

Abstract
BACKGROUND AND PURPOSE
This study was undertaken to characterize the ATP, adenosine and glutamate outflow evoked by depolarization with high K+ concentrations, in slices of rat hippocampus.
EXPERIMENTAL APPROACH
We utilized the microelectrode biosensor technique and extracellular electrophysiological recording for the real-time monitoring of the efflux of ATP, adenosine and glutamate.
KEY RESULTS
ATP, adenosine and glutamate sensors exhibited transient and reversible current during depolarization with 25 mM K+, with distinct kinetics. The ecto-ATPase inhibitor ARL67156 enhanced the extracellular level of ATP and inhibited the prolonged adenosine efflux, suggesting that generation of adenosine may derive from the extracellular breakdown of ATP. Stimulation-evoked ATP, adenosine and glutamate efflux was inhibited by tetrodotoxin, while exposure to Ca2+-free medium abolished ATP and adenosine efflux from hippocampal slices. Extracellular elevation of ATP and adenosine were decreased in the presence of NMDA receptor antagonists, D-AP-5 and ifenprodil, whereas non-NMDA receptor blockade by CNQX inhibited glutamate but not ATP and adenosine efflux. The gliotoxin fluoroacetate and P2X7 receptor antagonists inhibited the K+-evoked ATP, adenosine and glutamate efflux, while carbenoxolone in low concentration and probenecid decreased only the adenosine efflux.
CONCLUSIONS AND IMPLICATIONS
Our results demonstrated activity-dependent gliotransmitter release in the hippocampus in response to ongoing neuronal activity. ATP and glutamate were released by P2X7 receptor activation into extracellular space. Although the increased extracellular levels of adenosine did derive from released ATP, adenosine might also be released directly via pannexin hemichannels.
LINKED ARTICLE
This article is commented on by Sershen, pp. 1000–1002 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2012.02072.x
Keywords: ATP, glutamate, adenosine, release, pannexin, connexin, hippocampus, K+ depolarization, glia
Introduction
ATP and its extracellular breakdown product, adenosine, are important signalling molecules in the CNS under physiological and pathological conditions. ATP, which activates ionotropic P2X (P2X1-7) and metabotropic P2Y (P2Y1,2,4,6,11,12,13,14) receptors (nomenclature follows Alexander et al., 2011) acts as a fast transmitter and as a modulator of synaptic activity in several regions of the brain, including the hippocampus (North and Verkhratsky, 2006; Pankratov et al., 2006). Moreover, it also participates in long-term synaptic plasticity and regulates the survival of neurons and the following repair process under pathophysiological conditions, such as ischaemia and inflammation (Cunha and Ribeiro, 2000; Abbracchio et al., 2009). Adenosine, activating metabotropic adenosine receptors (A1, A2A, A2B, A3), has also a well-established role in the hippocampus, as a pre- and postsynaptic depressant of excitatory transmission, gaining significance under physiological conditions, metabolic stress and seizures (Cunha, 2001).
Accordingly, there is convincing evidence for the activity-dependent release of both ATP and adenosine from the hippocampus, under stimuli mimicking physiological neuronal activity such as electrical stimulation (Wieraszko et al., 1989; Cunha et al., 1996; Pankratov et al., 2006) or pathological challenge, such as combined oxygen–glucose deprivation (Juranyi et al., 1999; Frenguelli et al., 2007). However, due to their ubiquitous nature, the endogenous source of purines for synaptic modulation remained undefined under these conditions.
Recent studies highlighted that glial cells, in particular astrocytes, are active players in information processing in the brain and periphery (Araque et al., 2001; Haydon, 2001; Fellin and Carmignoto, 2004), and both adenine nucleotides and nucleosides as well as glutamate play a key role in the glia-neuron crosstalk (Nedergaard, 1994; Parpura et al., 1994; Fields and Burnstock, 2006; Halassa et al., 2009). Thus, ATP and glutamate coordinately activate astrocytes through the mobilization of their internal Ca2+, which in turn triggers the release of several neuroactive molecules from astrocytes, including ATP and glutamate themselves. These ‘gliotransmitters’ signal either to astrocytes, where they generate Ca2+ waves, or to neurons, where they modulate synaptic transmission and neuronal excitability (Cotrina et al., 1998; Guthrie et al., 1999; Fellin et al., 2009). In the hippocampus, stimulation of Schaffer collaterals leads to the glutamatergic activation of AMPA receptors located on astrocytes, which in turn releases ATP from astrocytes. This ATP, by itself, or by its metabolic degradation to adenosine elicits activity-dependent heterosynaptic depression of neighbouring pathways via activation of P2Y and adenosine A1 receptors, respectively (Koizumi et al., 2003; Zhang et al., 2003; Pascual et al., 2005; Serrano et al., 2006), which may lead to increased synaptic inhibition within intact hippocampal circuits (Bowser and Khakh, 2004). Although the astrocytic source of ATP for synaptic modulation has been demonstrated by a transgenic mouse model of the astrocyte-specific, inducible expression of the soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) instrumental for the release of gliotransmitters (Pascual et al., 2005), the physiological and pathological trigger for the release of ATP from astrocytes and its mechanism is still unknown.
Being a highly charged molecule, ATP by itself cannot permeate the cell membrane. Nevertheless, in previous studies, several release mechanisms has been identified for both ATP and adenosine. ATP is co-stored in synaptic vesicles, and its classical, Ca2+-dependent vesicular release has been demonstrated in a number of central synapses by both electrophysiological and neurochemical detection (Sperlagh and Vizi, 2000; Pankratov et al., 2006). Vesicular exocytosis of ATP has also been demonstrated from astrocytes (Coco et al., 2003; Pangrsic et al., 2007). In addition, other routes of ATP release have also been identified, such as P2X7 receptor channels (Ballerini et al., 1996; Pellegatti et al., 2005; Suadicani et al., 2006), gap junction hemichannels (e.g. Arcuino et al., 2002; Stout et al., 2002; Bao et al., 2004; Pearson et al., 2005; Anselmi et al., 2008), lysosome exocytosis (Zhang et al., 2007), volume sensitive anion channels (Darby et al., 2003; Okada et al., 2004) and the cystic fibrosis transmembrane conductance regulator protein (CFTR) (Kanno and Nishizaki, 2011). However, these latter mechanisms have been primarily described in non-neuronal or simplified experimental systems, such as cultured neurons and astrocytes, and less information is available whether these pathways also serve as a conduit of ATP release for purines involved in the modulation of synaptic signalling.
For adenosine, there are also a number of different mechanisms whereby it could accumulate in the extracellular space (Latini and Pedata, 2001; Wall and Dale, 2008). First, it can be generated from the released ATP through the actions of extracellular nucleotide catabolizing enzymes, such as E-NTPDases, E-NPPases, alkaline phosphatases and ecto-5′-nucleotidase. Second, it can also be released by its own right, via equilibrative or non-equilibrative transporters. Finally, although direct evidence is lacking, several studies support that adenosine can also be released in a vesicular fashion (Wall and Dale, 2008).
In spite of the key role of chemical signalling in neural function, only a few techniques are available to directly measure the concentrations of neurotransmitters and modulators in the extracellular space. Among them, the recently developed microelectrode biosensor technique allows the detection of the efflux of endogenous neurotransmitters with better resolution than conventional neurochemical methods (Llaudet et al., 2003; 2005; Tian et al., 2009) and also circumvents the limitations of widely used electrophysiological techniques, which indirectly detect the released transmitters. Using these multi-enzymatic microelectrode biosensors, it is possible to measure the concentration of different neurotransmitters and modulators with high temporal and spatial resolution and to detect paracrine and other non-classical release mechanisms as well (Dale et al., 2005).
In this study, we have characterized the efflux of the ATP, adenosine and glutamate in the rat hippocampus in vitro by the microelectrode biosensor technique. To activate the intact glia-neuron ensembles of the hippocampal slice (Sul et al., 2004), K+ depolarization was applied and we demonstrated and characterized such depolarization-evoked release of gliotransmitters in hippocampal slices from rats.
Methods
All animal care and experimental procedures complied with the principles and procedures outlined in the NIH Guide for the Care and Use of Laboratory animals and were approved by the local Animal Care Committee of the Institute of Experimental Medicine (Budapest, Hungary). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). Animals were kept under standard laboratory conditions (12 h light/12 h dark cycle) with food and water ad libitum. All efforts were made to minimize animal suffering and reduce the number of animals used. A total of 96 rats were used in these experiments.
Slice preparation
Young adult male Wistar rats (4 weeks of age, 85–110 g) were killed by cervical dislocation. After decapitation, the brain was rapidly removed and placed in ice-cold artificial CSF (aCSF) containing 11 mmol·L−1 Mg2+, and 400 µm coronal slices were prepared by a microtome with a vibrating blade (Microm HM 650 V, Microm International GmbH, Walldorf, Germany) as previously described (Dale et al., 2000). Slices were placed in an incubation chamber comprising a nylon mesh within a beaker of continuously circulating oxygenated (95% O2/5% CO2) standard aCSF (1.3 mmol·L−1 Mg2+) and kept at room temperature for at least 1 h before use. Standard aCSF contained (in mmol·L−1) NaCl, 127; KCl, 1.9; CaCl2, 2.4; NaHCO3, 26; KH2PO4, 1.2; d-glucose, 10; MgSO4, 1.3; pH 7.4 and was gassed with 95% O2/5% CO2. Glycerol (2 mmol·L−1) essential for ATP biosensor operation was added to the aCSF (see below).
Extracellular recording
A single slice was transferred to a recording chamber, fully submerged in aCSF and superfused at 6 mL·min−1 (34–36°C). Field epsp (fEPSPs) were extracellularly recorded in AC mode using a ISO-80 isolated bio-amplifier (Word Precision Instruments Inc, Sarasota, FL), with aCSF-filled glass microelectrode (1B150F-4, <2 MΩ; WPI), from the stratum radiatum of CA1 region in response to stimulation (at 3–7 V, 0.1 ms, 15 s intervals; parallel bipolar electrodes; FHC Inc, Bowdoin, ME) of the Schaffer collateral–commissural fibre pathway. Extracellular DC recordings were performed with the Multiclamp 700A (Molecular Devices Inc, Sunnyvale, CA) amplifier in DC mode using borosilicate glass pipettes (GC120F-10, 4–7 MΩ, Harvard Apparatus, Holliston, MA) filled with aCSF. Electrical signals from the electrode were acquired at 10 kHz (sampling frequency), using the pCLAMP 9 software package (Molecular Devices Inc) and displayed on the laboratory computer. Subtraction of the 50 Hz noise and successive Gauss filtering (2.5 kHz) were applied offline (MES, Femtonics Ltd, Budapest, Hungary). Signal analysis was performed using the Curve Analysis software package (Femtonics Ltd). Extracellular recordings were performed simultaneously with ATP and null sensor measurements in a set of experiments.
Biosensor recording using ATP, adenosine and glutamate sensors
The principles and operation of the Pt/Ir microelectrode biosensors for the purines, ATP and adenosine, have been described previously (Llaudet et al., 2003; 2005). The biosensors used in this study were obtained from Sarissa Biomedical (Coventry, UK). The ATP sensor comprised of two enzymes (glycerol kinase, EC 2.7.1.3 and glycerol-3-phosphate oxidase, EC 1.1.3.21), and the adenosine sensor comprised of three enzymes (adenosine deaminase, EC 3.5.4.4; nucleoside phosphorylase, EC 2.4.2; xanthine oxidase, EC 1.1.3.22) entrapped within a matrix around a fine platinum wire of 50 µm diameter and 0.5 mm long. Electrochemical sensor can respond not only to the analyte of interest but also to any electroactive species in the immediate environment. To check whether the sensors were responding to ATP, we used a dual recording configuration. To give a measure of net ATP concentrations, the signal from a NULL sensor (lacking enzymes, but otherwise identical) was used to measure background signals, which were then subtracted from the signals generated by the ATP biosensor. For the adenosine/inosine sensor (ADO/INO), a separate inosine sensor (INO), lacking adenosine deaminase, is required to yield a signal specific to adenosine (Frenguelli et al., 2003). Thus, the signal of the adenosine sensor can be described as that from both adenosine and inosine, and the net adenosine signal is achieved by subtracting the signal of inosine sensor from this value. Glutamate release was measured using a glutamate (GLU) sensor, comprising glutamate oxidase (EC 1.4.3.11) and using NULL sensor, as background. This GLU sensor is selective for glutamate over glutamine, aspartate, dopamine and 5-HT (Dale et al., 2005; Tian et al., 2009). All microbiosensors were operated at 500–700 mV (vs. Ag/AgCl) in a flow system for amperometric detection at 34–36°C temperature. In certain series of experiments, ATP, NULL, ADO/INO and INO recordings were performed simultaneously; whereas in other series of experiments, simultaneous ATP, GLU and NULL recordings were done in order to limit the maximal number of recording electrodes in the same measurement location. The time of the ATP peaks were not significantly different in the two sets of measurements (0.145 ± 0.131 s, P > 0.1, t-test), suggesting a negligible impact of the two different electrode arrangements on the measured data. Therefore, data from the two sets of measurements are shown simultaneously on the same time axis.
The microelectrode biosensors were each inserted through the 400 µm thickness of the CA1 region of the hippocampal slice such that most of the sensing part of the sensors was in intimate contact with the tissue. Previous studies showed that insertion of the sensor is not detrimental to the slice, exhibiting fEPSP activity in its place (Frenguelli et al., 2003). Before insertion and after removal, the sensors were calibrated with known concentrations of standards: ATP (10 µmol·L−1), adenosine (3 µmol·L−1), inosine (10 µmol·L−1) and glutamate (10 µmol·L−1), which allowed quantification of any rundown in sensor sensitivity over the duration of an experiment. Before experiments, a standard calibration curve was generated; and a high linear correlation was observed between the peak value of signal and the ATP, adenosine, inosine and glutamate concentration. To take into account that biosensor sensitivity varies during different experiments, we used the calibration of the sensor to normalize each signal. In addition, we tested all compounds for any interference with the sensitivity of the sensors, but none of them elicited any detectable change in the signal. Besides the sensors, the recording electrode was placed adjacent to the sensors. Continuous signal from the biosensor was acquired at 10 kHz using the pCLAMP 9 software package (Molecular Devices Inc) and displayed on a PC. For calculation, ‘net’ signals for each experiment were calculated subtracting the currents of the corresponding baseline, which were expressed in absolute units of concentration (µmol·L−1). In case of pharmacological treatments, their effects on the peaks of the responses of the biosensors were quantified. Unless otherwise stated, in case of ATP recording, when a biphasic signal was recorded, the effects of treatments on the peak of the second phase of the response were calculated; that is, the absolute maximum of the signal was taken into account. Calibration curves were constructed using the GraphPad software (GraphPad, La Jolla, CA, USA).
Experimental protocol
After insertion of biosensors, an approx. 20 min equilibration period was allowed to reach a steady-state baseline. Then, K+ depolarization was applied by subjecting the preparations to modified aCSF containing 26.9 mmol·L−1 K+ (Na+: 102 mmol·L−1) for 270 s. All drugs were applied to the perfusion solution, from 20 min before the beginning of K+ depolarization until the end of experiments, except sodium fluoroacetate (FAc), which was added 10 min before the onset of K+ stimulation. Drugs [ARL67156, AZ10606120, carbenoxolone, dipyridamole, FAc, Brilliant Blue G (BBG), probenecid, CNQX, D-AP-5, ifenprodil, tetrodotoxin (TTX), l-trans-pyrrolidine-2,4-dicarboxylic acid (L-trans-2,4-PDC)] were dissolved in fresh aCSF. Ca2+-free aCSF contained 1 mmol·L−1 EGTA in order to eliminate residual extracellular Ca2+.
Statistical analysis
Date are expressed as mean ± SEM with n= number of identical experiments. Student's t-test (pairwise comparisons) and one-way anova followed by Dunnett's test (multiple comparisons) were used as a statistical analysis, as appropriate. P-values of less than 0.05 were considered statistically significant.
Materials
Salts used in the aCSF were obtained from Reanal ZRT (Budapest, Hungary). The other drugs used were ATP, ARL671566 (N,N-diethyl-β-γ-dibromomethylene-D-adenosine-5′-triphosphate trisodium salt hydrate), carbenoxolone, BBG, dipyridamole, FAc, probenecid (all from Sigma-Aldrich Chemical Co., St. Louis, MO), adenosine, inosine (Merck, Darmstadt, Germany), AZ10606120 (N-[2-[[2-[(2-hydroxyethyl)amino]ethyl]amino]-5-quinolinyl]-2-tricyclodec-1-ylacetamide dihydrochloride), CNQX (6-cyano-7-nitroquinoxaline-2,3-dione disodium), D-AP-5 (D-(–)-2-amino-5-phosphonopentanoic acid), ifenprodil (1S*,2S*)-threo-2-(4-benzylpiperidino)-1-(4-hydroxyphenyl)-1-propanol hemitartrate, L-trans-2,4-PDC (all from Tocris Bioscience, Ellisville, MO), TTX (Alomone Labs, Jerusalem, Israel), L-glutamic acid (Pierce, Rockford, IL). All solutions were freshly prepared on the day of use.
Results
ATP, adenosine and glutamate released under basal conditions and after K+ depolarization in the hippocampal CA1 area
The selectivity of the biosensors used versus interferences found in the hippocampus allowed the basal levels of ATP, adenosine and glutamate in the slice to be estimated. As the biosensors have a background current, basal levels were measured with the following method: at first placing the sensors into the tissue chamber, allowing it to equilibrate for at least 20 min and then inserting the sensor into the slices, allowing it to equilibrate and measuring the changes and potential differences between these equilibrium states. To convert this difference to absolute concentration of ATP, adenosine and glutamate, we used null and inosine sensors as references (see Methods). Under basal conditions, the signals for extracellular ATP, adenosine and glutamate were close to the detection limit (ATP: 4.1 ± 0.8 pA, n= 10; ADO: 5.4 ± 0.7 pA, n= 10; GLU: 6.4 ± 0.8 pA, n= 10) and have been estimated to be as low as a few nmol·L−1 respectively.
When slices were subjected to K+ depolarization (25 mmol·L−1; 270 s), easily detectable signals were recorded on ATP, glutamate and adenosine sensors, respectively (Figure 1). All sensors showed a rapid response; however, the adenosine sensor detected an earlier and long-lasting signal than the ATP and glutamate sensor. The release of adenosine started 0.79 ± 0.01 min after the beginning of the K+ depolarization and reached its peak at 7.31 ± 0.01 min (Figure 1C, Table 1). The efflux of glutamate started 2.68 ± 0.04 min after K+ depolarization and reached its peak at 5.62 ± 0.06 min (Figure 1B, Table 1). Two phases of ATP release were detected, the first occurring at the same time when glutamate release started (Figure 1A, B), while the onset of the second phase coincided with the maximum peak of glutamate release (Figure 1A, B). ATP efflux started 2.56 ± 0.05 min after the beginning of the K+ stimulation and reached its absolute peak during the second phase at 8.08 ± 0.01 min (Figure 1A, Table 1). Null sensors that lacked enzymes in the polymer coating displayed only small fluctuations of the current around the baseline (not shown). Evoked signals of fEPSP (in AC mode) recording from stratum radiatum of rat hippocampus simultaneously with the ATP sensor were recovered after the K+ depolarization (Figure 1E), indicating that slices retained their viability during the experiments. During K+ depolarization, the gradual loss of biological electrophysiological activity was observed, as expected. fEPSP activity is then completely recovered in the next 15 min during wash out. High K+-induced spreading depression (SD; Schock et al., 2007) was detected at 3.81 ± 0.2 min (n= 6) after stimulation from the stratum radiatum of area CA1 (Figure 1F). This large negative deflection on the extracellular DC potential occurred immediately after the loss of fEPSP, , but before the peak of adenosine release ATP started to release before the beginning of SD, showed a small first peak at the time of SD (0.48 ± 0.16 µmol·L−1, n= 8) and continued to increase after the peak of SD. Likewise, glutamate release did not reach its plateau before the onset of SD (Figure 1B).
Figure 1.

(A–D) Real–time detection of the release of ATP, adenosine (ADO) and glutamate (GLU) from the rat hippocampus in vitro, during K+ (25 mmol·L−1; 270 s) depolarization. (A) Two phases of ATP release were detected, as indicated by the arrows on the top: the first occurring at the same time as glutamate release started, while the onset of the second phase coincided with the maximum peak of glutamate release (B). The adenosine sensor (C) detected an earlier and longer lasting signal than the ATP and glutamate sensors. ‘Net’ signal for each experiment was calculated subtracting the currents of the corresponding baseline. Net ATP and glutamate signals were obtained after subtraction of the simultaneously recorded null sensor, while net ADO signals (C) were obtained after subtraction of the simultaneously recorded inosine (INO) sensor (D) from the ADO/INO signal. (E) fEPSP recorded in AC mode before (i) and following K+ depolarization (ii). Note that full recovery of the fEPSP occurred (iii). Stimulus artifacts have been truncated. Bottom, Magnified examples of the fEPSP are shown at the times indicated on the continuous AC trace. (F) Extracellular DC potential recorded simultaneously with the ATP sensor signal showed SD (negative deflection on the DC trace) following K+ depolarization. Note that K+ (25 mmol·L−1; 270 s) depolarization-evoked purine efflux occurs before the SD (indicated by a continuous vertical line). ATP, ADO/INO, INO were recorded from one set of experiments and ATP and GLU from a separate set of experiments. All traces show means (black lines) ± SEM (grey bands) of eight independent experiments.
Table 1.
The effect of different treatments on the efflux of ATP, adenosine and glutamate from rat hippocampal slices
| Condition (µmol·L−1) | Net ATP (µmol·L−1) | n | Net Adenosine (µmol·L−1) | n | Net Glutamate (µmol·L−1) | n |
|---|---|---|---|---|---|---|
| Control | 1.23 ± 0.23 | 8 | 10.28 ± 1.41 | 8 | 3.49 ± 0.84 | 8 |
| Ca2+ free and EGTA | 0.21 ± 0.01 | 6 | 0.22 ± 0.02 | 6 | 3.64 ± 0.15 | 6 |
| P < 0.01 | P < 0.01 | P > 0.05 | ||||
| TTX (3) | 0.07 ± 0.05 | 6 | 0.08 ± 0.04 | 6 | 0.12 ± 0.03 | 6 |
| P < 0.01 | P < 0.01 | P < 0.01 | ||||
| FAc (1000) | 0.21 ± 0.14 | 8 | 3.08 ± 0.89 | 8 | 0.11 ± 0.07 | 7 |
| P < 0.01 | P < 0.01 | P < 0.01 | ||||
| CBX (100) | 0.21 ± 0.02 | 6 | 0.22 ± 0.02 | 6 | 0.013 ± 0.02 | 6 |
| P < 0.01 | P < 0.01 | P < 0.01 | ||||
| CBX (20) | 2.22 ± 0.37 | 5 | 1.02 ± 0.26 | 5 | – | – |
| P < 0.05 | P < 0.01 | |||||
| BBG (0.1) | 0.11 ± 0.14 | 5 | 4.60 ± 0.74 | 5 | 0.029 ± 0.06 | 7 |
| P < 0.01 | P < 0.01 | P < 0.01 | ||||
| Probenecid (150) | 2.70 ± 0.11 | 7 | 0.22 ± 0.01 | 7 | – | – |
| P < 0.05 | P < 0.01 | |||||
| ARL67156 (100) | 8.62 ± 0.76 | 6 | 0.15 ± 0.04 | 6 | 2.43 ± 0.63 | 6 |
| P < 0.01 | P < 0.01 | P > 0.05 | ||||
| D-AP-5 (10) | 0.21 ± 0.03 | 5 | 0.15 ± 0.01 | 5 | – | – |
| P < 0.01 | P < 0.01 | |||||
| Ifenprodil (10) | 0.13 ± 0.06 | 5 | 0.11 ± 0.02 | 5 | – | – |
| P < 0.01 | P < 0.01 | |||||
| CNQX (10) | 1.17 ± 0.12 | 5 | 8.43 ± 0.53 | 5 | 0.09 ± 0.06 | 5 |
| P > 0.05 | P > 0.05 | P < 0.01 | ||||
| AZ10606120 (0.1) | 0.17 ± 0.02 | 6 | 0.12 ± 0.05 | 6 | – | – |
| P < 0.01 | P < 0.01 | |||||
| Dipyridamole (50) + ARL67156(100) | – | – | 2.71 ± 0.23 | 5 | – | – |
| P < 0.01 | ||||||
| FAc (1000) + TTX (3) | – | – | 0.22 ± 0.1 | 6 | – | – |
| P < 0.01 | ||||||
| L-trans-2,4-PDC (300) | – | – | 1.24 ± 0.66 | 7 | ||
| P < 0.05 |
The net ATP, adenosine and glutamate efflux was calculated according to the procedures described in Methods. Data show the mean ± SEM with n= number of identical experiments. P-values indicate significant differences from respective controls (25 mmol·L−1 K+; 270 s), calculated by one-way anova followed by Dunnett's test.
Inhibition of extracellular ATP metabolism increases ATP and inhibits adenosine accumulation
The presence of ecto-nucleotidases that can metabolize ATP to ADP, AMP and adenosine in the hippocampus is well documented (Zimmermann, 2000; Goding et al., 2003). These enzymes could play a role in the production of adenosine during K+ depolarization (25 mmol·L−1; 270 s). We therefore tested the contribution of the hydrolysis of ATP to extracellular adenosine accumulation by the selective ecto-ATPase inhibitor ARL67156. ARL67156 was applied in a concentration (100 µmol·L−1), which, according to previous studies (Frenguelli et al., 2007; Sperlagh et al., 2007) elicits a substantial, although not complete blockade of extracellular breakdown of ATP in the hippocampus.
Inhibition of extracellular ATP metabolism by ARL67156 (100 µmol·L−1) enhanced the extracellular level of ATP following K+ depolarization (Figure 2A; Table 1). Moreover, K+ depolarization evoked ATP efflux appeared earlier than in the absence of ecto-ATPase inhibitor (1.69 ± 0.012 min after the beginning of the K+ stimulation). On the other hand, ARL67156 (100 µmol·L−1) had no significant effect on the glutamate efflux (Figure 2B, Table 1). The elevation in glutamate efflux in this case started coincidently with the elevation of ATP efflux (Figure 2B). In contrast, adenosine release during the stimulation was inhibited in the presence of ARL67156 (Figure 2A). These results show that ATP efflux detected by the ATP biosensor is subject to extracellular metabolism and contributes to the generation of adenosine that is detected by the adenosine biosensor. When the slices were preperfused with ARL67156 (100 µmol·L−1) and the adenosine transport inhibitor dipyridamole (50 µmol·L−1), the adenosine efflux was higher than detected in the presence of the ecto-ATPase inhibitor alone (n= 5, P < 0.001 vs. ARL67156 alone, Table 1). These findings indicate a minor additional direct adenosine release in response to K+ depolarization, which is not derived from the released ATP.
Figure 2.

Effect of the inhibition of extracellular ATP metabolism with ARL67156 (100 µmol·L−1) on ATP (A, B), adenosine (ADO; A) and glutamate (GLU; B) efflux. The treatment enhanced ATP release (A, B) but had no significant effect on glutamate release (B) following K+ depolarization. By contrast, ADO efflux during the stimulation was inhibited in the presence of ARL67156 (A). Net ATP and glutamate signals were obtained after subtraction of the simultaneously recorded null sensor, while net ADO signals were obtained after subtraction of the simultaneously recorded INO sensor from the ADO/INO signal. Traces are means ± SEM of six independent experiments.
Differential susceptibility of ATP, adenosine and glutamate release to extracellular Ca2+
Because ATP can be released in a [Ca2+]o-dependent manner (Sperlagh and Vizi, 2000; Pankratov et al., 2006), we asked whether the efflux of ATP during K+ depolarization (25 mmol·L−1; 270 s) was [Ca2+]o-dependent. To examine the Ca2+-dependency of purine release during K+ depolarization, we omitted Ca2+ from the perfusion medium, and 1 mmol·L−1 EGTA was added to chelate any residual extracellular Ca2+. Perfusion of Ca2+-free/EGTA and 25 mmol·L−1 K+ containing aCSF caused a significant decrease in ATP and adenosine (Table 1, Figure 3) efflux during K+ depolarization. On the other hand, glutamate efflux did not significantly change in response to stimulation (Table 1, Figure 3), when rat hippocampal slices were perfused with Ca2+-free/EGTA aCSF solution.
Figure 3.

The effects of different treatments on K+ depolarization-induced efflux of ATP, adenosine and glutamate from rat hippocampal slices. Inhibition of the voltage-dependent Na+ channels by TTX (3 µmol·L−1) totally inhibited the ATP (A) and adenosine (ADO; B) efflux following the K+ depolarization (25 mmol·L−1; 270 s). Similarly, glutamate release (GLU; C) was reduced in the presence of TTX after high K+ stimulation. Removal of extracellular Ca2+ (Ca2+ free) differentially affected ATP, adenosine (A, B) and glutamate (C) efflux. The hemichannel blocker CBX (high: 100 µmol·L−1) almost completely inhibited ATP, adenosine and glutamate efflux (A, B, C). Release of ATP (A), adenosine (B) and glutamate (C) following the K+ stimulation substantially decreased in the presence of FAc (1 mmol·L−1; 10 min). TTX (3 µmol·L−1; 20 min) almost completely inhibited adenosine efflux in the presence of FAc (1 mmol·L−1; 10 min) (B). **P < 0.01, significantly different from the respective control; one-way anova, Dunnett's test (n= 5–6).
Involvement of ongoing neuronal activity in ATP, adenosine and glutamate release
To test the involvement of the sodium channel-mediated axon potential propagation, the effect of TTX (3 µmol·L−1) was examined. Inhibition of the voltage-dependent Na+ channels by TTX inhibited ATP and adenosine accumulation following K+ depolarization (Table 1, Figure 3). Similarly, glutamate release was reduced in the presence of TTX after high K+ stimulation (Table 1, Figure 3). The Na+-channel blocker also caused a complete inhibition of the fEPSP activity (data not shown). These findings indicate that ATP, adenosine and glutamate released in response to K+ stimulation is associated with ongoing axonal activity. Next, we investigated the involvement of glutamatergic excitatory transmission in K+ depolarization evoked ATP, glutamate and adenosine efflux by testing the effect of glutamate receptor antagonists. When the slices were perfused with the non-NMDA glutamate receptor antagonist CNQX (10 µmol·L−1), it almost abolished glutamate efflux but did not significantly change ATP and adenosine efflux in response to 25 mmol·L−1 K+ stimulation (Table 1). The NMDA receptor antagonist D-AP-5 (10 µmol·L−1) and the NR2B-selective NMDA receptor antagonist ifenprodil (10 µmol·L−1) was applied before and during K+ depolarization (25 mmol·L−1; 270 s). Elevation of extracellular ATP levels was significantly decreased and accumulation of adenosine was inhibited in the presence of D-AP-5 and ifenprodil (Figure 4); Table 1).
Figure 4.

Effect of the NMDA glutamate receptor antagonist D-AP-5 on adenosine and ATP efflux evoked by K+ depolarization. D-AP-5 (10 µmol·L−1) decreased ATP and adenosine efflux in the rat hippocampal slices. Black bars denote the period of K+ depolarization and D-AP-5 perfusion. These individual recordings of the normalized signals (shown as pA) of ATP and adenosine (ADO) biosensors are representative of at least five independent experiments.
ATP, adenosine and glutamate are released from glial cells in response to K+ depolarization in CA1 area
Next, we investigated whether the source of detected extracellular purines and glutamate is glial or neuronal. Previous reports showed that fluoroacetate (FAc), a mitochondrial gliotoxin, selectively impairs the oxidative metabolism of glial cells (Clarke, 1991; Canals et al., 2008). The specificity of FAc in the concentration used is due to its selective uptake by acetate transporter present only in glial cells. In addition, we used a relatively short time for FAc perfusion period to further ensure glia selectivity (N. Hájos, pers. comm.). FAc perfusion (1 mmol·L−1, n= 6) was added 10 min before the beginning of K+ depolarization. Inhibition of oxidative metabolism in glial cells resulted in almost complete inhibition of the detected increase in the second phase of the efflux of ATP (Figure 5A; Table 1), whereas the first phase of the ATP efflux did not significantly change (ATP: 0.48 ± 0.16 µmol·L−1, n= 8, and 0.54 ± 0.20 µmol·L−1, n= 8, in the absence and presence of FAc, respectively, P > 0.05; Figure 5A). Glutamate efflux was also inhibited by the FAc perfusion (Figure 5B; Table 1). Adenosine accumulation was decreased by FAc treatment, although it was still detectable and appeared later than in the absence of FAc (Figure 5C: Table 1). The extracellular level of adenosine detected in the presence of FAc (1 mmol·L−1) was further inhibited in the presence of TTX (3 µmol·L−1; P < 0.05 vs. FAc alone; Figure 3B; Table 1). The electrical stimulation could evoke fEPSPs in the presence of FAc and the signal remained largely unchanged before and after stimulation, indicating that the treatment mostly affected the glial population (Figure 5D). Nevertheless, a slight reduction of the amplitude of the response could be observed (Figure 5D) when compared with signals recorded from control slices (Figure 1E), which might reflect the contribution of glia to the modulation of fEPSP activity or a minor effect of FAc on neuronal survival. These results show that glial and not neuronal cells are the main source of extracellular purines and glutamate under our experimental conditions, and this transmitter efflux might affect measured fEPSP amplitudes in the CA1 area (Figure 5).
Figure 5.

Effect of perfusion with the mitochondrial gliotoxin FAc (1 mmol·L−1) on ATP (A) glutamate (GLU; B) and adenosine (ADO; C) efflux, evoked by K+ depolarization. FAc was added 10 min before the beginning of K+ depolarization. Inhibition of oxidative metabolism in glial cells resulted in almost complete inhibition of the detected increase in the efflux of ATP and glutamate. (D) Recording of the evoked fEPSP (in AC mode) before (i) and following K+ depolarization (ii). Note the recovery of the fEPSP during wash out (iii). Stimulus artifacts have been truncated. Bottom, Magnified examples of the fEPSP (average of five responses) shown at the times indicated on the continuous AC trace. ATP and ADO signals were recorded from one set of experiments and GLU from a separate set of experiments. All data show means (black line) ± SEM (grey band) of six independent experiments.
Mechanism of hippocampal ATP, adenosine and glutamate release
In the next experiments, inhibitors of ion channels, known to mediate the release of ATP and/or glutamate, were tested in order to identify the underlying mechanism of their transmembrane efflux under our experimental conditions. At first, we examined the role of P2X7 receptor ion channels (Pellegatti et al., 2005; Anselmi et al., 2008), which are expressed in the hippocampus (Sperlagh et al., 2002) and which are known to mediate the release of glutamate and ATP from nerve terminals (Alloisio et al., 2008; Marcoli et al., 2008), as well as from cultured astrocytes (Ballerini et al., 1996; Duan et al., 2003). To investigate the involvement of P2X7 receptors in ATP, adenosine and glutamate efflux from the hippocampus, the effects of two antagonists of these receptors, BBG (0.1 µmol·L−1) and AZ10606120 (0.1 µmol·L−1), were tested. Extracellular elevation of ATP was inhibited in the presence of BBG and AZ10606120 (Figure 6; Table 1) after depolarization. On the other hand, BBG exerted partial inhibition, whereas AZ10606120 (Figure 7Table 1) caused complete inhibition of adenosine release in response to K+ stimulation. The efflux of glutamate was also inhibited by the same concentration of BBG (Table 1). Next, the effect of carbenoxolone was tested, which is known as a wide spectrum gap junction inhibitor (Bruzzone et al., 2005), but also inhibits P2X7 receptors (Suadicani et al., 2006). At a high concentration (100 µmol·L−1) carbenoxolone completely inhibited the ATP, adenosine and glutamate efflux in response to 25 mmol·L−1 K+ stimulation (Figure 3Table 1). Because it is known that carbenoxolone in low concentration inhibits only the pannexin Panx1-evoked current and P2X7 receptors co-immunoprecipitate with Panx1 (Pelegrin and Surprenant, 2006), we also examined a lower, pannexin-selective, concentration of carbenoxolone (20 µmol·L−1). This treatment completely abolished the adenosine efflux during K+ depolarization (Figure 7; Table 1). Administration of probenecid (150 µmol·L−1), another pannexin inhibitor, also caused a marked decrease in the extracellular concentration of adenosine (Figure 7; Table 1). In contrast, the extracellular level of ATP was significantly increased in the presence of low concentration carbenoxolone (20 µmol·L−1);and in the presence of probenecid (150 µmol·L−1; Figure 6; Table 1). These data demonstrate that the mechanism of the efflux of ATP and adenosine was partly different: although the efflux of adenosine did arise from previously released ATP, through the actions of ecto-nucleotidase, it might also be released directly via pannexin hemichannels. In contrast, ATP was released through activation of P2X7 receptors into the extracellular space, before its rapid breakdown to adenosine.
Figure 6.
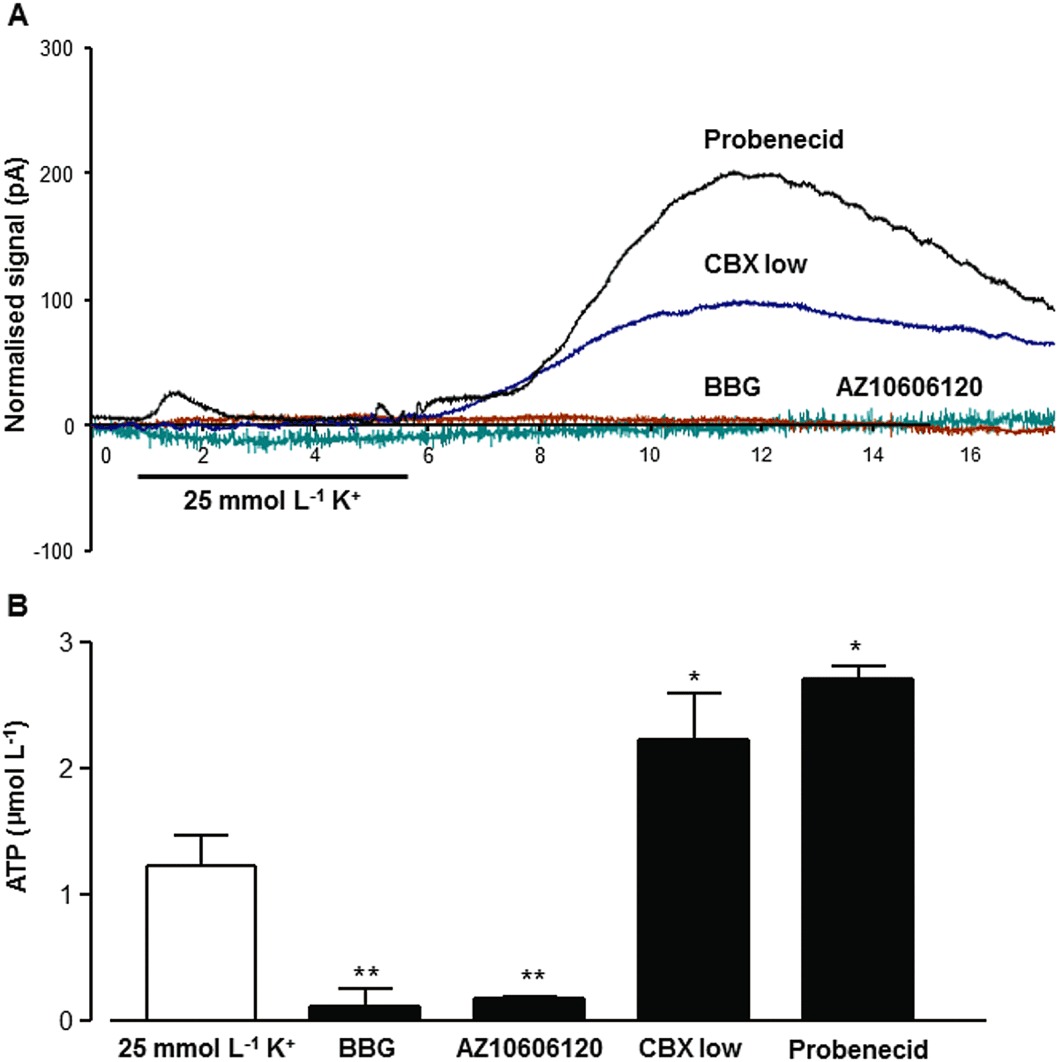
The effect of P2X7 receptor antagonists and pannexin inhibitors on ATP efflux evoked by K+ depolarization. In (A), individual recordings of the normalized signals (shown as pA) of the ATP biosensor are shown. BBG (100 nmol·L−1) and AZ10606120 (100 nmol·L−1) inhibited ATP efflux, whereas inhibition of pannexin hemichannels by CBX (20 µmol·L−1) significantly increased the extracellular level of ATP during K+ stimulation (25 mmol·L−1; 270 s), which was further increased in the presence of probenecid (150 µmol·L−1). In (B), summary data from five independent experiments are shown. *P < 0.05, **P < 0.01, significantly different from the respective control; one-way anova, Dunnett's test (n= 5).
Figure 7.

The effect of P2X7 receptor antagonists and pannexin inhibitors on adenosine efflux evoked by K+ depolarization. In (A), individual recordings of the normalized amperometric signals (shown as pA) of the ADO biosensor are shown. BBG (100 nmol·L−1) and AZ10606120 (100 nmol·L−1) inhibited adenosine efflux. Inhibition of pannexin hemichannels by CBX (20 µmol·L−1) greatly reduced adenosine efflux during K+ stimulation (25 mmol·L−1; 270 s). Administration of probenecid (150 µmol·L−1) also profoundly decreased the extracellular level of adenosine. In (B), summary data from five independent experiments are shown. **P < 0.01; significantly different from the respective control, ns, P > 0.05; one-way anova, Dunnett's test (n= 5).
As for the underlying mechanism of glutamate efflux, because it was Ca2+-independent, the participation of excitatory amino acid transporters (EAAT1-5), present on neuronal and glial cell membranes, was assessed with the wide spectrum EAAT inhibitor, L-trans-2,4-PDC, 300 µmol·L−1. This inhibitor significantly decreased glutamate efflux evoked by K+ depolarization (Table 1).
Discussion and conclusions
In our experiments, amperometric biosensors were utilized for the direct real-time measurement of extracellular purine and glutamate levels in the hippocampus under basal conditions and in response to K+ depolarization. Under unstimulated conditions, only low basal ATP, adenosine and glutamate levels were detected, which is in rough agreement with other previous studies in the hippocampus (Frenguelli et al., 2007) and cerebellum (Wall et al., 2007; Wall and Dale, 2007).
K+ depolarization (25 mmol·L−1), a widely used depolarizing stimulus used to study purine release (cf. Latini and Pedata, 2001), elicited activity-dependent ATP, adenosine and glutamate efflux, with distinct kinetics and mechanisms. Although the physiological relevance of this kind of stimulation remains to be investigated, high [K+]o levels are detected during high frequency neuronal discharge and prolonged elevation of extracellular K+ is also implicated in pathological conditions, such as hypoxia and ischemia (Olsen and Sontheimer, 2008). The efflux of all three compounds as well as the recorded fEPSP activity was sensitive to sodium channel inhibition by TTX. Because in situ astrocytes and other glial cells do not exhibit sodium channel activity and are therefore resistant to the action of TTX, we can conclude that the primary signal that leads to the accumulation of ATP, adenosine and glutamate is most likely axonal activity under our experimental conditions. An alternative, although less likely possibility, is that K+ depolarization primarily evokes gliotransmitter release in a neuronal activity-independent manner, which gives rise to a subsequent, action potential-dependent, transmitter release from neurons.
High extracellular K+ concentrations are also well known to induce SD, a phenomenon that plays important role in the pathogenesis of migraine, stroke and traumatic brain injury (Lauritzen et al., 2011). Because SD per se elicits ATP release in the brain (Frenguelli et al., 2007; Schock et al., 2007), the ATP efflux detected in our study might also be the consequence of SD. Indeed, as shown on extracellular DC recording (Figure 1F), SD developed after K+ depolarization in our experiments as well,although the initial rise in the level of ATP appeared earlier than the SD. Therefore, it appears that the first early phase of the ATP elevation was largely independent from the development of SD, whereas the second larger elevation could be related to the SD. Supporting this idea, treatments, which inhibited the second phase of the efflux of ATP, are known to inhibit or postpone SD as well (e.g. TTX, NMDA receptor antagonists) (Somjen, 2001).
It has been firmly established that ATP is a constituent of synaptic vesicles (Gualix et al., 1999; Sperlagh and Vizi, 2000; Pankratov et al., 2006) and astrocyte lysosomes (Zhang et al., 2007), and is taken up into vesicles by a recently identified specific vesicular nucleotide transporter (VNUT) (Sawada et al., 2008). Removal of extracellular Ca2+ from the perfusion medium inhibited both ATP and adenosine signals detected upon K+ depolarization, which implies that ATP is released by vesicular exocytosis or it is secondarily released, in response to a compound which enters the extracellular space, by vesicular exocytosis. Depolarization-induced Ca2+-dependent ATP efflux has already reported previously from the hippocampus (e.g. Cunha et al., 1996), and our observation adds further support to these findings with enhanced temporal and spatial resolution demonstrating Ca2+-dependent extracellular ATP accumulation in the low micromolar level, which is adequate for the activation of most of P2X and P2Y receptors. Therefore, this release might underlie ATP-mediated synaptic transmission (Pankratov et al., 1998; 2006; Mori et al., 2001) or synaptic modulation (e.g. Sperlagh et al., 2002; Rodrigues et al., 2005) detected in this region. Moreover, ATP levels found in the effluent probably underestimate the concentration of ATP at the release sites because of the rapid metabolism of ATP in the extracellular space detected by the biosensors (see below). Adenosine release was also found to be dependent on extracellular Ca2+ in response to electrical stimulation in previous studies (Latini et al., 1997; Wall et al., 2007; Wall and Dale, 2007); whereas other stimuli, such as combined oxygen-glucose deprivation (Frenguelli et al., 2007), lead to Ca2+-independent release.
In contrast, glutamate accumulation in response to K+ depolarization failed to decrease under Ca2+-free conditions. Although glutamate, being the major excitatory transmitter of the CNS, is released during neuronal activity in a Ca2+-dependent way, in neurochemical studies, depolarization-induced glutamate release has been found to be partly or completely independent on extracellular Ca2+ in different studies (Bernath, 1992).
Interestingly, the appearance, peak and decay of ATP, adenosine and glutamate responses were not identical using the same stimulation paradigm. Because all three sensors responded equally rapidly to external application of the compounds during the calibration, these differences cannot be explained by the different response times of these sensors. Instead, they could reflect interactions between the released compounds. Thus, adenosine accumulation could be the consequence of ATP efflux. Indeed, elevation of ATP levels started earlier and was substantially higher in the presence of the ecto-ATPase inhibitor ARL67156, indicating the rapid and efficient metabolism of extracellular ATP detected by the ATP biosensor, which is concordant with earlier (Dunwiddie et al., 1997; Cunha et al., 1998) and more recent (Frenguelli et al., 2007) assumptions. At the same time, ARL67156 virtually prevented adenosine accumulation following K+ depolarization, indicating that the majority of adenosine accumulation detected by the adenosine sensor is derived from the breakdown of extracellular ATP. In contrast, Frenguelli et al. (2007) reported that ARL67156 did not affect adenosine accumulation in response to ischaemic-like conditions in the hippocampus. However, K+ depolarization and combined oxygen-glucose deprivation probably releases purines from different pools and these tow stimuli might also differentially affect the activity of ecto-nucleotidases. Thus, it appears that the extracellular metabolism of released ATP, and its contribution to adenosine efflux is different, depending on the stimulation (Cunha et al., 1996), and is higher, when more physiological stimuli are applied (Latini and Pedata, 2001). Nevertheless, our results also indicate an additional direct release of adenosine, which is independent from the extracellular catabolism of ATP. We detected this additional adenosine efflux when the breakdown of ATP was inhibited by ARL67156, but the further re-uptake of adenosine was also prevented by dipyridamole. We should also assume, however, that due to the incomplete blockade of the ecto-ATPases by ARL67156, a minor adenosine production from the released ATP might also contribute to this residual elevation.
On the other hand, it seems less likely that ATP efflux is the consequence of glutamate efflux detected by the biosensor, because the latter was completely inhibited by CNQX, the blocker of non-NMDA receptors, which did not affect ATP efflux. Moreover, an early elevation of the adenosine signal preceding the rise of the glutamate efflux could also be observed in response to high K+ in the absence of any drug treatment, which also supports this assumption. In contrast, ATP and adenosine release was subject to inhibition of NMDA-type glutamate receptors by D-AP-5 and ifenprodil, which implies that ATP was released in response to the activation of NR2B subunit-containing NMDA receptors. This observation is concordant with findings showing that NMDA receptor activation releases purines, including ATP from brain slices (Pedata et al., 1991; Craig and White, 1993), and from cultured astrocytes (Queiroz et al., 1997). However, we were unable to dissect this ‘trigger’ glutamate efflux within the resolution of the technique. Although NMDA receptors co-localize with AMPA receptors at the majority of central synapses, they are also targeted to extrasynaptic sites in the hippocampus (Traynelis et al., 2010), where AMPA receptors are not present. In particular, NR2B subunit-containing NMDA receptors has been reported to localize to extrasynaptic sites and participate in distinct forms of synaptic plasticity that do not require the activation of synaptic glutamate receptors (Tovar and Westbrook, 2002; Massey et al., 2004). Therefore, it seems likely that that those NMDA receptors, which mediate the release of ATP, are extrasynaptic or glial and are not identical to these NMDA receptors, which co-localize with AMPA receptors in excitatory synapses and which probably mediate the glutamate efflux detected by the biosensor. However, because NR2B subunit-containing NMDA receptors are also expressed on astrocytes (Lee et al., 2010), both neurons and astrocytes should be taken into account as a potential source of ATP efflux.
In order to further identify the source of ATP, adenosine and glutamate released in response to K+ depolarization, the widely used glia-selective toxin FAc (Szerb and Issekutz, 1987; Berg-Johnsen et al., 1993) was utilized. To ensure its glial selectivity, a relatively short perfusion time was applied, which inhibited the efflux of ATP and glutamate, whilst fEPSP activity was largely preserved. Although we cannot exclude a minor effect of FAc on neurons, the majority of ATP and glutamate accumulation detected by the biosensor most likely originated from glial cells. Hence, the K+ depolarization paradigm used in our study seems to be a signal whereby the neuron–glia network of the hippocampus could be challenged and simultaneous glutamate and ATP efflux could be evoked from glial cells in response to ongoing neuronal activity. Although this ATP and glutamate accumulation is not identical with synaptic neurotransmitter release responsible for fEPSP activity, it may contribute to its modulation, as the fEPSP signal was slightly attenuated in the presence of FAc. One should consider here that in case of fEPSP recording, a focal stimulation of the Schaffer collateral–commissural fibre pathway was applied, whereas elevated extracellular K+ level affected the whole hippocampal slice. These two stimulations do not necessarily elicit identical pattern of release. According to the present findings, gliotransmitter release is dominant during K+ depolarization but contributes to evoked fEPSPs only to a minor extent. As for the accumulation of adenosine, the early elevation in its level, presumably stemming from extracellular interconversion of released ATP, was almost completely inhibited in the presence of FAc. On the other hand, a late elevation of adenosine signal was still detected in the presence of the gliotoxin, which might represent a direct neuronal adenosine efflux. Supporting this assumption, the residual efflux of adenosine in the presence of FAc was fully sensitive to TTX.
In previous studies, the following mechanisms have been identified for ATP to enter the extracellular space: (i) exocytotic release, which can occur both from neurons (Sperlagh and Vizi, 2000; Pankratov et al., 2006) and astrocytes (Coco et al., 2003; Pangrsic et al., 2007; Zhang et al., 2007); (ii) P2X7 receptor (Ballerini et al., 1996; Suadicani et al., 2006), (iii) connexin (Cotrina et al., 1998; Stout et al., 2002), and (iv) pannexin-mediated release (Bao et al., 2004; Huang et al., 2007), which have been described primarily in non-neuronal cells including astrocytes; and (v) volume regulated anion channel-mediated release (Darby et al., 2003; Okada et al., 2004), which also have been identified in non-neuronal cells; (vi) CFTR-mediated release, which has been demonstrated in DRG neurons (Kanno and Nishizaki, 2011) and (vii) cytolytic release.
Because both ATP and adenosine efflux under K+ depolarization was Ca2+- dependent and TTX sensitive, which is inconsistent with gross cellular damage, the cytolytic origin of ATP and adenosine accumulation does not appear very likely. Also, because volume regulated anion channels are primarily activated under hypo-osmotic conditions, such as swelling, which did not occur under our conditions, the involvement of volume regulated anion channels in the transmitter efflux measured in the present study is less likely.
Instead, P2X7 receptors, which are expressed in the hippocampus (Sperlagh et al., 2002) and could mediate the release of both ATP (Ballerini et al., 1996) and glutamate (Sperlagh et al., 2002; Marcoli et al., 2008), were taken into consideration. BBG, which is a selective antagonist of P2X7 receptors in the concentration used (Jiang et al., 2000), almost completely inhibited elevation of ATP and glutamate levels in response to high K+ stimulation; and adenosine accumulation was also significantly decreased. These findings imply that P2X7 receptors are also activated in response to depolarization and participate in the efflux of ATP and glutamate. Activation of P2X7 receptors could elicit both vesicular and non-vesicular transmitter release, the latter is probably mediated by the receptor ion channel complex itself (Duan et al., 2003; Alloisio et al., 2008; Marcoli et al., 2008) or by the reversal of transporters (Sperlagh et al., 2002). The finding that glutamate, but not ATP release was independent of [Ca2+]o implies that P2X7 receptor-mediated glutamate release is non-vesicular under the conditions of the present study, whereas P2X7 receptor-mediated ATP efflux could be either vesicular or non-vesicular. In support for this assumption, glutamate efflux detected in our experiments was sensitive to L-trans-2,4-PDC, the wide spectrum EAAT inhibitor, implicating the reversal of the excitatory amino acid transporters as its underlying mechanism.
On the other hand, carbenoxolone, which is known as a wide-spectrum gap junction hemichannel inhibitor, but also inhibits P2X7 receptors (Suadicani et al., 2006), in high concentrations profoundly inhibited ATP, adenosine and glutamate accumulation upon K+ depolarization, which raised the possibility that hemichannels participate in their release mechanisms. However, given the low specificity of carbenoxolone and other available gap junction hemichannel inhibitors, we favour the explanation that carbenoxolone prevented transmitter efflux by its inhibitory action on P2X7 receptors. Connexins could be functional in the hemichannel form (see Stout et al., 2004) and can conduct the release of ATP and glutamate from astrocytes (Stout et al., 2002; Ye et al., 2003; Iglesias et al., 2009). However, they are activated typically at low [Ca2+]o (Pfahnl and Dahl, 1999; Valiunas and Weingart, 2000) (i.e. different conditions from the present study), although there are exceptions to this rule (Pearson et al., 2005).
Further pharmacological characterization, using pannexin-selective agents [low concentration of CBX (Davidson and Baumgarten, 1988; Bruzzone et al., 2005) and probenecid (Silverman et al., 2008)], revealed that in contrast to the efflux of ATP, adenosine accumulation was sensitive to the action of pannexin-selective inhibitors as well. These data argue for the heterogeneous origin of extracellular adenosine in our study, which is partly released by pannexins. Pannexins can be activated under physiological calcium concentrations (Bruzzone et al., 2003) and under conditions that are known as triggers of adenosine release, like epileptiform activity (Thompson et al., 2008), metabolic distress and ischaemia (Latini and Pedata, 2001; Thompson and MacVicar, 2008). Although adenosine concentration in the nerve terminals is in the low micromolar level, we applied a relatively strong depolarizing stimulus, which could result in an increased ATP consumption and a consequent increase in intraterminal adenosine level. This elevated adenosine level, in turn, might provide a driving force for a channel-mediated release. Because pannexins are expressed in both neurons and glia in the CNS (Thompson and MacVicar, 2008), the source of this release could be either neuronal or glial, consistent with the partial inhibition of adenosine efflux by FAc. Alternatively, pannexin1 channels are gated by P2X7 receptors (Pelegrin and Surprenant, 2006), and thereby both participate in the release mechanism. We also have to note that given the low specificity of the available inhibitors of pannexin channels, we cannot exclude other explanations either.
Interestingly, a recent study detected pannexin-mediated ATP release in the CA3 region of the hippocampus (Kawamura et al., 2010), which was not seen in our study. Moreover, we observed an augmentation of the efflux of ATP in the presence of pannexin inhibitors. However, in the study of Kawamura et al., a different stimulus (i.e. mild hypoglycaemia) was used to activate pannexin-mediated ATP release. A potential explanation to the increase in ATP efflux in the presence of pannexin inhibitors is the attenuation of adenosinergic inhibitory neuromodulation on the release of ATP or glutamate in the absence of pannexin-mediated adenosine release. Nevertheless, this possibility, as well as the participation of CFTR in release mechanism of ATP needs further exploration.
In conclusion, our study demonstrated the glial release of ATP, adenosine and glutamate from the hippocampus in response to depolarization of neuronal membrane. K+ depolarization initiated sodium channel activation along the nerve terminals, which elicited the Ca2+-dependent, exocytotic release of a trigger compound (probably glutamate or ATP), leading to the subsequent efflux of ATP from glial cells. ATP was then rapidly metabolized to adenosine through the ecto-nucleotidase cascade. The release mechanism of ATP and glutamate from glial cells involved P2X7 receptors, whilst a part of extracellular adenosine accumulation was independent from extracellular ATP breakdown. The release mechanism demonstrated in our study might be relevant to synaptic modulation and coordination of synaptic networks carried out by ATP, the major astrocytic gliotransmitter in vivo (Halassa et al., 2009).
Acknowledgments
The authors are grateful for Prof Nicholas Dale for teaching and initial guidance in the microelectrode biosensor technique. This study was supported by project grants of the Hungarian Research and Development Fund (grant no. NN79957), the Hungarian Medical Research Council (05–102/2009) and the Pfizer Hungary Ltd (VT44838809) to BS. BS was a recipient of a Royal Society short-term fellowship.
Glossary
- aCSF
artificial CSF
- ARL67156
6-N,N-diethyl-β-γ-dibromomethylene-d-adenosine-5′-triphosphate trisodium salt hydrate
- AZ10606120
dihydrochloride, N-[2-[[2-[(2-hydroxyethyl)amino]ethyl]amino]-5-quinolinyl]-2-tricyclodec-1-ylacetamide dihydrochloride
- BBG
Brilliant blue G
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione-disodium
- D-AP-5
d-(–)-2-amino-5-phosphonopentanoic acid
- dipyridamole
2,6-bis(diethanolamino)-4,8-dipiperidinopyrimido pyrimidine
- fEPSP
field epsp
- FAc
fluoroacetic acid sodium salt
- L-trans-2
4-PDC, l-trans-pyrrolidine-2,4-dicarboxylic acid
- SD
spreading depression
- SNARE
soluble N-ethylmaleimide–sensitive factor attachment protein receptor, TTX, tetrodotoxin
- VNUT
vesicular nucleotide transporter
Conflict of interest
None.
References
- Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H. Purinergic signalling in the nervous system: an overview. Trends Neurosci. 2009;32:19–29. doi: 10.1016/j.tins.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloisio S, Cervetto C, Passalacqua M, Barbieri R, Maura G, Nobile M, et al. Functional evidence for presynaptic P2X7 receptors in adult rat cerebrocortical nerve terminals. FEBS Lett. 2008;582:3948–3953. doi: 10.1016/j.febslet.2008.10.041. [DOI] [PubMed] [Google Scholar]
- Anselmi F, Hernandez VH, Crispino G, Seydel A, Ortolano S, Roper SD, et al. ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc Natl Acad Sci U S A. 2008;105:18770–18775. doi: 10.1073/pnas.0800793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG. Dynamic signaling between astrocytes and neurons. Annu Rev Physiol. 2001;63:795–813. doi: 10.1146/annurev.physiol.63.1.795. [DOI] [PubMed] [Google Scholar]
- Arcuino G, Lin JH, Takano T, Liu C, Jiang L, Gao Q, et al. Intercellular calcium signaling mediated by point-source burst release of ATP. Proc Natl Acad Sci U S A. 2002;99:9840–9845. doi: 10.1073/pnas.152588599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballerini P, Rathbone MP, Di Iorio P, Renzetti A, Giuliani P, D'Alimonte I, et al. Rat astroglial P2Z (P2X7) receptors regulate intracellular calcium and purine release. Neuroreport. 1996;7:2533–2537. doi: 10.1097/00001756-199611040-00026. [DOI] [PubMed] [Google Scholar]
- Bao L, Locovei S, Dahl G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004;72:65–68. doi: 10.1016/j.febslet.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Berg-Johnsen J, Paulsen RE, Fonnum F, Langmoen IA. Changes in evoked potentials and amino acid content during fluorocitrate action studied in rat hippocampal cortex. Exp Brain Res. 1993;96:241–246. doi: 10.1007/BF00227104. [DOI] [PubMed] [Google Scholar]
- Bernath S. Calcium-independent release of amino acid neurotransmitters: fact or artifact? Prog Neurobiol. 1992;38:57–91. doi: 10.1016/0301-0082(92)90035-d. [DOI] [PubMed] [Google Scholar]
- Bowser DN, Khakh BS. ATP excites interneurons and astrocytes to increase synaptic inhibition in neuronal networks. J Neurosci. 2004;24:8606–8620. doi: 10.1523/JNEUROSCI.2660-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone R, Hormuzdi SG, Barbe MT, Herb A, Monyer H. Pannexins, a family of gap junction proteins expressed in brain. Proc Natl Acad Sci U S A. 2003;100:13644–13649. doi: 10.1073/pnas.2233464100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone R, Barbe MT, Jakob NJ, Monyer H. Pharmacological properties of homomeric and heteromeric pannexin hemichannels expressed in Xenopus oocytes. J Neurochem. 2005;92:1033–1043. doi: 10.1111/j.1471-4159.2004.02947.x. [DOI] [PubMed] [Google Scholar]
- Canals S, Larrosa B, Pintor J, Mena MA, Herreras O. Metabolic challenge to glia activates an adenosine-mediated safety mechanism that promotes neuronal survival by delaying the onset of spreading depression waves. J Cereb Blood Flow Metab. 2008;28:1835–1844. doi: 10.1038/jcbfm.2008.71. [DOI] [PubMed] [Google Scholar]
- Clarke DD. Fluoroacetate and fluorocitrate: mechanism of action. Neurochem Res. 1991;16:1055–1058. doi: 10.1007/BF00965850. [DOI] [PubMed] [Google Scholar]
- Coco S, Calegari F, Pravettoni E, Pozzi D, Taverna E, Rosa P, et al. Storage and release of ATP from astrocytes in culture. J Biol Chem. 2003;278:1354–1362. doi: 10.1074/jbc.M209454200. [DOI] [PubMed] [Google Scholar]
- Cotrina ML, Lin JH, Alves-Rodrigues A, Liu S, Li J, Azmi-Ghadimi H, et al. Connexins regulate calcium signaling by controlling ATP release. Proc Natl Acad Sci U S A. 1998;95:15735–15740. doi: 10.1073/pnas.95.26.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig CG, White TD. N-methyl-D-aspartate- and non-N-methyl-D-aspartate-evoked adenosine release from rat cortical slices: distinct purinergic sources and mechanisms of release. J Neurochem. 1993;60:1073–1080. doi: 10.1111/j.1471-4159.1993.tb03256.x. [DOI] [PubMed] [Google Scholar]
- Cunha RA. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem Int. 2001;38:107–125. doi: 10.1016/s0197-0186(00)00034-6. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Ribeiro JA. ATP as a presynaptic modulator. Life Sci. 2000;68:119–137. doi: 10.1016/s0024-3205(00)00923-1. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Vizi ES, Ribeiro JA, Sebastiao AM. Preferential release of ATP and its extracellular catabolism as a source of adenosine upon high- but not low-frequency stimulation of rat hippocampal slices. J Neurochem. 1996;67:2180–2187. doi: 10.1046/j.1471-4159.1996.67052180.x. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Sebastiao AM, Ribeiro JA. Inhibition by ATP of hippocampal synaptic transmission requires localized extracellular catabolism by ecto-nucleotidases into adenosine and channeling to adenosine A1 receptors. J Neurosci. 1998;18:1987–1995. doi: 10.1523/JNEUROSCI.18-06-01987.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, Pearson T, Frenguelli BG. Direct measurement of adenosine release during hypoxia in the CA1 region of the rat hippocampal slice. J Physiol. 2000;526:143–155. doi: 10.1111/j.1469-7793.2000.00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, Hatz S, Tian F, Llaudet E. Listening to the brain: microelectrode biosensors for neurochemicals. Trends Biotechnol. 2005;23:420–428. doi: 10.1016/j.tibtech.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Darby M, Kuzmiski JB, Panenka W, Feighan D, MacVicar BA. ATP released from astrocytes during swelling activates chloride channels. J Neurophysiol. 2003;89:1870–1877. doi: 10.1152/jn.00510.2002. [DOI] [PubMed] [Google Scholar]
- Davidson JS, Baumgarten IM. Glycyrrhetinic acid derivatives: a novel class of inhibitors of gap-junctional intercellular communication. Structure-activity relationships. J Pharmacol Exp Ther. 1988;246:1104–1107. [PubMed] [Google Scholar]
- Duan S, Anderson CM, Keung EC, Chen Y, Swanson RA. P2X7 receptor-mediated release of excitatory amino acids from astrocytes. The. J Neurosci. 2003;23:1320–1328. doi: 10.1523/JNEUROSCI.23-04-01320.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Diao L, Proctor WR. Adenine nucleotides undergo rapid, quantitative conversion to adenosine in the extracellular space in rat hippocampus. J Neurosci. 1997;17:7673–7682. doi: 10.1523/JNEUROSCI.17-20-07673.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Carmignoto G. Neurone-to-astrocyte signalling in the brain represents a distinct multifunctional unit. J Physiol. 2004;559:3–15. doi: 10.1113/jphysiol.2004.063214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Halassa MM, Terunuma M, Succol F, Takano H, Frank M, et al. Endogenous nonneuronal modulators of synaptic transmission control cortical slow oscillations in vivo. Proc Natl Acad Sci U S A. 2009;106:15037–15042. doi: 10.1073/pnas.0906419106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, Burnstock G. Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci. 2006;7:423–436. doi: 10.1038/nrn1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenguelli BG, Llaudet E, Dale N. High-resolution real-time recording with microelectrode biosensors reveals novel aspects of adenosine release during hypoxia in rat hippocampal slices. J Neurochem. 2003;86:1506–1515. doi: 10.1046/j.1471-4159.2003.01957.x. [DOI] [PubMed] [Google Scholar]
- Frenguelli BG, Wigmore G, Llaudet E, Dale N. Temporal and mechanistic dissociation of ATP and adenosine release during ischaemia in the mammalian hippocampus. J Neurochem. 2007;101:1400–1413. doi: 10.1111/j.1471-4159.2006.04425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goding JW, Grobben B, Slegers H. Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim Biophys Acta. 2003;1638:1–19. doi: 10.1016/s0925-4439(03)00058-9. [DOI] [PubMed] [Google Scholar]
- Gualix J, Pintor J, Miras-Portugal MT. Characterization of nucleotide transport into rat brain synaptic vesicles. J Neurochem. 1999;73:1098–1104. doi: 10.1046/j.1471-4159.1999.0731098.x. [DOI] [PubMed] [Google Scholar]
- Guthrie PB, Knappenberger J, Segal M, Bennett MV, Charles AC, Kater SB. ATP released from astrocytes mediates glial calcium waves. J Neurosci. 1999;19:520–528. doi: 10.1523/JNEUROSCI.19-02-00520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. Tripartite synapses: roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology. 2009;57:343–346. doi: 10.1016/j.neuropharm.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG. GLIA: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Huang YJ, Maruyama Y, Dvoryanchikov G, Pereira E, Chaudhari N, Roper SD. The role of pannexin 1 hemichannels in ATP release and cell-cell communication in mouse taste buds. Proc Natl Acad Sci U S A. 2007;104:6436–6441. doi: 10.1073/pnas.0611280104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias R, Spray DC, Scemes E. Mefloquine blockade of Pannexin1 currents: resolution of a conflict. Cell Commun Adhes. 2009;5–6:131–137. doi: 10.3109/15419061003642618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang LH, Mackenzie AB, North RA, Surprenant A. Brilliant blue G selectively blocks ATP-gated rat P2X(7) receptors. Mol Pharmacol. 2000;58:82–88. [PubMed] [Google Scholar]
- Juranyi Z, Sperlagh B, Vizi ES. Involvement of P2 purinoceptors and the nitric oxide pathway in [3H]purine outflow evoked by short-term hypoxia and hypoglycemia in rat hippocampal slices. Brain Res. 1999;823:183–190. doi: 10.1016/s0006-8993(99)01169-5. [DOI] [PubMed] [Google Scholar]
- Kanno T, Nishizaki T. CFTR mediates noradrenaline-induced ATP efflux from DRG neurons. Mol Pain. 2011;7:72. doi: 10.1186/1744-8069-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura M, Jr, Ruskin DN, Masino SA. Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J Neurosci. 2010;30:3886–3895. doi: 10.1523/JNEUROSCI.0055-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi S, Fujishita K, Tsuda M, Shigemoto-Mogami Y, Inoue K. Dynamic inhibition of excitatory synaptic transmission by astrocyte-derived ATP in hippocampal cultures. Proc Natl Acad Sci U S A. 2003;100:11023–11028. doi: 10.1073/pnas.1834448100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latini S, Pedata F. Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem. 2001;79:463–484. doi: 10.1046/j.1471-4159.2001.00607.x. [DOI] [PubMed] [Google Scholar]
- Latini S, Pedata F, Pepeu G. The contribution of different types of calcium channels to electrically-evoked adenosine release from rat hippocampal slices. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:250–255. doi: 10.1007/pl00004939. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31:17–35. doi: 10.1038/jcbfm.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Ting KK, Adams S, Brew BJ, Chung R, Guillemin GJ. Characterisation of the expression of NMDA receptors in human astrocytes. PLoS ONE. 2010;5:e14123. doi: 10.1371/journal.pone.0014123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llaudet E, Botting NP, Crayston JA, Dale N. A three-enzyme microelectrode sensor for detecting purine release from central nervous system. Biosens Bioelectron. 2003;18:43–52. doi: 10.1016/s0956-5663(02)00106-9. [DOI] [PubMed] [Google Scholar]
- Llaudet E, Hatz S, Droniou M, Dale N. Microelectrode biosensor for real-time measurement of ATP in biological tissue. Anal Chem. 2005;77:3267–3273. doi: 10.1021/ac048106q. [DOI] [PubMed] [Google Scholar]
- Marcoli M, Cervetto C, Paluzzi P, Guarnieri S, Alloisio S, Thellung S, et al. P2X7 pre-synaptic receptors in adult rat cerebrocortical nerve terminals: a role in ATP-induced glutamate release. J Neurochem. 2008;105:2330–2342. doi: 10.1111/j.1471-4159.2008.05322.x. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, et al. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;8:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Heuss C, Gahwiler BH, Gerber U. Fast synaptic transmission mediated by P2X receptors in CA3 pyramidal cells of rat hippocampal slice cultures. J Physiol. 2001;535:115–123. doi: 10.1111/j.1469-7793.2001.t01-1-00115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- North RA, Verkhratsky A. Purinergic transmission in the central nervous system. Pflugers Arch. 2006;452:479–485. doi: 10.1007/s00424-006-0060-y. [DOI] [PubMed] [Google Scholar]
- Okada SF, O'Neal WK, Huang P, Nicholas RA, Ostrowski LE, Craigen WJ, et al. Voltage-dependent anion channel-1 (VDAC-1) contributes to ATP release and cell volume regulation in murine cells. J Gen Physiol. 2004;124:513–526. doi: 10.1085/jgp.200409154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Sontheimer H. Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J Neurochem. 2008;107:589–601. doi: 10.1111/j.1471-4159.2008.05615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangrsic T, Potokar M, Stenovec M, Kreft M, Fabbretti E, Nistri A, et al. Exocytotic release of ATP from cultured astrocytes. J Biol Chem. 2007;282:28749–28758. doi: 10.1074/jbc.M700290200. [DOI] [PubMed] [Google Scholar]
- Pankratov Y, Castro E, Miras-Portugal MT, Krishtal O. A purinergic component of the excitatory postsynaptic current mediated by P2X receptors in the CA1 neurons of the rat hippocampus. Eur J Neurosci. 1998;10:3898–3902. doi: 10.1046/j.1460-9568.1998.00419.x. [DOI] [PubMed] [Google Scholar]
- Pankratov Y, Lalo U, Verkhratsky A, North RA. Vesicular release of ATP at central synapses. Pflugers Arch. 2006;452:589–597. doi: 10.1007/s00424-006-0061-x. [DOI] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, et al. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Pearson RA, Dale N, Llaudet E, Mobbs P. ATP released via gap junction hemichannels from the pigment epithelium regulates neural retinal progenitor proliferation. Neuron. 2005;46:731–744. doi: 10.1016/j.neuron.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Pedata F, Pazzagli M, Pepeu G. Endogenous adenosine release from hippocampal slices: excitatory amino acid agonists stimulate release, antagonists reduce the electrically-evoked release. Naunyn Schmiedebergs Arch Pharmacol. 1991;344:538–543. doi: 10.1007/BF00170649. [DOI] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegatti P, Falzoni S, Pinton P, Rizzuto R, Di Virgilio F. A novel recombinant plasma membrane-targeted luciferase reveals a new pathway for ATP secretion. Mol Biol Cell. 2005;16:3659–3665. doi: 10.1091/mbc.E05-03-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfahnl A, Dahl G. Gating of cx46 gap junction hemichannels by calcium and voltage. Pflugers Arch. 1999;437:345–353. doi: 10.1007/s004240050788. [DOI] [PubMed] [Google Scholar]
- Queiroz G, Gebicke-Haerter PJ, Schobert A, Starke K, von Kugelgen I. Release of ATP from cultured rat astrocytes elicited by glutamate receptor activation. Neuroscience. 1997;78:1203–1208. doi: 10.1016/s0306-4522(96)00637-9. [DOI] [PubMed] [Google Scholar]
- Rodrigues RJ, Almeida T, Richardson PJ, Oliveira CR, Cunha RA. Dual presynaptic control by ATP of glutamate release via facilitatory P2X1, P2X2/3, and P2X3 and inhibitory P2Y1, P2Y2, and/or P2Y4 receptors in the rat hippocampus. J Neurosci. 2005;25:6286–6295. doi: 10.1523/JNEUROSCI.0628-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada K, Echigo N, Juge N, Miyaji T, Otsuka M, Omote H, et al. Identification of a vesicular nucleotide transporter. Proc Natl Acad Sci U S A. 2008;105:5683–5686. doi: 10.1073/pnas.0800141105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schock SC, Munyao N, Yakubchyk Y, Sabourin LA, Hakim AM, Ventureyra EC, et al. Cortical spreading depression releases ATP into the extracellular space and purinergic receptor activation contributes to the induction of ischemic tolerance. Brain Res. 2007;1168:129–138. doi: 10.1016/j.brainres.2007.06.070. [DOI] [PubMed] [Google Scholar]
- Serrano A, Haddjeri N, Lacaille JC, Robitaille R. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci. 2006;26:5370–5382. doi: 10.1523/JNEUROSCI.5255-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman W, Locovei S, Dahl G. Probenecid, a gout remedy, inhibits pannexin 1 channels. Am J Physiol Cell Physiol. 2008;295:C761–C767. doi: 10.1152/ajpcell.00227.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81:1065–1096. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- Sperlagh B, Vizi ES. Regulation of purine release. In: Abbracchio MP, Williams M, editors. Handbook of Experimental Pharmacology. Berlin: Springer; 2000. pp. 179–209. [Google Scholar]
- Sperlagh B, Kofalvi A, Deuchars J, Atkinson L, Milligan CJ, Buckley NJ, et al. Involvement of P2X7 receptors in the regulation of neurotransmitter release in the rat hippocampus. J Neurochem. 2002;81:1196–1211. doi: 10.1046/j.1471-4159.2002.00920.x. [DOI] [PubMed] [Google Scholar]
- Sperlagh B, Zsilla G, Baranyi M, Illes P, Vizi ES. Purinergic modulation of glutamate release under ischemic-like conditions in the hippocampus. Neuroscience. 2007;149:99–111. doi: 10.1016/j.neuroscience.2007.07.035. [DOI] [PubMed] [Google Scholar]
- Stout C, Goodenough DA, Paul DL. Connexins: functions without junctions. Curr Opin Cell Biol. 2004;16:507–512. doi: 10.1016/j.ceb.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- Suadicani SO, Brosnan CF, Scemes E. P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J Neurosci. 2006;26:1378–1385. doi: 10.1523/JNEUROSCI.3902-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sul JY, Orosz G, Givens RS, Haydon PG. Astrocytic Connectivity in the Hippocampus. Neuron Glia Biol. 2004;1:3–11. doi: 10.1017/s1740925x04000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szerb JC, Issekutz B. Increase in the stimulation-induced overflow of glutamate by fluoroacetate, a selective inhibitor of the glial tricarboxylic cycle. Brain Res. 1987;410:116–120. doi: 10.1016/s0006-8993(87)80030-6. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Macvicar BA. Connexin and pannexin hemichannels of neurons and astrocytes. Channels (Austin) 2008;2:81–86. doi: 10.4161/chan.2.2.6003. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Jackson MF, Olah ME, Rungta RL, Hines DJ, Beazely MA, et al. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science. 2008;322:1555–1559. doi: 10.1126/science.1165209. [DOI] [PubMed] [Google Scholar]
- Tian F, Gourine AV, Huckstepp RT, Dale N. A microelectrode biosensor for real time monitoring of L-glutamate release. Anal Chim Acta. 2009;645:86–91. doi: 10.1016/j.aca.2009.04.048. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. Mobile NMDA receptors at hippocampal synapses. Neuron. 2002;34:255–264. doi: 10.1016/s0896-6273(02)00658-x. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiunas V, Weingart R. Electrical properties of gap junction hemichannels identified in transfected HeLa cells. Pflugers Arch. 2000;440:366–379. doi: 10.1007/s004240000294. [DOI] [PubMed] [Google Scholar]
- Wall M, Dale N. Activity-dependent release of adenosine: a critical re-evaluation of mechanism. Curr Neuropharmacol. 2008;6:329–337. doi: 10.2174/157015908787386087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall MJ, Dale N. Auto-inhibition of rat parallel fibre-Purkinje cell synapses by activity-dependent adenosine release. J Physiol. 2007;581:553–565. doi: 10.1113/jphysiol.2006.126417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall MJ, Atterbury A, Dale N. Control of basal extracellular adenosine concentration in rat cerebellum. J Physiol. 2007;582:137–151. doi: 10.1113/jphysiol.2007.132050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieraszko A, Goldsmith G, Seyfried TN. Stimulation-dependent release of adenosine triphosphate from hippocampal slices. Brain Res. 1989;485:244–250. doi: 10.1016/0006-8993(89)90567-2. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci. 2003;23:3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, et al. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chen G, Zhou W, Song A, Xu T, Luo Q, et al. Regulated ATP release from astrocytes through lysosome exocytosis. Nat Cell Biol. 2007;9:945–953. doi: 10.1038/ncb1620. [DOI] [PubMed] [Google Scholar]
- Zimmermann H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:299–309. doi: 10.1007/s002100000309. [DOI] [PubMed] [Google Scholar]