

Abstract
Development of small molecule drug-like inhibitors blocking both nitric oxide synthase and NFκB could offer a synergistic therapeutic approach in the prevention and treatment of inflammation and cancer. During the course of evaluating the biologic potential of a commercial compound library, 2-phenylindole (1) displayed inhibitory activity against nitrite production and NF κB with IC50 values of 38.1±1.8 and 25.4±2.1 μM, respectively. Based on this lead, synthesis and systematic optimization have been undertaken in an effort to find novel and more potent nitric oxide synthase and NF κB inhibitors with antiinflammatory and cancer preventive potential. First, chemical derivatizations of 1 and 2-phenylindole-3-carboxaldehyde (4) were performed to generate a panel of N-alkylated indoles and 3-oxime derivatives 2–7. Second, a series of diversified 2-arylindole derivatives (10) were synthesized from an array of substituted 2-iodoanilines (8) and terminal alkynes (9) by applying one-pot palladium catalyzed Sonogashira-type alkynylation and base-assisted cycloaddition. Subsequent biological evaluations revealed 3-carboxaldehyde oxime and cyano substituted 2-phenylindoles 5 and 7 exhibited the strongest nitrite inhibitory activities (IC50 = 4.4±0.5 and 4.8±0.4 μM, respectively); as well as NFκB inhibition (IC50 = 6.9±0.8 and 8.5±2.0 μM, respectively). In addition, the 6′-MeO-naphthalen-2′-yl indole derivative 10at displayed excellent inhibitory activity against NFκB with an IC50 value of 0.6±0.2 μM.
Introduction
The indole heterocyclic ring constitutes an important structural scaffold in natural products and pharmaceutical molecules (Fig. 1).1–3 Among them, indolyl alkaloids represent a distinct class of bioactive molecules in chemical and medicinal fields. As examples, alkaloid reserpine was developed as one of the first drugs for the treatment of central nervous system (CNS) and cardiovascular system (CVS)-related diseases,4, 5 and vincristine, a member of Vinca alkaloids and a mitotic inhibitor, was discovered in the 1960s and clinically used for cancer chemotherapy.4 Since then, indole chemistry has received increasing interest in diverse drug discovery programs. Derivatives have been developed as antiinflammatory,6–8 antidepression,2 antihypertensive,9, 10 antiosteoporosis11 agents, among others. In addition, indole-based molecules have been used as molecular probes and pharmacological tools.12–14 In this regard, a great deal of effort has been expended for the development of novel and advanced synthetic methods for the preparation of indole and related chemical derivatives, such as using transition metal-catalyzed cross coupling reactions,15 multi-component reactions,16 microwave-assisted organic synthesis (MAOS),17 and solid-phase organic synthesis (SPOS).18, 19
Fig. 1.

Selected examples of drug molecules with an indole scaffold.
It is generally agreed that chronic inflammation is an important contributing factor in the process of carcinogenesis.20 Various transcriptional factors, including nuclear factor-κB (NFκB), interferon regulatory factor-1 (IRF-1), signal transducer and activator of transcription-1α (STAT-1α), activating protein-1 (AP-1), T-cell factor 4 (TCF-4), forkhead (Drosophila) homolog rhabdomyosarcoma-like 1 (FKHRL1), nuclear receptors, glucocorticoid receptor-α, and -β, and estrogen receptor- α and -β, have been reported to regulate inducible nitric oxide synthase (iNOS) expression.21 Nitric oxide (NO) is produced from L-arginine by NOS.22 This substance serves as a cellular signaling molecule and may be generated as an inflammatory response due to cellular stress and bacterial infection.23 However, overproduction can lead to tumor promotion and progression,24 so NOS inhibitors have therapeutic potential for the prevention and treatment of inflammation and cancer.
In addition, in part due to facilitating iNOS expression, NFκB has been recognized for a dominant role in inflammation and cancer.25–27 Inactivation of the NFκB pathway can decrease the probability of tumor initiation and progression, and activate apoptosis and decrease cell proliferation.28–34 Accordingly, investigations have been conducted to discover inhibitors of NFκB with potential as promising anticancer and cancer chemoprevention agents.35
With LPS-stimulated RAW 264.7 cells, NFκB binds to the promoter region of iNOS gene and plays an important role in iNOS expression and NO production.36 Thus, this cell line can be used as a model for studying inhibitory activity. In our ongoing effort of developing antiinflammatory and cancer preventive agents, we have employed natural product-inspired and high throughput screening approaches.37 Recently, during the course of evaluating the biologic potential of a commercial compound library, 2-phenylindole 1 was identified as an inhibitor of nitrite production and NFκB with modest IC50 values of 38.1±1.8 and 25.4±2.1 μM, respectively.
Thus far, no phenylindole derivatives have been described in the literature as inhibitors of NO production and NFκB regulation. Accordingly, on the basis of this 2-phenylindole lead, we sought to synthesize a diverse array of indole derivatives by systematically modifying the functional groups on the 2-phenylindole motif using one-pot palladium-catalyzed Sonogashira-type alkynylation and base-assisted cycloaddition. Here we report the synthesis and biological evaluation of 2-phenylindole derivatives.38
Results and discussion
Chemistry
Our investigation started with chemical derivatizations of 2-phenylindole 1. N-Alkylation of 1 took place in moderate to high yields on the treatment with NaH and DMF or THF at room temperature (Scheme 1).39 Deprotonation of 1 with NaH as the base, followed by reaction with MeI40 or MOMCl, afforded the N-alkylation derivatives 2a or 2c in 94% yields, respectively. In contrast, when benzyl, propargyl, and prenyl bromides were used as alkylating agents, lower yields were obtained due to the formation of their corresponding 1,3-disubstituted indole derivatives (2b′, 2d′, and 2e′). The reaction of 1 with (Boc)2O proceeded smoothly and afforded N-Boc-2-phenylindole (2f) in 81% yield.41 For comparison, the reduction of 1 was studied to examine if the aromatic indole system plays an important role in its biological activity. The 2-phenylindoline (3) was afforded in 72% yield under NaBH3CN in acetic acid (Scheme 1).42
Scheme 1.

Chemical derivatizations of 1.
Based on our initial screening data, compared with 1, the 2-phenylindole-3-carboxaldehyde 4 showed much less inhibitory activity in the nitrite and NFκB assays. Then we chose to synthesize 3-substituted 2-phenylindole derivatives to evaluate the effects of different substituents in the 3-position of the 2-phenylindole scaffold. Chemical transformations based on 4 were undertaken to generate a series of 3-functionalized derivatives, such as oxime and cyano derivatives (Scheme 2). Several substituted 2-phenyl-1H-indole-3-carboxaldehyde oximes 5 and 6a–b were then prepared by reacting 4 and the hydrochloride salt of hydroxylamine, methoxylamine, and O-benzylhydroxylamine, respectively. All reactions proceeded smoothly to produce the corresponding oxime-derivatives in excellent yields.43, 44 Under ultrasound irradiation for 2 h with Cu(OAc)2 as catalyst, the 3-cyano-2-phenylindole 7 could be obtained from 5 in 84% yield.45
Scheme 2.

Chemical transformations of 4.
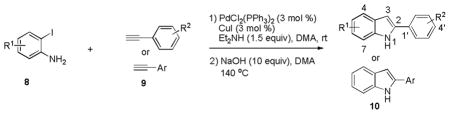
To further expand the chemical diversity of 2-phenylindole derivatives, a focused 2-phenylindole library 10 was next synthesized employing one-pot, two-step, Sonogashira-type alkynylation and base-assisted cycloaddition from an array of commercially available 2-iodoanilines 8 and terminal alkynes 9 (Table 1).46 The reaction was monitored by HPLC to make sure that the diaryl alkyne intermediate was transformed into the final cyclized indole product. Based on the optimized procedure, the scope and generality of Sonogashira-type alkynylation and base-assisted cycloaddition were examined.
Table 1.
Synthesis of indole derivatives 10 from 2-iodoanilines 8 and terminal alkynes 9a
| ||||
|---|---|---|---|---|
| Entry | R1 | R2 | Product | Yield (%)b |
| 1 | H | H |
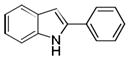 10aa |
70 |
| 2 | H | 2′-Me |
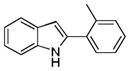 10ab |
67 |
| 3 | H | 4′-Me |
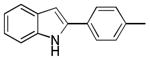 10ac |
68 |
| 4 | H | 3′,4′-Dichloro |
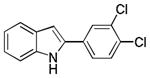 10ad |
59 |
| 5 | H | 3′,5′-Difluoro |
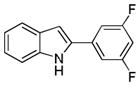 10ae |
70 |
| 6c | H | 3′,5′-Difluoro |
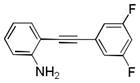 10ae′ |
95 |
| 7 | H | 4′-F |
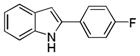 10af |
66 |
| 8 | H | 3′,5′-Dimethoxy |
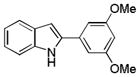 10ag |
67 |
| 9 | H | 4′-OMe |
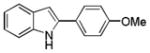 10ah |
59 |
| 10 | H | 3′-OMe |
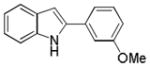 10ai |
70 |
| 11 | H | 4′-OCF3 |
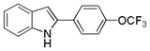 10aj |
71 |
| 12 | H | 4′-Br |
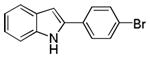 10ak |
56 |
| 13 | H | 4′-Pentyl |
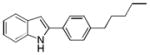 10al |
61 |
| 14 | H | 3′-COOH |
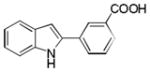 10am |
0 |
| 15 | H | 3′-NH2 |
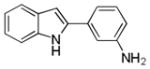 10an |
60 |
| 16 | H | 4′-NH2 |
 10ao |
52 |
| 17 | H | 4′-NMe2 |
 10ap |
40 |
| 18 | H | 3′-Pyridyl |
 10aq |
60 |
| 19 | H | 2′-Pyridyl |
 10ar |
0 |
| 20 | H | 3′-Thiophenyl |
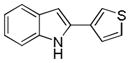 10as |
66 |
| 21 | H | 6′-Methoxy-naphthalen-2′-yl |
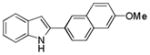 10at |
77 |
| 22 | 5-NO2 | H |
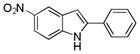 10ba |
79 |
| 23 | 5-NO2 | 2′-Me |
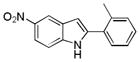 10 bb |
84 |
| 24 | 5-Cl | H |
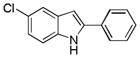 10ca |
64 |
| 25 | 6-Cl | H |
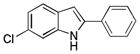 10da |
59 |
| 26 | 5-CN | H |
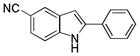 10ea |
84 |
| 27 | 5-Br | H |
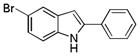 10fa |
83 |
| 28 | 6-Me | H |
 10ga |
25 |
| 29d | 5-NH2 | H |
 10ha |
67 |
Conditions: 0.5 mmol of substrate 8, 0.75 mmol of substrate 9, 3 mol % of PdCl2(PPh3)2, 3 mol % of CuI, 1.5 equiv of NHEt2, 10 equiv of NaOH, and 3 mL of DMA.
Isolated yield after column chromatography on silica gel.
Isolated alkynylation intermediate.
5-amino-2-phenyl-1H-indole 10ha was synthesized via reduction of 5-nitro-2-phenyl-1H-indole 10ba.
As shown in Table 1, in general, good functional group compatibility was observed among the terminal alkynes tested. Both electron-donating and electron-withdrawing substituents at the para, meta, or ortho positions of the 2-aryl ring were tolerated, producing the corresponding indole products 10aa-10at in moderate to good yields from 2-iodoaniline 8a (R1 = H) (entries 1–21, Table 1). Importantly, all the Sonogashira-type alkynylation reactions occurred smoothly within 1 h. With regarding to base-assisted cycloaddition, phenylacetylene with electron-withdrawing groups reacted relatively faster than those with electron-donating groups. Unfortunately, 3-ethynylbenzoic acid was found to be a poor substrate under these conditions (entry 14). Overall, terminal alkynes with heteroaromatic ring systems, such as pyridine and thiophene moieties, also reacted well, providing the corresponding indole derivatives in moderate yields (entries 18 and 20). However, the reaction of 2-ethynylpyridine was totally repressed compared with 3-ethynylpyridine, possibly due to the chelation of the 2′-nitrogen atom and the alkyne group with the palladium catalyst (entry 19).47 In contrast to the aromatic alkynes, when aliphatic terminal alkynes such as ethynylcyclohexane and ethynylcyclopentane were introduced in the reaction, no pure products could be isolated (data not shown). Meanwhile, the scope of substrate 8 was also briefly surveyed; different substituted 2-iodoanilines proved to be reactive under the same conditions (entries 22–28). It is important to note that electron-withdrawing groups on the substrates significantly facilitate the reaction, which is consistent with a previous report.46 For example, with the nitro or nitrile groups, the reaction proceeded to completion even at room temperature and the final products were obtained in high yields (entries 22, 23, and 26). The 5-amino derivative 10ha was also synthesized by reduction of 5-nitro-2-phenyl-1H-indole 10ba in 67% yield, employing Pd/C (10%) as catalyst under a hydrogen atmosphere (entry 29).48
Biological activities
All target compounds were tested as inhibitors of NO production and as the regulators of NFκB activity. Initial determinations were carried out at a compound concentration of 50 μM. If a compound displayed over 60% inhibition, further evaluations were then performed for the determination of IC50 values.
Nitrite inhibitory activity
As summarized in Table 2, the synthetic 2-phenylindole 10aa showed comparable inhibitory activity (IC50 = 24.7 μM) to commercial sample 1 (IC50 = 38.1 μM). To further evaluate the influence of the N-substituents, the alkylation derivatives of 2-phenylindole (2a–2e) and the N-Boc derivative 2f were synthesized and tested. Compound 2d (IC50 = 44.3 μM) with a propargyl substituent and compound 2f (IC50 = 34.2 μM) with a N-Boc group showed similar moderate inhibitory effects against nitrite production. However, the inhibitory effects of 2d and 2f were accompanied by cytotoxicity with IC50 values of 46.8 and 38.2 μM, respectively. Other alkylated indole products (2a–2c and 2e) were mainly inactive and exhibited less than 50% inhibition at a concentration of 50 μM. These data indicate an alkylation substituent at the N1 position may have a detrimental effect on NO inhibitory activity.
Table 2.
Evaluation of 2-arylindole derivatives for potential to inhibit iNOS activity and regulate NFκBa
| Compound | Nitrite assay | NFκB assay | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| % inhibition | % survivalb | IC50 (μM)c | Cytotoxic IC50 (μM)c | % inhibition | % survivalb | IC50 (μM)c | |
| 1 | 60.4±2.2 | 87.3±3.9 | 38.1±1.8 | 72.3±5 | 113.1±2.8 | 25.4±2.1 | |
| 2a | 33.6±4.9 | 74.3±6.2 | 0.0 | 105.5±12.3 | |||
| 2b | 0.0 | 84.6±2.0 | 0.0 | 96.3±8.9 | |||
| 2b′ | 0.0 | 101.0±6.1 | 0.0 | 124.1±6.2 | |||
| 2c | 33.8±4.9 | 67.3±6.2 | 21.5±3.3 | 106.7±2.8 | |||
| 2d | 96.9±1.4 | 35.8±3.2 | 44.3±2.1 | 46.8±1.9 | 46.2±2.1 | 105.2±5.3 | |
| 2e | 33.1±3.1 | 75.6±4.7 | 47.0±2.7 | 113.3±5.6 | |||
| 2e′ | 59.2±2.7 | 89.0±3.4 | 47.2±0.0 | 60.9±1.1 | 104.6±11.2 | 42.3±2.8 | |
| 2f | 92.4±4.0 | 27.6±2.4 | 34.2±1.5 | 38.2±5.0 | 0.0 | 107.1±16.3 | |
| 3 | 35.0±7.0 | 102.7±7.4 | 65.0±4.1 | 87.9±8.8 | NDd | ||
| 4 | 30.6±2.1 | 109.8±6.7 | 36.2±11.4 | 139.2±14.3 | |||
| 5 | 97.7±0.7 | 90.8±2.4 | 4.4±0.5 | 73.6±2.6 | 105.5±10.3 | 6.9±0.8 | |
| 6a | 97.2±2.4 | 48.3±7.8 | 22.5±1.2 | 48.0±5.1 | 48.8±8.0 | 51.8±23.0 | |
| 6b | 85.3±1.2 | 59.2±7.7 | 34.6±2.4 | 50.2±3.9 | 66.1±18.8 | ||
| 7 | 70.7±3.5 | 82.1±4.1 | 4.8±0.4 | 70.2±3.8 | 91.4±2.1 | 8.5±2.0 | |
| 10aa | 79.0±2.0 | 66.7±4.0 | 24.7±2.4 | 76.3±8.2 | 79.8±5.9 | 15.4±3.3 | |
| 10ab | 97.5±0.4 | 31.5±2.7 | 2.8±0.3 | 32.9±2.5 | 0.0 | 121.1±4.1 | |
| 10ac | 39.0±2.5 | 57.9±9.5 | 96.6±0.6 | 86.6±3.9 | 1.5±0.3 | ||
| 10ad | 55.8±2.7 | 67.4±2.8 | 45.0±2.5 | 68.2±6.3 | 115.1±2.1 | 40.2±3.5 | |
| 10ae | 23.5±4.6 | 86.5±9.4 | 62.3±8.3 | 107.9±10.0 | 48.3±1.5 | ||
| 10ae′e | 97.4±1.2 | 30.8±0.6 | 4.6±0.6 | 42.9±5.1 | 31.5±4.3 | 121.3±1.9 | |
| 10af | 58.0±5.9 | 69.7±3.7 | 17.9±3.1 | 77.9±1.4 | 84.3±8.8 | 16.5±3.7 | |
| 10ag | 62.6±3.4 | 109.5±2.4 | 29.6±3.8 | 56.4±2.3 | 66.16±4.1 | ||
| 10ah | 48.6±6.9 | 59.8±4.0 | 81.3±1.9 | 85.0±2.9 | 1.4±0.1 | ||
| 10ai | 50.7±0.9 | 92.8±6.4 | 49.7±0.2 | 85.5±3.0 | 65.8±15.0 | 5.8±1.1 | |
| 10aj | 31.2±5.7 | 105.7±3.5 | 79.7±8.8 | 74.8±6.6 | 6.5±0.1 | ||
| 10ak | 42.4±3.5 | 69.3±2.7 | 82.3±0.8 | 86.7±11.2 | 12.5±2.0 | ||
| 10al | 31.9±6.8 | 131.8±12.6 | 76.1±4.1 | 86.6±12.1 | 18.1±5.0 | ||
| 10an | 70.8±4.3 | 112.5±6.0 | 30.1±2.1 | 72.0±1.9 | 70.7±10 | 12.8±2.0 | |
| 10ao | 86.1±1.2 | 58.7±2.9 | 21.9±1.5 | 81.7±4.2 | 59.7±2.6 | 9.0±3.3 | |
| 10ap | 28.8±1.5 | 60.9±4.0 | 95.9±0.2 | 74.6±11.0 | 1.5±0.7 | ||
| 10aq | 46.6±3.4 | 107.0±7.0 | 12.3±4.4 | 71.4±5.5 | |||
| 10as | 66.3±4.6 | 90.6±2.6 | 20.4±1.8 | 35.6±3.1 | 76.0±16.6 | ||
| 10at | 13.9±3.6 | 58.0±7.7 | 94.4±1.6 | 93.5±2.8 | 0.6±0.2 | ||
| 10ba | 66.1±3.8 | 81.7±3.4 | 11.8±0.9 | 92.8±4.0 | 68.2±11.1 | 1.1±0.1 | |
| 10bb | 45.9±3.0 | 89.2±9.5 | 60.6±3.2 | 119.2±8.6 | NDd | ||
| 10ca | 49.3±2.1 | 77.8±2.9 | 2.3±0.9 | 112.6±12.1 | |||
| 10da | 36.3±2.6 | 74.0±11.9 | 91.5±1.2 | 89.4±15.7 | 9.8±2.3 | ||
| 10ea | 45.6±2.5 | 78.1±3.8 | 50.6±3.8 | 72.4±6.2 | |||
| 10fa | 42.4±4.0 | 91.8±8.7 | 92.2±2.7 | 86.1±6.8 | 2.4±0.3 | ||
| 10ga | 45.1±1.6 | 76.0±4.5 | 97.3±1.7 | 84.2±12.0 | 0.94±0.4 | ||
| 10ha | 67.4±3.3 | 100.2±6.7 | 21.1±5.7 | 78.4±12.1 | 73.8±19.6 | 24.6±3.6 | |
| 11f | 13.8±6.4 | 97.7±5.6 | 73.6±3.1 | 86.2±18 | 11.2±0.3 | ||
| Positive controlg | 22.1±0.2 | 3.8±0.6 | |||||
Concentration for screening: 50 μM.
Percentage of cell survival in comparison with vehicle-treated controls.
IC50 (μM): the half maximal inhibitory concentration (μM); if not listed, the IC50 values were >50 3M.
ND indicates the response was not dose-dependent.
10ae′: 2-[(3,5-difluorophenyl)ethynyl]aniline (the intermediate of 10ae).
2-phenylbenzimidazole 11 was purchased from Sigma-Aldrich.
IC50 values of control inhibitors in these assays; L-NG-monomethyl arginine for the nitrite assay and N-tosyl-L-phenylalanine-chloromethyl ketone for the κB assay.
Compound 3, the reduced variant of 1, was essentially inactive, indicating the importance of retaining the intact aromatic indole scaffold. From the 3-oxime series of 2-phenylindole, it was observed that 2-phenyl-1H-indole-3-carboxaldehyde oxime 5 showed the best inhibitory activity (IC50 = 4.4 μM). Compound 6a, with the N-methoxyl oxime functionality, exhibited a weaker response (IC50 = 22.5 μM) in comparison with 5. The bulky benzyl substituted product 6b was less effective. On the other hand, compound 7, with a 3-cyano group, exhibited inhibitory activity comparable with compound 5 (IC50 = 4.8 μM). These data suggest that both the size and electronic character of the substituent at the 3-position of 2-phenylindole have effect on the inhibitory activity.
When different functional substituents were introduced to investigate the influence of the 2-phenyl group on activity, variable results were obtained. Compared with 2-phenylindole 10aa, compound 10ab (2′-Me, IC50 = 2.8 μM) exhibited much more potent activity, whereas compound 10ac was essentially inactive (4′-Me, 39.0% at 50 μM). However, 10ab was also cytotoxic, yielding a cytotoxic IC50 value of 32.9 μM. This effect is likely due to the steric hindrance of the 2′-Me group and subsequent conformational change of the 2-phenylindole moiety.
We also investigated the effect of halogen atoms attached to the phenyl group. Derivative 10ad, bearing two chloro atoms, showed moderate inhibitory activity (IC50 = 45.0 μM). A reduction in activity was obtained by introducing two fluoro atoms (10ae, 23.5% inhibition at 50 μM). Interestingly, the intermediate 10ae′ (IC50 = 4.6 μM) showed much better inhibitory activity than the final cycloaddition product 10ae. Further improved activity was observed with compound 10af (4′-F group; IC50 = 17.9 μM). In contrast, the corresponding 4′-Br derivative (10ak) showed less activity (42.4% at 50 μM). Other groups, such as methoxyl (10ag-10ai), trifluoromethoxyl (10aj) and long alkyl chain (10al) were also tested; 10ag (IC50 = 29.6 μM) with 3′,5′-dimethoxyl groups demonstrated the most potent activity in this series. The mono-substituted methoxyl derivative (10ah, 4′-OMe, 48.6% inhibition at 50 μM; 10ai, 3′-OMe, IC50 = 49.7 μM) showed much weaker inhibitory activity.
By appending an amine group in the benzene ring, compound 10an with a 3′-amino group (IC50 = 30.1 μM) produced better inhibition of NO production than compound 10ao with a 4′-amino group. No improved activity could be achieved when the amino group was substituted with two methyl groups (10ap, 28.8% inhibition at 50 μM). Changing the phenyl into a heteroaromatic group, the thiophen-3-yl indole derivative showed greater activity (10as, IC50 = 20.4 μM) than the pyridine moiety (10aq, 46.6% at 50 μM).
Finally, the effect of substituents on the indole moiety was studied. It was observed that only 5-NO2-2-phenylindole (10ba) and its corresponding reduced variant, 5-NH2-2-phenylindole (10ha), showed good inhibitory activities with IC50 values of 11.8 and 21.1 μM, respectively. Compounds 10bb-10ga with Cl, CN, Br and Me substituents on the indole system were only marginally active.
For direct comparison, 2-phenylbenzimidazole 11 was assessed in this assay. Only marginal activity was observed (13.8% at 50 μM), which indicates that the replacement of the pyrrole ring with the imidazole ring is detrimental for inhibitory activity.
NFκB inhibitory activity
In the NFκB assay, 2-phenylindole 10aa displayed moderate inhibitory activity (IC50 = 15.4 μM), whereas the commercial sample 1 exhibited an IC50 value of 25.4 μM. From the N-alkylation and N-Boc series, all compounds (2a–f) were inactive at a concentration of 50 μM, except the diprenylsubstituted 2-phenylindole derivative (2e′, IC50 = 42.3 μM). In the oxime series (5 and 6a–b), only compound 5 with a N-hydroxyl group exhibited good NFκB inhibitory activity (IC50 = 6.9 μM). These data demonstrated the importance of the free N-hydroxyl group. Similar to the pattern observed in the nitrite assay, compound 7 with a cyano group in the 3-position also showed stronger inhibitory activity (IC50 = 8.5 μM). Taken together, it appears that small and polar substituents on the 3-position of 2-phenylindole scaffold may enhance the inhibitory effect.
In terms of different substitutents on the 2-phenyl ring, most compounds tested showed significant NFκB inhibitory activity. The substitution pattern also influenced activity. For instance, compound 10ac bearing a 4′-Me group produced potent activity (IC50 = 1.5 μM), while compound 10ab with a 2′-Me group was inactive. Introducing a mono-halogen atom into the benzene ring proved to be beneficial for activity compared with compound 10aa. For example, compound 10af with a 4′-F and 10ak with a 4′-Br on the benzene ring showed slightly improved activities (10af, IC50 = 16.5 μM; 10ak, IC50 = 12.5 μM). In contrast, both compounds 10ad with two chloro atoms and 10ae with two fluoro atoms showed a significant decrease in NFκB inhibition, with IC50 values of 40.2 and 48.3 μM, respectively. For comparison, the intermediate 10ae′ was also included in the assay and it displayed lower activity than the final cycloaddition product 10ae.
As presented in Table 2, it appears the methoxy group affected the NFκB inhibition in a manner similar to halogen atoms. Compound 10ag, bearing 3′,5′-dimethoxy on the phenyl ring, was inactive and only exhibited 56.4% inhibition at 50 μM. In contrast, when one methoxy group is attached, both compounds 10ah (4′-OMe) and 10ai (3′-OMe) showed much more potent inhibitory activities, with IC50 values of 1.4 and 5.8 μM, respectively. The most potent inhibitor obtained was compound 10at (IC50 = 0.6 μM), bearing one methoxy group on the naphthalene moiety. This was probably due to the enhanced and favorable lipophilicity profile.
Compared with the free amino-substituted compounds 10an (3′-NH2) and 10ao (4′-NH2), compound 10ap exhibited higher inhibition of NFκB with an IC50 of 1.5 μM. Less than 40% inhibition at 50 μM was produced when the phenyl group was replaced with heteroaromatic rings including the 3-pyridyl and 3-thiophenyl moieties. For substituents on the indole scaffold, compounds 10ba with a 5-NO2 group and 10ga with a 6-Me group demonstrated significant inhibitory activities with IC50 values of 1.1 and 0.94 μM, respectively. For the halogen substituted compounds, 10da and 10fa showed intermediate activities with IC50 values of 9.8 and 2.4 μM, respectively. In addition, 2-phenylbenzimidazole 11 mediated an inhibitory response with an IC50 value of 11.2 μM.
Correlation between NOS and NFκB and preliminary SAR considerations
Several factors can affect the production of NO including the cellular level of L-arginine as an iNOS substrate and the enzymatic activity or expression level of iNOS.49 In the assay employed for this investigation, increased production of NO results mainly from iNOS expression following stimulation with LPS. One potential mechanism of the inhibitory effect mediated by a test compound is inhibition of NFκB activity, one of transcription factors related to iNOS expression.21 In support of this suggestion, among the compounds tested with IC50 values under 50 μM in both the nitrite and NFκB assays (1, 2e′, 5, 7, 10aa, 10ad, 10af, 10an, 10ao, 10ba, and 10ha), linear regression analysis revealed a correlation coefficient of R2 = 0.76 (Fig. 2).
Fig. 2.

The correlation between inhibitory activities of eleven compounds (closed diamond) in nitrite and NFκB assays. Trend analysis using Microsoft Office Excel 2007: R2= 0.76. 10ai with an IC50 value of 49.7 μM (round number as 50 μM), is excluded.
However, a lack of correlation with other compounds is not surprising. First of all, the NFκB and nitrite assays were established using different experimental conditions, e.g., different stimuli (TNFα vs. LPS) with different signaling pathways (TNF receptor vs. Toll-like receptor 4) in different cell lines (HEK293 vs. RAW 264.7). Compounds exerting inhibitory effects on nitrite production without NFκB inhibition might suppress other signaling pathways such as JAK2/STAT1 and MAPK. On the other hand, compounds showing only NFκB inhibition might inhibit TNF receptor directly. In addition, despite structural similarities through sharing the phenylindole moiety, distinctly different mechanisms of actions may apply. For instance, it has been reported that resveratrol and an analog 4,4′-dihydroxy-trans-stilbene mediate an antiproliferative response in human fibroblasts via different mechanisms.50 In the final analysis, further studies are required to clarify underlying mechanisms.
In considering compounds mediating superior activity in one of our assay systems, cleavage of the pyrrole ring in 10ae led to loss of NFκB inhibitory activity but improved inhibition of nitrite production. The location and number of fluoro atoms at the 2-phenyl ring might be crucial for the dual activities. Compound 10ae with the 3′,5′-difluoro substituents showed NFκB inhibition while 10af with the 4′-F substituent exerted dual inhibition. Similarly, the location and number of methoxy (-OMe) groups affected the inhibitory activities (10ah, 10ai vs. 10ag).
Modifications of the oxime functionality (=N-OH) in 5 with a methyl or a benzyl group resulted in loss of NFκB activities, suggesting the crucial role of the free oxime functionality in phenylindoles for NFκB inhibition. Also, the introduction of the amino group (-NH2) at the 3′, 4′, or 5-position of the 2-phenylindole moiety (10an, 10ao, and 10ha) showed dual activities, while NMe2-substituted compound (10ap) lost inhibitory activity in nitrite production. Both hydroxyl (-OH) and amino (-NH2) groups can be involved in hydrogen bonding, therefore, the inhibitory effects might be associated with hydrogen bonds with these compounds. A preliminary SAR is summarized in Fig. 3.
Fig. 3.
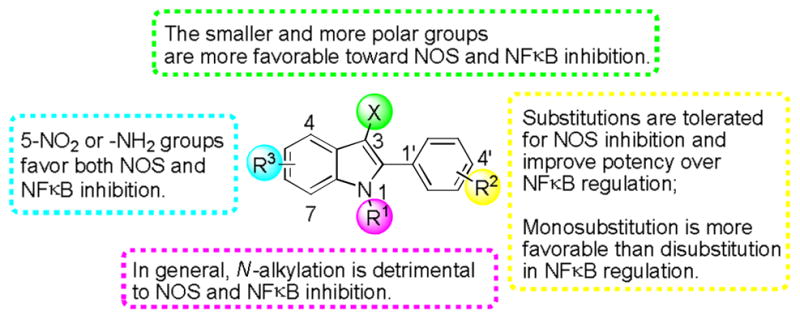
Preliminary SAR of 2-phenylindole derivatives.
Conclusions
In conclusion, a series of 2-phenylindole derivatives has been synthesized. The reactions employing one-pot palladium-catalyzed Sonogashira-type alkynylation and base-assisted cycloaddition feature broad substrate scope. This indole-based chemical library was subsequently evaluated for inhibition of nitrite production and NFκB activity. The 2-phenylindole-3-carboxaldehyde oxime 5 showed the most potent inhibition of nitrite production (IC50 = 4.4 μM) among this series of compounds without appreciable cytotoxicity (90.8±2.4% survival at the highest concentration of 50 μM tested). Stronger inhibition of NO production was observed with the 2′-tolyl indole derivative 10ab (IC50 = 2.8 μM). Although notable cytotoxicity was observed at 50 μM as 31.5±2.7% cell survival with an IC50 value of 32.9 μM, the cytotoxic effect of 10ab did not affect the inhibitory activity (e.g., the cell survival was 94.5% at the IC80 concentration, 9.3 μM). With NFκB, most 2-phenylindole derivatives exhibited good to excellent inhibitory activities, especially the 6′-MeO-naphthalen-2′-yl indole derivative 10at (IC50 = 0.6 μM). It is also important to note compounds 5 and 7 inhibit both NOS and NFκB with IC50 values ranging from 4.4 to 8.5 μM. Based on these promising results and existing SAR, further medicinal chemistry optimization is warranted to identify more potent and less toxic 2-arylindole dual NOS/NFκB inhibitors with antiinflammatory and anticancer therapeutic potential.
Experimental Section
Chemistry
All reagents and anhydrous solvents were purchased from commercial sources were used without further purification. All reactions were monitored either by thin-layer chromatography (TLC) or by analytical high performance liquid chromatography (HPLC) to detect the completion of reactions. Hydrogenation reactions were performed employing domnick hunter NITROX UHP-60H hydrogen generator, USA. Compounds were purified by flash column chromatography on silica gel using a Biotage Isolera One system and a Biotage SNAP cartridge. 1H and 13C NMR spectra were obtained on a Bruker Avance DRX-400 instrument with chemical shifts (δ, ppm) determined using TMS as internal standard. Coupling constants (J) are in hertz (Hz). ESI mass spectra in either positive or negative mode were provided by Varian 500-MS IT Mass Spectrometer. The purity of compounds was determined by analytical HPLC using a Gemini, 3 μm, C18, 110 Å column (50 mm × 4.6 mm, Phenomenex) and a flow rate of 1.0 mL/min. Gradient conditions: solvent A (0.1% trifluoroacetic acid in water) and solvent B (acetonitrile): 0–2.00 min 100% A, 2.00–7.00 min 0–100% B (linear gradient), 7.00–8.00 min 100% B, UV detection at 254 and 220 nm.
Representative procedure for the alkylation of 2-phenylindoles (2a–e)
A solution of 2-phenylindole (96.5 mg, 0.5 mmol) in DMF (1 mL) was added dropwise to a slurry of NaH (24 mg, 0.6 mmol, 60% dispersed in mineral oil) in DMF (2 mL) at 0 °C. After stirring for 1 h, neat iodomethane (71 mg, 0.5 mmol) was added dropwise to the reaction mixture. Upon disappearance of the starting material as indicated by TLC (1 h), the reaction mixture was poured into ice water and extracted with EtOAc (3 × 10 mL). The organic layer was combined and dried over Na2SO4 and the solvent was removed in vacuo. The crude material was purified by flash column chromatography on silica gel (hexane/EtOAc = 95/5) to yield 97.2 mg (94%) of 2-phenyl-N-methylindole (2a) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 8.0 Hz, 1H), 7.43 (d, J = 6.8 Hz, 2H), 7.37 (t, J = 7.6 Hz, 2H), 7.32-7.28 (m, 2H), 7.17 (t, J = 8.0 Hz, 1H), 7.07 (t, J = 8.0 Hz, 1H), 6.49 (s, 1H), 3.63 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 141.6, 138.5, 132.9, 129.4, 128.5, 128.1, 127.9, 121.7, 120.6, 119.9, 109.7, 101.7, 31.2; ESI-MS: calc. for C15H13N [M + H]+: 208.1, found: 208.1. HPLC purity: 98.7% (254 nm), tR: 7.47 min; 97.6% (220 nm), tR: 7.46 min.
2-Phenyl-N-benzylindole 2b: white solid; yield 64%; 1H NMR (400 MHz, CDCl3) δ 7.68 (s, 1H), 7.44-7.36 (m, 5H), 7.28-7.22 (m, 3H), 7.17-7.15 (m, 3H), 7.03-7.02 (m, 2H), 6.67-6.65 (m, 1H), 5.36 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 141.9, 138.3, 138.0, 132.8, 129.3, 128.8, 128.6, 128.4, 128.1, 127.2, 126.1, 122.0, 120.6, 120.3, 110.6, 102.4, 47.8; ESI-MS: calc. for C21H17N [M + H]+: 284.1, found: 284.2. HPLC purity: 100% (254 nm), tR: 7.83 min; 100% (220 nm), tR: 7.82 min.
2-Phenyl-3-benzyl-N-benzylindole 2b′: yellow solid; yield 28%; 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 8.0 Hz, 1H), 7.35 (t, J = 3.2 Hz, 3H), 7.30-7.28 (m, 2H), 7.23-7.11 (m, 10H), 7.06 (t, J = 6.8 Hz, 1H), 6.95 (d, J = 6.8 Hz, 2H), 5.25 (s, 2H), 4.09 (s, 2H); 13C NMR (400 MHz, CDCl3) δ 141.9, 138.9, 138.4, 137.0, 131.7, 130.5, 128.6, 128.4, 128.2, 127.0, 126.1, 125.6, 121.9, 119.6, 112.2, 110.3, 47.6, 30.7; ESI-MS: calc. for C28H23N [M + H]+: 374.2, found: 374.3. HPLC purity: 100% (254 nm), tR: 8.16 min; 100% (220 nm), tR: 8.15 min.
2-Phenyl-N-methoxymethylindole 2c: white solid; yield 94%; 1H NMR (400 MHz, CDCl3) δ 7.60 (t, J = 7.2 Hz, 3H), 7.49 (d, J = 8.0 Hz, 1H), 7.43 (t, J = 7.6 Hz, 2H), 7.37 (d, J = 7.2 Hz, 1H), 7.24 (t, J = 7.2 Hz, 1H), 7.16 (t, J = 7.6 Hz, 1H), 6.59 (s, 1H), 5.37 (s, 2H), 3.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 141.7, 138.3, 132.4, 129.4, 128.5, 128.3, 128.1, 122.3, 120.7, 120.5, 110.2, 103.4, 74.7, 55.8; ESI-MS: calc. for C16H15NO [M + H]+: 238.1, found: 238.2. HPLC purity: 100% (254 nm), tR: 7.37 min; 100% (220 nm), tR: 7.37 min.
2-Phenyl-N-propargylindole 2d: white solid; yield 70%; 1H NMR (400 MHz, CDCl3) δ 7.59 (dd, J = 8.0 and 7.2 Hz, 3H), 7.45 (dd, J = 8.8 and 8.4 Hz, 3H), 7.37 (t, J = 7.2 Hz, 1H), 7.27 (t, J = 8.0 Hz, 1H), 7.16 (t, J = 7.6 Hz, 1H), 6.57 (s, 1H), 4.76 (d, J = 2.4 Hz, 2H), 2.30 (t, J = 2.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 140.9, 137.6, 132.2, 129.2, 128.7, 128.3, 128.1, 122.1, 120.7, 120.5, 110.0, 102.5, 78.9, 72.7, 34.0; ESI-MS: calc. for C17H13N [M+ H]+: 232.1, found: 232.1. HPLC purity: 100% (254 nm), tR: 7.40 min; 100% (220 nm), tR: 7.40 min.
2-Phenyl-N-prenylindole 2e: oil; yield 8%; 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 8.0 Hz, 1H), 7.49 (d, J = 7.6 Hz, 2H), 7.45 (t, J = 7.2 Hz, 1H), 7.40-7.29 (m, 3H), 7.23 (t, J = 8.0 Hz, 1H), 7.20 (t, J = 8.0 Hz, 1H), 6.54 (s, 1H), 5.32-5.30 (m, 1H), 4.71 (d, J = 6.0 Hz, 2H), 1.69 (s, 3H), 1.33 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 141.3, 137.6, 134.2, 133.0, 129.5, 128.4, 128.2, 127.8, 121.5, 121.1, 120.5, 119.8, 110.2, 101.7, 42.6, 25.5, 17.9; ESI-MS: calc. for C19H19N [M + H]+: 262.2, found: 262.2. HPLC purity: 76.4% (254 nm), tR: 7.97 min; 81.1% (220 nm), tR: 7.97 min; with the remaining of 2e′.
2-Phenyl-3-prenyl-N-prenylindole 2e′: oil; yield 6%; 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 8.0 Hz, 1H), 7.44 (t, J = 7.2 Hz, 2H), 7.38 (t, J = 6.8 Hz, 3H), 7.30 (d, J = 8.0 Hz, 1H), 7.21 (t, J = 7.2 Hz, 1H), 7.12 (t, J = 7.2 Hz, 1H), 5.29 (t, J = 7.2 Hz, 1H), 5.20 (t, J = 6.4 Hz, 1H), 4.56 (d, J = 6.4 Hz, 1H), 3.35 (d, J = 6.8 Hz, 1H), 1.65 (s, 6H), 1.52 (d, J = 4.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 137.3, 136.4, 133.9, 132.3, 130.9, 130.3, 128.2, 127.9, 127.8, 124.4, 121.5, 121.1, 119.2, 119.1, 112.8, 109.8, 42.3, 25.6, 25.5, 23.9, 17.74, 17.71; ESI-MS: calc. for C24H27N [M + H] +: 330.5, found: 330.3. HPLC purity: 85.1% (254 nm), tR: 8.46 min; 86.4% (220 nm), tR: 8.46 min; with the remaining of 2e.
Synthesis of 2-phenyl-N-Boc-indole (2f)
Under nitrogen atmosphere, (Boc)2O (d 0.95 g/mL, 0.125 mL, 0.55 mmol) was added to a solution of 2-phenylindole (96.5 mg, 0.5 mmol) and 4-(N,N-dimethylamino)pyridine (DMAP) (1.8 mg, 0.015 mmol) in dry acetonitrile (3 mL) at room temperature. The solution was stirred at room temperature for 24 h, and then was evaporated under reduced pressure. After water was added, the resulting mixture was extracted twice with EtOAc (2 × 10 mL). The combined organic layer was washed with brine, dried with Na2SO4, and evaporated under reduced pressure. The residue was purified with flash column chromatography on silica gel (EtOAc/hexane = 1/50) to give the pure product 2f (118.4 mg, 81%) as a colorless solid. 1H NMR (400 MHz, CDCl3) δ 8.22 (d, J = 8.4 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.42-7.30 (m, 6H), 7.24 (t, J = 7.2 Hz, 1H), 6.54 (s, 1H), 1.29 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 150.2, 140.5, 137.4, 135.0, 129.2, 128.7, 127.7, 127.5, 124.2, 122.9, 120.4, 115.2, 109.9, 83.3, 27.5; ESI-MS: calc. for C19H19NO2 [M − H]−: 292.1, found: 292.3. HPLC purity: 100% (254 nm), tR: 7.98 min; 100% (220 nm), tR: 7.97 min.
Typical procedure for the reduction of 2-phenylindole to 2-phenylindoline 3
To a solution of 2-phenylindole (96.5 mg, 0.5 mmol) in AcOH (2 mL) was added NaBH3CN (189 mg, 3 mmol), and the resulting mixture was stirred until no starting material could be detected by TLC analysis (24 h). Then, 6 mL of H2O was added and additional NaOH pellets were added until pH > 12, the resulting solution was extracted with Et2O (3 × 10 mL). The organic phases were combined, dried over Na2SO4 and the solvent was evaporated under reduced pressure to give the residue, which was purified by flash column chromatography on silica gel to afford the pure product 2-phenylindoline 3 (70.5 mg, 72%). Oil; 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 7.2 Hz, 2H), 7.31 (t, J = 7.6 Hz, 2H), 7.24 (t, J = 7.6 Hz, 1H), 7.07-7.03 (m, 2H), 6.71 (t, J = 7.2 Hz, 1H), 6.63 (d, J = 7.6 Hz, 1H), 4.91 (t, J = 8.8 Hz, 1H), 3.41 (dd, J = 8.8 and 15.6 Hz, 1H), 2.96 (dd, J = 8.8 and 15.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 150.9, 144.6, 128.5, 128.0, 127.5, 127.3, 126.2, 124.5, 118.8, 108.8, 63.4, 39.5; ESI-MS: calc. for C14H13N [M + H] +: 196.1, found: 196.2. HPLC purity: 100% (254 nm), tR: 5.68 min; 100% (220 nm), tR: 5.68 min.
Synthesis of 2-phenylindole-3-carboxaldehyde oxime 5
Hydroxylamine hydrochloride (104 mg, 1.5 mmol) was added to a solution of 2-phenylindole-3-carboxaldehyde 4 (221 mg, 1 mmol) and sodium hydroxide (60 mg, 1.5 mmol) in methanol (5 mL). The reaction mixture was stirred at room temperature until no starting material was detected by TLC and HPLC analysis (3 h). The reaction mixture was evaporated in vacuo and EtOAc was added to the residue. The solution was washed with water, and the organic layer was dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography on silica gel to give the 2-phenylindole-3-carboxaldehyde oxime 5 (197.2 mg, 83%) as a white solid. 1H NMR (400 MHz, d6-DMSO) δ 11.75 (s, 1H), 10.73 (s, 1H), 8.30 (s, 1H), 8.11 (d, J = 7.6 Hz, 1H), 7.62 (t, J = 8.8 Hz, 2H), 7.56 (t, J = 7.6 Hz, 2H), 7.49-7.44 (m, 3H), 7.22 (t, J = 7.2 Hz, 1H), 7.12 (t, J = 7.2 Hz, 1H); 13C NMR (100 MHz, d6-DMSO) δ 144.4, 139.7, 136.4, 131.4, 129.0, 128.6, 125.6, 122.8, 121.9, 120.5, 111.5, 105.9; ESI-MS: calc. for C15H13N2O [M+ H] +: 237.1, found: 237.1. HPLC purity: 100% (254 nm), tR: 6.49 min; 100% (220 nm), tR: 6.49 min.
Representative procedure for the preparation of oximes 6a and 6b
Methoxylamine hydrochloride (50 mg, 0.6 mmol, 1.2 equiv) was suspended in 1 mL of absolute ethanol, and anhydrous pyridine (158 mg, 2 mmol, 4.0 equiv) was added quickly dropwise. Then 2-phenylindole-3-carboxaldehyde 4 (110.5 mg, 0.5 mmol) was added and the reaction was stirred at room temperature for 4 h. The ethanol was removed and the residue was redissolved in dichloromethane and washed with water. The organic layer was dried with Na2SO4, filtered and evaporated to give a crude oil, which was purified by flash chromatography to afford a clear oil as product 6a in 91% yield. 2-Phenyl-1H-indole-3-carboxaldehyde O-methyl oxime 6a: 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 8.38-8.36 (m, 2H), 7.46-7.44 (m, 5H), 7.37-7.36 (m, 1H), 7.30-7.28 (m, 2H), 4.06 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 145.2, 140.6, 136.1, 131.2, 129.1, 128.9, 128.8, 125.9, 123.7, 122.9, 121.6, 111.0, 106.6, 61.9; ESI-MS: calc. for C16H14N2O [M + H]+: 251.3, found: 251.2. HPLC purity: 100% (254 nm), tR: 7.19 min; 100% (220 nm), tR: 7.19 min.
2-Phenyl-1H-indole-3-carboxaldehyde O-benzyl oxime 6b: Oil, yield: 89%; 1H NMR (400 MHz, CDCl3) δ 8.40 (s, 1H), 8.28 (s, 1H), 8.18 (s, 1H), 7.46 (d, J = 7.2 Hz, 2H), 7.33-7.28 (m, 7H), 7.28-7.22 (m, 4H), 5.23 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 145.6, 140.7, 137.9, 136.0, 131.2, 129.0, 128.9, 128.7, 128.5, 127.9, 126.0, 123.6, 123.0, 121.6, 111.0, 106.6, 76.3; ESI-MS: calc. for C22H18N2O [M+ H]+: 327.4, found: 327.2. HPLC purity: 100% (254 nm), tR: 7.63 min; 100% (220 nm), tR: 7.63 min.
Synthesis of 2-phenylindole-3-carbonitrile 7
2-Phenylindole-3-carboxaldehyde oxime 5 (38.7 mg, 0.16 mmol), Cu(OAc)2 (1.5 mg, 5 mol %) and acetonitrile (1.0 mL) were added to a 10 mL tube. After being capped, the reaction mixture was under continuous ultrasound irradiation for 2 h. Following sonication, the reaction mixture was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel (EtOAc/hexane = 1/8) to afford 2-phenylindole-3-carbonitrile 7 (29.3 mg, 84%) as a white solid. 1H NMR (400 MHz, d6-DMSO) δ 12.59 (s, 1H), 7.98 (d, J = 7.6 Hz, 2H), 7.65-7.60 (m, 3H), 7.58-7.52 (m, 2H), 7.34-7.24 (m, 2H); 13C NMR (100 MHz, d6-DMSO) δ 144.7, 135.5, 129.9, 129.3, 128.2, 127.0, 123.9, 122.0, 118.3, 116.9, 112.6, 81.4; ESI-MS: calc. for C15H10N2 [M + H]+: 219.1, found: 219.1. HPLC purity: 95.3% (254 nm), tR: 6.87 min; 99.9% (220 nm), tR: 6.87 min.
Representative procedure for the synthesis of functionalized 2-phenylindole derivatives 10
A mixture of 2-iodoaniline (109.5 mg, 0.5 mmol), phenylacetylene (76.5 mg, 0.75 mmol), PdCl2(PPh3)2 (10.5 mg, 3 mol %), CuI (2.85 mg, 3 mol %) in DMA (3 mL) was stirred in a sealed tube at room temperature until no starting material was detected by HPLC (1 h). NaOH (200 mg, 5 mmol) was added and the reaction temperature was raised to 140 °C for several hours (the completion of the cyclization was monitored by HPLC). The reaction was cooled to room temperature, and EtOAc and water was added. The separated aqueous phase was extracted with EtOAc (3 × 10 mL) and the combined organic layers were washed with water (2 × 20 mL) and dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography (EtOAc/hexane = 1/20) to afford 2-phenyl-1H-indole 10aa (67.5 mg, 70%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 7.61 (d, J = 7.2 Hz, 3H), 7.40 (t, J = 7.2 Hz, 2H), 7.35 (dd, J = 0.8 and 8.4 Hz, 1H), 7.29 (t, J = 7.2 Hz, 1H), 7.21-7.18 (m, 1H), 7.16-7.09 (m, 1H), 6.80 (d, J = 0.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 137.9, 136.8, 132.4, 129.3, 128.9, 127.7, 126.1, 122.3, 120.6, 120.2, 110.9, 99.9; ESI-MS: calc. for C14H11N [M − H] −: 192.1, found: 192.3. HPLC purity: 100% (254 nm), tR: 7.12 min; 100% (220 nm), tR: 7.11 min.
2-o-Tolyl-1H-indole 10ab: brown solid; yield: 67%; 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 7.64 (d, J = 7.6 Hz, 1H), 7.48-7.45 (m, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.32-7.27 (m, 3H), 7.23-7.20 (m, 1H), 7.19 (t, J = 7.8 Hz, 1H), 6.61 (d, J = 1.2 Hz, 1H), 2.51 (d, J = 1.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 137.5, 136.23, 136.18, 132.7, 131.1, 129.1, 128.9, 128.0, 126.2, 122.1, 120.6, 120.1, 110.6, 103.0, 21.2; ESI-MS: calc. for C15H13N [M−H]−: 206.1, found: 206.3. HPLC purity: 100% (254 nm), tR: 7.23 min; 100% (220 nm), tR: 7.22 min.
2-p-Tolyl-1H-indole 10ac: yellow solid; yield: 68%; 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 7.62-7.59 (m, 1H), 7.50 (dd, J = 2.0 and 6.8 Hz, 2H), 7.34 (dd, J = 2.0 and 6.8 Hz, 1H), 7.22 (dd, J = 1.2 and 7.2 Hz, 2H), 7.19-7.16 (m, 1H), 7.15-7.08 (m, 1H), 6.77-6.76 (m, 1H), 2.37 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 138.2, 137.8, 136.8, 129.8, 129.7, 129.5, 125.2, 122.3, 120.7, 120.3, 111.0, 99.5, 21.4; ESI-MS: calc. for C15H13N [M + H]+: 208.1, found: 208.2. HPLC purity: 100% (254 nm), tR: 7.29 min; 100% (220 nm), tR: 7.28 min.
2-(3,4-Dichlorophenyl)-1H-indole 10ad: white solid; yield: 59%; 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 7.56 (d, J = 2.0 Hz, 1H), 7.52 (d, J = 7.6 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.30 (d, J = 2.0 Hz, 1H), 7.27 (dd, J = 2.0 and 8.0 Hz, 1H), 7.12 (t, J = 7.2 Hz, 1H), 7.05 (t, J = 7.2 Hz, 1H), 6.69 (d, J = 1.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 137.0, 135.3, 133.1, 132.3, 131.3, 130.9, 129.0, 126.7, 124.2, 123.0, 120.9, 120.6, 111.1, 101.2; ESI-MS: calc. for C14H9Cl2N [M + H]+: 262.0, found: 262.0. HPLC purity: 100% (254 nm), tR: 7.63 min; 100% (220 nm), tR: 7.64 min.
2-(3,5-Difluorophenyl)-1H-indole 10ae: white solid; yield: 70%; 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.25-7.21 (m, 1H), 7.16-7.12 (m, 3H), 6.83 (d, J = 1.2 Hz, 1H), 6.76-6.72 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 164.8, 164.7, 162.3, 162.2, 137.0, 135.5, 128.9, 123.3, 121.1, 120.7, 111.1, 108.0, 107.9, 107.8, 107.7, 103.0, 102.8, 102.5, 101.8; ESI-MS: calc. for C14H9F2N [M−H]−: 228.1, found: 228.2. HPLC purity: 100% (254 nm), tR: 7.29 min; 100% (220 nm), tR: 7.30 min.
2-((3,5-Difluorophenyl)ethynyl)aniline 10ae′: brown oil; yield: 95%; 1H NMR (400 MHz, CDCl3) δ 7.34 (t, J = 8.0 Hz, 1H), 7.18-7.14 (m, 1H), 7.04-6.99 (m, 2H), 6.81-6.70 (m, 3H), 4.20 (s, br, 2H); 13C NMR (100 MHz, CDCl3) δ 164.0, 163.9, 161.6, 161.5, 148.1, 132.4, 120.5, 126.1, 118.1, 114.6, 114.5, 114.3, 114.2, 106.9, 104.6, 104.3, 104.1, 92.53, 92.49, 92.46, 88.1; ESI-MS: calc. for C14H9F2N [M + H]+: 230.1, found: 230.2. HPLC purity: 100% (254 nm), tR: 7.19 min; 100% (220 nm), tR: 7.19 min.
2-(4-Fluorophenyl)-1H-indole 10af: white solid; yield: 66%; 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.62-7.58 (m, 2H), 7.38 (d, J = 8.0 Hz, 1H), 7.23 (t, J = 7.6 Hz, 1H), 7.18-7.12 (m, 3H), 6.76 (d, J = 2.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 163.9, 161.4, 137.2, 137.1, 129.5, 129.0, 128.9, 127.1, 127.0, 122.6, 120.8, 120.6, 116.3, 116.1, 111.1, 100.2; ESI-MS: calc. for C14H10FN [M + H]+: 212.2, found: 212.2. HPLC purity: 100% (254 nm), tR: 7.14 min; 100% (220 nm), tR: 7.14 min.
2-(3,5-Dimethoxyphenyl)-1H-indole 10ag: white solid; yield: 70%; 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.61 (d, J = 7.2 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.23-7.09 (m, 2H), 6.80 (s, 3H), 6.44 (s, 1H), 3.84 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 161.3, 137.8, 136.7, 134.3, 129.1, 122.4, 120.7, 120.2, 110.9, 103.6, 100.3, 99.7, 55.4; ESI-MS: calc. for C16H15O2N [M+ H]+: 254.1, found: 254.2. HPLC purity: 100% (254 nm), tR: 7.15 min; 100% (220 nm), tR: 7.14 min.
2-(4-Methoxyphenyl)-1H-indole 10ah: brown solid; yield: 59%; 1H NMR (400 MHz, d6-DMSO) δ 11.39 (s, 1H), 7.77 (d, J = 8.8 Hz, 2H), 7.48 (d, J = 7.6 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.05-6.97 (m, 4H), 6.73 (d, J = 2.0 Hz, 1H), 3.79 (s, 3H); 13C NMR (100 MHz, d6-DMSO) δ 159.0, 138.0, 137.1, 129.0, 126.6, 125.1, 121.3, 119.9, 119.5, 114.6, 111.3, 97.6, 55.4; ESI-MS: calc. for C15H13ON [M + H]+: 224.1, found: 224.2. HPLC purity: 97.8% (254 nm), tR: 7.06 min; 100% (220 nm), tR: 7.06 min.
2-(3-Methoxyphenyl)-1H-indole 10ai: white solid; yield: 70%; 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 7.60 (d, J = 7.6 Hz, 1H), 7.32-7.29 (m, 2H), 7.18-7.08 (m, 4H), 6.82 (dd, J = 2.0 and 7.6 Hz, 1H), 6.78 (d, J = 2.0 Hz, 1H), 3.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 160.2, 137.9, 136.9, 133.9, 130.2, 129.3, 122.5, 120.8, 120.4, 117.8, 113.2, 111.1, 111.0, 100.3, 55.4; ESI-MS: calc. for C15H13ON [M + H]+: 224.1, found: 224.2. HPLC purity: 100% (254 nm), tR: 7.11 min; 100% (220 nm), tR: 7.10 min.
2-(4-(Trifluoromethoxy)phenyl)-1H-indole 10aj: white solid; yield: 71%; 1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 7.65 (t, J = 9.2 Hz, 3H), 7.39 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 8.0 Hz, 2H), 7.23 (t, J = 7.2 Hz, 1H), 7.12 (t, J = 7.6 Hz, 1H), 6.81 (d, J = 1.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 148.6, 137.0, 136.5, 131.2, 129.8, 129.1, 126.4, 122.7, 121.6, 120.8, 120.5, 111.0, 100.7; ESI-MS: calc. for C15H10F3ON [M + H]+: 276.2, found: 276.2. HPLC purity: 100% (254 nm), tR: 7.49 min; 100% (220 nm), tR: 7.49 min.
2-(4-Bromophenyl)-1H-indole 10ak: yellow solid; yield: 56%; 1H NMR (400 MHz, CDCl3) δ 8.26 (s, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.54 (dd, J = 1.6 and 6.4 Hz, 2H), 7.49 (dd, J = 1.6 and 6.4 Hz, 2H), 7.36 (d, J = 7.6 Hz, 1H), 7.20 (t, J = 7.6 Hz, 1H), 7.12 (t, J = 7.2 Hz, 1H), 6.79 (d, J = 2.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 137.1, 136.8, 132.3, 131.5, 129.3, 126.7, 122.9, 121.7, 120.9, 120.6, 111.1, 100.7; ESI-MS: calc. for C14H10BrN [M+ H, 81Br]+: 274.0, found: 274.1; calc. for C14H10BrN [M+ H, 79Br]+: 272.0, found: 272.1. HPLC purity: 98.9% (254 nm), tR: 7.42 min; 96.6% (220 nm), tR: 7.42 min.
2-(4-Pentylphenyl)-1H-indole 10al: yellow solid; yield: 61%; 1H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 7.60 (d, J = 7.6 Hz, 1H), 7.55 (d, J = 8.0 Hz, 2H), 7.36 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 8.0 Hz, 2H), 7.17 (t, J = 8.0 Hz, 1H), 7.10 (t, J = 8.0 Hz, 1H), 6.77 (d, J = 1.2 Hz, 1H), 2.63 (t, J = 7.6 Hz, 2H), 1.66-1.60 (m, 2H), 1.36-1.32 (m, 4H), 0.90 (t, 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 142.8, 138.2, 136.8, 129.9, 129.4, 129.1, 125.2, 122.2, 120.6, 120.2, 110.9, 99.5, 35.7, 31.6, 31.1, 22.6, 14.1; ESI-MS: calc. for C19H21N [M + H]+: 264.2, found: 264.4. HPLC purity: 100% (254 nm), tR: 7.99 min; 100% (220 nm), tR: 7.97 min.
3-(1H-Indol-2-yl)aniline 10an: white solid; yield: 60%; 1H NMR (400 MHz, d6-DMSO) δ 11.37 (s, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.13-7.02 (m, 2H), 7.00-6.96 (m, 3H), 6.70 (t, J = 1.2 Hz, 1H), 6.56-6.53 (m, 1H), 5.15 (s, 2H); 13C NMR (100 MHz, d6-DMSO) δ 149.0, 138.7, 136.9, 132.8, 129.4, 128.6, 121.2, 119.8, 119.2, 113.5, 113.1, 111.2, 110.5, 97.9; ESI-MS: calc. for C14H12N2 [M + H]+: 209.2, found: 209.2. HPLC purity: 100% (254 nm), tR: 5.49 min; 100% (220 nm), tR: 5.49 min.
4-(1H-Indol-2-yl)aniline 10ao: brown solid; yield: 52%; 1H NMR (400 MHz, d6-DMSO) δ 11.19 (s, 1H), 7.52 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 8.0 Hz, 1H), 7.31 (d, J = 7.6 Hz, 1H), 7.00-6.93 (m, 2H), 6.63 (d, J = 8.4 Hz, 2H), 6.58 (s, 1H), 5.31 (s, 2H); 13C NMR (100 MHz, d6-DMSO) δ 148.5, 139.2, 136.7, 129.1, 126.1, 120.3, 119.9, 119.2, 119.0, 114.0, 110.8, 95.5; ESI-MS: calc. for C14H12N2 [M + H]+: 209.2, found: 209.2. HPLC purity: 100% (254 nm), tR: 5.51 min; 100% (220 nm), tR: 5.51 min.
4-(1H-Indol-2-yl)-N,N-dimethylaniline 10ap: yellow solid; yield: 40%;1H NMR (400 MHz, d6-DMSO) δ 11.26 (s, 1H), 7.66 (d, J = 7.2 Hz, 2H), 7.44 (d, J = 7.6 Hz, 1H), 7.34 (d, J = 7.6 Hz, 1H), 7.00 (t, J = 7.2 Hz, 1H), 6.96 (t, J = 7.6 Hz, 1H), 6.77 (d, J = 8.8 Hz, 2H), 6.63 (d, J = 1.6 Hz, 1H), 2.93 (s, 6H); 13C NMR (100 MHz, d6-DMSO) δ 149.9, 138.8, 136.9, 129.2, 126.1, 120.6, 120.2, 119.4, 119.2, 112.5, 111.0, 96.1, 40.1; ESI-MS: calc. for C16H16N2 [M + H]+: 237.3, found: 237.2. HPLC purity: 100% (254 nm), tR: 5.73 min; 100% (220 nm), tR: 5.72 min.
2-(Pyridin-3-yl)-1H-indole 10aq: white solid; yield: 60%; 1H NMR (400 MHz, CDCl3) δ 9.12 (s, 1H), 8.95 (d, J = 1.6 Hz, 1H), 8.51 (d, J = 4.4 Hz, 1H), 7.92-7.64 (m, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.30 (q, J = 4.8 Hz, 1H), 7.23-7.17 (m, 1H), 7.12 (t, J = 8.0 Hz, 1H), 6.85 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 148.4, 146.4, 137.6, 134.7, 132.7, 129.2, 128.9, 124.0, 123.1, 121.0, 120.7, 111.3, 101.5; ESI-MS: calc. for C13H10N2 [M + H]+: 195.2, found: 195.2. HPLC purity: 98.6% (254 nm), tR: 5.28 min; 98.2% (220 nm), tR: 5.27 min.
2-(Thiophen-3-yl)-1H-indole 10as: yellow solid; yield: 66%; 1H NMR (400 MHz, CDCl3) δ 8.11 (s, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.38 (s, 3H), 7.36-7.32 (m, 1H), 7.17 (dt, J = 1.2 and 8.0 Hz, 1H), 7.09 (dt, J = 0.8 and 8.0 Hz, 1H), 6.67 (d, J = 1.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 136.7, 134.4, 134.2, 129.4, 126.8, 125.9, 122.5, 120.8, 120.5, 119.3, 110.8, 100.2; ESI-MS: calc. for C12H9NS [M + H]+: 200.3, found: 200.1. HPLC purity: 100% (254 nm), tR: 6.99 min; 100% (220 nm), tR: 6.99 min.
2-(6-Methoxynaphthalen-2-yl)-1H-indole 10at: white solid; yield 77%; 1H NMR (400 MHz, d6-DMSO) δ 11.62 (s, 1H), 8.28 (s, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.87 (t, J = 8.0 Hz, 2H), 7.54 (d, J = 8.0 Hz, 1H), 7.43 (d, J = 8.0 Hz, 1H), 7.32 (d, J = 2.8 Hz, 1H), 7.20 (d, J = 2.8 and 8.0 Hz, 1H), 7.10 (t, J = 8.0 Hz, 1H), 7.00 (t, J = 7.2 Hz, 1H), 6.96 (d, J = 1.2 Hz, 1H), 3.88 (s, 3H); 13C NMR (100 MHz, d6-DMSO) δ 157.6, 138.0, 137.4, 133.8, 129.7, 129.0, 128.8, 127.6, 127.5, 124.4, 123.0, 121.7, 120.2, 119.6, 119.4, 111.4, 106.3, 99.0, 55.4; ESI-MS: calc. for C19H15NO [M + H]+: 274.3, found: 274.3. HPLC purity: 100% (254 nm), tR: 7.45 min; 100% (220 nm), tR: 7.45 min.
5-Nitro-2-phenyl-1H-indole 10ba: yellow solid; yield: 58%; 1H NMR (400 MHz, d6-DMSO) δ 12.30 (s, 1H), 8.52 (s, 1H), 8.00 (dd, J = 1.2 and 8.8 Hz, 1H), 7.89 (d, J = 7.6 Hz, 2H), 7.55 (d, J = 9.2 Hz, 1H), 7.49 (t, J = 7.2 Hz, 2H), 7.40 (t, J = 7.6 Hz, 1H), 7.16 (s, 1H); 13C NMR (100 MHz, d6-DMSO) δ 141.6, 141.1, 140.4, 131.1, 129.2, 128.6, 128.0, 125.5, 117.1, 111.8, 101.0; ESI-MS: calc. for C14H10N2O2 [M + H]+: 239.2, found: 239.2. HPLC purity: 100% (254 nm), tR: 7.11 min; 100% (220 nm), tR: 7.11 min.
5-Nitro-2-o-toyl-1H-indole 10bb: yellow solid; yield: 84%; 1H NMR (400 MHz, CDCl3) δ 8.64 (s, 1H), 8.59 (d, J = 1.6 Hz, 1H), 8.11 (dd, J = 2.4 and 9.2 Hz, 1H), 7.45 (dd, J = 6.4 and 9.2 Hz, 2H), 7.34-7.29 (m, 3H), 6.76 (s, 1H), 2.50 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 142.1, 140.8, 139.0, 136.3, 131.3, 128.9, 128.8, 128.2, 126.3, 117.7, 117.6, 110.7, 104.7, 20.9; ESI-MS: calc. for C15H12N2O2 [M + H]+: 253.1, found: 253.1. HPLC purity: 100% (254 nm), tR: 7.25 min; 100% (220 nm), tR: 7.25 min.
5-Chloro-2-phenyl-1H-indole 10ca: white solid; yield: 64%; 1H NMR (400 MHz, CDCl3) δ 8.33 (s, 1H), 7.60 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 1.6 Hz, 1H), 7.42 (t, J = 8.0 Hz, 2H), 7.32 (t, J = 7.6 Hz, 1H), 7.25 (t, J = 7.6 Hz, 1H), 7.11 (dd, J = 2.0 and 8.4 Hz, 1H), 6.72 (d, J = 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 139.3, 135.1, 131.8, 130.3, 129.1, 128.1, 125.8, 125.2, 122.5, 119.9, 111.8, 99.5; ESI-MS: calc. for C14H10ClN [M + H]+: 228.1, found: 228.2. HPLC purity: 100% (254 nm), tR: 7.40 min; 100% (220 nm), tR: 7.39 min.
6-Chloro-2-phenyl-1H-indole 10da: yellow solid; yield: 64%; 1H NMR (400 MHz, d6-DMSO) δ 11.71 (s, 1H), 7.83 (d, J = 7.6 Hz, 2H), 7.53 (d, J = 8.4 Hz, 1H), 7.48-7.42 (m, 3H), 7.33 (t, J = 7.6 Hz, 1H), 7.01 (dd, J = 2.0 and 7.6 Hz, 1H), 6.91 (d, J = 1.6 Hz, 1H); 13C NMR (100 MHz, d6-DMSO) δ 138.9, 137.6, 131.8, 129.1, 127.9, 127.5, 126.2, 125.2, 121.5, 119.9, 110.9, 98.9; ESI-MS: calc. for C14H10ClN [M + H]+: 228.1, found: 228.2. HPLC purity: 96.8% (254 nm), tR: 7.38 min; 96.4% (220 nm), tR: 7.38 min.
6-Carbonitrile-2-phenyl-1H-indole 10ea: white solid; yield: 84%; 1H NMR (400 MHz, d6-DMSO) δ 12.14 (s, 1H), 8.05 (s, 1H), 7.88 (d, J = 8.0 Hz, 2H), 7.55 (d, J = 8.4 Hz, 1H), 7.47 (t, J = 8.0 Hz, 2H), 7.44 (d, J = 8.4 Hz, 1H), 7.38 (t, J = 7.6 Hz, 1H), 7.04 (s, 1H); 13C NMR (100 MHz, d6-DMSO) δ 140.4, 138.9, 131.2, 129.1, 128.5, 128.4, 125.6, 125.5, 124.4, 120.8, 112.6, 101.5, 99.4; ESI-MS: calc. for C15H10N2 [M+ H] +: 219.2, found: 219.2. HPLC purity: 100% (254 nm), tR: 6.95 min; 100% (220 nm), tR: 6.95 min.
5-Bromo-2-phenyl-1H-indole 10fa: white solid; yield: 83%; 1H NMR (400 MHz, d6-DMSO) δ 11.78 (s, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.71 (s, 1H), 7.47 (t, J = 8.0 Hz, 2H), 7.36 (t, J = 7.6 Hz, 2H), 7.20 (d, J = 8.8 Hz, 1H), 6.88 (s, 1H); 13C NMR (100 MHz, d6-DMSO) δ 139.2, 135.8, 131.7, 130.5, 129.0, 127.9, 125.2, 124.0, 122.2, 113.3, 111.9, 98.3; ESI-MS: calc. for C14H10BrN [M + H, 81Br]+: 274.2, found: 274.1; C14H10BrN [M + H, 79Br]+: 272.2, found: 272.2. HPLC purity: 100% (254 nm), tR: 7.47 min; 100% (220 nm), tR: 7.46 min.
6-Methyl-2-phenyl-1H-indole 10ga: white solid; yield: 25%; 1H NMR (400 MHz, d6-DMSO) δ 11.36 (s, 1H), 7.83 (d, J = 7.6 Hz, 2H), 7.46-7.40 (m, 3H), 7.29 (t, J = 7.6 Hz, 1H), 7.19 (s, 1H), 6.83 (s, 2H), 2.40 (s, 3H); 13C NMR (100 MHz, d6-DMSO) δ 137.6, 137.0, 132.4, 130.7, 128.9, 127.2, 126.5, 124.8, 121.2, 119.8, 111.1, 98.6, 21.5; ESI-MS: calc. for C15H13N [M + H]+: 208.3, found: 208.2. HPLC purity: 98.8% (254 nm), tR: 7.28 min; 100% (220 nm), tR: 7.28 min.
Synthesis of 5-amino-2-phenylindole 10ha
A mixture of 5-nitro-2-phenylindole 10ba (33 mg, 0.13 mmol) and 10% Pd/C (1.5 mg) in 2.5 mL of absolute ethanol was hydrogenated at room temperature. The reaction was detected by HPLC and was found to be completed after 4 h. The catalyst was filtered off and the solvent was evaporated. The residue was purified by flash column chromatography to afford 18 mg (67%) of 5-amino-2-phenylindole 10ha as a brown solid. 1H NMR (400 MHz, CDCl3) δ 8.16 (s, 1H), 7.62 (d, J = 7.2 Hz, 2H), 7.40 (t, J = 7.2 Hz, 2H), 7.32-7.27 (m, 1H), 7.22 (s, 1H), 6.92 (d, J = 2.0 Hz, 1H), 6.66-6.63 (m, 2H), 3.2 (br, 2H); 13C NMR (100 MHz, CDCl3) δ 140.0, 138.5, 132.6, 131.9, 130.3, 129.0, 127.6, 125.1, 113.3, 111.5, 105.5, 99.2; ESI-MS: calc. for C14H12N2 [M+ H]+: 209.2, found: 209.2. HPLC purity: 99.2% (254 nm), tR: 5.32 min; 99.1% (220 nm), tR: 5.30 min.
Biological assays
Measurement of the production of NO in LPS-stimulated RAW 264.7 murine macrophage cells (nitrite assay)
The level of NO in the cultured media was estimated by measuring the level of nitrite due to the instability of NO and its subsequent conversion to nitrite. The nitrite assay was performed as previously described.51 Briefly, RAW 264.7 cells were incubated in 96-well culture plates at 37 °C, 5% CO2 in a humidified air incubator for 24 h. Then cells were treated with serially diluted compounds for 15 min, followed by treatment with or without LPS (1 μg/mL) for an additional 20 h. After the incubation, nitrite released in the cultured media was measured using Griess regent [1:1 mixture (v/v) of 1% sulfanilamide in 5% H3PO4 and 0.1% N-(1-naphthyl)ethylenediamine dihydrochloride solution], and absorbance was measured at 540 nm. The concentration of nitrite was calculated using a standard curve created with known concentration of sodium nitrite. Under the same experimental conditions, sulforhodamine B (SRB) assay was performed to evaluate the cytotoxic effect of tested compounds on RAW 264.7 cells. After transferring 100 μL of the cultured media, cells were fixed with 10% trichloroacetic acid, and stained with 0.4% SRB solution in 1% acetic acid. The protein-bound SRB was dissolved in 10 mM Tris base solution, and the absorbance was measured at 515 nm. Percentage of cell survival was calculated in comparison with LPS-treated control.52 Initial screening was performed at a concentration of 50 μM. Compounds which exhibited over 50% inhibition of nitrite production, and under 50% cell survival, were considered as active and cytotoxic, respectively. Only active inhibitors of nitrite production lacking appreciable cytotoxicity were examined in greater detail to determine whether the inhibitory effect was derived from a false positive cytotoxic effect, and to compare the relative potency of the compounds (SAR).
NFκB luciferase assay
Human embryonic kidney cells 293 were used to monitor any changes occurring along the NFκB pathway. This cell line contains chromosomal integration of a luciferase reporter construct regulated by the NFκB response element. Transcription factors can bind to the response element when stimulated by certain agents, allowing transcription of the luciferase gene. Following an incubation period of 6 h with TNFα and test compounds, cells were analyzed for luciferase activity using the Luc assay system from Promega Corporation (Madison, WI). Results were expressed as a percentage, relative to control (TNFα-treated with DMSO) samples, and dose-response curves were constructed for the determination of IC50 values, which were generated from the results of five serial dilutions of test compounds and were the mean of two different experiments.53 N-Tosyl-L-phenylalanyl chloromethyl ketone (TPCK) was used as a positive control (IC50 = 3.8±0.6 μM).
Supplementary Material
Acknowledgments
This project was supported in part by grants from the National Center for Research Resources (5P20RR016467-11) and the National Institute of General Medical Sciences (8P20GM103466-11) from the National Institutes of Health. The content is solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. We also thank UH Hilo CoP for start-up funds.
Footnotes
Electronic Supplementary Information (ESI) available: [copies of 1H, 13C NMR spectra, and HPLC chromatographs of target compounds]. See DOI: 10.1039/b000000x/
References
- 1.Thomas EJ. “Indole and its Derivatives” in Science of Synthesis: Houben-Weyl Methods of Molecular Transformation. Thieme; Stuttgart: 2000. [Google Scholar]
- 2.Kochanowska-Karamyan AJ, Hamann MT. Chem Rev. 2010;110:4489–4497. doi: 10.1021/cr900211p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gul W, Hamann MT. Life Sci. 2005;78:442–453. doi: 10.1016/j.lfs.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bandini M, Eichholzer A. Angew Chem Int Ed. 2009;48:9608–9644. doi: 10.1002/anie.200901843. [DOI] [PubMed] [Google Scholar]
- 5.Chen F-E, Huang J. Chem Rev. 2005;105:4671–4706. doi: 10.1021/cr050521a. [DOI] [PubMed] [Google Scholar]
- 6.Ambros R, Von Angerer S, Wiegrebe W. Arch Pharm. 1988;321:481–486. doi: 10.1002/ardp.19883210811. [DOI] [PubMed] [Google Scholar]
- 7.Winters G, Di Mola N, Berti M, Arioli V. Farmaco. 1979;34:507–517. doi: 10.1002/chin.197949235. [DOI] [PubMed] [Google Scholar]
- 8.Saundane AR, Ranganath SH, Prayagraj G, Rudresh K, Satyanarayana ND. Orient J Chem. 1998;14:251–254. [Google Scholar]
- 9.Grasso S, Molica C, Monforte AM, Monforte P, Zappala M, Monforte MT, Trovato A. Farmaco. 1995;50:113–117. [PubMed] [Google Scholar]
- 10.Scheiper B, Matter H, Steinhagen H, Stilz U, Böcskei Z, Fleury V, McCort G. Bioorg Med Chem Lett. 2010;20:6268–6272. doi: 10.1016/j.bmcl.2010.08.092. [DOI] [PubMed] [Google Scholar]
- 11.Hopkins CR, O’Neil SV, Laufersweiler MC, Wang Y, Pokross M, Mekel M, Evdokimov A, Walter R, Kontoyianni M, Petrey ME, Sabatakos G, Roesgen JT, Richardson E, Demuth TP., Jr Bioorg Med Chem Lett. 2006;16:5659–5663. doi: 10.1016/j.bmcl.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Dolušić E, Kowalczyk M, Magnus V, Sandberg G, Normanly J. Bioconjugate Chem. 2001;12:152–162. doi: 10.1021/bc000035o. [DOI] [PubMed] [Google Scholar]
- 13.Hill TA, Gordon CP, McGeachie AB, Venn-Brown B, Odell LR, Chau N, Quan A, Mariana A, Sakoff JA, Chircop M, Robinson PJ, McCluskey A. J Med Chem. 2009;52:3762–3773. doi: 10.1021/jm900036m. [DOI] [PubMed] [Google Scholar]
- 14.Sugden D, Pickering H, Teh M-T, Garratt PJ. Biol Cell. 1997;89:531–537. doi: 10.1016/s0248-4900(98)80009-9. [DOI] [PubMed] [Google Scholar]
- 15.Vicente R. Org Biomol Chem. 2011;9:6469–6480. doi: 10.1039/c1ob05750b. [DOI] [PubMed] [Google Scholar]
- 16.Shiri M. Chem Rev. 2012;112:3508–3549. doi: 10.1021/cr2003954. [DOI] [PubMed] [Google Scholar]
- 17.Patil SA, Patil R, Miller DD. Curr Med Chem. 2011;18:615–637. doi: 10.2174/092986711794480195. [DOI] [PubMed] [Google Scholar]
- 18.Patil SA, Patil R, Miller DD. Curr Med Chem. 2009;16:2531–2565. doi: 10.2174/092986709788682010. [DOI] [PubMed] [Google Scholar]
- 19.Bräse S, Gil C, Knepper K. Bioorg Med Chem. 2002;10:2415–2437. doi: 10.1016/s0968-0896(02)00025-1. [DOI] [PubMed] [Google Scholar]
- 20.Kundu JK, Surh Y-J. Mutat Res, Rev Mutat Res. 2008;659:15–30. doi: 10.1016/j.mrrev.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. Nitric Oxide. 2010;23:75–93. doi: 10.1016/j.niox.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 22.Alderton WK, Cooper CE, Knowles RG. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hofseth LJ. Cancer Lett. 2008;268:10–30. doi: 10.1016/j.canlet.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aktan F. Life Sci. 2004;75:639–653. doi: 10.1016/j.lfs.2003.10.042. [DOI] [PubMed] [Google Scholar]
- 25.Gilmore TD. Oncogene. 1999;18:6842–6844. doi: 10.1038/sj.onc.1203237. [DOI] [PubMed] [Google Scholar]
- 26.Gélinas CRB. Oncogene. 1999;18:6938. doi: 10.1038/sj.onc.1203221. [DOI] [PubMed] [Google Scholar]
- 27.Karin M. Cold Spring Harb Perspect Biol. 2009;1:a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beg AA, Baltimore D. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 29.Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M. Mol Cell Biol. 1999;19:2690–2698. doi: 10.1128/mcb.19.4.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karin M, Cao Y, Greten FR, Li Z-W. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 31.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 32.Wang C-Y, Cusack JC, Liu R, Baldwin AS. Nat Med. 1999;5:412–417. doi: 10.1038/7410. [DOI] [PubMed] [Google Scholar]
- 33.Wang C-Y, Mayo MW, Baldwin AS. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 34.Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS. Mol Cell Biol. 1999;19:5785–5799. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luqman S, Pezzuto JM. Phytother Res. 2010;24:949–963. doi: 10.1002/ptr.3171. [DOI] [PubMed] [Google Scholar]
- 36.Goldring CEP, Narayanan R, Lagadec P, Jeannin JF. Biochem Biophys Res Commun. 1995;209:73–79. doi: 10.1006/bbrc.1995.1472. [DOI] [PubMed] [Google Scholar]
- 37.Shen L, Park E-J, Kondratyuk TP, Guendisch D, Marler L, Pezzuto JM, Wright AD, Sun D. Bioorg Med Chem. 2011;19:6182–6195. doi: 10.1016/j.bmc.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Part of this work has been presented at the following ACS meeting, see: Yu X, Park E-J, Kondratyuk TP, Pezzuto JM, Sun D. 243rd ACS National Meeting & Exposition. American Chemical Society; San Diego, CA, United States: Mar 25–29, 2012. MEDI-101.
- 39.Merlic CA, You Y, McInnes DM, Zechman AL, Miller MM, Deng Q. Tetrahedron. 2001;57:5199–5212. [Google Scholar]
- 40.Shi Z, Ding S, Cui Y, Jiao N. Angew Chem Int Ed. 2009;48:7895–7898. doi: 10.1002/anie.200903975. [DOI] [PubMed] [Google Scholar]
- 41.Kuwano R, Kashiwabara M. Org Lett. 2006;8:2653–2655. doi: 10.1021/ol061039x. [DOI] [PubMed] [Google Scholar]
- 42.Gotor-Fernández V, Fernández-Torres P, Gotor V. Tetrahedron: Asymmetry. 2006;17:2558–2564. [Google Scholar]
- 43.Alonso R, Caballero A, Campos PJ, Rodríguez MA. Tetrahedron. 2010;66:8828–8831. [Google Scholar]
- 44.MacNevin CJ, Moore RL, Liotta DC. J Org Chem. 2008;73:1264–1269. doi: 10.1021/jo7018202. [DOI] [PubMed] [Google Scholar]
- 45.Jiang N, Ragauskas AJ. Tetrahedron Lett. 2010;51:4479–4481. [Google Scholar]
- 46.Sanz R, Guilarte V, Pérez A. Tetrahedron Lett. 2009;50:4423–4426. [Google Scholar]
- 47.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taliani S, Da Pozzo E, Bellandi M, Bendinelli S, Pugliesi I, Simorini F, La Motta C, Salerno S, Marini AM, Da Settimo F, Cosimelli B, Greco G, Novellino E, Martini C. J Med Chem. 2010;53:4085–4093. doi: 10.1021/jm100100q. [DOI] [PubMed] [Google Scholar]
- 49.Suschek CV, Schnorr O, Kolb-Bachofen V. Curr Mol Med. 2004;4:763–775. doi: 10.2174/1566524043359908. [DOI] [PubMed] [Google Scholar]
- 50.Savio M, Coppa T, Bianchi L, Vannini V, Maga G, Forti L, Cazzalini O, Lazzè MC, Perucca P, Prosperi E, Stivala LA. Int J Biochem Cell Biol. 2009;41:2493–2502. doi: 10.1016/j.biocel.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 51.Hoshino J, Park E-J, Kondratyuk TP, Marler L, Pezzuto JM, van Breemen RB, Mo S, Li Y, Cushman M. J Med Chem. 2010;53:5033–5043. doi: 10.1021/jm100274c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee SK, Cui B, Mehta RR, Kinghorn AD, Pezzuto JM. Chem Biol Interact. 1998;115:215–228. doi: 10.1016/s0009-2797(98)00073-8. [DOI] [PubMed] [Google Scholar]
- 53.Kondratyuk TP, Park EJ, Yu R, van Breemen RB, Asolkar RN, Murphy BT, Fenical W, Pezzuto JM. Mar Drugs. 2012;10:451–464. doi: 10.3390/md10020451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.