

Abstract
Isolated protein motifs that are involved in interactions with their binding partners can be used to inhibit these interactions. However, peptides corresponding to protein fragments tend to have no defined secondary or tertiary structures in the absence of scaffolding by the rest of protein molecule. This results in low potency of corresponding inhibitors. NMR and CD spectroscopy studies of lipopeptide inhibitors of the Hedgehog pathway revealed that membrane anchoring allows the cell membrane to function as a scaffold facilitating folding of short peptides. In addition, lipidation enhances cell permeability and increases the local concentration of the compounds near the membrane thus facilitating potent inhibition. General applicability of this rational approach was further confirmed by generation of selective antagonists of insulin-like growth factor 1 receptor with GI50 values in the nanomolar range. Lipopeptides corresponding to protein fragments were found to serve as potent and selective inhibitors of a number of non-druggable molecular targets.
Keywords: protein-protein interaction, insulin-like growth factor receptor, lipopeptide, cancer, retro-inverso
Introduction
Protein-protein interactions control practically all biological processes. The ability to manipulate these interactions is crucial tor finding cures for a majority of human diseases[1–3]. Conventional high-throughput screens for small molecule blockers of protein-protein interactions produce a disappointingly small number of lead compounds[4]. Although there have been notable successes in blocking “hot spots” of large interacting surfaces with small molecules [5, 6], such “hot spots” have proven elusive in many protein complexes. However, specific motifs from the interfaces of the interacting proteins have been successfully mimicked for inhibition of the target interactions[7–10] Since peptides corresponding to fragments of protein primary structures tend to have little or no defined conformation, the major effort in mimicking the interface is directed towards making the mimicking peptide or peptidomimetic as rigid as feasible. Strategies have been developed that allow simulation of reverse turns, β-sheets and alpha-helixes (reviewed in[10, 11]). Cyclization is the most frequently used approach to affect stabilization of both β-turns and α-helices. However, for inhibitors of intracellular protein-protein contacts, there remains the problem of cell permeability. Hydrocarbon-stapled α-helix peptidomimetics have demonstrated improved cell penetration [7, 12], but this method is applicable only to helical fragments of proteins. Palmitoylated peptide analogs of protein regions that are positioned adjacent to the cell membrane have been used to generate inhibitors of the corresponding membrane proteins[13, 14]. When applied to the seven transmembrane protein Smoothened (SMO), a critical component of the Hedgehog (HH) signaling pathway this strategy allowed generation of subnanomolar inhibitors of HH signalling[15]. We have now found that palmitoylation not only improves cell permeability of peptides but also facilitates their folding upon membrane anchoring and thus improves dramatically their biological activity. We tested the approach on several classes of membrane and intracellular proteins and have found that it can be applied to a broad range of molecular targets. The approach allows for straightforward generation of potent and selective inhibitors of the target proteins. Lipopeptides represent a new type of potential therapeutics with a wide range of applications.
Results and Discussion
Membrane anchoring changes conformation of Hedgehog pathway peptide antagonist
Circular dichroism (CD) spectroscopy has revealed that a palmitoylated peptide corresponding to the N-terminal half of the second intracellular loop (i2) of SMO, SMOi2-43 (Ac-(ε-Pal)-KLTYAWHTSFKAL-NH2), which is a potent inhibitor of the Hedgehog pathway[15], adopts mostly a β-type conformation in aqueous solution (Figure 1b, THIS SHOULD BE 1A). In contrast, solutions of SMOi2-43 in membrane-mimicking dodecylphosphocholine micelles show a fold that is predominantly α-helical. Removal of the fatty acid (SMOi2-9, AcLTYAWHTSFKAL-NH2) causes a reversion to a random conformation in aqueous solutions. Addition of membrane-mimicking micelles facilitates partial folding of the non-lipidated peptide (Figure 1a), but it is not as efficient as in the case of the palmitoylated homolog. The non-lipidated peptide is also inactive in inhibiting SMO function.
Figure 1.
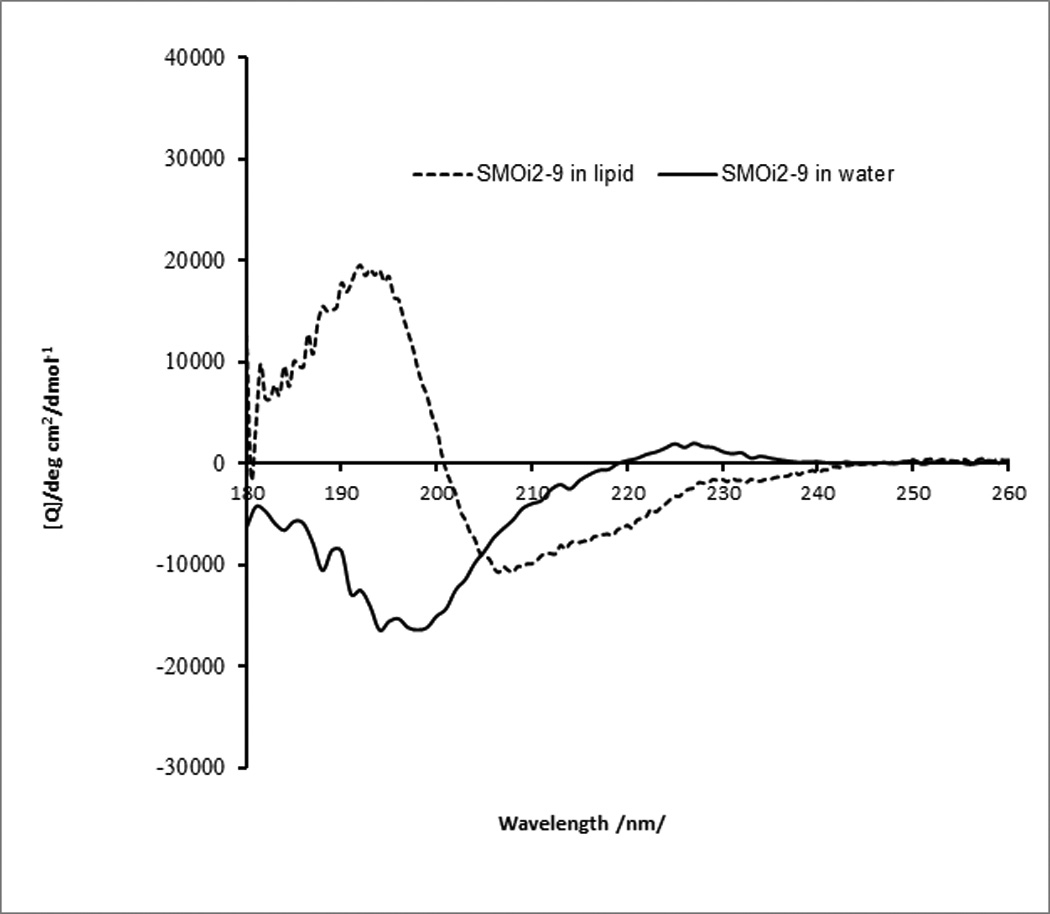
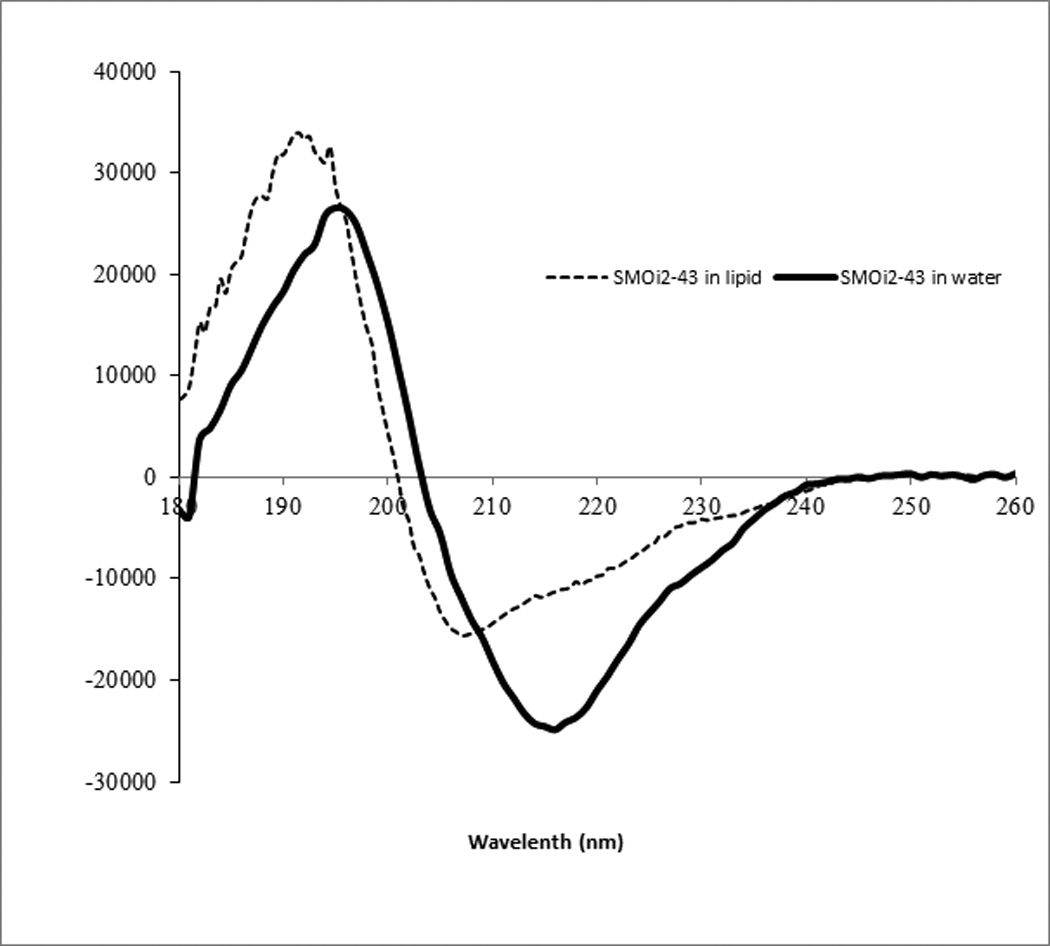

Structural transitions in lipopeptide inhibitors of the Hedgehog pathway upon interaction with membrane-mimicking lipid micelles. (a) CD spectra of SMO intracellular loop derivative SMOi2-9 (Ac-LTYAWHTSFKAL-NH2).and (b) CD spectra of a palmitoylated peptide SMOi2-43 (Ac-ε-Palmitoyl-KLTYAWHTSFKAL) in water and in dodecyl phosphocholine micelles. (c) Overlay of the 13Cα and 13Cβ (Ser and Thr only) regions of 13C-HSQC spectra of SMOi2-9 (blue), SMOi-9 in lipid micelles (red), and SMOi2-43 (green) in lipid micelles. Two alternative conformations of Ser are marked. The arrows indicate the direction of chemical shift changes
NMR analysis of SMOi2-9 and SMOi2-43 further confirmed stabilization of the lipidated peptide structure upon incorporation into micelles. The 13C-HSQC spectrum of SMOi2-9 in water revealed the presence of the expected 12 Cα signals (Figure 1c, blue spectrum). The 13Cα and 13Cβ chemical shifts do not significantly deviate from the expected random coil values as defined by the comprehensive BMRB database (www.bmrb.wisc.edu) and are indicative of an extended conformation of the SMOi2-9 peptide in the aqueous environment. Upon addition of dodecylphosphocholine- d38 micelles, the 13Cα nuclei experience a downfield chemical shift change whereas the 1Hα nuclei move upfield, suggesting a transition of the peptide structure to a helical conformation. In the presence of the lipid micelles, the total number of observed 13Cα signals for all residues and Cβ signals for the single Ser and two Thr residues increases to 18, suggesting the presence of two distinct conformations of the peptide (Figure 1c, red spectrum). The β-methylene protons of Ser8 in this spectrum become non-equivalent and clearly display a second minor conformation (Figure 1c). The presence of an alternative conformation may indicate that the peptide interacts with the micelles in more than one way. For instance, it may insert into the lipid micelles or it may bind to the surface of the micelles.
Addition of the palmitoyl group to the peptide structure (compound SMOi2-43) changes the 13Cα and 1Hα chemical shifts toward the values indicative of a more stable helical conformation in the presence of micelles, in good agreement with CD data. The total number of observed 13Cα and 13Cβ (Ser and Thr) signals in the spectrum of SMOi2-43 (Figure 1c, green spectrum) increases to 28 suggesting alternative modes of interaction with the micelles, compared with non-lipidated peptide. Our NMR data suggest that the peptide derived from the second intracellular loop of SMO can spontaneously associate with membrane-mimicking lipid micelles. This association is accompanied by transition from a a stretched to a partially helical fold. Palmitoylation facilitates peptide-lipid association and further stabilizes peptide tertiary structure.
Lipidation facilitates peptides cell entry
Although micelles are the best available mimics of the cell membrane for biophysical studies, their physical and chemical properties do differ significantly from real membranes. We used confocal laser scanning microscopy of live cells to evaluate the degree of association of palmitoyl-peptides with membranes of live cells and intracellular localization of the inhibitor. HH lipopeptide antagonist (Pal-CLTYAWHTSFKAL-NH2) was labeled with rhodamine red by reacting the cysteine residue with rhodamine maleimide. The label was introduced on the N-terminal part of the peptide because modifications of the C-terminus were detrimental to the activity of the inhibitor [15]. Microscopy of cells treated with fluorescent HH lipopeptide revealed that the peptide concentrated on the outer cell membrane within minutes after application and saturated intracellular membranes upon longer exposure (Figure 2), while non-palmitoylated fluorescent peptide of the same sequence did not enter the cells and did not concentrate on the membranes, although a loose association with lipid micelles was detected by NMR.
Figure 2.

Lipopeptide inhibitor of Hedgehog pathway fuses with cell membrane spontaneously and saturates intracellular membranes upon prolonged incubation. Confocal laser scanning microscopy of rhodamine red-labeled lipopeptide inhibitor of Hedgehog pathway. A) Peptide fluorescence localizes to intracellular membranes; B), Nomaski image of the cells. For generation of the fluorescent antagonist, Cys-contaning peptide (Pal-CLTYAWHTSFKAL-NH2) was labeled with rhodamine red by reacting cysteine residue with rhodamine maleimide. SK-Mel-2 melanoma cells were exposed to 100 nM solution of the peptide in medium containing Hoechst-33342 for 2 hour and observed under laser scanning confocal microscope (LSCM 510, Zeiss). Rhodamine red fluorescence was excited with 561 nm laser and 575-615 filter was used for emission detection.
Membrane anchoring was shown previously to increase potency of small molecules targeting membrane proteins by increasing their local membrane concentration [16]. Microscopy studies with fluorescent HH lipopeptide antagonists also showed efficient concentrating of the palmitoylated peptide not only in the cellular, but intracellular membranes. Since many signalling events take place in the space adjacent to cellular membranes, intracellular membranes or cellular organelles, increasing concentration of the drug in these vicinities may be advantageous not only for inhibition of integral membrane protein function, but for the function of many intracellular proteins. We have previously reported on lipopeptide inhibitors of STAT transcription factors that retained the original fold of the helix they were derived from and were effective in inhibition of transcriptional activity of these cytoplasmic proteins[17, 18].
Lipopeptide analogs of the juxtamembrane domain are potent and selective inhibitors of insulin-like growth factor 1 receptor
To test if the utilization of lipidated protein fragments has broad applications in the rational design of membrane protein inhibitors, we applied the approach for the development of antagonists for an important class of drug targets, receptor tyrosine kinases (RTKs)[19]. We started from insulin-like growth factor 1 receptor (IGF1R) because it is an established molecular target for the treatment of many tumor types (reviewed in [20]). IGF1R is a challenging target and the development of selective small molecule IGF1R tyrosine kinase inhibitors is complicated by the fact that the kinase domain of the IGF1R shares 85% identity with that of the insulin receptor (IR), and the ATP binding cleft is 100% conserved. All currently available small molecule IGF1R antagonists also cause significant inhibition of insulin receptor (IR)[21, 22]. The latter results in hyperglycaemia and other unwanted side effects[22, 23]. In addition, IGF1R and IR form functional heterodimers, which further complicates the generation of selective inhibitors. Juxtamembrane (JM) segments connecting the transmembrane domain with the kinase domain of many RTKs were shown to be involved in autoinhibition of kinase activity [24]. The IGF1R JM is highly conserved among the species (Figure 3a) and thus, is likely to play an essential role in receptor function. However, the primary structures of JMs differ significantly for different RTKs, including IGF1R and IR (Figure 3b). As we have predicted, construction of a small library of palmitoylated synthetic analogs of IGF1R JM yielded potent inhibitors of IGF1R as well as breast cancer cell growth (Table 1).
Figure 3.

High degree of evolutional conservation of the juxtamembrane region of IGF1R that suggests this part of structure is important to the function of the receptor. (A) Comparison of the primary structures of the Juxtamembrane regions of IGF-1R (in grey) from different species. (B) Comparison of the JM regions (highlighted in grey) of human IGF-1R and insulin receptor shows
Table 1.
The effects of IGF1R juxtamembrane domain analogues on the growth of MCF-7 breast cancer cells.
| Peptide | Peptide Sequence | GI50 [µM][[a]] |
TGI [µM] |
|---|---|---|---|
| 1 | Pal-HRKRNNSRLGNG-NH2 | 1.8±0.05 | N/A |
| 2 | Pal-HRKRNNSRLG-NH2 | 1.3±0.05 | N/A |
| 3 | Pal-HRKRNNSRLGNGVLYASVN-NH2 | 1.0±0.05 | 1.55±0.1 |
| 4 | Pal-HRKRNNSRLGNGVLYASVNPEYFSAA-NH2 | 1.0±0.05 | N/A |
| 23 | Pal-HRKRNNSRLGNGVLYASVNP-NH2 | 0.1±0.05 | N/A |
| 5 | Pal-HRKRNNSRLGNGVLYASV-NH2 | 0.45±0.05 | 1.45±0.3 |
| 6 | Pal-VHRKRNNSRLGNGVLYASV-NH2 | 0.25±0.03 | 1.05±0.1 |
| 7 | Pal-HRKRNNSRLGNGVLYAS-NH2 | 1.45±0.1 | 4.1±0.3 |
| 8 | Pal-HRKRNNSRLGNGVLYA-NH2 | 1.4±0.1 | N/A |
| 9 | Pal-HRKRNNSRLGNGVLY-NH2 | 1.65±0.1 | N/A |
| 10 | Pal-HRKRNNSRLGNGVL-NH2 | 1.8±0.1 | N/A |
| 11 | Pal-HRKRNNSRLGNGV-NH2 | 2.5±0.1 | N/A |
| 24 | Pal-VFHRKRNNSRLGNGVLYASVN-NH2 | 0.1±0.05 | 1.1±0.1 |
| 12 | Pal-RKRNNSRLGNGVLYASVN-NH2 | 1.2±0.1 | 2.7±0.3 |
| 13 | Pal-KRNNSRLGNGVLYASVN-NH2 | 0.08±0.007 | 0.5±0.08 |
| 14 | Pal-RNNSRLGNGVLYASVN-NH2 | 0.1±0.004 | 0.4±0.02 |
| 27 | Pal-KRNNSRLGNGVLY-NH2 | 1.1±0.06 | 2.5±0.5 |
| 16 | Pal-RNNSRLGNGVLY-NH2 | 0.07±0.005 | 0.5±0.02 |
| 15 | Pal-NNSRLGNGVLY-NH2 | 0.1±0.01 | 1.5±0.05 |
| 17 | Pal-NSRLGNGVLY-NH2 | 0.2±0.01 | 1.5±0.1 |
| 19 | Pal-SRLGNGVLY-NH2 | 0.15±0.05 | 2.0±0.1 |
| 20 | Pal-RLGNGVLY-NH2 | 0.2±0.006 | N/A |
| 18 | Ac-YLVGNGLRSNNRK-(ε-Pal)[All D-] | 0.04±0.001 | 0.4±0.02 |
| 22 | Pal-YLVGNGLRSNNR-NH2[All D-] | 0.04±0.001 | 0.5±0.025 |
| 21 | Pal-VSAYLVGNGLRSNNR-NH2[All D-] | 0.04±0.001 | 0.1±0.005 |
| IR-2 | Pal-RQPDGPLGPLY-NH2 | 0.06±0.001 | 0.4±0.05 |
Cells were exposed to compounds for 48 hours and cell number was determined with the help of MTT assay. GI50 corresponds to concentrations causing 50% reduction in growth rate. TGI is a concentration causing total growth inhibition.
For preliminary assessment of the inhibitory properties of the structural analogs of the JM region of IGF-1R, we synthesized peptide 4 corresponding to the entire JM region of IGF1R (959–984), and three truncated versions (1, 2 and 3). Initially, truncation was introduced at Gly and Pro residues since those are most likely to occur at the ends of secondary structure elements (Table 1). Peptide 3, which corresponds to sequence 959–977 had the highest impact on breast cancer cell survival. Subsequent systematic truncation of 3 from either the C or N-terminus lead to the most active analogue 16, which corresponds to residues 962–973 of IGF1R and has GI50 = 70 nM in inhibition of the growth of MCF-7 breast cancer cells. Even shorter variants, viz. 17, 19 and 20 are still active, but have progressively lower potency (Table 1). As in the case with Hedgehog pathway antagonists, the retro-inverso variant of 16, 18 constructed from all-D amino acids is more potent than the natural sequence and has GI50=40 nM. However, the gain in activity is not as dramatic as it is in Hedgehog antagonists, where the retro-inverso analogue was more than an order of magnitude more potent than the parent all-L peptide [15]. In the Hedgehog antagonists, positioning of the palmitoyl group on the terminus that is adjacent to the membrane in the native protein is critical for the activity[15]. On the contrary, for the IGF1R antagonists, a peptide with an N-terminal palmitoyl group (22) is equipotent to the one with the C-terminal modification (18).
CD spectroscopy revealed that a derivative of 16 without the palmitoyl residue, 28 (Ac-RNNSRLGNGVLY-NH2) is disordered both in water and in dodecylphosphocholine micelles, while the palmitoylated peptide is ordered both in water and in micelles (Figure 4a). As predicted, addition of the micelles causes significant structural rearrangement of the palmitoylated peptide (Figure 4b). CD spectra are in agreement with a mixed stretched plus helix conformation in the lipid. The retro-inverso analogue 22 has higher molar ellipticity than the parent all-L peptide suggesting that the improvement in activity is caused by a higher population of a stable three-dimensional fold.
Figure 4.
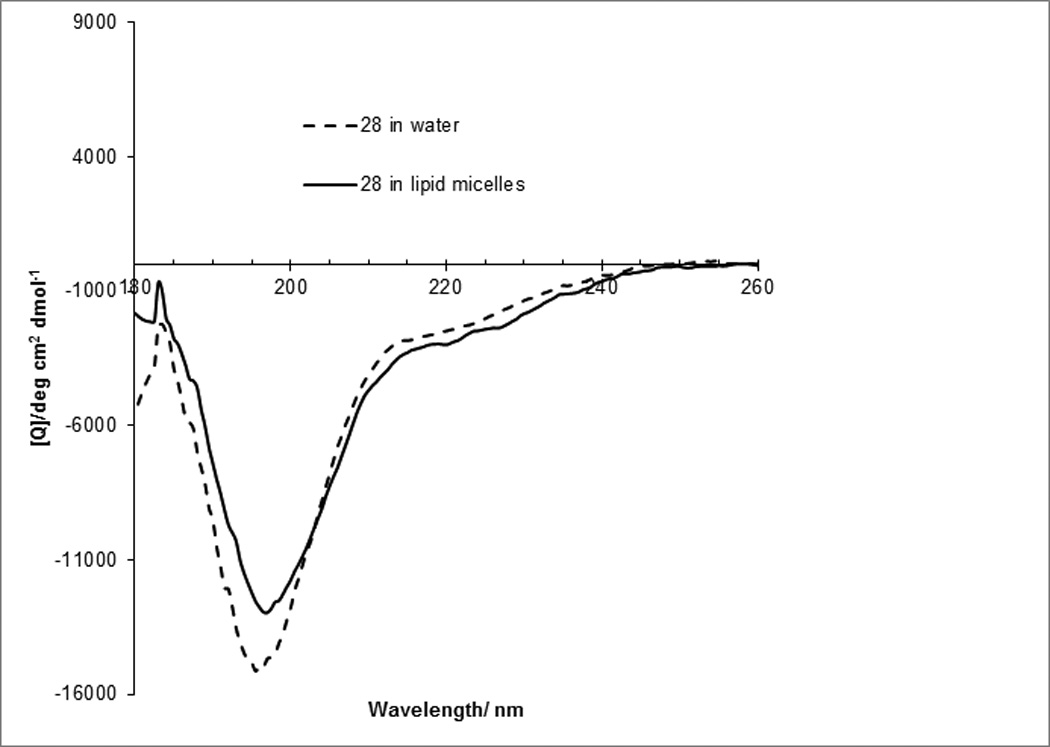
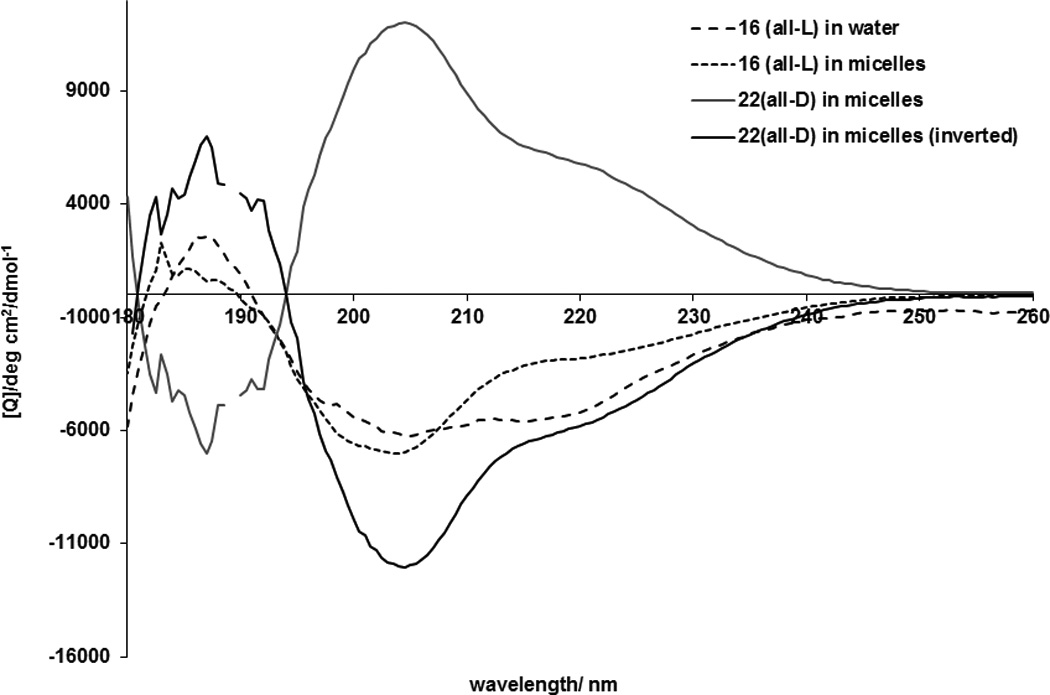
Palmitoylation facilitates folding of IFG1R JM analogs. (A) CD spectra of a non-palmitoylated analog of 16, 28 (Ac-RNNSRLGNGVLY-NH2) reveals unordered structure both in water and in dodecylphosphocholine micelles. (B) CD spectra of 16 (Pal-RNNSRLGNGVLY-NH2) reveal significant differences in peptide fold in dodecylphosphocholine micelles compared to water. Compound 22, the retro-inverso version of 16, had a stronger CD signal.
To evaluate the selectivity of inhibitory effects of IGF1R JM analogues, a proliferation assay was performed in serum-free media using human recombinant IGF-1 as the only growth stimulant. Overall growth inhibitory effects were similar to the ones observed in serum-containing media thus confirming that cell toxicity effects were caused by inhibition of IGF-1- mediated signalling (Supplementary Figure S2).
IGF1R activation is known to result in AKT activation. Inhibition of AKT kinase activity upon stimulation of cells with IGF-1 was used to assess the selectivity of new inhibitors. IGF1R JM analogue 16 inhibited IGF-1 induced activity of AKT in a dose-dependent manner (Figure 5) and was significantly less effective in inhibiting insulin-induced activation of AKT. Neither palmitoylated nor non-lipidated JM analogs were able to inhibit recombinant IGF1R kinase in a cell-free environment, strongly suggesting that the cellular membrane is essential for fully functional inhibitors (Supplementary Figure S4).
Figure 5.
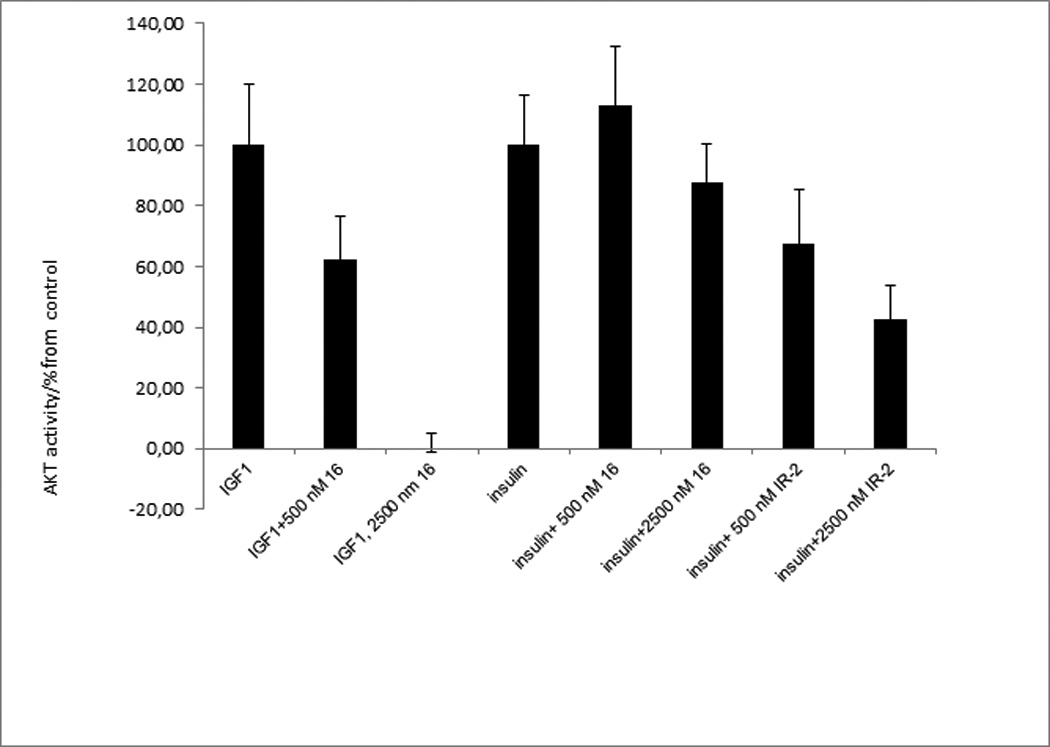
IGF1R juxtamembrane domain analogs selectively inhibited IGF-1, but not insulin-induced AKT activation in breast cancer cells. AKT activity in cell lysates was determined with the help of the Omni fluorogenic peptide substrate after 20 min incubation with IGF-1 (25 ng/ml) or insulin. A non-optimized analogue of insulin receptor juxtamembrane domain IR-2 (Pal-RQPDGPLGPLY-NH2) was used as a positive control in inhibition of insulin-induced AKT activation.
To exclude the possibility that cell growth inhibition and cell kill by lipopeptides inhibitors is caused by non-selective cell lysis, we tested the integrity of membranes of treated cells with the help of an assay that measures lactate dehydrogenase (LDH) released from cells with damaged membranes (Supplementary Figure S5). No cell lysis could be detected even with 2.5 µM concentrations of compounds.
Lipopeptides antagonist can be delivered in vivo
The antibiotic daptomycin remains to date the only lipopeptide approved for clinical use[25] and consequently the pharmacological properties of lipopeptides as a therapeutic class are poorly characterized. To further evaluate the applicability of lipopeptides for in vivo use, we have characterized the ability of tritiated HH antagonist SMOi2-17 (Ac-AKFSTHWAYTLK-(ε-Pal), all-D) to penetrate different organs and tissues when administered through different routes. Upon subcutaneous, topical, peritoneal or oral administration, the lipopeptide remained in the tissue adjacent to the application site (Supplementary Figure S6) and did not enter the circulation. The compound appeared to be skin permeable because detectable amounts were found in the muscles under the site of topical application. The tendency to adhere to the tissue at the application site may be a valuable property of these potential drugs because it limits the action of the compounds to certain organs thus decreasing systemic toxicity. Intravenous administration allowed delivering the lipopeptide efficiently into all organs except the brain. The brain showed about 10 nM concentration of the peptide, while all other organs had amounts comparable to the blood, i.e., ~ 400 nM. Since in vitro activity of the HH antagonist was in the subnanomolar range[15], it may be possible to achieve therapeutic concentrations even in the brain upon intravenous administration. Biodistribution studies suggest that lipopeptides can be effectively delivered in vivo and allow for both systemic and local administration (Figures 6 and S6).
Figure 6.
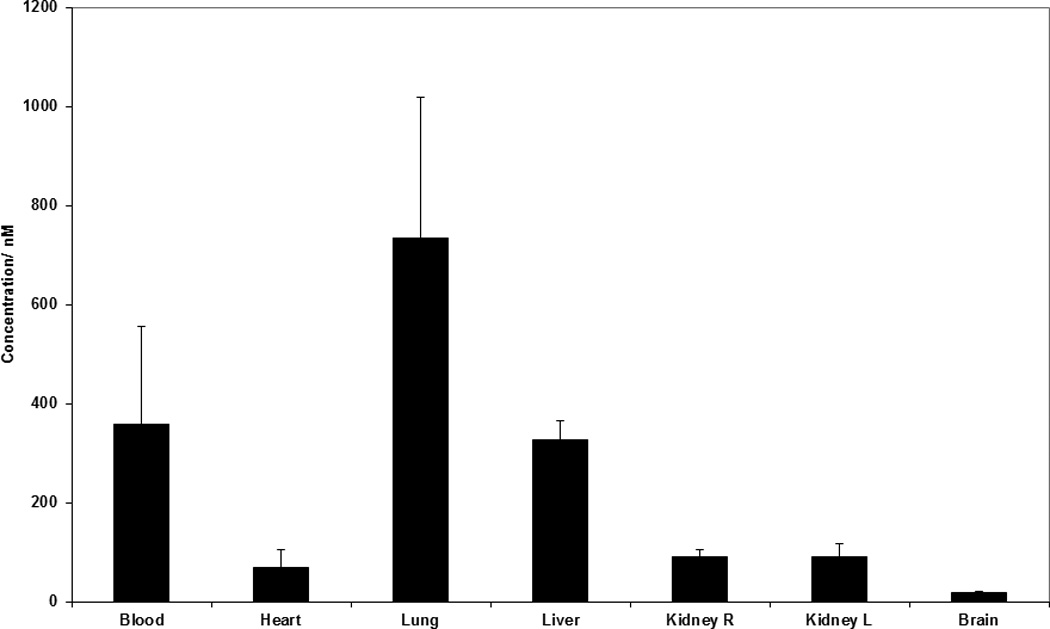
Biodistribution of Hedgehog lipopeptide antagonist SMOi2-17 upon intravenous administration in mice. Distribution was evaluated with the help of 3H-labeled compound upon injections of 2µCi per animal or 1.1 mg/kg (610 nmol/kg). The tritiated compound was generated by AmbiosLabs by catalytic substitution of protons of hydrocarbon chains of the peptide (http://ambioslabs.com/label.php). Mice were sacrificed 30 min after administration. Organs were collected, tissue was homogenized with razor blades and radioactivity counted in scintillation counter. The data is an average for samples collected from groups of three animals.
Discussion
Many intracellular proteins and protein-protein interactions remain out-of-reach targets for the development of small molecule therapeutic agents, antibodies and chemical biology probes. Discovery of inhibitors even for druggable targets remains costly and time-consuming. Consequently, methods that allow for rational, straightforward and inexpensive ways of developing selective inhibitors for these targets can be highly beneficial for studying their biology and for drug development. Conversion of protein fragments into potent, selective and cell-permeable inhibitors presents such an opportunity, but only if we can preserve the original fold of the fragment and can make the corresponding peptide cell-permeable. We have found that synthetically accessible modification of peptides such as lipidation can dramatically improve structural rigidity, cell permeability and biological activity and thus can be used for the rational design of protein inhibitors.
Post-translational lipid modification of proteins is a frequent event. Lipidation has been shown to regulate protein folding, stability, trafficking and interactions with specific membranes or membrane domains[26–30]. Presented results along with published data suggest that similar to proteins, lipidation of short peptides can have profound effects on their structure and function. Solid-state NMR studies of lipidated peptides have shown that the hydrophobic side chains as well as lipid moieties do insert into the hydrophobic core of the membrane, while polar residues and backbone amides tend to localize in the lipid-water interface[26–28, 31]. The inner leaflet of the plasma membrane has a negative surface charge. Thus, positively charged residues of peptides can be involved in electrostatic interactions with the membrane. These interactions can restrict random movement of the peptide “freezing” it in a certain folded state. We have observed stabilization of both α-helical conformation (Hedgehog antagonists and inhibitors of STAT transcription factors[17]) and predominantly stretched or β-type folding (IGF1R inhibitors). It is logical to assume that juxtamembrane domains of membrane proteins are frequently involved in interaction with the membrane. Experimental confirmation of such interactions was obtained for EGF receptor[32] and Ras proteins[31]. Consequently, membrane–tethered protein fragments are likely to mimic well the parts of protein structures involved in interactions with the membrane and are likely to fold in a conformation resembling their native state. The effects of membrane tethering on the structure of protein fragments that are distant from the membrane are less obvious. Amphiphilicity of peptides was shown to contribute significantly to interactions of the peptide with the membrane[33] and thus structural stabilization of amphiphilic peptides by membrane anchoring is likely to be very effective even if they are derived from a protein that is not involved in direct interaction with the membrane, like in the case with fragments of STAT N-terminal domains[17, 18]. Thus, many factors can define the effectiveness of peptide structure stabilization by membrane tethering. The nature of the lipid is one of such factors. We have found that shortening of HH antagonist acyl chain by substitution of palmitoyl group (C16) with myristoyl residue (C14) results in 2.5 fold reduction in biological activity[15]. The affect can be attributed to less efficient membrane anchoring, since palmitoyl group was shown to have 15-fold higher membrane affinity than the myristoyl one[34]. Since the peptide itself can also contribute significantly to stable association with the membrane, the nature of the optimal lipid anchor can be different for different peptides. Poor solubility of lipopeptides can limit the use of long hydrocarbon chains and of such effective membrane anchors as cholesterol. Membranes are unlikely to serve as a universal scaffold for promoting folding of any peptide but appear to work well in many cases. Lipidation was shown to improve the biological activity of HIV-1 fusion inhibitors [35], G-protein coupled receptor antagonists [14, 36, 37] and STAT transcription factors [17, 18]. Screening of a library of 52 palmitylated peptides corresponding to conserved juxtamembrane regions of membrane proteins highly expressed in platelets allowed identification of 22 inhibitors of platelet aggregation[13], thus confirming that lipidated protein fragments can serve as effective inhibitors of many proteins. The presented data shows that enhancement in potency of peptide inhibitors caused by lipidation is largely due to facilitation of peptide folding. In addition, negative surface charge of the membrane was shown to define subcellular targeting of proteins containing cationic motifs[38]. The same type of interactions can also provide for additional targeting of lipopeptide inhibitors to certain parts of cellular membrane and cell organelles. The choice of the fragment for the construction of inhibitors is greatly simplified when the tertiary structure of the complex of interacting proteins is available. However, even in the absence of structural data for the target proteins, clues as to the conformational motifs present in specific proteins can be obtained from the evolutionary degree of sequence conservation as well as from mutational data. We had a high rate of success while relying on the sequence alignment of protein orthologues for the identification of fragments critical for protein function and using them as lipopeptide dominant-negative inhibitors [15, 39]. As in the case with IGF1R antagonists, active compounds are frequently generated from the very first attempt (4, Table 1). However, peptide length has profound influence on both folding and activity with longer peptides being in some cases less folded and less active (4 and 23, Table 1). Thus, inhibitor length needs optimization in the absence of the tertiary structure information for the target protein or protein-protein interaction. Lipopeptide inhibitors are not only easier and quicker to develop than small molecule antagonists, they are also much more selective, as was evident from the potent inhibition of IFG1R with juxtamembrane analogs but not the insulin receptor that is 85% identical to IGF1R in the kinase domain (Figure 5). Previous studies have shown that retro-inverso peptide analogues constructed from all-D amino acids can adopt more rigid solution structures as compared to their all-L counterparts[40]. This correlates well with our observations. Retro-inverso analogues have been successfully used as immunogens and inhibitors of protein-protein interactions [41, 42]. However, retro-inverso analogues have significantly reduces biological activity compared to the parent peptide in several cases and thus may not be universally applicable [41, 42]. CD and NMR data of hedgehog antagonists and IGF1R inhibitors strongly suggest that retro-inverso analogues are not plain mirror images of the parent peptides. Retro-inverso versions of lipopeptides that we have tested thus far were both more structurally defined and more biologically active. Remarkably, the increase in activity frequently amounted to several orders of magnitude[15, 17]. The structural basis for this increase in potency requires further studies. The positioning of the fatty acid on the C- or N-terminus terminus did matter for the majority of inhibitors, (for example, the Hedgehog antagonists), but had no influence on the activity of the others (IGF1R inhibitors 18 and 22, Table 1). Independence of activity from the palmitoyl group position can be attributed to stretched or β-type conformation of the IGF1R inhibitor. In many cases, better folding and improved activity was achieved by placing the fatty acid residue on the side chain of a sequence-expanded amino acid (e.g., the ε-amino group of a Lys residue added to one of the termini), rather than on a backbone amino group. However, in some cases, (IGF1R inhibitors), palmitoylation of the amino-terminus produced inhibitors that were as potent as the ones with a palmitoyl residue on a side chain.
Conclusion
We have found that lipopeptides can serve as effective inhibitors for intracellular targets that cannot be inhibited by small molecules, or for which selective small molecule inhibitors cannot be developed. The method permits development of inhibitors in a rational and straightforward way even for proteins with unknown tertiary structures. Lipidation enhances the potency of peptide inhibitors via three mechanisms: 1) stabilization of peptide folding through interactions with cell membrane, 2) enhancement of cell permeability and 3) increasing the local concentration of the compounds near the membrane. We have found that lipopeptide inhibitors can be used as effective chemical probes for studying the function of the target proteins both in vitro and in vivo. They are likely to become a new class of therapeutic agents that can be effective against a wide variety of diseases[15, 17, 39, 43].
Experimental Section
Peptide Synthesis and Purification
The peptides were synthesized on a 433A Peptide Synthesizer (Applied Biosystems) using Fmoc chemistry. The peptides were cleaved from the resin and deprotected with a mixture of 90.0% (v/v) trifluoroacetic acid (TFA) with 2.5% water, 2.5% triisopropyl-silane, and 5% thioanisol. The resin and deprotection mixture were pre-chilled to −5°C and reacted for 15 minutes at −5°C with stirring. The reaction was allowed to continue on at room temperature for 1 hour and 45 minutes. The resin was filtered off and the product was precipitated with cold diethyl ether. The resin was washed with neat TFA. Peptide suspended in diethyl ether was centrifuged at −20 °C and the precipitate was washed with diethyl ether four more times and left to dry in a vacuum overnight. The dried crude peptide was dissolved in DMSO and purified on a preparative (25mm × 250mm) Atlantis C18 reverse phase column (Agilent Technologies) in a 90 minute gradient of 0.1% (v/v) trifluoroacetic acid in water and 0.1% trifluoroacetic acid in acetonitrile, with a 10 mL/min flow rate. The fractions containing peptides were analysed on Agilent 1100 LC/MS spectrometer with the use of a Zorbax 300SB-C3 Poroshell column and a gradient of 5% acetic acid in water and acetonitrile. Fractions that were more than 95% pure were combined and freeze dried. Retro-inverso peptide made of all-D amino acids was synthesized using essentially the same protocol, except that palmitic acid had to be introduced in the side chain. Resin preloaded with α-Fmoc-ε-palmytoil-D-Lys was prepared as described [15].
Circular Dichroism Spectroscopy
For spectral analysis, 0.5 mg/ml solutions of peptides in deionized water have been prepared. UV spectra were used to determine the exact concentration of peptides in solutions. Extinction coefficients at 280 nm were determined with the use of ProtParam software (http://expasy.org/tools/protparam.html). For studying the effects of lipid micelles, peptide solutions were supplemented with 100-fold molar excess of dodecylphosphocholine. The solutions were transferred to a 1mm cuvette for analysis on the Circular Dichroism Spectrometer Model 202 (AVIV Instruments Inc.) The data was acquired and processed using CDs Software (AVIV BioMedical, Inc.).
NMR spectroscopy
All data were acquired at natural isotope abundance using a Bruker 600 MHz spectrometer at 25 °C. Peptides were dissolved in H2O/D2O (9:1) or 300mM dodecylphosphocholine in H2O/D2O to give final concentration of 5 mM. All spectra were processed with nmrPipe[44].
Cell Toxicity Assay
The cells (MCF-7 (breast cancer), T47D (breast cancer), Colo 205 (colon cancer), JM-1 (rat hepatoma), Sk Mel-2 (melanoma), PLC (human hepatoma), and HepG2 (human hepatoma)) were obtained from American Type Cell Culture Collection. MCF-7 cells were grown in RPMI medium supplemented with 10% Fetal Bovine Serum. The rest of the cell lines were grown in DMEM medium supplemented with 10% Fetal Bovine Serum. For the assay, cells were seeded into 96 well plates in medium containing 1% Fetal Bovine Serum and 100µL of a cell suspension containing 5000 cells per well were used for each well. Cell growth was evaluated utilizing MTT ((3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium ). The absorbance of the wells at 544 nm was determined by a FLUOstar/POLARstar Galaxy (BMG Lab Technologies GmbH) microplate reader.
In an alternative assay to test the inhibition specifically of IGF-1-induced growth a cell suspension was made to approximately 5000 cells per every 100 µL of suspension in a 5% FBS RPMI medium and seeded into 96 count wells and incubated overnight. After one day’s incubation, the 5% FBS medium was aspirated and replaced with 100 µL of serum-free RPMI medium containing 1 mg Bovine Serum Albumin (BSA) per 1 mL RPMI. The compounds were prepared to the desired dilutions in no serum medium containing 1mg BSA/mL. 75 µL of compounds were added to the wells and the cells were allowed to incubate for 15–20 minutes as per the suggested time, after which 25 µL of human recombinant IGF-1 (Peprotech) solution was added to the wells to attain final concentrations of 10 ng IGF-1/ mL medium. The activity was calculated from the data using the formula: 100 × [(T - T0)/(C - T0)] for T > T0 and 100 × [(T - T0)/T0] for T < T0. T0 corresponds to cell density at the time of compound addition.
AKT downstream activation through IGF Pathway
Omnia fluorogenic AKT Kinase Activity Assay Kit (BioSource through Invitrogen) was used for the measurement of AKT activity in cell lysates. For the cell treatment, MCF-7 cells were seeded into 4 wells on a 6 well plate in RPMI medium supplemented with 10% Fetal Bovine Serum to aid the cell attachment. The cells were incubated for 24 hours, and the 10% serum medium was aspirated and replaced with 2mL of serum-free medium and cells were starved overnight. On the third day, the medium was replaced and different dilutions of compounds in serum-free medium were added to the four wells. One hour after the addition of the compounds, cells were stimulated with 25ng IGF-1/ mL for 20 min[45]. The cells were then placed on ice to aid them in detaching and scraped from the bottom of the wells, then rinsed with cold PBS. The cell suspension was collected and centrifuged for 5 minutes at 1500 rpm at 4°C and the medium aspirated. The cells were lysed with 5X the volume of the cell pellet with Omnia Cell Extraction Buffer (Biosource, Kinase Activity Assay Kit, Catalog #KNZ0011). Cell lysis buffer was supplemented with 25µL of 100X protease inhibitor cocktail (Sigma- Aldrich) and 25µL of a 100X phosphotase inhibitor cocktail (Sigma-Aldrich) per 2.5 mL mixture. The cells were briefly sonicated and centrifuged at 13000 rpm and 4°C for 10 min to remove cell debris, to create clear cell extract. Master Mix of AKT fluorogenic substrate was prepared according to the protocol provided by BioSource. 10 µl of cell extracts were placed in a white 96-well plate. 50µL of the master mix were added to each well containing extracts and the measurement of the kinetics of the reaction was started immediately. The fluorescence was excited at 360 nm and read at 485 nm for a length of 60 minutes every thirty seconds on a FLUOstar/POLARstar Galaxy (BMG Lab Technologies GmbH) microplate reader.
Supplementary Material
Acknowledgements
We are indebted to Dr. Joseph Kalen, Lisa Riffle and Avrum Leeder (NCI-Frederick Small Animal Imaging Program) for the help with biodistribution studies, to Carrie J. Saucedo (Molecular Target Development Program, NCI-Frederick) for recombinant human IGF1R kinase domain, and to Mary Starich (Structural Biophysics Laboratory, NCI-Frederick) for the help with collecting NMR data. LJ and JR have been supported by the Cancer Research Training Awards for undergraduate students of the National Cancer Institute. The research was sponsored by the Drug Development Committee of the National Cancer Institute. The research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author
References
- 1.Stumpf MP, Thorne T, de SE, Stewart R, An HJ, Lappe M, Wiuf C. Proc. Natl. Acad. Sci. U. S. A. 2008;105(19):6959–6964. doi: 10.1073/pnas.0708078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verdine GL, Walensky LD. Clin. Cancer Res. 2007;13(24):7264–7270. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- 3.Hoshi N, Langeberg LK, Gould CM, Newton AC, Scott JD. Mol. Cell. 2010;37(4):541–550. doi: 10.1016/j.molcel.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watt PM. Nat. Biotechnol. 2006;24(2):177–183. doi: 10.1038/nbt1190. [DOI] [PubMed] [Google Scholar]
- 5.Che Y, Marshall GR. J Med. Chem. 2006;49(1):111–124. doi: 10.1021/jm0507072. [DOI] [PubMed] [Google Scholar]
- 6.Wells JA, McClendon CL. Nature. 2007;450(7172):1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 7.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305(5689):1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viaud J, Zeghouf M, Barelli H, Zeeh JC, Padilla A, Guibert B, Chardin P, Royer CA, Cherfils J, Chavanieu A. Proc. Natl. Acad. Sci. U. S. A. 2007;104(25):10370–10375. doi: 10.1073/pnas.0700773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sillerud LO, Larson RS. Curr. Protein Pept. Sci. 2005;6(2):151–169. doi: 10.2174/1389203053545462. [DOI] [PubMed] [Google Scholar]
- 10.de Vega MJ, Martin-Martinez M, Gonzalez-Muniz R. Curr. Top. Med. Chem. 2007;7(1):33–62. doi: 10.2174/156802607779318325. [DOI] [PubMed] [Google Scholar]
- 11.Che Y, Marshall GR. Expert. Opin. Ther. Targets. 2008;12(1):101–114. doi: 10.1517/14728222.12.1.101. [DOI] [PubMed] [Google Scholar]
- 12.Verdine GL, Walensky LD. Clin. Cancer Res. 2007;13(24):7264–7270. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- 13.Edwards RJ, Moran N, Devocelle M, Kiernan A, Meade G, Signac W, Foy M, Park SD, Dunne E, Kenny D, Shields DC. Nat. Chem. Biol. 2007;3(2):108–112. doi: 10.1038/nchembio854. [DOI] [PubMed] [Google Scholar]
- 14.Covic L, Gresser AL, Talavera J, Swift S, Kuliopulos A. Proc. Natl. Acad. Sci. U. S. A. 2002;99(2):643–648. doi: 10.1073/pnas.022460899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Remsberg JR, Lou H, Tarasov SG, Dean M, Tarasova NI. J Med. Chem. 2007;50(18):4534–4538. doi: 10.1021/jm0705657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, Jennings G, Knolker HJ, Simons K. Science. 2008;320(5875):520–523. doi: 10.1126/science.1156609. [DOI] [PubMed] [Google Scholar]
- 17.Timofeeva OA, Gaponenko V, Lockett SJ, Tarasov SG, Jiang S, Michejda CJ, Perantoni AO, Tarasova NI. ACS Chem. Biol. 2007;2(12):799–809. doi: 10.1021/cb700186x. [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Yang Y, Sharma N, Tarasova NI, Timofeeva OA, Winkler-Pickett RT, Tanigawa S, Perantoni AO. Cell Signal. 2010 doi: 10.1016/j.cellsig.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubbard SR, Miller WT. Curr. Opin. Cell Biol. 2007;19(2):117–123. doi: 10.1016/j.ceb.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim SY, Wan X, Helman LJ. Bull. Cancer. 2009;96(7):E52–E60. doi: 10.1684/bdc.2009.0915. [DOI] [PubMed] [Google Scholar]
- 21.Gualberto A, Pollak M. Curr. Drug Targets. 2009;10(10):923–936. doi: 10.2174/138945009789577945. [DOI] [PubMed] [Google Scholar]
- 22.Hewish M, Chau I, Cunningham D. Recent Pat Anticancer Drug Discov. 2009;4(1):54–72. doi: 10.2174/157489209787002515. [DOI] [PubMed] [Google Scholar]
- 23.Chapuis N, Lacombe C, Tamburini J, Bouscary D, Mayeux P. Cancer Res. 2010 doi: 10.1158/0008-5472.CAN-10-0136. [DOI] [PubMed] [Google Scholar]
- 24.Hubbard SR. Nat. Rev. Mol. Cell Biol. 2004;5(6):464–471. doi: 10.1038/nrm1399. [DOI] [PubMed] [Google Scholar]
- 25.Weis F, Beiras-Fernandez A, Schelling G. Curr. Opin. Investig. Drugs. 2008;9(8):879–884. [PubMed] [Google Scholar]
- 26.Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO, Thompson JX, Roth AF, Drisdel RC, Mastro R, Green WN, Yates JR, III, Davis NG, El-Husseini A. Nature. 2008;456(7224):904–909. doi: 10.1038/nature07605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linder ME, Deschenes RJ. Nat. Rev. Mol. Cell Biol. 2007;8(1):74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 28.Pylypenko O, Schonichen A, Ludwig D, Ungermann C, Goody RS, Rak A, Geyer M. J Mol. Biol. 2008;377(5):1334–1345. doi: 10.1016/j.jmb.2008.01.099. [DOI] [PubMed] [Google Scholar]
- 29.Charollais J, Van Der Goot FG. Mol. Membr. Biol. 2009;26(1):55–66. doi: 10.1080/09687680802620369. [DOI] [PubMed] [Google Scholar]
- 30.Greaves J, Prescott GR, Gorleku OA, Chamberlain LH. Mol. Membr. Biol. 2009;26(1):67–79. doi: 10.1080/09687680802620351. [DOI] [PubMed] [Google Scholar]
- 31.Brunsveld L, Waldmann H, Huster D. Biochim. Biophys. Acta. 2009;1788(1):273–288. doi: 10.1016/j.bbamem.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 32.Sato T, Pallavi P, Golebiewska U, McLaughlin S, Smith SO. Biochemistry. 2006;45(42):12704–12714. doi: 10.1021/bi061264m. [DOI] [PubMed] [Google Scholar]
- 33.Fernandez-Vidal M, Jayasinghe S, Ladokhin AS, White SH. J Mol. Biol. 2007;370(3):459–470. doi: 10.1016/j.jmb.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peitzsch RM, McLaughlin S. Biochemistry. 1993;32(39):10436–10443. doi: 10.1021/bi00090a020. [DOI] [PubMed] [Google Scholar]
- 35.Wexler-Cohen Y, Shai Y. PLoS. Pathog. 2009;5(7):e1000509. doi: 10.1371/journal.ppat.1000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang E, Boire A, Agarwal A, Nguyen N, O'Callaghan K, Tu P, Kuliopulos A, Covic L. Cancer Res. 2009;69(15):6223–6231. doi: 10.1158/0008-5472.CAN-09-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Covic L, Misra M, Badar J, Singh C, Kuliopulos A. Nat Med. 2002;8(10):1161–1165. doi: 10.1038/nm760. [DOI] [PubMed] [Google Scholar]
- 38.Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. Science. 2008;319(5860):210–213. doi: 10.1126/science.1152066. [DOI] [PubMed] [Google Scholar]
- 39.Tarasova NI, Trinchieri G, Yuong H, Perantoni A, Stewart CA. Peptide-based ihnibitors of interleukin 10 or interferon gamma signalling. US61/333512. 2010 5-11-2010. Ref Type: Patent.
- 40.Petit MC, Benkirane N, Guichard G, Du AP, Marraud M, Cung MT, Briand JP, Muller S. J Biol. Chem. 1999;274(6):3686–3692. doi: 10.1074/jbc.274.6.3686. [DOI] [PubMed] [Google Scholar]
- 41.Fischer PM. Curr. Protein Pept. Sci. 2003;4(5):339–356. doi: 10.2174/1389203033487054. [DOI] [PubMed] [Google Scholar]
- 42.Chorev M, Goodman M. Trends Biotechnol. 1995;13(10):438–445. doi: 10.1016/S0167-7799(00)88999-4. [DOI] [PubMed] [Google Scholar]
- 43.Tarasova NI, Tarasov SG. Synthetic analogs of the juxtamembrane domain of IGF1R and uses thereof. US61/040203. 2008 3-28-2008. Ref Type: Patent.
- 44.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol. NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 45.Maloney EK, McLaughlin JL, Dagdigian NE, Garrett LM, Connors KM, Zhou XM, Blattler WA, Chittenden T, Singh R. Cancer Res. 2003;63(16):5073–5083. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.