.001 in both directions) and the 95% HPD intervals ranged from 0 to 1.148 and 0 to 0.495 in the direction of I. bullockii and I. abeillei, respectively. Similarly, the three-population analysis did not detect any gene flow between the highly allopatric I. galbula and I. abeillei (Fig. 4c), with gene flow estimates peaking near zero and 95% HPD intervals ranging from 0 to 0.631 and 0 to 0.719 in the direction of I. galbula and I. abeillei, respectively. There was no evidence of historical gene flow between the I. galbula lineage and the ancestral I. abeillei/I. bullockii lineage.
IMa2 provided very concise estimates of the timing of divergence events within the group. The analysis based on the nuclear species tree suggested that the initial divergence between the I. galbula lineage and the ancestor of I. abeillei and I. bullockii appears to have occurred approximately 350,000 years ago (95% HPD = 215,838–568,125), whereas I. abeillei and I. bullockii split much more recently approximately 95,000 years ago (41,865–159,087, Fig. 5a). In contrast, the analysis based on the mtDNA population tree (I. bullockii, (I. abeillei, I. galbula)) resulted in perfectly overlapping estimates of the two splitting events around 215–250,000 years ago, with nearly perfectly overlapping HPD intervals (Fig. 5b). Effective population sizes (Ne) were approximately 398,000 black-backed orioles, 557,000 Baltimore orioles, and 921,000 Bullock's orioles (Fig. 6).
Figure 5.
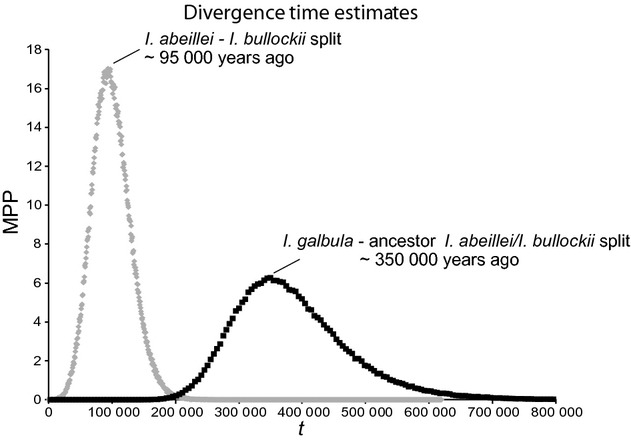
Posterior probability plot of divergence (splitting) times within the northern oriole group. According to IMa2, the initial split between Icterus galbula and the ancestor of I. abeillei and I. bullockii occurred roughly 350,000 years ago, whereas the more recent split between I. abeillei and I. bullockii occurred approximately 95,000 years ago.
Figure 6.
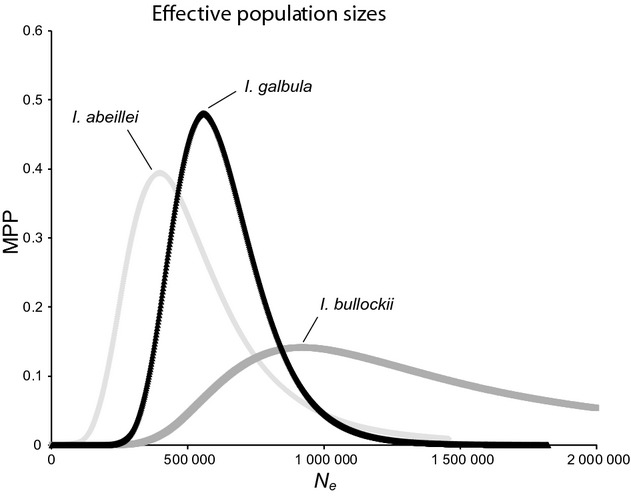
Posterior probability plot of effective population sizes (Ne) of extant populations of I. abeillei, I. bullockii, and I. galbula. Peak estimates of Ne are very similar for all three populations, with highly overlapping confidence intervals (95% HPDs).
Two-population IMa2 analyses
A summary of the three separate two-population analyses is presented in Table 3. Estimates of gene flow were mostly consistent between the pairwise two-population analyses and the three-population analysis. However, the two-population analysis of I. abeillei and I. galbula suggested a small but significant amount of introgression from I. abeillei into I. galbula (2NmI. abeillei > I. galbula = 0.477 (Fig. 4f). Although the 95% HPD interval included zero (see Table 5), nested model testing conducted in IMa2 indicated that a reduced model with zero migration was a significantly worse fit to the data than the full model allowing for migration (results not shown). Furthermore, the two-population analysis of I. galbula and I. bullockii indicated lower but equal rates of introgression in both directions (2NmI. bullockii > I. galbula = 1.331 and 2NmI. galbula > I. bullocki = 1.275, see Fig. 4d).
Table 5.
Summary of pairwise two-population IMa2 analyses of black-backed oriole (Icterus abeillei), Bullock's oriole (I. bullockii), and Baltimore oriole (I. galbula). Given are highest posterior parameter estimate (HiPt) and lower (HPD95Lo) and upper (HPD95Hi) bounds of 95%HPD intervals for the effective population size of population 1 (N1), population 2 (N2), and ancestral population (Na), splitting time (t), and population migration rate from population 2 into population 1 (2Nm1 > 2) and from population 1 into population 2 (2Nm2 > 1) forward in time. Population sizes are given in 1000 individuals and time since divergence in 1000 years
| N1 | N2 | Na | t | 2Nm1 > 2 | 2Nm2 > 1 | |
|---|---|---|---|---|---|---|
| I. abeillei (1) vs. I. galbula (2) | ||||||
| HiPt | 328 | 951 | 169 | 411 | 0.001 | 0.477 |
| HPD95Lo | 200 | 532 | 66 | 216 | 0 | 0 |
| HPD95Hi | 527 | 1,937 | 332 | 750 | 0.351 | 1.486 |
| I. abeillei (1) vs. I. bullockii (2) | ||||||
| HiPt | 517 | 1,198 | 225 | 163 | 0.005 | 0.025 |
| HPD95Lo | 252 | 528 | 118 | 79 | 0 | 0 |
| HPD95Hi | 1,184 | 4,732 | 373 | 299 | 2.404 | 7.461 |
| I. bullockii (1) vs. I. galbula (2) | ||||||
| HiPt | 595 | 688 | 163 | 424 | 1.275 | 1.331 |
| HPD95Lo | 350 | 397 | 62 | 245 | 0 | 0.069 |
| HPD95Hi | 1,005 | 1,215 | 313 | 749 | 3.206 | 4.25 |
The divergence time estimates from the pairwise two-population analyses were comparable to those produced by the three-population IMa2 analysis, except that both divergence events (I. galbula–I. abeillei/I. bullockii split, and I. abeillei–I. bullockii split) were estimated to have occurred roughly 70,000 years earlier than that indicated by the three-population analysis (see previous section, Fig. 5). In agreement with the nuclear species tree of this group (Jacobsen and Omland 2011), the most recent divergence in this group between I. abeillei and I. bullockii was estimated at approximately 163,000 years ago, well within the 95% HPD interval of the three-population estimate (see previous section). Similarly, the older split between the I. galbula lineage and the sister taxa I. abeillei and I. bullockii were estimated at 411,000 and 424,000 years ago, respectively.
Effective population size (Ne) estimates from the pairwise two-population analyses were also roughly similar to those estimated by the three-population analysis. The peak estimates were slightly higher in the two-population analyses, but the 95% HPD intervals overlap greatly with the estimates from the three-population analysis. The most striking deviations in population size estimates arose when the non-sister species, I. galbula and I. abeillei, were analyzed separately, which resulted in overestimation of NI. galbula and a slight underestimation of NI. abeillei (Table 5). Similarly, NI. bullockii was substantially smaller when paired with the non-sister I. galbula than when correctly paired with its sister species I. abeillei (Table 5, Fig. 6).
Discussion
The recent development of a multispecies coalescent framework for the study of divergence and gene flow among three or more taxa simultaneously in the program IMa2 (Hey 2010b) has proven useful for understanding the population dynamics of complex taxonomic groups consisting of several recently diverged species (e.g., Hey 2010a; Illera et al. 2011; Schoville et al. 2011; So et al. 2011; Li et al. 2012). In this study, we were able to estimate levels of nuclear introgression between allopatric populations of the three species in the northern oriole group while also accounting for their recent shared ancestry.
The hybrid zone between the non-sister species, I. galbula and I. bullockii, represents perhaps one of the most blatant case examples for the need of a multipopulation framework to disentangle the demographic history of larger groups of taxa. The reproductive barriers that so effectively prevent mtDNA and phenotypes of both I. galbula and I. bullockii from introgressing beyond the stable hybrid zone appear to be “leaky”, allowing near-neutral nuclear alleles to introgress and successfully spread in a heterospecific genetic background without erasing the diagnostic species differences. Such a substantial rate of introgression evident in populations several hundred miles away from the nearest known areas of sympatry has major implications for our understanding of the population dynamics of all three species within the group. Limited mtDNA introgression (Allen 2002; Carling et al. 2011) but extensive nDNA introgression (Allen 2002; this study) follows the predictions of Haldane's rule (Haldane 1922), wherein reduced female hybrid fitness prevents maternally inherited mtDNA, but not bi-parentally inherited nDNA from introgressing (Brookfield 1993; Turelli and Orr 1995; Orr 1997). Sex-biased (i.e., male driven) gene flow due to female heterogamety and the effects of Haldane's rule have increasingly been documented in a wide range of avian groups (e.g., Borge et al. 2005; Carling and Brumfield 2008; Carling et al. 2010; Storchová et al. 2010; Backström and Väli 2011).
A question of interest to researchers studying divergence and gene flow is to determine the timing of migration events between diverging populations. If possible, knowing when gene flow occurred in the speciation process would be of great importance to distinguish between an allopatric speciation model with recent secondary contact following range expansion (gene flow late) and a parapatric speciation model where divergence occurred in the presence of gene flow with subsequent decrease, then cessation of gene flow once reproductive barriers evolved. However, it is now clear that IMa2 and similar methods are unable to distinguish between these different modes of speciation (Sousa et al. 2011; Strasburg and Rieseberg 2011). We therefore cannot infer with confidence whether the currently hybridizing I. galbula and I. bullockii diverged in allopatry and only recently came back into secondary contact following postglacial range expansion, or if they diverged in parapatry while still exchanging genes throughout the early stages of divergence. Nonetheless, the fact that these two orioles are nearly 5% divergent in mtDNA (Omland et al. 1999) indicates that they have been evolving independently for an extended period of time, consistent with allopatric speciation followed by one or more episodes of gene flow during the last glacial cycles of the Pleistocene epoch. Pliocene or Pleistocene divergence followed by postglacial expansion has been proposed to explain the current distribution of many parapatric species found in North America and Eurasia (Klicka and Zink 1999; Hewitt 2004; Johnson and Cicero 2004). A recent exchange of nuclear variation through introgressive hybridization during secondary contact in the late Pleistocene would explain the wide gap between the mtDNA and nDNA divergence time estimates for these two species, even though the TMRCA for a single locus (e.g., mtDNA) will necessarily always predate the divergence between two species (Edwards and Beerli 2000).
In contrast, the IMa2 analyses revealed no evidence of introgression between I. abeillei and I. bullockii. The region of parapatry in Durango, Mexico where hybridization between these two species is alleged to have taken place was unfortunately not represented in this study. It is therefore possible that some hybridization could occur within this region, but restricted enough to prevent widespread introgression into the core ranges of either species. Thus, the ubiquitous nuclear haplotype sharing observed between these sister species most likely represents ancestral polymorphism retained in both descendent lineages since their recent split in the late Pleistocene (Hudson and Turelli 2003)
The low rate of unidirectional introgression from I. abeillei into I. galbula that was indicated by the two-population IMa2 analysis is somewhat puzzling, given that no evidence of gene flow between these two species was revealed in the three-population analysis. Previous studies on these two species using IM based on mtDNA and two nuclear introns also found no evidence of gene flow between these two mitochondrial sister taxa (Kondo et al. 2004, 2008). The fact that they are reciprocally monophyletic at the mtDNA level separated by a single fixed substitution also strongly suggests that there is no recent or ongoing mitochondrial gene flow between these highly allopatric species (Kondo et al. 2008).
Rather, we suspect that this signature of gene flow is a spurious outcome arising when violating the assumption of the IM model that no genetic exchange has occurred between the two focal populations and ghost populations (Beerli 2004; Slatkin 2005). As I. abeillei shares a more recent evolutionary history with I. bullockii than with I. galbula (Jacobsen et al. 2010; Jacobsen and Omland 2011), retained ancestral polymorphisms shared between I. abeillei and I. bullockii could introgress into I. galbula via I. bullockii over the extensive hybrid zone. This study revealed just how easily neutral nuclear alleles can cross this species boundary and freely introgress across the entire range of the other species. By excluding I. bullockii from the analysis, such shared ancestral alleles could mistakenly be inferred by IMa2 to have introgressed directly from I. abeillei into I. galbula (Fig. 7). Furthermore, the many alleles introgressing from I. bullockii into I. galbula have probably contributed to the high nucleotide diversity in I. galbula, causing IMa2 to overestimate the current effective population size of I. galbula in analyses that do not account for I. bullockii.
Figure 7.
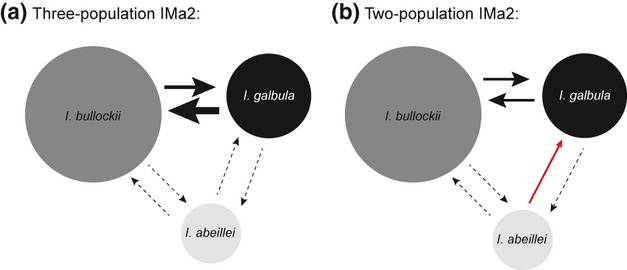
Patterns of gene flow among the three species in the northern oriole group estimated using IMa2. Circle sizes reflect relative Ne estimates. Relative rates of gene introgression are indicated by the width of solid arrows. Dashed arrows indicate little or no introgression. (a) The full three-population analysis inferred gene flow only between the hybridizing Icterus galbula and I. bullockii, whereas (b) two-population analyses also indicated a low level of introgression (highlighted in red) from I. abeillei into I. galbula. The introgression between these highly allopatric species is best ascribed to ancestral polymorphisms shared between I. abeillei and I. bullockii introduced to I. galbula across the I. galbula–I. bullockii hybrid zone. This study clearly illustrates how introgression from other taxa not included in the analysis (in this case I. bullockii) can bias gene flow estimation if not accounted for.
Thus, exclusion from the analysis of other taxa that have exchanged genes with either of the two focal taxa through shared ancestry or gene flow can bias estimation of population sizes and gene flow in IM, and the severity of the bias depends on how large the genetic contribution from the third taxon has been (Strasburg and Rieseberg 2010).
Conclusions
Our study reveals the complexity of evolutionary processes (recent divergence, slow sorting of ancestral variation, and introgressive hybridization) that have contributed to produce the observed pattern of rampant allele sharing among the three species within the northern oriole group. Furthermore, our findings highlight the importance of accounting for all potentially interbreeding taxa when interpreting the divergence history of a group of taxa, and the extent to which gene flow may retard the rate of divergence at neutral loci. Thus, this study provides an interesting perspective on the broad-scale impacts of introgressive hybridization, which has important implications for everything from understanding adaptive evolution to inferring species trees. The ease with which genome-scale datasets can now be obtained will soon enable researchers to study even more complex clades with four or more taxa that may interbreed.
Acknowledgments
We thank the curatorial staff at ANSP, LSUMNS, MZFC, BMNH, FMNH, UWBM, and H. L. Gibbs for providing the tissue samples used in this study. We especially thank A. G. Navarro-Sigüenza, B. E. Hernández-Baños, and M. Honey for help in collecting in Mexico. We further thank D. Kenny for laboratory assistance, and J. L. Peters for assistance with coalescent analyses. This work was funded by National Science Foundation grants to K. E. O. (DEB-0347083 and DEB-1119506), and Maryland Ornithological Society Avian Research Grants and an American Ornithologist's Union Research Grant to F. J.
Appendix 1.
Taxonomic sampling
| Species | Museuma | ID | Sex | Collection Localityb |
|---|---|---|---|---|
| Icterus abeillei | MZFC | 2339 | M | Mexico, Morelos, Carretera |
| Icterus abeillei | MZFC | 5381 | M | Mexico, Guerrero, Apaxtla |
| Icterus abeillei | MZFC | 3468 | M | Mexico, Guanajuato, San Pedro |
| Icterus abeillei | MZFC | KEO27 | F | Mexico: Guanajuato, Salvatierra |
| Icterus abeillei | MZFC | KEO28 | M | Mexico: Guanajuato, Salvatierra |
| Icterus abeillei | MZFC | KEO29 | M | Mexico: Guanajuato, Salvatierra |
| Icterus abeillei | MZFC | 1087 | M | Mexico, Guanajuato, Yuriria |
| Icterus abeillei | MZFC | 5441 | M | Mexico, Guanajuto, Yuriria |
| Icterus abeillei | MZFC | KEO30 | M | Mexico: Guanajuato, Yuriria |
| Icterus abeillei | MZFC | KEO31 | M | Mexico: Guanajuato, Yuriria |
| Icterus abeillei | MZFC | 3467 | M | Mexico, Jalisco, Laguna de Chapala |
| Icterus abeillei | MZFC | 7966 | M | Mexico: Jalisco |
| Icterus abeillei | MZFC | 10733 | M | Mexico: Jalisco |
| Icterus abeillei | MZFC | 11483 | M | Mexico: Jalisco |
| Icterus abeillei | MZFC | KEO32 | M | Mexico: Jalisco, Chapala |
| Icterus abeillei | MZFC | KEO33 | M | Mexico: Jalisco, Chapala |
| Icterus abeillei | MZFC | KEO34 | M | Mexico: Jalisco, Chapala |
| Icterus abeillei | MZFC | KEO35 | F | Mexico: Jalisco, Chapala |
| Icterus abeillei | MZFC | KEO37 | F | Mexico: Jalisco, Chapala |
| Icterus abeillei | MZFC | KEO45 | M | Mexico: Michoacán, Briseñas |
| Icterus abeillei | MZFC | KEO46 | M | Mexico: Michoacán, Briseñas |
| Icterus abeillei | MZFC | KEO48 | F | Mexico: Michoacán, Briseñas |
| Icterus abeillei | MZFC | KEO49 | M | Mexico: Michoacán, Briseñas |
| Icterus bullockii | MVZ | 176515 | M | USA: NM, Mora Co. |
| Icterus bullockii | LSUMNS | 3874 | F | USA: CA, Inyo Co. |
| Icterus bullockii | LSUMNS | 3875 | F | USA: CA, Inyo Co. |
| Icterus bullockii | LSUMNS | 3876 | M | USA: CA, Inyo Co. |
| Icterus bullockii | LSUMNS | 3877 | F | USA: CA, Inyo Co. |
| Icterus bullockii | LSUMNS | 3878 | M | USA: CA, Inyo Co. |
| Icterus bullockii | LSUMNS | 3879 | M | USA: CA, Inyo Co. |
| Icterus bullockii | LSUMNS | 3880 | M | USA: CA, Inyo Co. |
| Icterus bullockii | FMNH | 330029 | M | USA: CA, San Bernardino Co. |
| Icterus bullockii | FMNH | 341933 | M | USA: CA, Imperial Co. |
| Icterus bullockii | FMNH | 341934 | M | USA: CA, Monterrey Co. |
| Icterus bullockii | FMNH | 341937 | M | USA: CA, Monterrey Co. |
| Icterus bullockii | FMNH | 341938 | M | USA: CA, Monterrey Co. |
| Icterus bullockii | UWBM | 55942 | M | USA: WA, Grant Co. |
| Icterus bullockii | UWBM | 55951 | F | USA: WA, Douglas Co. |
| Icterus bullockii | UWBM | 55975 | M | USA: WA, Douglas Co. |
| Icterus bullockii | UWBM | 55976 | F | USA: WA, Douglas Co. |
| Icterus bullockii | UWBM | 55978 | F | USA: WA, Douglas Co. |
| Icterus bullockii | UWBM | 59055 | F | USA: WA, Asotin Co. |
| Icterus bullockii | UWBM | 59056 | M | USA: WA, Asotin Co. |
| Icterus bullockii | UWBM | 59058 | M | USA: WA, Asotin Co. |
| Icterus galbula | LSUMNS | 131146 | M | USA: LA, Cameron Co. |
| Icterus galbula | ANSP | 10126 | M | USA: PA, Bucks Co. |
| Icterus galbula | ANSP | 10134 | M | USA: PA, Bucks Co. |
| Icterus galbula | ANSP | 10148 | F | USA: PA, Bucks Co. |
| Icterus galbula | USFWS | 17860 | M | USA: MD, Baltimore Co. |
| Icterus galbula | USFWS | 17861 | M | USA: MD, Baltimore Co. |
| Icterus galbula | USFWS | 17862 | M | USA: MD, Baltimore Co. |
| Icterus galbula | USFWS | 17864 | M | USA: MD, Baltimore Co. |
| Icterus galbula | USFWS | 17866 | M | USA: MD, Baltimore Co. |
| Icterus galbula | USFWS | 17867 | M | USA: MD, Baltimore Co. |
| Icterus galbula | USFWS | 17869 | M | USA: MD, Baltimore Co. |
| Icterus galbula | USFWS | 17870 | M | USA: MD, Baltimore Co. |
| Icterus galbula | UWBM | 67201 | M | USA: MD, Prince Georges Co. |
| Icterus galbula | BMNH | 42547 | M | USA: MN, Becker Co. |
| Icterus galbula | BMNH | X7630 | M | USA: MN, Hennepin Co. |
| Icterus galbula | BMNH | X7763 | F | USA: MN, Chisago Co. |
| Icterus galbula | BMNH | X7799 | M | USA: MN, Chisago Co. |
| Icterus galbula | FMNH | 350604 | M | USA: IL, Cook Co. |
| Icterus galbula | FMNH | 395866 | F | USA: IL, Cook Co. |
| Icterus galbula | HLG | LG03 | M | Canada: Manitoba |
| Icterus galbula | HLG | LG04 | M | Canada: Manitoba |
| Icterus galbula | HLG | LG05 | M | Canada: Manitoba |
| Icterus galbula | HLG | LG06 | F | Canada: Manitoba |
| Sturnella neglecta | NMNH | 586115 |
Museums are abbreviated as follows: ANSP = Academy of Natural Sciences, Philadelphia; BMNH = J. F. Bell Museum of Natural History, University of Minnesota; FMNH = Field Museum of Natural History; HLG = H. Lisle Gibbs, blood samples; LSUMNS = Louisiana State University, Museum of Natural Science; MVZ = Museum of Vertebrate Zoology, University of California, Berkeley; MZFC = Museo de Zoología, Facultad de Ciencias, Universidad Nacional Autónoma de México; NMNH = National Museum of Natural History; UWBM = University of Washington, Burke Museum and USFWS = United States Fish and Wildlife Service band numbers.
San Pedro is the short form of San Pedro de Los Naranjos, and Briseñas is the short form of Briseñas de Matamoros.
Appendix 2.
GenBank accession numbers for DNA sequences obtained from previous studies on Icterus (Kondo et al. 2008; Jacobsen et al. 2010; Jacobsen and Omland 2011)
| Species | GenBank Accession Nos. |
|---|---|
| I. abeillei | DQ898412-DQ898433, DQ898358-DQ898385, GU972843, GU972875, JN169143-JN169144, JN169230-JN169239, JN169317-JN169326, JN169404-JN169413, JN169491-JN169500, JN169578-JN169587, JN169665-JN169674 |
| I. bullockii | JN169159-JN169170, JN169246-JN169257, JN169333-JN169344, JN169422-JN169431, JN169507-JN169518, JN169594-JN169605, JN169681-JN169692 |
| I. galbula | DQ898316-DQ898331, DQ898344-DQ898347, DQ898356-DQ898357, DQ898386-DQ898392, DQ898407, GU972855, GU972887, JN169191- JN169196, JN169274-JN169283, JN169361-JN169370, JN169448-JN169457, JN169535-JN169544, JN169622-JN169631, JN169709- JN169718 |
| S. neglecta | GU972873, GU972905 |
Conflict of Interest
None declared.
References
- Allen ES. Long-term hybridization and the maintenance of species identity in orioles (Icterus) Bloomington: Indiana University; 2002. Ph.D diss. [Google Scholar]
- AOU. Thirty-second supplement to the American Ornithologist's Union check-list of North American birds. Auk. 1973;90:411–419. [Google Scholar]
- AOU. Fortieth supplement to the American Ornithologist's Union check-list of North American birds. Auk. 1995;112:819–830. [Google Scholar]
- Arnold M. Evolution through genetic change. Oxford, U.K: Oxford Univ. Press; 2006. [Google Scholar]
- Avise JC. Molecular markers, natural history, and evolution. Sunderland, MA: Sinauer Associates; 2004. [Google Scholar]
- Axelsson E, Smith NGC, Sundström H, Berlin S, Ellegren H. Male-biased mutation rate and divergence in autosomal, Z-linked and W-linked introns of chicken and turkey. Mol. Biol. Evol. 2004;21:1538–1547. doi: 10.1093/molbev/msh157. [DOI] [PubMed] [Google Scholar]
- Backström N, Väli Ü. Sex- and species-biased gene flow in a spotted eagle hybrid zone. BMC Evol. Biol. 2011;11:100. doi: 10.1186/1471-2148-11-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backström N, Brandström M, Gustafsson L, Qvarnström A, Cheng H, Ellegren H. Genetic mapping in a natural population of collared flycatchers (Ficedula albicollis): conserved synteny but gene order rearrangements on the avian Z chromosome. Genetics. 2006;174:377–386. doi: 10.1534/genetics.106.058917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backström N, Forstmeier W, Schielzeth H, Mellenius H, Nam K, Bolund E, et al. The recombination landscape of the zebra finch Taeniopygia guttata genome. Genome Res. 2010;20:485–495. doi: 10.1101/gr.101410.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Barker FK. Monophyly and relationships of wrens (Aves: Troglodytidae): a congruence analysis of heterogeneous mitochondrial and nuclear DNA sequence data. Mol Phyl Evol. 2004;31:486–504. doi: 10.1016/j.ympev.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Beerli P. Effect of unsampled populations on the estimation of population sizes and migration rates between sampled populations. Mol. Ecol. 2004;13:827–836. doi: 10.1111/j.1365-294x.2004.02101.x. [DOI] [PubMed] [Google Scholar]
- Borge T, Webster MT, Andersson G, Sætre G-P. Contrasting patterns of polymorphism and divergence on the Z chromosome and autosomes on two Ficedula flycatcher species. Genetics. 2005;171:1861–1873. doi: 10.1534/genetics.105.045120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookfield J. Haldane's rule is significant. Evolution. 1993;47:1885–1888. doi: 10.1111/j.1558-5646.1993.tb01278.x. [DOI] [PubMed] [Google Scholar]
- Bureš S, Nádvorník P, Sætre G-P. Hybridization and apparent hybridization between meadow pipit (Anthus pratensis) and water pipit (A. spinoletta. Hereditas. 2002;136:254–256. doi: 10.1034/j.1601-5223.2002.1360313.x. [DOI] [PubMed] [Google Scholar]
- Butcher GS. Mate choice in female northern orioles with a consideration fo the role of the black male coloration in female choise. Condor. 1991;93:82–88. [Google Scholar]
- Carling MD, Brumfield RT. Haldane's rule in an avian system: using cline theory and divergence population genetics to test for differential introgression of mitochondrial, autosomal, and sex-linked loci across the Passerina bunting hybrid zone. Evolution. 2008;62:2600–2615. doi: 10.1111/j.1558-5646.2008.00477.x. [DOI] [PubMed] [Google Scholar]
- Carling MD, Lovette IJ, Brumfield RT. Historical divergence and gene flow: coalescent analyses of mitochondrial, autosomal and sex-linked loci in Passerina buntings. Evolution. 2010;64:1762–1772. doi: 10.1111/j.1558-5646.2010.00954.x. [DOI] [PubMed] [Google Scholar]
- Carling MD, Serene LG, Lovette IJ. Using historical DNA to characterize hybridization between Baltimore (Icterus galbula) and Bullock's Orioles. Auk. 2011;128:61–68. [Google Scholar]
- Corbin KW, Sibley CG, Ferguson A. Genic changes associated with the establishment of sympatry in orioles of the genus Icterus. Evolution. 1979;33:624–633. doi: 10.1111/j.1558-5646.1979.tb04715.x. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SV, Beerli P. Gene divergence, population divergence, and the variance in coalescence time in phylogenetic studies. Evolution. 2000;54:1839–1854. doi: 10.1111/j.0014-3820.2000.tb01231.x. [DOI] [PubMed] [Google Scholar]
- Ellegren H. Molecular evolutionary genomics of birds. Cytogenet. Genome Res. 2007;117:120–130. doi: 10.1159/000103172. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- Flockhart DTT, Wiebe KL. Absence of reproductive consequences of hybridization in the Northern Flicker (Colaptes auratus) hybrid zone. Auk. 2009;126:351–358. [Google Scholar]
- Flood NJ. Adaptive significance of delayed plumage maturation in male Northern Orioles. Evolution. 1984;38:267–279. doi: 10.1111/j.1558-5646.1984.tb00285.x. [DOI] [PubMed] [Google Scholar]
- Fu Y-X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrick R, Sunnucks P, Dyer R. Nuclear gene phylogeography using PHASE: dealing with unresolved genotypes, lost alleles, and systematic bias in parameter estimation. BMC Evol. Biol. 2010;10:118. doi: 10.1186/1471-2148-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS. Sex ration and unisexual sterility in animal hybrids. Genetics. 1922;12:101. [Google Scholar]
- Hewitt GM. Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R Soc. London Ser. B Biol. Sci. 2004;359:183–195. doi: 10.1098/rstb.2003.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J. Recent advances in assessing gene flow between diverging populations and species. Curr. Opin. Genet. Dev. 2006;16:592–596. doi: 10.1016/j.gde.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Hey J. The divergence of chimpanzee species and subspecies as revealed in multipopulation isolation-with-migration analyses. Mol. Biol. Evol. 2010a;27:921–933. doi: 10.1093/molbev/msp298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J. Isolation with migration models for more than two populations. Mol. Biol. Evol. 2010b;27:905–920. doi: 10.1093/molbev/msp296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167:747–760. doi: 10.1534/genetics.103.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Integrating within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc. Natl. Acad. Sci. USA. 2007;104:2785–2790. doi: 10.1073/pnas.0611164104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Turelli M. Stochasticity overrules the “three-times rule”: genetic drift, genetic draft, and coalescence times for nuclear loci versus mitochondrial DNA. Evolution. 2003;57:182–190. doi: 10.1111/j.0014-3820.2003.tb00229.x. [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguadé M. A test of neutral molecular evolution based on nucleotide data. Genetics. 1987;116:153–159. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illera JC, Koivula K, Broggi J, PÄCkert M, Martens J, Kvist L. A multi-gene approach reveals a complex evolutionary history in the Cyanistes species group. Mol. Ecol. 2011;20:4123–4139. doi: 10.1111/j.1365-294X.2011.05259.x. [DOI] [PubMed] [Google Scholar]
- Jacobsen F, Omland KE. Species tree inference in a recent radiation of orioles (genus Icterus): multiple markers and methods reveal cytonuclear discordance in the northern oriole group. Mol. Phyl. Evol. 2011;61:460–469. doi: 10.1016/j.ympev.2011.06.017. [DOI] [PubMed] [Google Scholar]
- Jacobsen F, Friedman NR, Omland KE. Congruence between nuclear and mitochondrial DNA: Combination of multiple nuclear introns resolves a well-supported phylogeny of New World orioles (Icterus. Mol Phyl Evol. 2010;56:419–427. doi: 10.1016/j.ympev.2010.03.035. [DOI] [PubMed] [Google Scholar]
- Jaramillo A, Burke P. New World blackbirds: the Icterids. Princeton, NJ: Princeton University Press; 1999. [Google Scholar]
- Johnson NK, Cicero C. New mitochondrial DNA data affirm the importance of Pleistocene speciation in North American birds. Evolution. 2004;58:1122–1130. doi: 10.1111/j.0014-3820.2004.tb00445.x. [DOI] [PubMed] [Google Scholar]
- Klicka J, Zink RM. Pleistocene effects on North American songbird evolution. Proc. R Soc. London Ser. B Biol. Sci. 1999;266:695–700. [Google Scholar]
- Kliman RM, Andolfatto P, Coyne JA, Depaulis F, Kreitman M, Berry AJ, et al. The population genetics of the origin and divergence of the Drosophila simulans complex species. Genetics. 2000;156:1913–1931. doi: 10.1093/genetics/156.4.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo B, Baker JM, Omland KE. Recent speciation between the Baltimore Oriole and the Black-backed Oriole. Condor. 2004;106:674–680. [Google Scholar]
- Kondo B, Peters JL, Rosensteel BB, Omland KE. Coalescent analyses of multiple loci support a new route to speciation in birds. Evolution. 2008;62:1182–1191. doi: 10.1111/j.1558-5646.2008.00345.x. [DOI] [PubMed] [Google Scholar]
- Li Z, Zou J, Mao K, Lin K, Li H, Liu J, et al. Population genetic evidence for complex evolutionary histories of four high altitude juniper species in the Qinghai-Tibetan Plateau. Evolution. 2012;66:831–845. doi: 10.1111/j.1558-5646.2011.01466.x. [DOI] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Machado CA, Kliman RM, Markert JA, Hey J. Inferring the history of speciation from multilocus dna sequence data: the case of Drosophila pseudoobscura and close relatives. Mol. Biol. Evol. 2002;19:472–488. doi: 10.1093/oxfordjournals.molbev.a004103. [DOI] [PubMed] [Google Scholar]
- Mettler RD, Spellman GM. A hybrid zone revisited: molecular and morphological analysis of the maintenance, movement, and evolution of a Great Plains avian (Cardinalidae: Pheucticus) hybrid zone. Mol. Ecol. 2009;18:3256–3267. doi: 10.1111/j.1365-294X.2009.04217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WD. Lists of birds collected in northwestern Durango, Mexico, by F. H. Batty, during 1903. Bull. Am. Mus. Nat. Hist. 1906;22:161–183. [Google Scholar]
- Milne I, Wright F, Rowe G, Marshall DF, Husmeier D, McGuire G. TOPALi: software for automatic identification of recombinant sequences within DNA multiple alignments. Bioinformatics. 2004;20:1806–1807. doi: 10.1093/bioinformatics/bth155. [DOI] [PubMed] [Google Scholar]
- Moore WS. Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution. 1995;49:718–726. doi: 10.1111/j.1558-5646.1995.tb02308.x. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Wakeley J. Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics. 2001;158:885–896. doi: 10.1093/genetics/158.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosil P. Speciation with gene flow could be common. Mol. Ecol. 2008;17:2103–2106. doi: 10.1111/j.1365-294X.2008.03715.x. [DOI] [PubMed] [Google Scholar]
- Omland KE, Lanyon SM, Fritz SJ. A molecular phylogeny of the New World orioles (Icterus): the importance of dense taxon sampling. Mol Phyl Evol. 1999;12:224–239. doi: 10.1006/mpev.1999.0611. [DOI] [PubMed] [Google Scholar]
- Orr HA. Haldane's rule. Annu. Rev. Ecol. Syst. 1997;28:195–218. [Google Scholar]
- Peters JL, Zhuravlev Y, Fefelov I, Logie A, Omland KE. Nuclear loci and coalescent methods support ancient hybridization as cause of mitochondrial paraphyly between gadwall and falcated duck (Anas spp.) Evolution. 2007;61:1992–2006. doi: 10.1111/j.1558-5646.2007.00149.x. [DOI] [PubMed] [Google Scholar]
- Petit RJ, Excoffier L. Gene flow and species delimitation. Trends Ecol. Evol. 2009;24:386–393. doi: 10.1016/j.tree.2009.02.011. [DOI] [PubMed] [Google Scholar]
- Pinho C, Hey J. Divergence with gene flow: models and data. Annu. Rev. Ecol. Evol. Syst. 2010;18:215–230. [Google Scholar]
- Primmer CR, Borge T, Lindell J, Sætre G-P. Single-nucleotide polymorphism characterization in species with limited available sequence information: high nucleotide diversity revealed in the avian genome. Mol. Ecol. 2002;11:603–612. doi: 10.1046/j.0962-1083.2001.01452.x. [DOI] [PubMed] [Google Scholar]
- Ramos-Onsins SE, Rozas J. Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol. 2002;19:2092–2100. doi: 10.1093/oxfordjournals.molbev.a004034. [DOI] [PubMed] [Google Scholar]
- Remington CL. Suture-zones of hybrid interaction between recently joined biotas. In: Dobzhansky T, Hecht MK, Steere WC, editors. Evolutionary biology. New York: Plenum Press; 1968. pp. 321–428. [Google Scholar]
- Richardson DS, Burke T. Extra-pair paternity in relation to male age in Bullock's oriole. Mol. Ecol. 1999;8:2115–2126. doi: 10.1046/j.1365-294x.1999.00832.x. [DOI] [PubMed] [Google Scholar]
- Ridgely RS, Allnutt TF, Brooks T, McNicol DK, Mehlman DW, Young BE, et al. Digital distribution maps of the birds of the Western Hemisphere, version 3.0. Arlington, VA: NatureServe; 2007. [Google Scholar]
- Rising JD. A comparison of metabolism and evaporative water loss of baltimore and bullock orioles. Comp. Biochem. Physiol. 1969;31:915–925. [Google Scholar]
- Rising JD. Morphological variation and evolution in some North American orioles. Syst. Zool. 1970;19:315–351. [Google Scholar]
- Rising JD. Morphological variation and status of the orioles, Icterus galbulaI. bullockii, and I. abeillei, in the northern Great Plains and in Durango, Mexico. Can. J. Zool. 1973;51:1267–1273. [Google Scholar]
- Rising JD. The progress of Oriole hybridization in Kansas. Auk. 1983;100:885–897. [Google Scholar]
- Rising JD. The stability of the oriole hybrid zone in Western Kansas. Condor. 1996;98:658–663. [Google Scholar]
- Rising JD, Flood NJ. Baltimore Oriole (Icterus galbula. In: Poole A, editor. The Birds of North America. Ithaca, NY: Cornell Lab of Ornithology; 1998. [Google Scholar]
- Rising JD, Williams PL. Bullock's Oriole (Icterus bullockii) In: Poole A, editor. The Birds of North America. Ithaca, NY: Cornell Lab of Ornithology; 1999. [Google Scholar]
- Rohwer S, Johnson MS. Scheduling differences of molt and migration for Baltimore and Bullock's Orioles persist in a common environment. Condor. 1992;94:992–994. [Google Scholar]
- Rohwer S, Manning J. Differences in timing and number of molts for Baltimore and Bullock's orioles: implications to hybrid fitness and theories of delayed plumage maturation. Condor. 1990;92:125–140. [Google Scholar]
- Sæther B-E, Lande R, Engen S, Weimerskirch H, Lillegard M, Altwegg R, et al. Generation time and temporal scaling of bird population dynamics. Nature. 2005;436:99–102. doi: 10.1038/nature03666. [DOI] [PubMed] [Google Scholar]
- Schoville SD, Tustall TS, Vredenburg VT, Backlin AR, Gallegos E, Wood DA, et al. Conservation genetics of evolutionary lineages of the endangered mountain yellow-legged frog, Rana muscosa (Amphibia: Ranidae), in southern California. Biol. Conserv. 2011;144:2031–2040. [Google Scholar]
- Sibley CG, Short LLJ. Hybridization in the Orioles of the Great Plains. Condor. 1964;66:130–150. [Google Scholar]
- Slatkin M. Seeing ghosts: the effect of unsampled populations on migration rates estimated for sampled populations. Mol. Ecol. 2005;14:67–73. doi: 10.1111/j.1365-294X.2004.02393.x. [DOI] [PubMed] [Google Scholar]
- So J, Uthicke S, Hamel J-F, Mercier A. Genetic population structure in a commercial marine invertebrate with long-lived lecithotrophic larvae: Cucumaria frondosa (Echinodermata: Holothuroidea) Mar. Biol. 2011;158:859–870. [Google Scholar]
- Sousa VC, Grelaud A, Hey J. On the nonidentifiability of migration time estimates in isolation with migration models. Mol. Ecol. 2011;20:3956–3962. doi: 10.1111/j.1365-294x.2011.05247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am. J. Hum. Genet. 2005;76:449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchová R, Reif J, Nachman MW. Female heterogamety and speciation: reduced introgression of the Z chromosome between two species of nightingales. Evolution. 2010;64:456–471. doi: 10.1111/j.1558-5646.2009.00841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasburg JL, Rieseberg LH. How robust are “isolation with migration” analyses to violations of the IM model? a simulation study. Mol. Biol. Evol. 2010;27:297–310. doi: 10.1093/molbev/msp233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasburg JL, Rieseberg LH. Interpreting the estimated timing of migration events between hybridizing species. Mol. Ecol. 2011;20:2353–2366. doi: 10.1111/j.1365-294X.2011.05048.x. [DOI] [PubMed] [Google Scholar]
- Sutton GM. Oddly plumaged orioles from western Oklahoma. Auk. 1938;55:1–6. [Google Scholar]
- Sutton GM. Oriole hybridization in Oklahoma. Bull Ok Ornit Soc. 1968;1:1–7. [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli M, Orr HA. The dominance theory of Haldane's rule. Genetics. 1995;140:389–402. doi: 10.1093/genetics/140.1.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltari E, Edwards SV. Evolutionary dynamics of intron size, genome size, and physiological correlates in Archosaurs. Am Nat. 2002;160:539–552. doi: 10.1086/342079. [DOI] [PubMed] [Google Scholar]