

Abstract
2,3,7,8-Tetrachlorodibenzo-ρ-dioxin (TCDD) increases fatty acid (FA) transport and FA levels resulting in hepatic steatosis in mice. Diet as a source of lipids was investigated using customized diets, stearoyl-CoA desaturase 1 (Scd1) null mice, and 14C-oleate (18:1n9) uptake studies. C57BL/6 mice fed with 5, 10, or 15% fat or 50, 60 or 70% carbohydrate diets exhibited increased relative liver weight following gavage with 30 µg/kg TCDD for 168 h. Hepatic lipid extract analysis from mice fed with 5, 10, and 15% fat diets identified a dose-dependent increase in total FAs induced by TCDD. Mice fed with fat diet also exhibited a dose-dependent increase in the dietary essential linoleic (18:2n6) and α-linolenic (18:3n3) acids. No dose-dependent FA increase was detected on carbohydrate diets, suggesting dietary fat as a source of lipids in TCDD-induced steatosis as opposed to de novo lipogenesis. TCDD also induced oleate levels threefold in Scd1 null mice that are incapable of desaturating stearate (18:0). This is consistent with oleate representing > 90% of all monounsaturated FAs in rodent chow. Moreover, TCDD increased hepatic 14C-oleate levels twofold in wild type and 2.4-fold in Scd1 null mice concurrent with the induction of intestinal and hepatic lipid transport genes (Slc27a, Fabp, Ldlr, Cd36, and Apob). In addition, computational scanning identified putative dioxin response elements and in vivo ChIP-chip analysis revealed regions of aryl hydrocarbon receptor (AhR) enrichment in lipid transport genes differentially regulated by TCDD. Collectively, these results suggest the AhR mediates increased uptake of dietary fats that contribute to TCDD-elicited hepatic steatosis.
Key Words: NALFD, diet, metabolic disease.
Activation of the aryl hydrocarbon receptor (AhR), a basic helix-loop-helix Per-AhR nuclear translocator (ARNT)-Sim transcription factor, elicits a broad spectrum of species-specific effects (Denison et al., 2011). Epidemiological and rodent studies have linked exposure to 2,3,7,8-tetrachlorodibenzo-ρ-dioxin (TCDD) and related compounds to dyslipidemia and disrupted energy balance (Lee et al., 2006; Swedenborg et al., 2009). Briefly, ligands bind to the cytosolic AhR, causing a conformational change, dissociation of chaperone proteins, and translocation to the nucleus where it heterodimerizes with ARNT (Hankinson 1995; Pollenz et al., 1994). The complex binds dioxin response elements (DREs) to modulate gene transcription, although DRE-independent binding to DNA has also been reported (Dere et al., 2011b; Murray et al., 2010).
The role of the AhR in hepatotoxicity and lipid metabolism has not been fully elucidated. TCDD-induced hepatic steatosis is characterized by increases in total fatty acids (TFAs), triglycerides (TAGs), vacuolization, inflammatory cell infiltration, and serum alanine aminotransferase levels in C57BL/6 mice (Boverhof et al., 2006; Kopec et al., 2010) that are absent in AhR null mice. AhR also mediates mobilization of peripheral fat (Albro et al., 1978; Lakshman et al., 1991; Pohjanvirta and Tuomisto, 1990), inhibition of FA oxidation (Lakshman et al., 1991; Lee et al., 2010), repression of very low-density lipoprotein (VLDL) secretion (Lee et al., 2010), and alterations in hepatic lipid composition (Angrish et al., 2011).
In this report, we examine diet as a lipid source in TCDD-elicited hepatic steatosis. Dose-dependent increases in hepatic fat accumulation, including essential dietary FAs, suggest dietary fat rather than carbohydrate is an important lipid source in TCDD-elicited steatosis. Increases in hepatic monounsaturated fatty acid (MUFA) levels in Scd1 null mice and hepatic 14C levels provide further evidence that AhR activation increases dietary fat processing that contributes to TCDD-elicited hepatic steatosis. Complementary gene expression analysis integrated with computational DRE search (Dere et al., 2011a) and ChIP-chip (Dere et al., 2011b) data indicate the AhR mediates intestinal and hepatic responses that enhanced dietary fat processing and transport, resulting in hepatic steatosis. Collectively, these results suggest continuous AhR activation may contribute to diseases associated with hepatic steatosis.
MATERIALS AND METHODS
Animal handling. C57BL/6 ovariectomized (ovx) female mice were obtained from Charles Rivers Laboratories (Portage, MI) on postnatal day (PND) 25 with body weights within 10% of the mean body weight upon arrival. B6.129-Scd1 tm1Myz/J heterozygous mice (Miyazaki et al., 2001) (Jackson Laboratory, Ben Harbor, ME) had free access to chow or custom diets and water upon arrival and throughout the study. On PND 21, mice were genotyped and weaned. Mice were maintained on a 12-h light/dark cycle and housed in standard cages containing Aspen woodchips. B6.129-Scd1 tm1Myz/J heterozygous mice were fed Harlan Teklad 7964 F6 Rodent diet (chow). All procedures were carried out with All-University Committee on Animal Use and Care approval.
Diet compositions. Custom diets consisted (by weight) of 19.6% protein, 5, 10, and 15% fat with decreasing carbohydrate (68, 56, and 44%) for an isocaloric intake of 3.7 kcal/g (Table 1). Carbohydrate-adjusted diets consisted (by weight) of constant fat and protein (6.6 and 19.6%, respectively) with increasing total carbohydrate content of 50, 60, and 70% for total caloric intakes of 3.3, 3.7, and 4.1 kcal/g, respectively (Table 1). Custom diets use different ingredients compared with standard chow, which confounds comparisons to other studies. Specifically, custom diet formulations use purified ingredients. In contrast, chow (31% protein, 19% fat, and 50% carbohydrate calories by weight) consists of a proprietary blend of soybean meal, ground corn, wheat, fishmeal, soybean oil, whey, brewer’s yeast, and other vitamins and minerals.
Table 1.
Nutrient and Energy Composition of Custom Diets
| Chow | Isocaloric, fat-adjusted diets | Constant fat, carbohydrate-adjusted diets | |||||
| % Fat | % Carbohydrate | ||||||
| 5% | 10% | 15% | 50% | 60% | 70% | ||
| Metabolizable energy (kcal/g) | 3.1 | 3.7 | 3.7 | 3.7 | 3.3 | 3.7 | 4.1 |
| % by weight | |||||||
| Fat | 6.4 | 5 | 10 | 15 | 6.6 | 6.6 | 6.6 |
| Protein | 24.3 | 18.3 | 18.3 | 18.3 | 18.3 | 18.3 | 18.3 |
| Carbohydrate | 38.7 | 63.6 | 52.4 | 41.1 | 48.6 | 59 | 68.6 |
| Fiber | 11.3 | 3 | 10.5 | 18 | 18 | 7 | — |
Diet study in vivo treatment. On PND 28, mice fed with custom diets (n = 5) were gavaged with 0.1ml of sesame oil (vehicle control) or 30 µg TCDD (Dow Chemical Company, Midland, MI) per kg body weight. Immature ovx mice were used to facilitate comparisons with other data sets, as well as to minimize potential interactions with estrogens from maturing ovaries. The dose was chosen to elicit moderate hepatic effects while avoiding overt toxicity. Animals were sacrificed at 24 and 168 h postdose, weighed, and blood was collected via submandibular vein puncture before sacrifice. Tissue samples were removed, weighed, flash frozen in liquid nitrogen, and stored at −80°C.
Gas chromatography mass spectrometry fatty acid methyl ester hepatic lipid profiling. Hepatic lipid analysis was performed as previously described (Angrish et al., 2011) and as described in Supplementary data. Briefly, liver lipids were extracted by Folch method, dried down under nitrogen, converted to methyl esters and resuspended in hexane. Samples were separated and analyzed by gas chromatography mass spectrometry (GC-MS). 19:1n9 free FA (FFA) and 19:0 TAG were added as extraction efficiency controls, and 17:1n1 FAME (Nu-chek, Elysian, MN) was spiked in as a loading control. Data were analyzed with QuanLynx software and reported as µmol/g liver tissue. FA levels are based on peak areas from total ion chromatograms and µmol/g is obtained from a linear calculation of a calibration curve normalized to sample weight.
14 C-Oleate studies. On PND 28, Scd1 wild type and null mice (n = 5) were gavaged with sesame oil or 30 µg/kg TCDD. 4 h before sacrifice at 120 h, mice were gavaged with 2 µCi 14C-oleate (0.1ml of 20 µCi/ml in sesame oil; ARC 0297; American Radiolabeled Chemicals, St Louis, MO). Blood was collected from the saphenous vein at 0.5, 1, and 2 h after 14C-oleate gavage or the submandibular vein at 4 h. Tissues were harvested, weighed, flash frozen in liquid nitrogen, and stored at −80°C. Duodenum (~3.5cm) and jejunum (~6cm) sections were collected, flushed with phosphate buffered saline, and cut longitudinally. Intestinal epithelium were scraped into vials containing ~1.0ml of TRIzol (Invitrogen, Carlsbad, CA), snap-frozen in liquid nitrogen, and stored at −80°C. For fecal pellet analysis, mice were gavaged with 2 µCi 14C-oleate 120 h post-TCDD dose, and all fecal matter was collected until 48 h after 14C-oleate gavage.
Liver, parametrial adipose, and muscle samples were homogenized in Folch solution (2:1 chloroform:methanol), 0.2ml 40% methanol was added, vortexed, and centrifuged at 10,000 × g for 5min. The organic phase was dried under nitrogen and resuspended in hexane. Samples were directly added to 10ml liquid scintillation fluid (Safety-Solve, RPI, Mount Prospect, IL), and 14C levels counted on a Packard Tri Carb Liquid Scintillation Counter (PerkinElmer; Waltham, MA). Each sample was spiked with 17:1n1 FAME (Nu-chek; Elysian, MN) to control for extraction efficiency and quantified by GC-MS. Liver and adipose samples were normalized to sample weight × whole organ weight. Muscle samples were normalized to sample weight. For 14C levels in fecal pellets, ~30 µg dried pellets were ground to a fine powder with mortar and pestle and added to 10ml liquid scintillation fluid. Fecal samples were normalized to total dry fecal pellet weight. For 14C levels in serum, 5 µl of serum was added directly to scintillation cocktail. Samples were normalized to the average body weight of each mouse.
Quantitative real-time PCR. RNA was isolated from frozen liver samples and intestinal scrapings, and quantitative real-time PCR (QRTPCR) expression was performed as previously described (Boverhof et al., 2006). The copy number of each sample was standardized to the geometric mean of glyceraldehyde-3-phosphate dehydrogenase (Gapdh), hypoxanthine guanine phosphoribosyl transferase (Hprt), and beta actin (Actb) to control for differences in RNA loading, quality, and cDNA synthesis (Vandesompele et al., 2002). Data are reported as the fold change of standardized treated over standardized vehicle values (Supplementary table 5 provides a complete list of gene names and abbreviations).
Statistical analysis. Data were analyzed by analysis of variance (ANOVA) followed by Tukey’s post hoc test in SAS V9.2 (SAS Institute, Cary, NC). Differences between treatment groups were considered significant when p < 0.05.
RESULTS
Body and Liver Weights
Mice fed with fat-adjusted diets had increased relative liver weights (RLWs) (see Supplementary table 1) at 24 and 168 h following a single oral gavage of 30 µg/kg TCDD. In contrast, mice fed with carbohydrate-adjusted diets exhibited increased RLW at 168 h only. There were no significant alterations in body weight or body weight gain throughout the study, suggesting treatment had no effect on feed consumption.
Hepatic Lipid Content in Mice Fed With an Isocaloric, Fat-Adjusted Diet
Vehicle-treated mice exhibited a dose-dependent decrease in TFAs primarily due to decreases in MUFAs (Table 2A). However, TCDD increased TFAs 1.4-, 1.7-, and 2.0-fold in mice fed with 5, 10, and 15% fat diets, respectively (Fig. 1A), compared with diet-matched vehicles. More specifically, absolute levels of saturated FA (SFA), MUFA, and polyunsaturated FA (PUFA) increased with palmitic (16:0) and oleic (18:1n9) acids representing 80–90% of all hepatic SFAs and MUFAs, respectively (Supplementary table 2). Note that palmitic and oleic acids represent > 66 and > 98% of dietary SFAs and MUFAs, respectively, in the fat-adjusted diets (Supplementary table 3A), with absorption efficiencies of > 90% (Labonte et al., 2008).
Table 2.
Hepatic Lipid Levels in Mice Fed With Fat- or Carbohydrate-Adjusted Diets 168 h Post 30 µg/kg TCDD Dose
| A. Fat-adjusted diet | Treatment | % Fat | ||
| 5% | 10% | 15% | ||
| Total FA | Vehicle | 135.6±16.7 | 113.5±9.5 | 96.3±7.1**** |
| TCDD | 195.0±14.0* | 194.3±16.4* | 188.0±15.6* | |
| Total SFA | Vehicle | 40.9±3.3 | 41.9±3.8 | 36.6±2.0 |
| TCDD | 53.9±3.3*,** | 53.8±1.4* | 47.7±3.8* | |
| Total MUFA | Vehicle | 54.1±12.1 | 31.0±4.8*** | 18.2±2.8**** |
| TCDD | 92.6±12.0*,** | 66.0±11.0* | 54.8±7.3* | |
| Total PUFA | Vehicle | 33.2±2.7 | 40.6±2.6 | 41.5±2.9 |
| TCDD | 47.7±2.8*,**,*** | 74.4±4.4*,** | 85.5±7.2* | |
| B. Carbohydrate-adjusted diet | Treatment | % Carbohydrate | ||
| 50% | 60% | 70% | ||
| Total FA | Vehicle | 108.8±24.6 | 113.0±4.7 | 135.9±9.8 |
| TCDD | 156.8±7.6* | 148.6±22.3** | 193.0±34.8* | |
| Total SFA | Vehicle | 45.1±7.74 | 46.7±3.2 | 49.7±4.9 |
| TCDD | 54.3±2.7 | 51.0±5.4** | 64.5±8.7* | |
| Total MUFA | Vehicle | 28.4±11.6 | 31.0±2.6 | 51.2±6.1 |
| TCDD | 50.7±5.5** | 51.3±12.8 | 79.8±26.0* | |
| Total PUFA | Vehicle | 35.4±5.6 | 35.3±3.3 | 35.0±2.4 |
| TCDD | 51.8±2.5* | 46.3±4.9* | 48.7±5.0* | |
Note. A. Fat-adjusted diet: *p < 0.05 for TCDD compared with vehicle within a diet, **p < 0.05 for TCDD 15% fat compared with TCDD 5 or 10%, ***p < 0.05 for TCDD 5% fat compared with TCDD 10%, ****p < 0.05 for vehicle 5% compared with vehicle 10% or 15%, n = 5.
B. Carbohydrate-adjusted diet: *p < 0.05 for TCDD compared with vehicle within a diet, **p < 0.05 for TCDD 70% carbohydrate compared with TCDD 50 or 60% carbohydrate, n = 5.
FIG. 1.
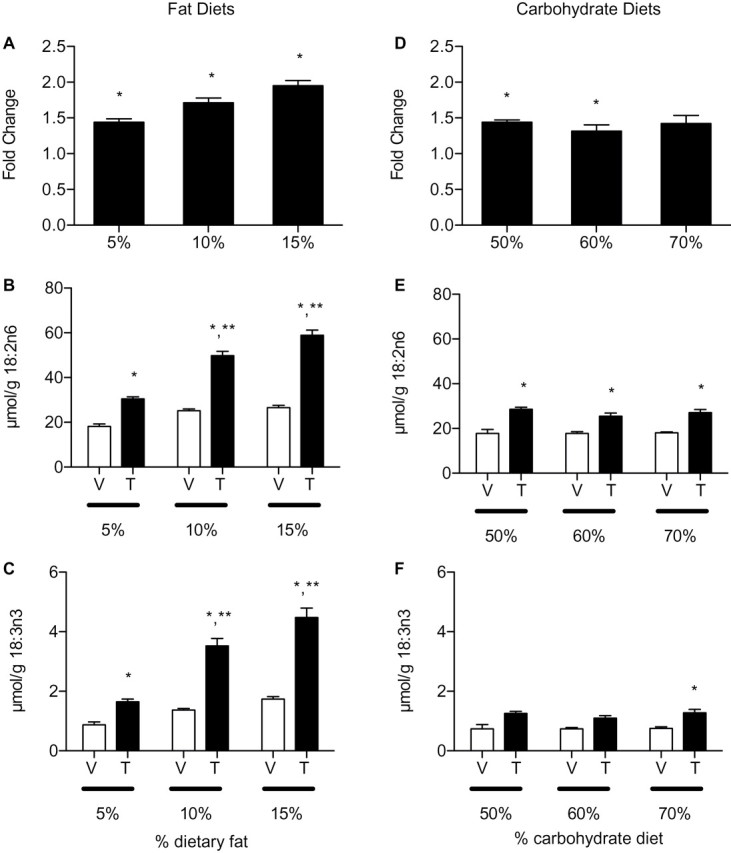
Hepatic absolute and essential FA levels in mice fed with increasing fat or carbohydrate diets treated with sesame oil vehicle (V) or 30 µg/kg TCDD (T) for 168 h. (A–C) Mice fed with 5, 10, and 15% fat diet, (A) total fatty acids (TFA), (B) α-linoleic acid (18:2n6), and (C) α-linolenic acid (18:3n3) levels. *p < 0.05 for TCDD compared with vehicle within a diet, **p < 0.05 for TCDD 15% fat or 10% fat compared with TCDD 5% fat. (D–F) Mice fed with 50, 60, and 70% carbohydrate diet, (D) TFAs, (E) 18:2n6, and (F) 18:3n3. *p < 0.05 for TCDD compared with vehicle within a diet; **p < 0.05 for TCDD 70% carbohydrate compared with TCDD 50% carbohydrate. (A–F) Bars represent mean ± standard error of the mean (SEM), n = 5.
Hepatic PUFAs also exhibited a dose-dependent increase in dietary fat (Table 2A) primarily due to linoleic acid (18:2n6) accumulation (Fig. 1B). 18:2n6, which represents ~65% of all hepatic PUFAs, is an essential FA that can only be acquired from the diet with a reported absorption efficiency of > 95% (Labonte et al., 2008). α-Linolenic acid (18:3n3), another dietary essential FA, exhibited similar hepatic increases with increasing dietary fat content that was further induced by TCDD (Fig. 1C). 18:2n6 and 18:3n3 represent > 99% of the PUFA content in isocaloric, fat-adjusted diets (Supplementary table 3A). These results suggest that AhR activation enhances dietary fat processing and/or transport that contributes to TCDD-elicited hepatic steatosis.
Hepatic Lipid Content in Mice Fed With a Constant Fat, Carbohydrate-Adjusted Diet
Mice fed with 50, 60, or 70% carbohydrate diets did not exhibit a dose-dependent increase in hepatic TFA following TCDD treatment. TCDD induced TFAs ~1.4-fold across all carbohydrate-adjusted diets (Fig. 1D). Absolute SFA and MUFA levels increased 1.3- and 1.6-fold, respectively, in mice fed with 70% carbohydrate diet compared with controls (Table 2B). 16:0, 18:1n9, and 18:2n6 were the predominant hepatic SFA, MUFA, and PUFA species, respectively, similar to the composition in mice fed with fat-adjusted diets (Supplementary table 4). However, 18:2n6 and 18:3n3 levels, the essential dietary FAs, remained constant across TCDD-treated mice on carbohydrate diets (Figs. 1E and 1F), suggesting dietary carbohydrate is not a significant contributor to AhR-mediated steatosis.
14C-Oleate Studies
Lipid accumulation in the liver was investigated in Scd1 wild type and null mice gavaged with 2 µCi 14C-oleate 5 days postdose with 30 µg/kg TCDD. Scd1 performs the rate-limiting step in MUFA synthesis. Therefore, null mice are incapable of desaturating 18:0 to 18:1n9, yet 18:1n9 is still available via the diet (Supplementary table 3). 14C-levels in hepatic lipid extracts increased twofold and 2.4-fold in TCDD-treated wild type and Scd1 null mice, respectively (Fig. 2A). This increase is consistent with the approximately twofold oleate increase in mice fed with fat- or carbohydrate-adjusted diets (Supplementary tables 2 and 4), and the approximately threefold increase in 18:1n9 levels in TCDD-treated Scd1 wild type and null mice (Fig. 2B) (Angrish et al., 2011). The absorption efficiency of oleate is reported to be > 95% (Labonte et al., 2008). Furthermore, oleate represents > 90% of total MUFAs in treated mouse livers (Fig. 2C) (Angrish et al., 2011). TCDD increased serum (1.4-fold) and muscle (1.2-fold) and decreased adipose (–1.3-fold) and stool (–1.5-fold) 14C levels, although statistical significance was not achieved (Supplementary figs. 1A–D).
FIG. 2.

Hepatic 14C and lipid levels in Scd1 wild type and null mice 120 h postdose with 30 µg/kg TCDD. (A) 14C-levels were measured in lipid extracts by liquid scintillation counting after gavage with 2 µCi 14C-oleate 4 h prior to sacrifice. Gas chromatography mass spectrometry analysis of hepatic oleate (18:1n9) (B) and monounsaturated fatty acid (C) levels (expressed in nmol/g) in Scd1 wild type and null mice 168 h after oral gavage with 30 µg/kg TCDD (Angrish et al., 2011). *p < 0.05 for TCDD compared with vehicle, **p < 0.05 for Scd1 wild type TCDD compared with Scd1 null mice TCDD. Bars represent mean ± SEM, n = 5.
Differential Intestinal and Hepatic Gene Expression
To further examine the effect of AhR activation on dietary lipid uptake, intestinal and hepatic gene expression was examined. Of the genes examined, > 80% contained a putative, functional DRE (matrix similarity score > 0.847) and/or had hepatic AhR enrichment in ChIP-chip analysis (Table 3) (Dere et al., 2011a,b).
Table 3.
DREsa and Regions of AhR Enrichmentb in TCDD Responsive Genes Associated With Lipid and Carbohydrate Transport and Metabolism
| Gene ID | Gene symbol | Liver | Duodenum | Jejunum | ** of DREsa | ChIP peaks 2 hb | Functionc | Regulated byc |
| Xenobiotic metabolism | ||||||||
| 13076 | Cyp1a1 | 5799* | 222* | 22.7* | 7 | 4 | Xenobiotic metabolism | AhR |
| Fatty acid and triglyceride synthesis | ||||||||
| 14104 | Fasn | −2.8* | NC | NC | 4 | 1 | Fatty acid synthesis | SREBP, TR, LXR, cAMP, AMPK |
| 153674 | Acly | −2.6*** | nd | nd | 0 | 0 | Citrate metabolism | Oxaloacetate, ATP |
| 107476 | Acaca | −1.4*** | nd | nd | 3 | 5 | Malonyl CoA acylation | glucagon |
| 68393 | Mogat1 | 3.6* | NC | NC | 1 | 0 | Triglyceride synthesis | PPAR, CEBP |
| 233549 | Mogat2 | 3.0* | NC | 1.4* | 4 | 3 | ||
| 13350 | Dgat1 | 2.0 | 1.4 | 1.6* | 4 | 0 | ||
| 67800 | Dgat2 | 1.3 | 1.4 | 1.6 | 2 | 7 | ||
| Fatty acid transport | ||||||||
| 238055 | Apob | 2.5* | NC | 1.8* | 2 | 0 | VLDL and chylomicron assembly | APOBEC-1 |
| 16835 | Ldlr | 3.2* | 2.3* | NC | 2 | 4 | Lipoprotein uptake | LXR |
| 12491 | Cd36 | 4.4* | 2.7* | NC | 0 | 1 | Fatty acid uptake | PPAR, CEBPα, AMPK |
| 26457 | Slc27a1 | −2.9* | 2.3* | −5.8** | 2 | 0 | Mitochondrial β-oxidation | PPARα, PPARγ |
| 26568 | Slc27a3 | 2.7* | NC | NC | 1 | 0 | Unknown | |
| 26569 | Slc27a4 | 3.0* | 1.4** | 1.4** | 0 | 0 | Peroxisomal β-oxidation,CHOL-ester synthesis | PPARγ, SREBP1c |
| 14080 | Fabp1 | 2.9* | 1.4* | 1.7* | 2 | 4 | TAG synthesis, β-oxidation | PPARα, HNF4α |
| 11770 | Fabp4 | 1.4** | 1.5 | 2.5 | 0 | 0 | Chylomicron assembly | cJun, PPARγ |
| 117147 | Acsm1 | −2.0*** | nd | nd | 3 | 0 | Medium-chain fatty acid transport (mitochondrial β-oxidation) | Acetyl-CoA, malonyl-CoA, NADPH, NADH |
| 233799 | Acsm2 | −1.5*** | nd | nd | 0 | 0 | ||
| 20216 | Acsm3 | −1.6*** | nd | nd | 2 | 0 | ||
| 233801 | Acsm4 | −1.2*** | nd | nd | 0 | 0 | ||
| 14081 | Acsl1 | −1.4*** | nd | nd | 6 | 5 | Long-chain fatty acid transport (mitochondrial β-oxidation) | |
| 74205 | Acsl3 | −2.3*** | nd | nd | 0 | 0 | ||
| 50790 | Acsl4 | −1.3*** | nd | nd | 0 | 2 | ||
| Fatty acid metabolism | ||||||||
| 16956 | Lpl | 3.2*** | nd | nd | 2 | 0 | TAG metabolism of lipoproteins and chylomicrons | Insulin, glucagon, epinephrine |
| 109791 | Clps | 2.6*** | nd | nd | 0 | 0 | ||
| 18946 | Pnliprp1 | 3.7*** | nd | nd | 0 | 0 | ||
| 11343 | Mgll | 1.4*** | nd | nd | 9 | 0 | Monoglyceride metabolism | PPARα |
| 116939 | Pnpla3 | −3.3*** | nd | nd | 0 | 1 | TAG metabolism | |
| Glycolysis/Gluconeogenesis/Glycogen synthesis | ||||||||
| 18534 | Pck1 | −2.0*** | nd | nd | 0 | 1 | Gluconeogenesis | Insulin, glucagon, cAMP |
| 14377 | G6pc | −2.7*** | nd | nd | 1 | 5 | Insulin, glucose | |
| 18563 | Pcx | −1.3*** | nd | nd | 5 | 4 | ATP | |
| 103988 | Gck | −1.5*** | nd | nd | 3 | 2 | Glucose metabolism | G6P, insulin, glucagon |
| 212032 | Hk3 | 2.2*** | nd | nd | 1 | 0 | ||
| 232493 | Gys2 | −1.5*** | nd | nd | 2 | 4 | ||
Note. NC, no change; nd, not detected.
aDRE distributions were previously determined (Dere et al., 2011a). Only DREs satisfying a matrix similarity score of ≥ 0.85 were included.
bAhR enrichment was previously determined (Dere et al., 2011b).
cData from Boron and Boulpaep (2008), Hardwick et al. (2009), and Watkins (2008).
*p < 0.05 or **p < 0.01 for TCDD compared with vehicle, QRTPCR data at 24 h postdose. QRTPCR data were analyzed by Dunnett’s t-test, n = −5. *** for P1(t) ≥ 0.999 for microarray data at 168 h postdose (Dere et al., 2011a).
Dietary FAs (> 16C) hydrolyzed from TAG by gastric and pancreatic lipases in the intestinal lumen are actively transported into enterocytes before export into the lymphatic and systemic circulation (Iqbal and Hussain, 2009). In the duodenum, TCDD induced low-density lipoprotein receptor (Ldlr, 2.3-fold), Cd36 antigen (Cd36, 2.7-fold), and solute carrier family 27 (FA transporter), member 4 (Slc27a4, 1.4-fold), as well as FA binding protein 1, liver (Fabp1, 1.4-fold), and FA binding protein 4, adipocyte (Fabp4, 1.4-fold) (Table 3). Although implicated in mitochondrial β-oxidation (Hardwick et al., 2009; Watkins, 2008) and insulin sensing (Wu et al., 2006), solute carrier family 27 (FA transporter), member 1 (Slc27a1) expression (2.3-fold in duodenum, −5.8-fold in jejunum) occurs primarily in muscle and adipose tissue making its role in other tissues uncertain.
Endothelial lipases hydrolyze serum lipids absorbed by the intestine. The resulting products are taken up by the liver via facilitated transport or receptor-mediated endocytosis. TCDD induced hepatic long-chain FA uptake of family members Cd36 (4.4-fold), solute carrier family 27 (FA transporter), member 3 (Slc27a3, 2.7-fold), Slc27a4 (3.0-fold), and Ldlr (3.2-fold) (Table 3). Hydrolytic cleavage of TAG by cytosolic lipases further adds FAs to the hepatic pool. TCDD induced lipoprotein lipase (Lpl, 3.2-fold), pancreatic colipase (Clps, 2.6-fold), pancreatic lipase-related protein 1 (Pnliprp1, 3.7-fold), and monoglyceride lipase (Mgll, 1.4-fold), but repressed patatin-like phospholipase domain containing 3 (Pnpla3, 3.3-fold) (Table 3). Interestingly, sequence variations in PNPLA3 are associated with hepatic TAG content and nonalcoholic fatty liver disease (NAFLD) in humans (Romeo et al., 2008).
Intracellular FAs are directed to TAG biosynthetic and β-oxidation pathways by Fabps. Fabp1 and 4 were induced 2.9- and 1.4-fold, respectively. However, TCDD repressed hepatic mitochondrial medium- and long-chain acyl-CoA synthetase genes (Acsm1-4 and Acsl1, 3-4, repressed 1.3 to 2.3-fold) that activate FAs for transport into the inner membrane space for subsequent β-oxidation (Table 3). These gene expression changes are consistent with the reported inhibition of mitochondrial β-oxidation by TCDD (Lakshman et al., 1991; Lee et al., 2010). Furthermore, TCDD induced the expression of hepatic TAG biosynthesis genes (monoacylglycerol O-acyltransferase 1 and 2 [Mogat1 and Mogat2] and diacylglycerol O-acyltransferase 1 and 2 [Dgat1 and Dgat2] induced 3.6-, 3.0-, 1.6-, and 1.3-fold, respectively), consistent with hepatic TAG accumulation (Angrish et al., 2011; Kopec et al., 2010).
Hepatic differential gene expression is also consistent with TCDD-elicited disruption of carbohydrate catabolism (Table 3). TCDD induced hexokinase 3 (Hk3) 2.2-fold, which catalyzes the irreversible phosphorylation of glucose to glucose-6-phosphate (G6P). In contrast, genes involved in gluconeogenesis (pyruvate carboxylase [Pcx] −1.3-fold, phosphoenolpyruvate carboxykinase 1, cytosolic [Pck1] −2.0-fold, and glucose-6-phosphatase, catalytic [G6pc] −2.7-fold) and glycogen synthesis (glucokinase [Gck] −1.5-fold and glycogen synthase 2 [Gys2] −1.5-fold) were repressed. Similarly, genes that provide FA synthesis substrates, such as ATP-citrate lyase (Acly −2.6-fold) and acetyl-CoA carboxylase (Acaca −1.4-fold), and fatty acid synthetase (Fasn, −2.8-fold) were repressed (Table 3). These changes are consistent with the lack of a dose-dependent increase in hepatic TFAs through de novo lipogenesis in mice fed with carbohydrate-adjusted diets. Furthermore, reported changes in hepatic gene expression are consistent with hepatic TCDD reported in the same model (Boverhof et al., 2006).
DISCUSSION
Hepatic steatosis can result from the disruption of multiple processes involved in lipid and carbohydrate uptake, metabolism, and efflux. Our studies provided evidence that dietary fat, rather than carbohydrate, is an important lipid source in TCDD-elicited steatosis in mice. Computational DRE search, ChIP-chip, and gene expression (Dere et al., 2011b) data indicate AhR activation results in a coordinated response involving the digestive, circulatory, and hepatic systems (Fig. 3). This suggests any ligand (e.g., chemical, drug, endogenous substance, and natural product) capable of activating the AhR may enhance dietary fat processing and transport, although continuous exposure may be required.
FIG. 3.

AhR-mediated increase in dietary lipid in TCDD-elicited hepatic steatosis. Steps 1–4: Fat absorption by the intestinal epithelium and export to the circulatory system. Steps 5–11: Enhanced hepatic fatty acid uptake and storage. Inhibition of efflux and β-oxidation-mediated degradation pathways. Steps 12–17: Hepatic glucose metabolism including glycogen synthesis, gluconeogenesis and fatty acid synthesis are inhibited. Lines with arrowheads indicate reaction/pathway direction. Lines with blunted ends indicate reactions/pathways that are inhibited. Red boxes indicate induced gene expression. Green boxes indicate repressed gene expression. A more detailed description is provided in the Discussion section.
Increases in hepatic 14C levels clearly demonstrated diet as a source of lipids in TCDD-elicited hepatic steatosis. Previous studies have implicated the mobilization of peripheral adipose tissue based on increased serum 16:0, 18:1, 18:2, and 18:3 FFA levels and their abundance in adipose tissue (Albro et al., 1978; Lakshman et al., 1991; Pohjanvirta et al., 1990). However, these FAs also represent the primary lipids in chow (Supplementary table 3). Furthermore, increases in oleate, the primary MUFA in rodent chow, and hepatic 14C levels in Scd1 null mice provide further evidence of a role for dietary fat in AhR-mediated steatosis.
Complementary gene expression analysis is consistent with a role for the AhR in mediating hepatic accumulation of dietary lipids (Fig. 3). FFAs hydrolyzed by pancreatic and gastric lipases, colipases, and bile salts passively diffuse (FA < 16C) and are actively transported (FA > 16C) into enterocytes by Cd36 and Slc27a4 that were induced by TCDD (steps 1–3). A role for Cd36 in intestinal lipid clearance and FFA uptake has been demonstrated in null mice (Drover et al., 2005), whereas Slc27a4 (Fatp4) is associated with obesity in humans (Gertow et al., 2004). Once intracellular, FA binding proteins (Fabps) sequester FAs to prevent their transport back into the intestinal lumen and target them to specific organelles (Smathers and Petersen, 2011). TCDD inducible Fabp1, unlike other family members, binds two rather than one FA, as well as other small hydrophobic ligands (Storch and Thumser, 2010), and is involved in intestinal FA processing and chylomicron maturation (step 4) (Neeli et al., 2007; Storch and McDermott, 2009). Although intestinal lipid absorption is highly efficient (Labonte et al., 2008; Simon et al., 2011,), AhR activation may enhance intestinal lipid processing and efflux, consistent with TCDD-induced increases in serum FFAs and TAGs (Boverhof et al., 2006).
The concurrent induction of several hepatic genes associated with lipid transport, processing, and metabolism further promotes steatosis (Fig. 3). Ldlr, Cd36, and Slc27a actively transport increased circulating FFAs, chylomicrons, and lipoprotein remnants into the liver (step 5). TCDD also induced Lpl, monoglyceride lipase Mgll, Pnliprp1, and pancreatic colipase (Clps) that hydrolyze intracellular lipoprotein remnants to further increase the intracellular FA pool (step 6). Induced Fabp1 binds and sequesters intracellular FAs and targets them for TAG synthesis (Atshaves et al., 2010). Mogat1/2 and Dgat1/2, induced by TCDD, then facilitate hepatic TAG biosynthesis (steps 7 and 8). TAGs are stored in lipid droplets (step 9), or incorporated into VLDLs (step 10). However, TCDD inhibits VLDL secretion (step 10) (Lee et al., 2010), consistent with AhR-mediated increases in hepatic TAG and vacuolization (Angrish et al., 2011; Miyazaki et al., 2000).
Fabp1 also targets FAs for mitochondrial β-oxidation (Atshaves et al., 2010). TCDD inhibits FA oxidation (Lakshman et al., 1991; Lee et al., 2010) possibly by inhibiting transport into mitochondria, further adding to hepatic FA accumulation. More specifically, TCDD inhibits medium- and long-chain FA mitochondrial acyl-CoA synthetase gene expression (Acsm1-4, Ascl1, and 3-4) (step 11) that is required for transport across the mitochondrial matrix via carnitine for subsequent β-oxidation. Yet, TCDD induced ketone body accumulation in vitro (Lakshman et al., 1991), suggesting an intact carnitine pathway. Nonetheless, plasma ketones do not increase in response to TCDD exposure in vivo (Pohjanvirta and Tuomisto, 1994) and requires additional investigation.
The inability of carbohydrate diets to enhance hepatic steatosis appears to involve TCDD dysregulation of carbohydrate metabolism gene expression (step 12). For example, anabolic pathways typically dominate in fed animals, yet TCDD decreased glucokinase (Gck) and glycogen synthase (Gys2), suggesting suppression of hepatic glycogen synthesis (step 13). Glucokinase is the predominant enzyme regulating hepatic glucose metabolism in response to nutritional states such as refeeding and insulin stimulation (Agius, 2008). Although TCDD induced hexokinase (Hk3), this enzyme is expressed at low levels in hepatocytes, yet exhibits compensatory induction following glucokinase repression, as found in liver cirrhosis (Lowes et al., 1998). TCDD also inhibited mitochondrial pyruvate carboxylase (Pcx) that converts pyruvate into oxaloacetate, suggesting flux toward glycerol production to support hepatic TAG production leading to greater sequestration of hepatic FA. Metabolomic studies also report TCDD increases hepatic glycerol levels (Forgacs et al., 2012) and that TCDD treatment prevents glycerol ketogenesis without affecting esterification to TAG (Lakshman et al., 1991).
TCDD suppressed phosphoenolpyruvate carboxykinase (Pck1) and glucose-6 phosphatase (G6pc) expression, key regulators of gluconeogenesis (steps 14 and 16). These genes are commonly regulated by peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α (PGC-1α) (Burgess et al., 2006) that is functionally impaired by AhR-mediated induction of TCDD-inducible poly(ADP-ribose) polymerase 7, PARP7 (TiPARP) (Diani-Moore et al., 2010). TCDD also repressed ATP-citrate lyase (Acly), which converts citrate to oxaloacetate and acetyl-CoA (step 15). Acetyl CoA is critical for de novo FA synthesis by Fasn (step 17), which was dose dependently repressed by TCDD, TCDF, and PCB126 (Kopec et al., 2011).
These gene expression changes are consistent with TCDD-mediated inhibition of gluconeogenesis (Viluksela et al., 1999), de novo lipogenesis (Lakshman et al., 1988), and FA oxidation, but roughly concordant with DRE distributions and AhR ChIP-chip peaks (Table 3). This may be partially explained by AhR interactions with other signaling pathways involved in hepatic glucose and FA metabolism regulation including PPARs (Table 3), Pgc1α (Diani-Moore et al., 2010), Forkhead box O1 (Foxo1) (Matsumoto et al., 2007), and hepatocyte nuclear factor 4, alpha (HNF4α) (Hardwick et al., 2009). Evidence suggests AhR and PPAR signaling pathways interact (Alexander et al., 1998; Liu and Matsumura, 1995; Remillard and Bunce, 2002) to alter PPAR expression (Wang et al., 2011). Other studies identified overrepresentation of PPAR and HNF4α binding motifs in ChIP-chip regions of AhR enrichment that lack DRE cores, suggesting AhR binding to DNA independent of DREs (Dere et al., 2011b; Murray et al., 2010). For example, AhR interacts with chicken ovalbumin upstream promoter transcription factor (COUP-TF) (Klinge et al., 2000), and COUP-TF is reported to antagonize HNF4α-mediated responses by binding to HNF4α response elements (Mietus-Snyder et al., 1992). Consequently, AhR-COUP-TF complexes binding to HNF4α response elements may inhibit HNF4α-regulated lipid transport, metabolism, and gene expression and contribute to TCDD-elicited steatosis (Dere et al., 2011b). Interestingly, hepatic steatosis has been reported in HNF4α null mice (Hayhurst et al., 2001).
Collectively, our data indicate that TCDD-mediated hepatic steatosis involves enhanced uptake of dietary fat suggesting a novel endogenous role for the AhR. Other ligands including endogenous metabolites (indoles, tetrapyrroles, and arachidonic acid metabolites), and natural products (e.g., vegetable-, fruit-, and tea-derived indole and flavonoid metabolites) (Denison and Nagy, 2003; Nguyen and Bradfield, 2008) also activate the AhR providing a possible selective evolutionary advantage that optimizes fat absorption to maximize energy intake. Interactions with other nuclear receptors and transcription factors can further impact energy homeostasis and lipid metabolism, transport, and deposition. However, persistent AhR activation in combination with the consumption of a high-fat diet may also have adverse health implications for fatty liver and its associated diseases including NAFLD, metabolic syndrome, and diabetes. Preliminary studies indicate AhR activation increases TFA levels in human primary hepatocytes (data not shown), although further studies are needed to elucidate species-specific differences in AhR-mediated effects including steatosis.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institute of Environmental Health Sciences Superfund Research Program (P42ES04911).
ACKNOWLEDGMENTS
We thank Claudia Dominici for technical support and Dr Anna Kopec, Rance Nault, and Agnes Forgacs for critical review.
REFERENCES
- Albro P. W., Corbett J. T., Harris M.,, Lawson L. D. (1978). Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on lipid profiles in tissue of the Fischer rat Chem. Biol. Interact. 23 315–330 [DOI] [PubMed] [Google Scholar]
- Alexander D. L., Ganem L. G., Fernandez-Salguero P., Gonzalez F.,, Jefcoate C. R. (1998). Aryl-hydrocarbon receptor is an inhibitory regulator of lipid synthesis and of commitment to adipogenesis J. Cell Sci. 111(Pt 22)3311–3322 [DOI] [PubMed] [Google Scholar]
- Angrish M. M., Jones A. D., Harkema J. R.,, Zacharewski T. R. (2011). Aryl hydrocarbon receptor-mediated induction of stearoyl-CoA desaturase 1 alters hepatic fatty acid composition in TCDD-elicited steatosis Toxicol. Sci. 124 299–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atshaves B. P., Martin G. G., Hostetler H. A., McIntosh A. L., Kier A. B.,, Schroeder F. (2010). Liver fatty acid-binding protein and obesity J. Nutr. Biochem. 21 1015–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boron W., Boulpaep E. Medical Physiology. Saunders,; Philadelphia, PA: (2008). [Google Scholar]
- Boverhof D. R., Burgoon L. D., Tashiro C., Sharratt B., Chittim B., Harkema J. R., Mendrick D. L., Zacharewski T. R. (2006). Comparative toxicogenomic analysis of the hepatotoxic effects of TCDD in Sprague Dawley rats and C57BL/6 mice Toxicol. Sci. 94 398–416 [DOI] [PubMed] [Google Scholar]
- Burgess S. C., Leone T. C., Wende A. R., Croce M. A., Chen Z., Sherry A. D., Malloy C. R., Finck B. N. (2006). Diminished hepatic gluconeogenesis via defects in tricarboxylic acid cycle flux in peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1alpha)-deficient mice J. Biol. Chem. 281 19000–19008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison M. S., Nagy S. R. (2003). Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals Annu. Rev. Pharmacol. Toxicol. 43 309–334 [DOI] [PubMed] [Google Scholar]
- Denison M. S., Soshilov A. A., He G., DeGroot D. E.,, Zhao B. (2011). Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor Toxicol. Sci. 124 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dere E., Forgacs A. L., Zacharewski T. R.,, Burgoon L. D. (2011a). Genome-wide computational analysis of dioxin response element location and distribution in the human, mouse, and rat genomes Chem. Res. Toxicol. 24 494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dere E., Lo R., Celius T., Matthews J.,, Zacharewski T. R. (2011b). Integration of genome-wide computation DRE Search, AhR ChIP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver BMC Genomics 12 365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diani-Moore S., Ram P., Li X., Mondal P., Youn D. Y., Sauve A. A., Rifkind A. B. (2010). Identification of the aryl hydrocarbon receptor target gene TiPARP as a mediator of suppression of hepatic gluconeogenesis by 2,3,7,8-tetrachlorodibenzo-p-dioxin and of nicotinamide as a corrective agent for this effect J. Biol. Chem. 285 38801–38810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drover V. A., Ajmal M., Nassir F., Davidson N. O., Nauli A. M., Sahoo D., Tso P., Abumrad N. A. (2005). CD36 deficiency impairs intestinal lipid secretion and clearance of chylomicrons from the blood J. Clin. Invest. 115 1290–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgacs A. L., Kent M. N., Makley M. K., Mets B., DelRaso N., Jahns G. L., Burgoon L. D., Zacharewski T. R., Reo N. V. (2012). Comparative metabolomic and genomic analyses of TCDD-elicited metabolic disruption in mouse and rat liver Toxicol. Sci. 125 41–55 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertow K., Pietilainen K. H., Yki-Jarvinen H., Kaprio J., Rissanen A., Eriksson P., Hamsten A., Fisher R. M. (2004). Expression of fatty-acid-handling proteins in human adipose tissue in relation to obesity and insulin resistance Diabetologia 47 1118–1125 [DOI] [PubMed] [Google Scholar]
- Hankinson O. (1995). The aryl hydrocarbon receptor complex Annu. Rev. Pharmacol. Toxicol. 35 307–340 [DOI] [PubMed] [Google Scholar]
- Hardwick J. P., Osei-Hyiaman D., Wiland H., Abdelmegeed M. A.,, Song B. J. PPAR/RXR regulation of fatty acid metabolism and fatty acid omega-hydroxylase (CYP4) isozymes: Implications for prevention of lipotoxicity in fatty liver disease. PPAR Res. (2009);2009:952734. doi: 10.1155/2009/952734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayhurst G. P., Lee Y. H., Lambert G., Ward J. M.,, Gonzalez F. J. (2001). Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis Mol. Cell Biol. 21 1393–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal J., Hussain M. M. (2009). Intestinal lipid absorption Am. J. Physiol. Endocrinol. Metab. 296 E1183–E1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge C. M., Kaur K.,, Swanson H. I. (2000). The aryl hydrocarbon receptor interacts with estrogen receptor alpha and orphan receptors COUP-TFI and ERRalpha1 Arch. Biochem. Biophys. 373 163–174 [DOI] [PubMed] [Google Scholar]
- Kopec A. K., Burgoon L. D., Ibrahim-Aibo D., Mets B. D., Tashiro C., Potter D., Sharratt B., Harkema J. R., Zacharewski T. R. (2010). PCB153-elicited hepatic responses in the immature, ovariectomized C57BL/6 mice: Comparative toxicogenomic effects of dioxin and non-dioxin-like ligands Toxicol. Appl. Pharmacol. 243 359–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec A. K., D’Souza M. L., Mets B. D., Burgoon L. D., Reese S. E., Archer K. J., Potter D., Tashiro C., Sharratt B., Harkema J. R., et al. (2011). Non-additive hepatic gene expression elicited by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and 2,2’,4,4’,5,5’-hexachlorobiphenyl (PCB153) co-treatment in C57BL/6 mice Toxicol. Appl. Pharmacol 256 154–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labonte E. D., Camarota L. M., Rojas J. C., Jandacek R. J., Gilham D. E., Davies J. P., Ioannou Y. A., Tso P., Hui D. Y., Howles P. N. (2008). Reduced absorption of saturated fatty acids and resistance to diet-induced obesity and diabetes by ezetimibe-treated and Npc1l1-/- mice. Am. J. Physiol. Gastrointest. Liver Physiol. 295 G776–G783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshman M. R., Campbell B. S., Chirtel S. J.,, Ekarohita N. (1988). Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on de novo fatty acid and cholesterol synthesis in the rat Lipids 23 904–906 [DOI] [PubMed] [Google Scholar]
- Lakshman M. R., Ghosh P.,, Chirtel S. J. (1991). Mechanism of action of 2,3,7,8-tetrachlorodibenzo-p-dioxin on intermediary metabolism in the rat J. Pharmacol. Exp. Ther. 258 317–319 [PubMed] [Google Scholar]
- Lee C. C., Yao Y. J., Chen H. L., Guo Y. L.,, Su H. J. (2006). Fatty liver and hepatic function for residents with markedly high serum PCDD/Fs levels in Taiwan J. Toxicol. Environ. Health A 69 367–380 [DOI] [PubMed] [Google Scholar]
- Lee J. H., Wada T., Febbraio M., He J., Matsubara T., Lee M. J., Gonzalez F. J., Xie W. (2010). A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis Gastroenterology 139 653–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P. C., Matsumura F. (1995). Differential effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the “adipose- type” and “brain-type” glucose transporters in mice Mol. Pharmacol. 47 65–73 [PubMed] [Google Scholar]
- Lowes W., Walker M., Alberti K. G.,, Agius L. (1998). Hexokinase isoenzymes in normal and cirrhotic human liver: Suppression of glucokinase in cirrhosis Biochim. Biophys. Acta 1379 134–142 [DOI] [PubMed] [Google Scholar]
- Matsumoto M., Pocai A., Rossetti L., Depinho R. A.,, Accili D. (2007). Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver Cell Metab. 6 208–216 [DOI] [PubMed] [Google Scholar]
- Mietus-Snyder M., Sladek F. M., Ginsburg G. S., Kuo C. F., Ladias J. A., Darnell J. E., Jr, Karathanasis S. K. (1992). Antagonism between apolipoprotein AI regulatory protein 1, Ear3/COUP-TF, and hepatocyte nuclear factor 4 modulates apolipoprotein CIII gene expression in liver and intestinal cells Mol. Cell Biol. 12 1708–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M., Kim Y. C., Gray-Keller M. P., Attie A. D.,, Ntambi J. M. (2000). The biosynthesis of hepatic cholesterol esters and triglycerides is impaired in mice with a disruption of the gene for stearoyl-CoA desaturase 1 J. Biol. Chem. 275 30132–30138 [DOI] [PubMed] [Google Scholar]
- Miyazaki M., Man W. C.,, Ntambi J. M. (2001). Targeted disruption of stearoyl-CoA desaturase1 gene in mice causes atrophy of sebaceous and meibomian glands and depletion of wax esters in the eyelid J. Nutr. 131 2260–2268 [DOI] [PubMed] [Google Scholar]
- Murray I. A., Morales J. L., Flaveny C. A., Dinatale B. C., Chiaro C., Gowdahalli K., Amin S., Perdew G. H. (2010). Evidence for ligand-mediated selective modulation of aryl hydrocarbon receptor activity Mol. Pharmacol. 77 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeli I., Siddiqi S. A., Siddiqi S., Mahan J., Lagakos W. S., Binas B., Gheyi T., Storch J., Mansbach C. M., II. (2007). Liver fatty acid-binding protein initiates budding of pre-chylomicron transport vesicles from intestinal endoplasmic reticulum J. Biol. Chem. 282 17974–17984 [DOI] [PubMed] [Google Scholar]
- Nguyen L. P., Bradfield C. A. (2008). The search for endogenous activators of the aryl hydrocarbon receptor Chem. Res. Toxicol. 21 102–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohjanvirta R., Sankari S., Kulju T., Naukkarinen A., Ylinen M.,, Tuomisto J. (1990). Studies on the role of lipid peroxidation in the acute toxicity of TCDD in rats Pharmacol. Toxicol. 66 399–408 [DOI] [PubMed] [Google Scholar]
- Pohjanvirta R., Tuomisto J. (1994). Short-term toxicity of 2,3,7,8- tetrachlorodibenzo-p-dioxin in laboratory animals: Effects, mechanisms, and animal models Pharmacol. Rev. 46 483–549 [PubMed] [Google Scholar]
- Pollenz R. S., Sattler C. A.,, Poland A. (1994). The aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator protein show distinct subcellular localizations in Hepa 1c1c7 cells by immunofluorescence microscopy Mol. Pharmacol. 45 428–438 [PubMed] [Google Scholar]
- Remillard R. B., Bunce N. J. (2002). Linking dioxins to diabetes: Epidemiology and biologic plausibility Environ. Health Perspect. 110 853–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo S., Kozlitina J., Xing C., Pertsemlidis A., Cox D., Pennacchio L. A., Boerwinkle E., Cohen J. C., Hobbs H. H. (2008). Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease Nat. Genet. 40 1461–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon T., Cook V. R., Rao A.,, Weinberg R. B. (2011). Impact of murine intestinal apolipoprotein A-IV expression on regional lipid absorption, gene expression, and growth J. Lipid Res. 52 1984–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smathers R. L., Petersen D. R. (2011). The human fatty acid-binding protein family: Evolutionary divergences and functions Hum. Genomics 5 170–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch J., McDermott L. (2009). Structural and functional analysis of fatty acid-binding proteins J. Lipid Res. 50 S126–S131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch J., Thumser A. E. (2010). Tissue-specific functions in the fatty acid-binding protein family J. Biol. Chem. 285 32679–32683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedenborg E., Ruegg J., Makela S.,, Pongratz I. (2009). Endocrine disruptive chemicals: Mechanisms of action and involvement in metabolic disorders J. Mol. Endocrinol. 43 1–10 [DOI] [PubMed] [Google Scholar]
- Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. (2002);3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viluksela M., Unkila M., Pohjanvirta R., Tuomisto J. T., Stahl B. U., Rozman K. K., Tuomisto J. (1999). Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on liver phosphoenolpyruvate carboxykinase (PEPCK) activity, glucose homeostasis and plasma amino acid concentrations in the most TCDD-susceptible and the most TCDD-resistant rat strains Arch. Toxicol. 73 323–336 [DOI] [PubMed] [Google Scholar]
- Wang C., Xu C. X., Krager S. L., Bottum K. M., Liao D. F.,, Tischkau S. A. (2011). Aryl hydrocarbon receptor deficiency enhances insulin sensitivity and reduces PPAR-alpha pathway activity in mice Environ. Health Perspect. 119 1739–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins P. A. (2008). Very-long-chain acyl-CoA synthetases J. Biol. Chem. 283 1773–1777 [DOI] [PubMed] [Google Scholar]
- Wu Q., Ortegon A. M., Tsang B., Doege H., Feingold K. R.,, Stahl A. (2006). FATP1 is an insulin-sensitive fatty acid transporter involved in diet-induced obesity Mol. Cell Biol. 26 3455–3467 [DOI] [PMC free article] [PubMed] [Google Scholar]