

Abstract
Almost 30 years ago, neuropeptide Y (NPY) was discovered as a sympathetic co-transmitter and one of the most evolutionarily conserved peptides abundantly present all over the body. Soon afterward, NPY’s multiple receptors were characterized and cloned, and the peptide’s role in stress was first documented. NPY has proven to be pivotal for maintaining many stress responses. Most notably, NPY is known for activating long-lasting vasoconstriction in many vascular beds, including coronary arteries. More recently, NPY was found to play a role in stress-induced accretion of adipose tissue which many times can lead to detrimental metabolic changes. It is however due to its prominent actions in the brain, one of which is its powerful ability to stimulate appetite as well as its anxiolytic activities that NPY became a peptide of importance in neuroscience. In contrast, its actions in the rest of the body, including its role as a stress mediator, remained, surprisingly underappreciated and not well understood. Our research has focused on that other, “peripheral” side of NPY. In this review, we will discuss those actions of NPY on the cardiovascular system and metabolism, as they relate to adaptation to stress, and attempt to both distinguish NPY’s effects from and integrate them with the effects of the classical stress mediators, glucocorticoids, and catecholamines. To limit the bias of someone (ZZ) who has viewed the world of stress through the eyes of NPY for over 20 years, fresh insight (DH) has been solicited to more objectively assess NPY’s contributions to stress-related diseases and the body’s ability to adapt to stress.
Keywords: Neuropeptide Y, Stress, Cardiovascular, Metabolism, Adrenergic system, Purines, Glucocorticoids, Sympathetic nerve activity
Introduction
In 1982, Tatemoto and Mutt (Carlquist et al. 1982), the two chemists responsible for designing a method for identifying bioactive peptides through amidation, extracted neuropeptide Y (NPY) from porcine brain. In addition, they are also credited with identifying the other members of the NPY peptide family, peptide YY (PYY) and pancreatic polypeptide (PP), both of which are largely derived from the gut (Tatemoto 1982). All three peptides share a high level of homology and are highly evolutionarily conserved, with up to 92% conservation in structure between non-vertebrates and vertebrates (Colmers and Wahlestedt 1993). PYY shares 75% homology with NPY and thus, is able to activate the same receptors, Y1, Y2, Y4, Y5, and Y6, all of which are Gi-coupled receptors (Larhammar et al. 1993).
Due to the multiplicity of receptors and ligands, some of which are generated through a proteolytic cleavage of NPY and PYY to shorter forms, it is not surprising that this peptide family displays a plethora of activities. Of its three members, however, it is NPY, due to its location in the nervous system, both centrally and peripherally, that has been most implicated during stress. In this review, we will focus on the role of NPY as a mediator of stress-induced adaptation and maladaptation of the cardiovascular and metabolic systems, and we will discuss how these changes are able to trigger or accelerate diseases such as atherosclerosis and obesity. Considering the complexity of the body’s ability to adapt to various types of environmental stressors, no single mediator would be expected to be solely responsible for the body’s response to stress. With this in mind, we will not only review the direct actions of NPY, but also its interactions with other classical stress mediators such as the sympathetic neurotransmitters: catecholamines and purines, and hormones from the hypothalamic–pituitary–adrenal (HPA) axis. To limit the bias of someone (ZZ) who has viewed the world of stress through the eyes of NPY for over 20 years, fresh insight (DH) has been solicited. It is the authors’ hope that with such an approach, we will provide a fresh look on how NPY works within the stress response, and thus, improve the understanding of its physiological involvement in the regulation of stress adaptation and stress-related diseases. Although many excellent reviews provide abundant information on NPY’s actions within the brain (Yulyaningsih et al. 2011; Morales-Medina et al. 2010; Hökfelt et al. 2008; Brothers and Wahlestedt 2010; Thorsell 2010; Vezzani and Sperk 2004), this review will focus on the activities of the peptide’s release from the peripheral pools: sympathetic nerves and other non-neuronal sources. We will start with a short discussion regarding the current views on stress and the stress response.
What is Stress?
In order to survive, every organism has developed sophisticated mechanisms, which allow it to cope with changing environments. The challenges that a changing environment poses on the body is what is commonly referred to as stress. However, a clear scientific definition is lacking. One view that perhaps best describes stress is one proposed by David Goldstein (Goldstein 2010). In his definition, he breaks down the term “stress” into three main components: stressors, which are stimuli that disrupt homeostasis, stress, which is what the brain perceives when faced with a disturbance of homeostasis, and the stress response, which is elicited by the brain to amend the disturbance. Using the example of a thermostat (Goldstein 2010) he argues that just like a thermostat is able to sense changes in environmental temperature; the body also senses detrimental changes within its physical environment. Whether the danger is real or perceived, the body will mount a stress response in an attempt to return back to homeostasis. The stress response stops once the stressor is removed and the brain no longer senses changes beyond its normal set point.
It is understood that most times acute stress should not result in harming the organism, instead, through physiological adaptation the organism should become capable of functioning in the midst of this altered environment. However, it is during chronic or unusually intense bouts of stress that organisms experience its negative side effects. Hans Seyle, whom many regard as the father of stress, was the first to document physical changes within animals exposed to acute and chronic stress and was the first to develop the idea of stress-related diseases (Selye 1998). While this idea is still valid, specific disease mechanisms resulting from exposure stress have evolved greatly over the years.
Classically, it is understood that the stress response primarily involves two groups of hormones/neurotransmitters: glucocorticoids (cortisol in humans, corticosterone in rodents) and catecholamines (norepinephrine, NE and epinephrine, Epi). Under normal conditions the levels of these hormones rise and fall with the body’s natural circadian rhythm. However, following a stressful stimulus, a flood of catecholamines and glucocorticoids are released throughout the body. In response, heart rate begins to climb, cardiac output increases, respiration is accelerated, catabolism is enhanced, short term energy stores are used, and blood-flow to major organs such as the brain, heart, and muscles increases—a reaction known as the “fight-or-flight” response. It is well accepted that this stress response is mediated by two principal endocrine systems: the HPA axis and the sympatho-adrenomedullary system (SAS). Sympathetic stimulation is known to activate the SAS which results in both the release of NE from the nerve endings onto target tissues, as well as Epi (“adrenaline”) and NE secretion from the adrenal medulla. Activation of the HPA axis begins with the release of corticotropin-releasing hormone (CRH) together with arginine vasopressin (AVP) into the hypophyseal portal blood (Crowley 2004), which in turn, stimulates the release of adrenocorticotropic hormone (ACTH) from the anterior pituitary (Crowley 2004; Buckingham 2000). Circulating ACTH goes on to activate the adrenal cortex, leading to the release of the primary glucocorticoid, cortisol.
A common view still expressed in a large body of scientific literature is that stress can be “measured” by either plasma or salivary levels of cortisol, and/or plasma catecholamines as signs of activation of the HPA axis or SAS, respectively. In addition, a second common oversimplification is that “sympatho-adrenomedullary” activation is considered synonymous with “adrenergic” activation. Although plasma levels of cortisol and/or catecholamines are a valued measurement of stress, these values do not always correctly characterize the stress response. First, each neurotransmitter released into the body decays different rates. Therefore, there are specific windows of time for which each neurotransmitter can act before it becomes inactivated. For example, it is known that catecholamines are released and degraded within minutes (Benedict et al. 1978) and on the other hand, that NPY, has a much longer half-life (Ahlborg et al. 1992) of up to 39 min. Understanding, the rate at which these chemicals will be broken down within the body is crucial for understanding the physiological actions of these peptides. In addition, it is known that platelets are capable of taking in and storing various peptides from the circulation during their lifetime of up to 2 weeks (Daimon 1991; Zukowska-Grojec and Neuropeptide 1995). Moreover, not all stressors evoke the same stress response. For example, restraint stress in mice (Abe et al. 2010) and immobilization stress in rats (Zukowska-Grojec et al. 1988) results in high levels of NE release from nerve terminals without significant elevations in NPY. However, more intense stressors, such as exposure to foot shocks (Zukowska-Grojec et al. 1988) or cold stress (Kuo et al. 2007) leads to a large increase in NPY release, while NE release does not change. This becomes even more evident during chronic stress, when often both catecholamines (Kuo et al. 2007) and glucocorticoid secretion is down-regulated (Rasmusson et al. 2010). Together these findings suggest that there is no single, traditional or new, stress mediator that can be used as an absolute indicator of stress. However, perhaps a more accurate way to assess the intensity of the stress would be to measure a combination of different neurohormones. As elegantly shown and discussed in an early paper by Fischer-Colbrie et al. (1988), the “secretory cocktail” of stress mediators that are released from the nerve terminal following differential sympathetic stimuli could be the key to understanding multiple forms of stress and their effects on the body.
Thus, we believe, that after 30 years since the discovery of sympathetic co-transmission, which includes NPY, it is about time that a monolithic view of the SAS is abandoned. Stress still remains difficult to measure, but our ability can improve with the assessment of not one, but many stress mediators such as ACTH, vasopressin, oxytocin, cortisol and the sympathetic neurotransmitters-NE, Epi, and NPY in plasma as well as within circulating cells such as platelets.
History of NPY: Synthesis, Structure, and Functions
Soon after NPY was first isolated from porcine brain by Carlquist et al. (1982) and Tatemoto et al. (1982) its discovery as one of the most structurally conserved peptides emerged. NPY has 92% sequence homology between cartilaginous fish and today’s mammals (Colmers and Wahlestedt 1993; Larhammar et al. 1993; Larsson et al. 2008), indicating that it has remained almost completely conserved within the genome for over 400 million years. This high level of conservation suggests that NPY must serve important physiological roles within the body. Indeed, in support of this notion, throughout the animal kingdom, from C. elegans to Drosophila and zebrafish, NPY was found to subserve similar functions in regulating behavior and adaptation of the organism during environmental challenges such as starvation, infection or predator attack (Cohen et al. 2009; Sokolowski 2003). In addition, other peptides such as peptide YY (PYY) and pancreatic polypeptide (PY) have been found to share a high degree of homology with NPY. Today these peptides (PYY, PY, and NPY) are all categorized as the NPY family (Larhammar et al. 1993). Depending on the length, these peptides can act on multiple G-protein coupled receptors: Y1, Y2, Y4, Y5, and Y6 which is known to be non-functional in humans (Yulyaningsih et al. 2011; Brothers and Wahlestedt 2010; Merten and Beck-Sickinger 2006; Michel 2004). Table 1 describes the expression and functions of each receptor both centrally and peripherally.
Table 1.
Neuropeptide Y family of receptors, their preferred ligands, receptor distribution, and physiological functions
| Y1 | Y2 | Y4 | Y5 | |
|---|---|---|---|---|
| Preferred ligand |
• NPY • PYY |
• NPY • PYY • NPY 3-36 • PYY 3-36 |
• PP |
• NPY • PYY • NPY 3-36 • PYY 3-36 |
| Expression of receptor | ||||
| Central vs. peripheral |
Central Cerebral cortex, brainstem and thalamus Peripheral Smooth muscle of blood vessels, immune cells, osteoblasts |
Central Hippocampus, brainstem and hypothalamus Peripheral Autonomic nerves, gastrointestinal tract endothelial cells, adipocytes |
Central Paraventricular nucleus and hypothalamus Peripheral Colon, small intestine and prostate |
Central Hippocampus, plexiform cortex of the olfactory bulb, suprachiasmatic nucleus and the arcuate nucleus |
| Pre-junctional vs. post-junctional expression | Post-junctional | Pre-junctional | Post-junctional | Post-junctional |
| Tissue with highest expression | Smooth muscle of vessels innervated by the SNS | Central and peripheral neurons | Gut | Hypothalamus |
| Physiological function (central vs. peripheral) |
Central Regulation of food intake, anxiolytic Peripheral Vasoconstriction, regulation of neurotransmitter release |
Central Inhibition of neurotransmitter release (glutamate). Improved learning and memory Peripheral Inhibition of norepinephrine release (pre-junctional); angiogenesis, adipogenesis (post-junctional) |
Central Luteinizing hormone secretion? |
Central Stimulation of food intake; anti-epileptic |
NPY is synthesized within the endoplasmic reticulum as a large precursor protein. The peptide, once synthesized, then moves to the Golgi apparatus, which is followed by its translocation into the trans-Golgi network (TGN) where the peptide is stored until further activation. The majority of NPY is stored in large dense-core vesicles (Michel 2004). When NPY is needed, the precursor protein undergoes several post-translational modifications along with several enzymatic cleavages before it is exocytosed into the extracellular space. Upon entry into the extracellular space, NPY can undergo further proteolysis, which cleaves it into different sized peptides. Aminopeptidase P (APP) and dipeptidyl peptidase IV (DPPIV) are two major amino peptidases responsible for cleaving NPY to NPY2–36 and NPY3–36, respectively (Hörsten et al. 2004). This proteolysis however, does not inactivate NPY, but rather it abolishes its Y1 receptor affinity and maintains the peptide’s affinity for the Y2 and Y5 receptors. This way, peptidases, and in particular DPPIV, which is abundantly present in endothelial, epithelial, and immune cells, act as a converting enzyme, determining the kinds of activities that NPY-like peptides can exert.
History of NPY: Where Is It Found?
NPY is the most, or one of the most, abundant peptides in the brain. NPY is found in highest concentration within the hypothalamic arcuate nucleus, brainstem, and anterior pituitary. At these sites, NPY is thought to have two major functions: stimulate food intake and modulate the stress response. NPY is the most potent orexigenic (Crowley 2004; Stanley and Leibowitz 1984) peptide known to date and its powerful effects can best be seen when injected into the paraventricular nucleus (Fig. 1). Other studies have shown that, injection into the paraventricular nucleus can trigger the secretion of ACTH (Crowley 2004), a hormone that initiates a cascade of events resulting in the release of cortisol, through activation of CRH. Many of NPY’s central actions are important during stress and stress-related diseases. In contrast to the periphery where NPY—in the face of stress—amplifies the stress response, a reaction which can lead to detrimental effects on the body, in the brain, NPY’s actions moderate and improve the body’s ability to cope with stress. Thus, within the brain, NPY acts as a “brake” to inhibit excessive activation of the stress response.
Fig. 1.
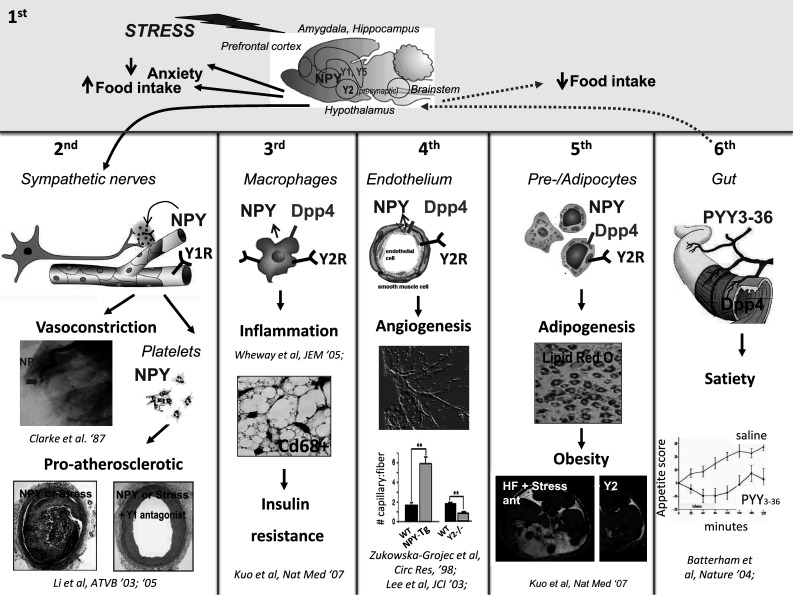
Sources and actions of NPY in the brain and the periphery. Pools 1–6 delineate peripheral and central regions within the body where NPY is found. In the 1st pool, the brain, NPY and its receptors are expressed post-synaptically (Y1, Y5) and pre-synaptically (Y2) in many stress-sensitive regions (the hypothalamus and hippocampus) where the peptide exerts potent orexigenic and anxiolytic activities. In the periphery, pools 2nd–6th, there are neuronal (2nd Pool) and non-neuronal sources of expression of NPY (3rd–6th Pools). In the cardiovascular system (secreted from the sympathetic nerves and platelets—2nd Pool), NPY mediates stress-induced vasoconstriction, vascular hyperplasia, and atherosclerosis via its Y1 receptor. In the immune cells (3rd Pool), NPY is inducible and can mediate either pro- or anti-inflammatory actions through its Y1 receptor on macrophages. In the autocrine/paracrine systems of the endothelial cells (4th Pool), NPY, converted by DPPIV to NPY3–36, a Y2 receptor-selective agonist, promotes angiogenesis and vascularization of ischemic and non-ischemic tissues. NPY when up-regulated in transgenic mice increases the basal level of capillary fibers in the hind limb, whereas Y2rKO mice show a decreased number of capillary fibers as compared to WT (4th Pool). NPY, DPPIV, and Y2Rs are also inducible in adipocytes (5th Pool) where they mediate stress-and-diet-induced adipogenesis and obesity. Finally, both NPY and PYY are expressed in the gastrointestinal system, but it is the gut PYY (6th Pool) when converted to PYY3–36 by DPPIV, which is known to induce satiety by acting on the hypothalamic presynaptic Y2 receptor to suppress food intake
Centrally, NPY is anti-epileptic (Xapelli et al. 2006), anxiolytic (Thorsell 2010) and it is associated with physiological resilience and increased performance levels during military training (Rasmusson et al. 2010). In addition, central increases in levels of NPY in patients suffering from post-traumatic stress disorder (PTSD) have been associated with a decrease in PTSD symptoms (Rasmusson et al. 2010). Last, increased NPY has been shown to improve memory (Flood et al. 1987), increase neurogenesis (Scharfman and Gray 2006) and to modulate peripheral sympathetic outflow (Egawa et al. 1991).
In the periphery, NPY can be found in three main pools: sympathetic nerves, the adrenal medulla, and platelets (Fig. 1). Immunocytochemistry has been used to confirm the existence of NPY in postganglionic sympathetic nerve fibers throughout the body. Further staining indicated that NPY exists within these terminals alongside NE and ATP (Morris 1999). Moreover, isolated, in situ and in vivo stimulation of organs by sympathetic activation have demonstrated the co-release of NPY, NE, and ATP from axon terminals (Morris 1999; Lundberg et al. 1990). However, it is important to note that NPY, NE, and ATP are not always released in similar quantities. Instead, the release of each transmitter is dependent on the intensity and the pattern of sympathetic nerve activation (Fig. 2c). Clinically, increases in plasma NPY have been documented in diseases such as hypertension (Wocial et al. 1995), chronic heart failure (Callanan et al. 2007; McDermott and Bell 2007), and renal failure (Bald et al. 1997) where sympathetic outflow is increased (Fig. 2d).
Fig. 2.
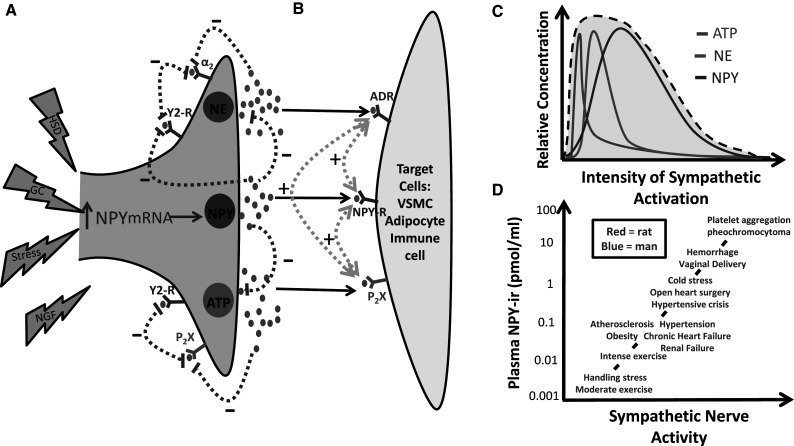
Schematic representation of the interactions between the neurotransmitters released following sympathetic activation and their actions both pre- and post-synaptically. Part a depicts how activation of the sympathetic nervous system (SNS) by glucocorticoids (GC), High Fat Sugar Diet (HFS), Nerve Growth Factor (NGF) or stress results in increased NPY mRNA expression in the nerve terminal. Part a also shows that activation of the SNS induces the release of three neurotransmitters: ATP, NE, and NPY. Each neurotransmitter is capable of having both pre- a and post-synaptic b effects. Pre-synaptically (a), NPY can inhibit the release of NE, ATP and itself. Both NE and ATP are also capable of inhibiting NPY’s release as well their own release. Post-synaptically (b) each neurotransmitter is capable of potentiating each other’s effects. Part c shows the relationship between sympathetic activation and the relative release of ATP, NE, and NPY. Part d describes the relationship between plasma NPY levels and multiple pathological states during varying degrees of sympathetic nerve activity
NPY is also known to exist within the chromaffin cells in the adrenal medulla (Rosmaninho-Salgado et al. 2007) as shown in studies using perfused bovine adrenal glands (Hexum et al. 1987) and cultured chromaffin cells (Kataoka et al. 1985). Interestingly, rats following adrenalectomy show no changes in circulating NPY levels within the body at rest. This observation led researchers to believe that under normal conditions, NPY within the adrenal medulla did not play a systemic role (Hörsten et al. 2004). Some of the highest levels of circulating NPY have been measured in patients with pheochromocytoma (Grouzmann et al. 1989). This potentially fatal, neuroendocrine tumor originating from the adrenal medulla results in increased release of catecholamines as well as NPY into the plasma. Although the trigger for NPY release is not well understood, it is thought that angiotensin II may be involved (Grouzmann et al. 1994).
In addition, NPY is expressed in platelets and their precursor, megakaryocytes residing in the bone marrow (Ericsson et al. 1987; Myers et al. 1988). However, the amount of NPY found in platelets varies greatly among species. Rats seem to display the highest levels of NPY in their platelets, although some strains of mice, rabbits, and humans display NPY in their platelets as well (Ericsson et al. 1987; Myers et al. 1988). Interestingly, under normal physiological conditions, NPY may not be expressed in platelets and megakaryocytes. Instead, it is thought that expression can be induced by disease, and perhaps by chronic stress. Interestingly, support for this idea can be seen in patients with peripheral vascular disease (Li et al. 2011) and depression (Nilsson et al. 1996), who show increased NPY in their platelets. Former studies by our group led us to propose the idea that increased NPY levels within platelets may be implicated in the acceleration of atherosclerosis and other stress-related diseases. Platelet NPY may also be a better marker of chronic stress than plasma levels of this peptide, or other stress mediators, since they are cleared more quickly from the circulation, whereas platelets can maintain their NPY stores for approximately 2 weeks.
NPY as a Sympathetic Co-transmitter
Within the sympathetic nerves, NPY is co-localized with the classical neurotransmitter, NE and another non-adrenergic transmitter, ATP. While NE and ATP are stored in a larger pool of small dense-core vesicles, NPY is contained within a smaller population of so called large, dense-core vesicles, which are also known to contain NE (Hörsten et al. 2004). Thus, although multiple transmitters may exist within one vesicle, the concentration of each transmitter may differ between vesicles within one axon terminal. This differential intra-cellular localization implies differences in the mode of their release. Indeed, each neurotransmitter shows differential release depending on the intensity and pattern of sympathetic nerve activation (Westfall 2004) (Fig. 2). For example, studies using guinea pig vas deferens have shown that although ATP and NE are co-stored, the amount of ATP and NE within each vesicle may not be uniform and furthermore, that differential release of each vesicle is dependent on the intensity of stimulation by the SNS (Burnstock 1995; Ralevic 2009; Donoso et al. 2004) (Fig. 2).
It appears that during very low frequency nerve activation, ATP is the dominant co-transmitter released (Todorov et al. 1994). Upon its release within the tunica media of blood vessels, ATP binds to its P2X1 purinoceptors and induces a very brief twitch-like contraction of its target smooth muscle cell (Burnstock 1990) (Fig. 2b). NE is also released during low nerve frequency activation and continues to be released throughout a wide range of nerve activities (Fig. 2c). NE acts on postsynaptic adrenergic receptors, which in blood vessels, are predominantly of the α1-type (although α2 receptors exist post-junctionally as well), whereas in cardiac myocytes and adipocytes the receptors are of the β1–3 type (Fig. 2b). Activation of an α1 receptor results in release of Ca++ from intracellular compartments which induces contraction of smooth muscle even during mild sympathetic stimulation (Burnstock 1990). Interestingly, NE and ATP are also capable of regulating each other’s effects post-junctionally (Burnstock 1990). The release of NE potentiates the effects of ATP, while at the same time, the release of ATP exacerbates the effects of NE post-junctionally (Burnstock 1990) (Fig. 2b).
The third neurotransmitter released from sympathetic nerves is NPY which exerts both pre- and post-junctional effects. Unlike ATP and NE, NPY is released predominately during bursts of intense and prolonged high-frequency sympathetic activation (Lundberg et al. 1986) (Fig. 2c). NPY’s vasoconstrictive effects are mediated primarily through activation of its Y1 receptor located post-junctionally (Westfall 2004) (Fig. 2b). Activation of the Y1 receptor results in a slow-onset, but long lasting smooth muscle contraction even in the midst of α-adrenoceptors desensitization when NE is no longer effective (Westfall 2004). Several studies using NPY-Y1 (Y1) receptor antagonists have highlighted the powerful effects of NPY as a direct mediator of vasoconstriction (Morris 1999; Buckwalter et al. 2005; Pernow and Lundberg 1989) in guinea pig vena cava (Malmstrom and Lundberg 1995a, b), pig hind limb, spleen, kidney (Malmstrom et al. 1997), and the rat mesenteric arterial bed (Zukowska-Grojec et al. 1996; Han et al. 1998).
Although SNS activation can induce wide-spread release of NPY within the body, its vasoconstrictive effects are perhaps showcased best within the coronary arteries of the human heart. A study in humans performed by Clarke et al. (1987) revealed the potent effects of NPY when injected directly into the coronary artery (Fig. 1, 2nd Pool). NPY when infused directly into the artery during coronary angiography produced a marked increase in blood pressure of approximately 20 mmHg within minutes of administration. Moreover, many patients felt pain in their chest related to angina along with a temporary bradycardia, both of which are markers of a severe ischemic event (Clarke et al. 1987). Shockingly, the infusion of NPY led to vasospasms resulting in almost complete vasoconstriction of the coronary artery (Clarke et al. 1987) (Fig. 1, 2nd Pool).
Extensive research has been put forth to better understand the effects of NPY release from nerve terminals. In addition to directly activating Y1 receptors post-junctionally to induce vasoconstriction, NPY also has many indirect effects as well. The type of effects varies with cell types and NPY is capable of either inhibiting or potentiating the actions of ATP and NE (Fig. 2b). Acting through its Y1 receptor post-junctionally, NPY potentiates the vasomotor contractile responses of both ATP and NE. However, in adipocytes, by acting through an unidentified NPY receptor, the peptide can completely oppose the lipolytic actions of β-adrenergic activation (Turtzo et al. 2001). In contrast, NPY appears to have uniformly strong inhibitory effects on neurotransmitter release. When NPY acts on its Y2 receptor found pre-junctionally, NPY inhibits the release of NE, ATP as well as itself (Westfall 2004) (Fig. 2b).
Thus, it is becoming clear that the effects of sympathetic nerve activation are dependent not only on the frequency of activation, but also on the pre-existing content of the neurotransmitters within their respective vesicles as well as the effects they exert on each other. Post-synaptically, NPY contributes directly to vasoconstriction by activation of its Y1 receptors, as well as indirectly by potentiating the effects of ATP and NE. The contribution of NPY to sympathetic vasoconstriction is to prolong the response and to maintain it in the face of declining or desensitized adrenergic effects (Wahlestedt et al. 1990). In contrast, pre-synaptically via the Y2 receptors, NPY limits sympathetic activation by reducing NE and ATP and its own release (Fig. 2b). In addition, NPY’s Y1,Y2, and Y5 receptors appear to inhibit synthesis of catecholamines (Westfall 2004). Such pre- and post-synaptic interactions among the three sympathetic neurotransmitters minimize the amount of a single transmitter released per nerve impulse, protecting against depletion of their stores, while at the same time, maximizing the amplitude and duration of the response evoked following activation of the nerve. A good illustration of the benefits of having NPY within the sympathetic neuro-effector junction can be seen in the stress responses of the rats over expressing NPY. Although NPY over-expression led to higher basal plasma levels of the peptide, it protected these rats from excessive stress-induced catecholamine release, lowered their cardiovascular responses to stress and significantly increased their lifespan (Michalkiewicz et al. 2003). Figure 2 outlines the relative contributions of NPY, NE, and ATP following differential activation of the sympathetic nervous system.
NPY and the HPA Axis
NPY also interacts with the HPA axis, both centrally and peripherally. However, the HPA axis, through the actions of glucocorticoids, is also capable of regulating the release of NPY as well. Centrally, NPY expression and its presumable release are known to increase following exposure to an acute stressor within many stress-sensitive areas of the brain (Thorsell 2010). In the hypothalamus, increases in NPY have been suggested to be involved in triggering the HPA axis activation during stress resulting in the initial release of CRH from the paraventricular nucleus. Other studies investigating changes in prepro-NPY expression levels, following a restraint stress test of 1 h, found that various regions of the brain respond to stress differently in regards to mRNA levels of prepro-NPY produced (Sweerts et al. 2001). Some regions of the brain showed increased expression of prepro-NPY while others showed no change (Sweerts et al. 2001). Interestingly, they found that prepro-NPY levels trended to increase in some strains of rats more than others following acute and chronic restraint exposures (Sweerts et al. 2001). Additionally, several other studies performed in rats, sheep and dogs have shown that central administration of NPY resulted in increased release of CRH (Crowley 2004). In rats, administration of NPY into the ventricles immediately produced increases in ACTH and corticosterone (Leibowitz et al. 1988; Sainsbury et al. 1996). Moreover, central administration of NPY also results in increases in CRH immunoreactivity as well as mRNA expression within the paraventricular nucleus (Haas and George 1989). In addition, an extensive network of NPY positive neurons has been shown to exist within the vicinity of CRH positive cell bodies (Li et al. 2000).
On the other hand, glucocorticoids are capable of up-regulating the expression of NPY mRNA in the arcuate nucleus following their release, thus exacerbating the stress response (Fig. 2a). Support for glucocorticoid-dependent release of NPY comes from the fact that several of the NPY-positive neurons within the arcuate nucleus are also positive for glucocorticoid receptors (Hisano et al. 1988; Harfstrand et al. 1989). Although not extensive, these experiments support the possibility that NPY plays an important role in central triggering and propagation of the stress response. Interestingly, similar interactions between NPY and the glucocorticoid system are present in the periphery as well (Kuo et al. 2007). In the periphery, glucocorticoids potently up-regulate the expression of NPY within sympathetic neurons and the expression of the Y2 receptor in adipocytes, thus increasing the effects of NPY in stimulating adipose tissue growth.
Such positive interactions between NPY and the HPA axis can well explain why under many forms of chronic stress, both of these systems show similar signs of activation. For example in war veterans suffering from PTSD, both NPY and cortisol levels are lower than in combat veterans without the disease (Rasmusson et al. 2010), and this is thought to be a reflection of the over-activation and exhaustion of these systems. In contrast, in stress-induced obesity, glucocorticoid, and NPY levels within the plasma and locally in the fat tissue, was found to be elevated and together it is thought that they synergistically contribute to the pathology (Kuo et al. 2007).
NPY and Cardiovascular Responses to Stress
Due to its release during more intense and prolonged sympathetic activation, NPY has been found to be primarily involved in chronic cardiovascular adaptations to stress. In mice, some of the strongest stressors that evoke the release of NPY are intense cold or an exposure to an aggressive intruder, both of which are conditions that threaten an animals’ chance of survival and impose on them high metabolic demands (Kuo et al. 2007). Both in rodents (Kuo et al. 2007; Zukowska-Grojec and Neuropeptide 1995; Li et al. 2005) and in humans (Lewandowski et al. 1996) a laboratory stressor known as the cold pressor test, a stressor which involves immersing the hands and feet in ice-cold water, induces marked elevation of NPY in plasma and in platelets, along with increased blood pressure and tachycardia. Interestingly, both the NPY and the cardiovascular responses are gender-specific with males showing increased responsiveness as compared to females (Zukowska-Grojec and Vaz 1988; Zukowska-Grojec et al. 1991). In rats, this was found to be due to the strong effects of androgens on NPY expression, an effect which estrogens were lacking (Zukowska-Grojec et al. 1991). In contrast, others found estrogens capable of inhibiting both NPY expression and the Y1-mediated vascular responses in rat skeletal muscles (Jackson et al. 2010).
The increased NPY responsiveness to cold stress observed in males is also associated with an exaggerated and prolonged post-stress pressor effect. The pressor response was found to be primarily due to prolonged increases in mesenteric vascular resistance (MVR). Up to 80% of stress-induced MVR was prevented by administration of a Y1 receptor antagonist, which also eliminated the post-stress lingering elevation in blood pressure. Y1 receptor inhibitor was also found to be effective in reducing exercise-induced increases in blood pressure in patients with coronary heart disease (Gullestad et al. 2003), even if they were treated with the β-blocker atenolol (Gullestad et al. 2012). However, acutely, it did not improve their ischemic parameters (Gullestad et al. 2003). Conversely, in pigs, the β-blocker metoprolol reduced stress-induced NPY release and its prolonged pressor effects (Ablad et al. 2010). These finding underscore the importance of interactions between adrenergic and NPY-ergic transmission and reveals a variety of targets for NPY. They also suggest that the release of NPY contributes significantly to vascular tone and regulation of blood pressure during stressful exposures. Whether or not NPY directly, or by interacting with catecholamines, plays a role in maintaining basal vascular tone remains controversial considering Y1 receptor antagonists do not lower resting blood pressure in normotensive (Zukowska-Grojec et al. 1996) or hypertensive rats (Zhang et al. 1997). However, NPY and its Y1 receptors were shown to be important in maintaining vascular tone in the rat hind limb by potentiating the α1-adrenergic responses in addition to direct vasoconstriction (Jackson et al. 2004). Thus, while the involvement of Y1 receptors in the maintenance of resting blood pressure may not be striking, it greatly increases during stress particularly in the mesenteric, coronary, and skeletal muscle vascular beds. NPY’s vasoconstrictive activities markedly increase under conditions of elevated adrenergic tone such as during exposure to stress, or following infusion of adrenergic agonists.
NPY and Chronic Stress: From Physiological Adaptation to Development of Cardiovascular Diseases
Once the first NPY receptor antagonist was found to have little hypotensive effect, interest in the NPY system, in terms of its cardiovascular physiological significance, plummeted. However, for our lab and a few other groups, it marked the beginning of research into the chronic effects of NPY, which eventually lead to the discovery of NPY’s powerful growth promoting activities and its role in the development of multiple diseases. Acting in a receptor specific manner, NPY was found to be a potent growth factor in a number of cell types. NPY, through its Y1 and Y2 receptor has been shown to have powerful mitogenic effects. Through activation of the Y1 receptor, NPY is mitogenic in vascular smooth muscle cells (VSMC) (Zukowska-Grojec et al. 1998a; Zukowska-Grojec et al. 1993; Erlinge et al. 1994) and via the Y2 receptor, it is mitogenic for endothelial cells (Zukowska-Grojec et al. 1998b; Ekstrand et al. 2003) (Fig. 1) and sympathetic neuroblasts (Kitlinska et al. 2005). On the other hand, activation of the Y1 receptor (and possibly Y2 and Y5 receptors) in cardiomyocytes induced hypertrophy (McDermott and Bell 2007; Pellieux et al. 2000). The switch from Y1 receptor-mediated actions, vasoconstriction and vascular hyperplasia, to Y2 and Y5 receptor-mediated effects requires activation of DPPIV, the endothelial and immune cell derived protease that forms NPY3–36 (Ghersi et al. 2001; Mentlein 1999) (Fig. 1).
Several injurious stimuli such as hypoxia can lead to marked up-regulation of NPY, its Y2 receptor and DPPIV in endothelial cells where the peptide promotes angiogenesis (Lee et al. 2003; Zukowska et al. 2003). These angiogenic effects are direct, but the peptide can also interact with catecholamines and other growth factors as well. For example, adrenergic and NPY-ergic activation can stimulate vascular hyperplasia, possibly through postsynaptic potentiation, or synergistically by activating other growth factors such as epidermal or fibroblastic growth factors (Zhang et al. 2004). Which of the sympathetic neurotransmitter takes the lead in modulating these actions is not known and likely, would vary depending on conditions, as well as the individual. Angiogenic activity of NPY has been well established in hind limb ischemia (Lee et al. 2003), retinopathy (Koulu et al. 2004), wound healing (Ghersi et al. 2001), cancers (Kitlinska et al. 2005) and recently in the adipose tissue (Kuo et al. 2007).
In addition, NPY can also play a deleterious role in the development of atherosclerosis by mediating the effects of stress. Previous research from the Zukowska group has shown that administration of NPY, locally into the rat carotid artery (Li et al. 2003) leads to marked exacerbation of the vascular lesions following angioplasty of this vessel. Interestingly in rats, it was discovered that exposure to chronic stress (a daily cold pressor test) following angioplasty mimicked the effect that local administration of NPY had on injured rat carotid arteries (Li et al. 2005)(Fig. 1, 2nd Pool). Both stress and NPY led to complete occlusion of the rat artery with neo-intima containing macrophages, neo-vessels, thrombus, and lipids (Li et al. 2003). Strikingly, in animals undergoing daily stress, administration of a Y1 or a Y5 receptor antagonist, individually or combined, prior to or following angioplasty-induced injury, completely prevented lesion formation and reduced neo-intima (Li et al. 2003). On the other hand, administration of these antagonists individually or together in rats which only received angioplasty reduced neo-intima formation by 40% suggesting that even under non-stressed conditions, endogenous NPY plays a role in angioplasty-induced neo-intima formation (Li et al. 2003). In addition, this was confirmed by findings that the “stress” of angioplasty alone was capable of elevating NPY levels, primarily within platelets. Moreover, these angioplasty-induced increases in NPY were further exacerbated by cold stress (Li et al. 2003, 2005).
Taken together, these studies have revealed a novel role for NPY as a mediator of chronic stress, leading to the development of restenosis and acceleration of atherosclerosis.
NPY and Metabolic Adaptations to Chronic Stress
While studying the angiogenic properties of NPY in various tissues, the Zukowska lab has discovered another peripherally mediated role for the NPY system in the growth of adipose tissue, and hence, in stress-induced obesity and metabolic syndrome. Traditionally, stress has been viewed as a condition which leads to weight loss, as most of the rodent studies, particularly in rats, indeed point in this direction. Moreover, the mediator of this stress-induced reduction in fat and weight was originally thought to be driven by the sympathetic nervous system through the activation of beta-adrenergic receptors which are known for their lipolytic and thermogenic activities (Collins et al. 2004). Interestingly, obesity is often associated with an increase in adrenergic activity that appears to be insufficient to restore normal weight (Straznicky et al. 2010). Moreover, in humans, stress has varying effects on body weight which, in different people, can lead to either weight loss or gain. In addition, it was assumed that these fluctuations in weight during stress were dependent on food intake.
Having found that NPY and its angiogenic system—the Y2 receptor and DPPIV–are highly expressed in growing adipose tissue, Kuo et al. (2007) hypothesized that stress induced coping mechanisms such as the ingestion of foods rich in sugar and fats could lead to alteration of the neurochemistry within the sympathetic nerve terminals. Therefore, it was thought that instead of NE, NPY may be the primary neurotransmitter released upon sympathetic activation. If true, this neurotransmitter profile change would lead to a down-regulation of the weight loss-promoting, beta-adrenergic activities and instead up-regulate the anti-lipolytic and fat promoting effects mediated by NPY (Kuo et al. 2008). Using mice, either stressed or not, and fed normal or a high fat sugar diet, Kuo et al. (2007) provided extensive evidence in support of this hypothesis.
As anticipated, chronic stress, when combined with a high fat sugar diet (HFS) led to an up- regulation of NPY and its Y2 receptor within the abdominal fat (Kuo et al. 2007). As shown by others, stress alone in mice fed a normal diet resulted in no change in weight and under some conditions it resulted in weight loss. Most strikingly, stress and-HFS up-regulated NPY and its “angiogenic” system: Y2R and DPPIV locally within the visceral adipose tissue, and this system was shown to be not only angiogenic, but also adipogenic. Thus, NPY increased pre-adipocyte proliferation and adipogenesis in vitro. In vivo, NPY increased adipose tissue expansion in lean and obese ob/ob mice, and stimulated the growth of a human fat xenograft in nude mice. Both increases in adipose tissue were associated with increased vascularization of the fat pads, that could be inhibited by a Y2 receptor antagonist (Kuo et al. 2007).
Predominantly within the abdominal area, stress-induced up-regulation of the NPY-Y2 receptor system which resulted in doubling the amount of fat accumulated within the abdominal area as compared to animals consuming a HFS diet alone (Kuo et al. 2007). The morphology of the visceral adipose tissue contained increased number of neo-vessels, small adipocytes, macrophages and cell proliferation, while at the same time apoptosis was reduced. Within 3 months, stress accelerated the development of gross abdominal obesity and metabolic syndrome, with animals showing signs of glucose intolerance, insulin resistance, hyperleptinemia and hypertension in addition to fatty liver and skeletal muscles. Strikingly, local intra-fat inhibition of Y2 receptors, pharmacologically or genetically through delivery of a viral vector with Cre-recombinase to Y2lox/lox mice, fully prevented stress-induced obesity (Kuo et al. 2007). Importantly, neither acceleration of obesity by stress nor its prevention by Y2 receptor antagonist was associated with changes in food intake or discernible changes in thermogenesis as measured by expression of UCP1, 2 and 3 in brown and white adipose tissue and skeletal muscles (Kuo et al. 2007).
Overall, this indicated that stress-induced obesity was due to local alteration in metabolism and growth of some or all of the cells present in the adipose tissue due to activation of Y2 receptors following NPY release. It is thought that the cells altered in this model could involve the sympathetic nerves, adipose stem cells, adipocytes, and immune cells. However, the cells that are primarily responsible for the stress induced NPY effect, are currently being investigated.
Glucocorticoids were also implicated in this model of obesity. Both stress-and a HFS diet elevated corticosterone levels in plasma and even more so in the abdominal adipose tissue (Kuo et al. 2007). The latter was likely due to stress-induced up-regulation of 11β-Hydroxysteroid dehydrogenase (HSD11B), an enzyme, which converts inactive corticoids into their active form (Buckingham 2000). Due to its preferential localization in the visceral fat, it is thought that the HSD11B could contribute to localized increases in glucocorticoids where they could then prime pre-adipocytes for growth and differentiation (Kuo et al. 2007). In addition, Kuo et al. (2007) found that glucocorticoids are strong inducers of NPY expression and are also capable of up-regulating the Y2 receptor in adipocytes. Therefore, albeit indirectly, glucocorticoids could be involved in stress-induced obesity via the NPY pathway (Kuo et al. 2007).
In addition to NPY and the effects of glucocorticoids, in this model of stress-induced obesity the levels of catecholamines in plasma and adipose tissue were reduced compared to un-stressed mice indicating a decrease in adrenergic activity (Kuo et al. 2007). Several mechanisms could have led to this phenomenon. As previously mentioned, NPY released from nerve terminals can also exert a strong inhibitory effects on the release of NE upon exposure to stress. This inhibition of NE reduces the stimulation of β-adrenergic receptors, which induce lipolysis. In addition, while glucocorticoids stimulate NPY expression, they have no effect on catecholamines. Thus, down-regulation of the adrenergic system, combined with an up-regulation of glucocorticoids in addition to the pro-adipogenic and angiogenic effects of over-activation of NPY and its Y2 receptor appear to be responsible for the acceleration of diet-induced obesity and metabolic syndrome resulting from chronic stress.
Overall, these studies demonstrate the powerful metabolic and growth-regulatory activities of NPY, and the central role it plays in metabolic adaptation to stress and over-feeding.
Immune Functions of NPY
Our brief discussion of NPY’s roles in stress would be incomplete without mentioning its immune functions. During the late 80s it became well known that increases in sympathetic nerve activity leads to a decrease in natural killer cell activity (Reder et al. 1989). The discovery that sympathetic nerves releasing NPY were found in the vicinity of the spleen and other lymphocytes fueled interest in understanding the possible role of NPY within the immune system (Romano et al. 1991). Natural killer cells are important lymphocytes within the immune system that are specialized in initiating a host response to fight off the spread of tumors and other cells that have been infected with viruses. Studies in vitro have shown that the addition of NPY induces a dose dependent inhibition of natural killer cell activity (Nair et al. 1993). In vivo work looking at the effects of people with depression and caretakers of spouses with Alzheimer’s disease found increased levels of NPY and decreased levels of natural killer cell activity (Irwin et al. 1991). At this point, further understanding was needed to seek out the receptors involved in mediating this effect.
One of the first reports published addressing these issues was a study using lymphocytes. Petitto et al. (1994) found that rat splenic lymphocytes expressed the Y1 receptor. In addition, this receptor displayed 100% sequence homology with the Y1 receptor found in the rat brain (Petitto et al. 1994). Since then, additional experiments have corroborated these results and other studies have revealed NPY receptor expression within the thymus of multiple species as well (Silva et al. 2006). NPY has also been implicated in activating many different immune cells both within the innate and adaptive branches of the immune system. Interestingly, increases in NPY expression have been found following activation of T-cells, B-cells, and macrophages (Schwarz et al. 1994).
Interestingly, it appears that NPY can have bimodal effects on immune cells (Wheway et al. 2005). On one hand, the peptide exerts pro-inflammatory actions on antigen presenting cells (APC) (Wheway et al. 2005) and polymorphonuclear cells (Bedoui et al. 2008) while on the other hand, NPY exerts inhibitory effects on T-cell activation (Wheway et al. 2005). In a seminal study performed by Wheway et al. (2005), T-cells isolated from Y1 receptor knockout mice (Y1rKO) became hyper-reactive upon transfer into a lymphopenic mouse and were capable of inciting severe colitis (Wheway et al. 2005). However, these studies also revealed a novel role for NPY in APCs. Y1 receptor knockout mice showed decreased T-helper-type-1 cell-mediated inflammatory responses. This observation which curiously opposed the authors’ first observation in Y1rKO mice (Wheway et al. 2005) of increased immune response can be explained by a defect in the APC. Wheway et al. (2005) describe these opposing immune effects as the body’s way to limit the immune response. Nerve derived NPY, when released, acts on Y1 receptors located on T-cells to inhibit their proliferation, while on the other hand, NPY can also act on APC’s to initiate an immune response (Wheway et al. 2005). Therefore, NPY appears to act as a finely tuned modulator of the immune response by selectively activating specific cells within the immune system, while at the same time inhibiting others in an attempt to protect the host.
NPY, as previously mentioned, also induces bimodal effects on human neutrophils in a receptor specific manner. Activation of the Y5 receptor increases oxidative bursts while on the other hand, activation of the Y1 and Y2 receptor results in increased phagocytosis (Bedoui et al. 2008). Elevated levels of NPY have also been shown to increase chemotaxis, phagocytosis, adherence, and secretion of pro-inflammatory cytokines in macrophages, primarily via NPY’s Y1 receptor (De la Fuente et al. 1993, 2001; Dimitrijevic et al. 2005) (Fig. 2, 3rd Pool). Additional evidence from the Zukowska lab has shown that both stress and NPY are capable of increasing macrophage infiltration in atherosclerotic plaques (Li et al. 2011) and obesity (Kuo et al. 2007) and moreover, that the NPY receptors found on these macrophages are up-regulated (Abe et al. 2010).
Conclusions
Over the past 30 years NPY has been implicated in a growing number of stress-related activities and diseases. As a ubiquitous sympathetic neurotransmitter, present both within and outside the nervous system, NPY exerts its effects on nearly every tissue and regulatory system in the body. Centrally, NPY is a stress-responsive anxiolytic and acts as the most powerful hunger stimulant within the body. It has also been proposed that NPY, acting centrally, could be responsible for initiating the cascade of events leading to the release of cortisol in response to stress. In addition, the central effects of NPY have revealed a potential role in mood altering disorders such as anxiety, depression, and post-traumatic stress syndrome where NPY levels are known to be reduced.
Peripherally, via its Y1 receptor, NPY potentiates the effects of catecholamines to produce potent and sustained vasoconstriction in addition to independently regulating long-term vasoconstriction in the face of prolonged activation of the sympathetic nervous system. NPY is also a stimulator of angiogenesis via its Y2 and Y5 receptor and additionally plays several significant roles within the immune system, most likely through its Y1 receptor. Y1 receptor activation, resulting from elevated NPY levels, have also been implicated in diseases such as atherosclerosis where increases in NPY due to exogenous or endogenous administration following exposure to chronic stress can render vessels hypersensitive to angioplasty and can induce occlusive lesions that resemble advanced atherosclerotic plaques. In addition, NPY3–36, a product of cleaved NPY, has been shown to promote angiogenesis, inflammation, and adipogenesis in the visceral abdominal fat via its Y2 receptor.
Not surprising, these mechanistically complex actions of NPY are still perplexing researchers today. In regards to NPY’s possible beneficial role as a growth factor, agonists could be useful in accelerating the rate of wound healing and tissue re-vascularization. Moreover, following its release, NPY acts not only to potentiate the actions of purines and catecholamines, but acts independently and is preferentially released over NE during chronic stress. Figure 1 depicts some of the currently known roles for NPY within different pools in the body.
In summary, NPY appears to be a stress mediator that is activated under conditions of chronic stress and during bouts of intense metabolic demands such as starvation, oxygen deprivation, or following a stress which threatens survival. The evolutionary benefit of having increased levels of NPY appear to be under conditions of cold climate and in situations where food is sparse, as the peptide stimulates hunger and food-seeking behavior, and regulates cardiovascular, neuro-endocrine and immune adaptations. Interestingly, a gain-in-function polymorphism, Leu7Pro7, in the NPY gene was found to be very common in Northern European populations (Kallio et al. 2001; Karvonen et al. 2001), where colder conditions are more prevalent. On the other hand, this mutation is absent in warmer climates such as southern Europe, Africa, and Asia (Kuo and Zukowska 2007). In todays’ world, with people in the most northern regions living in well-heated houses and having more than adequate food supply, the adaptive value of high NPY levels have turned maladaptive, and within these same populations, the NPY polymorphism is now associated with hyperlipidemia (Kallio et al. 2001; Karvonen et al. 2000), accelerated atherosclerosis (Karvonen et al. 2001) and vascular complications resulting from diabetes (Koulu et al. 2004).
Throughout the last 30 years of research on NPY remarkable progress has been made in understanding its central and peripherally mediated roles in disease. As a result the peptide and its specific receptors are becoming attractive therapeutic targets for obesity, mood disorders, and epilepsy. Ironically, in spite of the fact that NPY is as ubiquitously present in the central nervous system as it is in the peripheral nervous system, the knowledge of its peripheral functions still lags behind. It is the hope of the authors that this review will contribute to a better understanding of those peripheral actions of NPY and the role the peptide plays among other mediators in adapting to stress and in the development of stress-related diseases.
Acknowledgments
This research was supported by grants from NHLBI to ZZ: R37HL055310 and HL067357.
Abbreviations
- NPY
Neuropeptide Y
- NE
Norepinephrine
- Epi
Epinephrine
- PYY
Peptide YY
- PP
Pancreatic polypeptide
- AVP
Vasopressin
- SAS
Sympatho-adrenomedullary system
- HPA
Hypothalamic–pituitary–adrenal
- ACTH
Adrenocorticotropic hormone
- CRH
Corticotrophin-releasing hormone
- APP
Aminopeptidase P
- DPPIV
Dipeptidyl peptidase IV
- HSD11B
11β-Hydroxysteroid dehydrogenase
- VSMC
Vascular smooth muscle cell
- MVR
Mesenteric vascular resistance
- PTSD
Post-traumatic stress disorder
- Y1RKO
Y1 receptor knockout
- APC
Antigen presenting cell
- NGF
Nerve Growth Factor
- HFS
High Fat Sugar Diet
- GC
Glucocorticoids
- ADR
Adrenergic Receptor
References
- Abe K, Kuo L, Zukowska Z (2010) Neuropeptide Y is a mediator of chronic vascular and metabolic maladaptations to stress and hypernutrition. Exp Biol Med 235(10):1179–1184 [DOI] [PubMed] [Google Scholar]
- Ablad B et al (2010) Metoprolol, but not atenolol, reduces stress induced neuropeptide Y release in pigs. Scand Cardiovasc J 44(5):273–278 [DOI] [PubMed] [Google Scholar]
- Ahlborg G et al (1992) Splanchnic and renal vasoconstrictor and metabolic responses to neuropeptide Y in resting and exercising man. Acta Physiol Scand 145(2):139–149 [DOI] [PubMed] [Google Scholar]
- Bald M, Gerigk M, Rascher W (1997) Elevated plasma concentrations of neuropeptide Y in children and adults with chronic and terminal renal failure. Am J Kidney Dis 30(1):23–27 [DOI] [PubMed] [Google Scholar]
- Bedoui S et al (2008) Neuropeptide Y receptor-specifically modulates human neutrophil function. J Neuroimmunol 195(1–2):88–95 [DOI] [PubMed] [Google Scholar]
- Benedict CR, Fillenz M, Stanford C (1978) Changes in plasma noradrenaline concentration as a measure of release rate. Br J Pharmacol 64(2):305–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brothers SP, Wahlestedt C (2010) Therapeutic potential of neuropeptide Y (NPY) receptor ligands. EMBO Molecular Medicine 2(11):429–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham JC (2000) Glucocorticoids, effects of stress on. In: Fink G (ed) Encyclopedia of stress. Academic Press, San Diego, pp 229–237 [Google Scholar]
- Buckwalter JB, Hamann JJ, Clifford PS (2005) Neuropeptide Y1 receptor vasoconstriction in exercising canine skeletal muscles. J Appl Physiol 99(6):2115–2120 [DOI] [PubMed] [Google Scholar]
- Burnstock G (1990) Noradrenaline and ATP as cotransmitters in sympathetic nerves. Neurochem Int 17(2):357–368 [DOI] [PubMed] [Google Scholar]
- Burnstock G (1995) Noradrenaline and ATP: cotransmitters and neuromodulators. J Physiol Pharmacol 46(4):365–384 [PubMed] [Google Scholar]
- Callanan EY et al (2007) Renal and cardiac neuropeptide Y and NPY receptors in a rat model of congestive heart failure. American journal of physiology. Renal physiology 293(6):F1811–F1817 [DOI] [PubMed] [Google Scholar]
- Carlquist M et al (1982) A porcine brain polypeptide is identical to the vasoactive intestinal polypeptide. Gastroenterology 83(1 Pt 2):245–249 [PubMed] [Google Scholar]
- Clarke JG et al (1987) Coronary artery infusion of neuropeptide Y in patients with angina pectoris. Lancet 1(8541):1057–1059 [DOI] [PubMed] [Google Scholar]
- Cohen M et al (2009) Coordinated regulation of foraging and metabolism in C. elegans by RFamide neuropeptide signaling. Cell Metabolism 9(4):375–385 [DOI] [PubMed] [Google Scholar]
- Collins S, Cao W, Robidoux J (2004) Learning new tricks from old dogs: β-adrenergic receptors teach new lessons on firing up adipose tissue metabolism. Mol Endocrinol 18(9):2123–2131 [DOI] [PubMed] [Google Scholar]
- Colmers WF, Wahlestedt C (eds) (1993) The biology of neruopeptide Y and related peptides. Humana Press, Totowa, p 564 [Google Scholar]
- Crowley WR (2004) In: Michel MC (ed) Neuroendocrine actions of neuropeptide Y. Springer, Berlin, pp 185–220 [Google Scholar]
- Daimon T (1991) Autoradiographic analysis of vasoactive neuropeptide uptake by rabbit megakaryocytes and platelets. J Anat 176:105–112 [PMC free article] [PubMed] [Google Scholar]
- De la Fuente M et al (1993) Stimulation of murine peritoneal macrophage functions by neuropeptide Y and peptide YY. Involvement of protein kinase C. Immunology 80(2):259–265 [PMC free article] [PubMed] [Google Scholar]
- De la Fuente M, Del Rio M, Medina S (2001) Changes with aging in the modulation by neuropeptide Y of murine peritoneal macrophage functions. J Neuroimmunol 116(2):156–167 [DOI] [PubMed] [Google Scholar]
- Dimitrijevic M et al (2005) Neuropeptide Y and its receptor subtypes specifically modulate rat peritoneal macrophage functions in vitro: counter regulation through Y1 and Y2/5 receptors. Regul Pept 124(1–3):163–172 [DOI] [PubMed] [Google Scholar]
- Donoso MV et al (2004) Neuropeptide Y is released from human mammary and radial vascular biopsies and is a functional modulator of sympathetic cotransmission. J Vasc Res 41(5):387–399 [DOI] [PubMed] [Google Scholar]
- Egawa M, Yoshimatsu H, Bray GA (1991) Neuropeptide Y suppresses sympathetic activity to interscapular brown adipose tissue in rats. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology 260(2):R328–R334 [DOI] [PubMed] [Google Scholar]
- Ekstrand AJ et al (2003) Deletion of neuropeptide Y (NPY) 2 receptor in mice results in blockage of NPY-induced angiogenesis and delayed wound healing. Proc Natl Acad Sci 100(10):6033–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson A et al (1987) Detection of neuropeptide Y and its mRNA in megakaryocytes: enhanced levels in certain autoimmune mice. Proc Nat Ac Sci 84(16):5585–5589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlinge D, Brunkwall J, Edvinsson L (1994) Neuropeptide Y stimulates proliferation of human vascular smooth muscle cells: cooperation with noradrenaline and ATP. Regul Pept 50(3):259–265 [DOI] [PubMed] [Google Scholar]
- Fischer-Colbrie R, Iacangelo A, Eiden LE (1988) Neural and humoral factors separately regulate neuropeptide Y, enkephalin, and chromogranin A and B mRNA levels in rat adrenal medulla. Proc Natl Acad Sci USA 85(9):3240–3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood JF, Hernandez EN, Morley JE (1987) Modulation of memory processing by neuropeptide Y. Brain Res 421(1–2):280–290 [DOI] [PubMed] [Google Scholar]
- Ghersi G et al (2001) Critical role of dipeptidyl peptidase IV in neuropeptide Y-mediated endothelial cell migration in response to wounding. Peptides 22(3):453–458 [DOI] [PubMed] [Google Scholar]
- Goldstein DS (2010) Adrenal responses to stress. Cell Mol Neurobiol 30(8):1433–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grouzmann E, Comoy E, Bohuon C (1989) Plasma neuropeptide Y concentrations in patients with neuroendocrine tumors. J Clin Endocrinol Metab 68(4):808–813 [DOI] [PubMed] [Google Scholar]
- Grouzmann E et al (1994) Angiotensin-II mediates norepinephrine and neuropeptide-Y secretion in a human pheochromocytoma. J Clin Endocrinol Metab 79(6):1852–1856 [DOI] [PubMed] [Google Scholar]
- Gullestad L et al (2012) Differential effects of metoprolol and atenolol to neuropeptide Y blockade in coronary artery disease. Scand Cardiovasc J 46(1):23–31 [DOI] [PubMed] [Google Scholar]
- Gullestad L et al (2003) The effect of a neuropeptide Y Y1 receptor antagonist in patients with angina pectoris. Eur Heart J 24(12):1120–1127 [DOI] [PubMed] [Google Scholar]
- Haas DA, George SR (1989) Neuropeptide Y-induced effects on hypothalamic corticotropin-releasing factor content and release are dependent on noradrenergic/adrenergic neurotransmission. Brain Res 498(2):333–338 [DOI] [PubMed] [Google Scholar]
- Han S et al (1998) Direct evidence for the role of neuropeptide Y in sympathetic nerve stimulation-induced vasoconstriction. Am J Physiol 274(1 Pt 2):H290–H294 [DOI] [PubMed] [Google Scholar]
- Harfstrand A et al (1989) Regional differences in glucocorticoid receptor immunoreactivity among neuropeptide Y immunoreactive neurons of the rat brain. Acta Physiol Scand 135(1):3–9 [DOI] [PubMed] [Google Scholar]
- Hexum TD et al (1987) Neuropeptide Y release from the adrenal medulla after cholinergic receptor stimulation. J Pharmacol Exp Ther 243(3):927–930 [PubMed] [Google Scholar]
- Hisano S et al (1988) Localization of glucocorticoid receptor in neuropeptide Y-containing neurons in the arcuate nucleus of the rat hypothalamus. Neurosci Lett 95(1–3):13–18 [DOI] [PubMed] [Google Scholar]
- Hökfelt T et al (2008) NPY and its involvement in axon guidance, neurogenesis, and feeding. Nutrition 24(9):860–868 [DOI] [PubMed] [Google Scholar]
- Hörsten S et al (2004) In: Michel MC (ed) PP, PYY and NPY: synthesis, storage, release and degradation. Neuropeptide Y and related peptides. Springer, Berlin, pp 23–44 [Google Scholar]
- Irwin M et al (1991) Neuropeptide Y and natural killer cell activity: findings in depression and Alzheimer caregiver stress. FASEB J 5(15):3100–3107 [DOI] [PubMed] [Google Scholar]
- Jackson DN, Noble EG, Shoemaker JK (2004) Y1- and α1-receptor control of basal hindlimb vascular tone. Am J Physiol - Regul Integr Comp Physiol 287(1):R228–R233 [DOI] [PubMed] [Google Scholar]
- Jackson DN, Ellis CG, Shoemaker JK (2010) Estrogen modulates the contribution of neuropeptide Y to baseline hindlimb blood flow control in female Sprague-Dawley rats. Am J Physiol - Regul Integr Comp Physiol 298(5):R1351–R1357 [DOI] [PubMed] [Google Scholar]
- Kallio J et al (2001) Altered intracellular processing and release of neuropeptide Y due to leucine 7 to proline 7 polymorphism in the signal peptide of preproneuropeptide Y in humans. FASEB J 15:1242–1244 [PubMed] [Google Scholar]
- Karvonen MK et al (2000) Leucine 7 to Proline 7 polymorphism in the preproneuropeptide Y is associated with birth weight and serum triglyceride concentration in preschool-aged children. J Clin Endocrin Metab 85(4):1455–1460 [DOI] [PubMed] [Google Scholar]
- Karvonen MK et al (2001) Leucine7 to proline7 polymorphism in the preproneuropeptide Y is associated with the progression of carotid atherosclerosis, blood pressure and serum lipids in Finnish men. Atherosclerosis 159(1):145–151 [DOI] [PubMed] [Google Scholar]
- Kataoka Y, Majane EA, Yang HY (1985) Release of NPY-like immunoreactive material from primary cultures of chromaffin cells prepared from bovine adrenal medulla. Neuropharmacology 24(7):693–695 [DOI] [PubMed] [Google Scholar]
- Kitlinska J et al (2005) Differential effects of neuropeptide Y on the growth and vascularization of neural crest-derived tumors. Cancer Res 65(5):1719–1728 [DOI] [PubMed] [Google Scholar]
- Koulu M et al (2004) Neuropeptide Y and Y2-receptor are involved in development of diabetic retinopathy and retinal neovascularization. Ann Med 36(3):232–240 [DOI] [PubMed] [Google Scholar]
- Kuo LE, Zukowska Z (2007) Stress, NPY and vascular remodeling: Implications for stress-related diseases. Peptides 28(2):435–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo LE et al (2007) Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat Med 13(7):803–811 [DOI] [PubMed] [Google Scholar]
- Kuo LE et al (2008) Chronic stress, combined with a high-fat/high-sugar diet, shifts sympathetic signaling toward neuropeptide Y and leads to obesity and the metabolic syndrome. Ann N Y Acad Sci 1148:232–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larhammar D, Blomqvist AG, Soderberg C (1993) Evolution of neuropeptide Y and its related peptides. Comp Biochem Physiol 106(3):743–752 [DOI] [PubMed] [Google Scholar]
- Larsson T et al (2008) Early vertebrate chromosome duplications and the evolution of the neuropeptide Y receptor gene regions. BMC Evol Biol 8(1):184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EW et al (2003) Neuropeptide Y induces ischemic angiogenesis and restores function of ischemic skeletal muscles. J Clin Investig 111(12):1853–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz SF et al (1988) Neuropeptide Y, epinephrine and norepinephrine in the paraventricular nucleus: stimulation of feeding and the release of corticosterone, vasopressin and glucose. Brain Res Bull 21(6):905–912 [DOI] [PubMed] [Google Scholar]
- Lewandowski J, Pruszczyk P, Wocial B, Ignatowska-Switalska H, Romejko-Wolniewicz E, Madej-Firek A (1996) Sex hormone modulation of neruopeptide Y and cardiovascular responses to stress in humans. In: McCarty R (ed) Stress, vol 2. Gordon and Breach Science Publishers, New York, NY
- Li C, Chen P, Smith MS (2000) Corticotropin releasing hormone neurons in the paraventricular nucleus are direct targets for neuropeptide Y neurons in the arcuate nucleus: an anterograde tracing study. Brain Res 854(1–2):122–129 [DOI] [PubMed] [Google Scholar]
- Li L et al (2003) Neuropeptide Y-induced acceleration of postangioplasty occlusion of rat carotid artery. Arterioscler Thromb Vasc Biol 23(7):1204–1210 [DOI] [PubMed] [Google Scholar]
- Li L et al (2005) Chronic stress induces rapid occlusion of angioplasty-injured rat carotid artery by activating neuropeptide Y and its Y1 receptors. Arterioscler Thromb Vasc Biol 25(10):2075–2080 [DOI] [PubMed] [Google Scholar]
- Li L et al (2011) Of mice and men: neuropeptide y and its receptors are associated with atherosclerotic lesion burden and vulnerability. Journal of Cardiovascular Translational Research 4(3):351–362 [DOI] [PubMed] [Google Scholar]
- Lundberg JM et al (1986) Frequency- and reserpine-dependent chemical coding of sympathetic transmission: differential release of noradrenaline and neuropeptide Y from pig spleen. Neurosci Lett 63(1):96–100 [DOI] [PubMed] [Google Scholar]
- Lundberg JM et al (1990) Pharmacology of noradrenaline and neuropeptide tyrosine (NPY)-mediated sympathetic cotransmission. Fundam Clin Pharmacol 4(4):373–391 [DOI] [PubMed] [Google Scholar]
- Malmstrom RE, Lundberg JM (1995a) Endogenous NPY acting on the Y1 receptor accounts for the long-lasting part of the sympathetic contraction in guinea-pig vena cava: evidence using SR 120107A. Acta Physiol Scand 155(3):329–330 [DOI] [PubMed] [Google Scholar]
- Malmstrom RE, Lundberg JM (1995b) Neuropeptide Y accounts for sympathetic vasoconstriction in guinea-pig vena cava: evidence using BIBP 3226 and 3435. Eur J Pharmacol 294(2–3):661–668 [DOI] [PubMed] [Google Scholar]
- Malmstrom RE, Balmer KC, Lundberg JM (1997) The neuropeptide Y (NPY) Y1 receptor antagonist BIBP 3226: equal effects on vascular responses to exogenous and endogenous NPY in the pig in vivo. Br J Pharmacol 121(3):595–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott BJ, Bell D (2007) NPY and cardiac diseases. Curr Top Med Chem 7(17):1692–1703 [DOI] [PubMed] [Google Scholar]
- Mentlein R (1999) Dipeptidyl-peptidase IV (CD26)-role in the inactivation of regulatory peptides. Regul Pept 85(1):9–24 [DOI] [PubMed] [Google Scholar]
- Merten N, Beck-Sickinger A (2006) In: Zukowska Z, Feuerstein G (eds) Molecular ligand–receptor interaction of the NPY/PP peptide family. Birkhäuser, Basel, pp 35–62 [DOI] [PubMed] [Google Scholar]
- Michalkiewicz M et al (2003) Hypotension and reduced catecholamines in neuropeptide Y transgenic rats. Hypertension 41(5):1056–1062 [DOI] [PubMed] [Google Scholar]
- Michel MC (2004) Neuropeptide Y and related peptides. In: Starke K, Br FI (eds) Handbook of experimental pharmacology. Springer, New York, p 555 [Google Scholar]
- Morales-Medina JC, Dumont Y, Quirion R (2010) A possible role of neuropeptide Y in depression and stress. Brain Res 1314:194–205 [DOI] [PubMed] [Google Scholar]
- Morris JL (1999) Cotransmission from sympathetic vasoconstrictor neurons to small cutaneous arteries in vivo. Am J Physiol 277(1 Pt 2):H58–H64 [DOI] [PubMed] [Google Scholar]
- Myers AK et al (1988) Release of immunoreactive-neuropeptide by rat platelets. Biochem Biophys Res Commun 155(1):118–122 [DOI] [PubMed] [Google Scholar]
- Nair MP et al (1993) Effect of neuropeptide Y on natural killer activity of normal human lymphocytes. Brain Behav Immun 7(1):70–78 [DOI] [PubMed] [Google Scholar]
- Nilsson C et al (1996) Differences in the neuropeptide Y-like immunoreactivity of the plasma and platelets of human volunteers and depressed patients. Peptides 17(3):359–362 [DOI] [PubMed] [Google Scholar]
- Pellieux C et al (2000) Neuropeptide Y (NPY) potentiates phenylephrine-induced mitogen-activated protein kinase activation in primary cardiomyocytes via NPY Y5 receptors. Proc Natl Acad Sci 97(4):1595–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernow J, Lundberg JM (1989) Release and vasoconstrictor effects of neuropeptide Y in relation to non-adrenergic sympathetic control of renal blood flow in the pig. Acta Physiol Scand 136(4):507–517 [DOI] [PubMed] [Google Scholar]
- Petitto JM, Huang Z, McCarthy DB (1994) Molecular cloning of NPY-Y1 receptor cDNA from rat splenic lymphocytes: evidence of low levels of mRNA expression and [125I]NPY binding sites. J Neuroimmunol 54(1–2):81–86 [DOI] [PubMed] [Google Scholar]
- Ralevic V (2009) Purines as neurotransmitters and neuromodulators in blood vessels. Curr Vasc Pharmacol 7(1):3–14 [DOI] [PubMed] [Google Scholar]
- Rasmusson AM et al (2010) Adaptation to extreme stress: post-traumatic stress disorder, neuropeptide Y and metabolic syndrome. Exp Biol Med (Maywood) 235(10):1150–1162 [DOI] [PubMed] [Google Scholar]
- Reder A, Checinski M, Chelmicka-Schorr E (1989) The effect of chemical sympathectomy on natural killer cells in mice. Brain Behav Immun 3(2):110–118 [DOI] [PubMed] [Google Scholar]
- Romano TA et al (1991) Neuropeptide-Y innervation of the rat spleen: another potential immunomodulatory neuropeptide. Brain Behav Immun 5(1):116–131 [DOI] [PubMed] [Google Scholar]
- Rosmaninho-Salgado J et al (2007) Intracellular signaling mechanisms mediating catecholamine release upon activation of NPY Y1 receptors in mouse chromaffin cells. J Neurochem 103(3):896–903 [DOI] [PubMed] [Google Scholar]
- Sainsbury A et al (1996) Acute intracerebroventricular administration of neuropeptide Y stimulates corticosterone output and feeding but not insulin output in normal rats. Neuroendocrinology 63(4):318–326 [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Gray WP (2006) Plasticity of neuropeptide Y in the dentate gyrus after seizures, and its relevance to seizure-induced neurogenesis. EXS 95:193–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz H et al (1994) Neuropeptide Y is an inducible gene in the human immune system. J Neuroimmunol 51(1):53–61 [DOI] [PubMed] [Google Scholar]
- Selye H (1998) A syndrome produced by diverse nocuous agents. 1936. J Neuropsychiatry Clin Neurosci 10(2):230–231 [DOI] [PubMed] [Google Scholar]
- Silva AB, Aw D, Palmer DB (2006) Evolutionary conservation of neuropeptide expression in the thymus of different species. Immunology 118(1):131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolowski MB (2003) NPY and the regulation of behavioral development. Neuron 39(1):6–8 [DOI] [PubMed] [Google Scholar]
- Stanley BG, Leibowitz SF (1984) Neuropeptide Y: stimulation of feeding and drinking by injection into the paraventricular nucleus. Life Sci 35(26):2635–2642 [DOI] [PubMed] [Google Scholar]
- Straznicky NE, Lambert GW, Lambert EA (2010) Neuroadrenergic dysfunction in obesity: an overview of the effects of weight loss. Curr Opin Lipidol 21(1):21–30 [DOI] [PubMed] [Google Scholar]
- Sweerts BW, Jarrott B, Lawrence AJ (2001) The effect of acute and chronic restraint on the central expression of prepro-neuropeptide Y mRNA in normotensive and hypertensive rats. J Neuroendocrinol 13(7):608–617 [DOI] [PubMed] [Google Scholar]
- Tatemoto K (1982) Isolation and characterization of peptide YY (PYY), a candidate gut hormone that inhibits pancreatic exocrine secretion. Proc Natl Acad Sci USA 79(8):2514–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatemoto K, Carlquist M, Mutt V (1982) Neuropeptide Y—a novel brain peptide with structural similarities to peptide YY and pancreatic polypeptide. Nature 296(5858):659–660 [DOI] [PubMed] [Google Scholar]
- Thorsell A (2010) Brain neuropeptide Y and corticotropin-releasing hormone in mediating stress and anxiety. Experimental Biology and Medicine 235(10):1163–1167 [DOI] [PubMed] [Google Scholar]
- Todorov LD, Bjur RA, Westfall DP (1994) Temporal dissociation of the release of the sympathetic co-transmitters ATP and noradrenaline. Clin Exp Pharmacol Physiol 21(11):931–932 [DOI] [PubMed] [Google Scholar]
- Turtzo LC, Marx R, Lane MD (2001) Cross-talk between sympathetic neurons and adipocytes in coculture. Proc Natl Acad Sci 98(22):12385–12390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Sperk G (2004) Overexpression of NPY and Y2 receptors in epileptic brain tissue: an endogenous neuroprotective mechanism in temporal lobe epilepsy? Neuropeptides 38(4):245–252 [DOI] [PubMed] [Google Scholar]
- Wahlestedt C et al (1990) Norepinephrine and neuropeptide Y: vasoconstrictor cooperation in vivo and in vitro. Am J Physiol 258(3 Pt 2):R736–R742 [DOI] [PubMed] [Google Scholar]
- Westfall TC (2004) In: Michel MC (ed) Prejunctional effects of neuropeptide Y and its role as a cotransmitter. Springer, Berlin, pp 137–183 [Google Scholar]
- Wheway J et al (2005) A fundamental bimodal role for neuropeptide Y1 receptor in the immune system. J Exp Med 202(11):1527–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wocial B et al (1995) Plasma neuropeptide Y and catecholamines in women and men with essential hypertension. Blood Press 4(3):143–147 [DOI] [PubMed] [Google Scholar]
- Xapelli S et al (2006) Neuropeptide Y as an endogenous antiepileptic, neuroprotective and pro-neurogenic peptide. Recent Pat CNS Drug Discov 1(3):315–324 [DOI] [PubMed] [Google Scholar]
- Yulyaningsih E et al (2011) NPY receptors as potential targets for anti-obesity drug development. Br J Pharmacol 163(6):1170–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Lundberg JM, Thorén P (1997) Neuropeptide Y Y1 receptor antagonist (BIBP 3226) attenuates stress evoked tachycardia in conscious spontaneously hypertensive rats. Cardiovas Drugs The 11(6):801–806 [DOI] [PubMed] [Google Scholar]
- Zhang H et al (2004) Transactivation of epidermal growth factor receptor mediates catecholamine-induced growth of vascular smooth muscle. Circ Res 95(10):989–997 [DOI] [PubMed] [Google Scholar]
- Zukowska Z, Grant DS, Lee EW (2003) Neuropeptide Y: a novel mechanism for ischemic angiogenesis. Trends in cardiovascular medicine 13(2):86–92 [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z, Neuropeptide Y (1995) A novel sympathetic stress hormone and more. Ann N Y Acad Sci 771:219–233 [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z, Vaz AC (1988) Role of neuropeptide Y (NPY) in cardiovascular responses to stress. Synapse 2(3):293–298 [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z, Konarska M, McCarty R (1988) Differential plasma catecholamine and neuropeptide Y responses to acute stress in rats. Life sciences 42(17):1615–1624 [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z et al (1991) Cardiovascular, neuropeptide Y, and adrenergic responses in stress are sexually differentiated. Physiol Behav 49(4):771–777 [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z et al (1993) Mitogenic effect of neuropeptide Y in rat vascular smooth muscle cells. Peptides 14(2):263–268 [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z et al (1996) Stress-induced mesenteric vasoconstriction in rats is mediated by neuropeptide Y Y1 receptors. Am J Physiol 270(2 Pt 2):H796–H800 [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z et al (1998a) Mechanisms of vascular growth-promoting effects of neuropeptide Y: role of its inducible receptors. Regul Pept 75–76:231–238 [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z et al (1998b) Neuropeptide Y: a novel angiogenic factor from the sympathetic nerves and endothelium. Circ Res 83(2):187–195 [DOI] [PubMed] [Google Scholar]