

Abstract
PIWI-family proteins and their associated small RNAs (piRNAs) act in an evolutionarily conserved innate immune mechanism that provides an essential protection for germ cell genomes against the activity of mobile genetic elements1. piRNA populations comprise a molecular definition of transposons that permits them to be distinguished from host genes and selectively silenced. piRNAs can be generated in two distinct ways. Primary piRNAs emanate from discrete genomic loci, termed piRNA clusters, and appear to be derived from long, single-stranded precursors2. The biogenesis of primary piRNAs involves at least two nucleolytic steps. An unknown enzyme cleaves piRNA cluster transcripts to generate monophosphorylated piRNA 5' ends. piRNA 3' ends are likely formed by exonucleolytic trimming, after a piRNA precursor is loaded into its PIWI partner1,3. Secondary piRNAs arise during the adaptive ping-pong cycle, with their 5' termini being formed by the activity of PIWIs themselves2,4. A number of proteins have been implicated genetically in primary piRNA biogenesis. One of these, Zucchini, is a member of the phospholipase D family of phosphodiesterases, which includes both phospholipases and nucleases5–7. We have produced a dimeric, soluble fragment of the mouse Zucchini homolog (mZuc/PLD6) and have shown that it possesses single strand-specific nuclease activity. A crystal structure of mZuc at 1.75 Å resolution indicates greater architectural similarity to PLD-family nucleases than to phospholipases. Considered together, our data suggest that the Zucchini proteins act in primary piRNA biogenesis as nucleases, perhaps generating the 5' ends of primary piRNAs.
Zucchini was first noted as a gene essential for female fertility in Drosophila. Multiple independent alleles caused fully penetrant sterility and the production of eggs with dorsoventral patterning defects7. Subsequent studies have traced the impacts of Zucchini on germ cell development to its function in the piRNA pathway. Animals lacking Zucchini fail to silence transposons and show a general collapse of primary piRNA populations6,8,9. Mutant animals also accumulate transcripts from piRNA clusters, indicating a failure to process these precursors into small RNAs5. Thus, Zucchini was strongly implicated as a factor promoting primary piRNA biogenesis.
Zuc belongs to the phospholipase D (PLD) family of phosphodiesterases, which share a common biochemical mechanism and a signature H(X)K(X4)D (HKD) motif within their active site (reviewed in Selvy et al.10). Notably, one of the zuc alleles that emerged from the original forward genetic screen was a point mutation that changed the catalytic histidine to a tyrosine. This produced a phenocopy of the presumed null allele, including similar effects on piRNA populations, strongly suggesting that the catalytic activity of Zucchini was critical for piRNA production or stability5,6.
Biochemical and genetic studies of the mouse Zucchini homolog, mZuc/PLD6, led to the conclusion that it acted as a phospholipase which affected mitochondrial fusion in a manner linked to its processing of the mitochondrial lipid, cardiolipin11–13. Thus, it was suggested that the impact of Zucchini on the piRNA pathway was indirect, through alterations in lipid levels or through changes in the structure of mitochondria. Yet, the proposed model of Zucchini activity required an enzyme that localized to the outer mitochondrial membrane and faced the cytoplasm to hydrolyze a lipid that is almost exclusively found on the inner mitochondrial membrane14. We therefore sought to discriminate between the two divergent hypotheses for Zucchini function in piRNA biogenesis, namely that it acts indirectly through its role as a phosphodiesterase or that it acts directly as a nuclease.
To enable biochemical analysis of mZuc, we expressed an amino-terminally truncated form of the protein in Sf9 cells (Fig. 1a). This produced a soluble enzyme by removal of the transmembrane domain that normally anchors it to the mitochondrial outer membrane. One of the hallmarks of the PLD family is the presence of two HKD motifs that are brought together to create the catalytic center10. This active site can arise from a single polypeptide that contains two copies of the motif, an arrangement typical of PLDs with phospholipid substrates. Alternatively, the catalytic center can be assembled as an intermolecular dimer, a form most common with the PLDs that act as nucleases (reviewed in Selvy et al.10). mZuc has only one HKD motif, but the protein formed a stable, ~41 kDa dimer in Sf9 cells, as indicated by both gel filtration chromatography and multi-angle light scattering (Fig. S1). For comparison, we also produced a mutant version of the protein, which lacks an intact catalytic motif (H153N).
Figure 1. mZuc acts as a nuclease but not a phospholipase in vitro.

a) The domain architecture of mZuc and Zuc are similar with an N-terminal transmembrane helix (TM, red) and a cytoplasmic domain, which contains the catalytic HKD motif (gold). The construct used for crystallization is indicated as a dashed box. Residue numbers delineating each domain indicated below each schematic. b) Phosphodiesterase activity for mZuc, its catalytic mutant (H153N), and a known phospholipase (Phospholipase D from Streptomyces chromofuscus, scPLD) were monitored by SRM-MS. Levels of the cardiolipin substrate and the expected phophatidic acid product (PA) are shown for each reaction. Error bars indicate standard deviation (n=3). c) DNase activity of recombinant mZuc was monitored using 50 nt 5'-32P-labeled DNA (red). Single-stranded (ss), double-stranded (ds), and partially double-stranded substrates were incubated with wild-type (wt) or catalytic mutant H153N enzyme (as indicated), and the resulting products separated by urea-PAGE. d) mZuc or the H153N mutant were incubated in the presence of 32PO43- followed by SDS-PAGE and transfer to a nitrocellulose membrane. e) The DNase of mZuc was measured in the presence of Na3VO4. scPLD is shown as control.
We used two assays to test whether recombinant mZuc could function as a phospholipase, liberating phosphatidic acid (PA) from cardiolipin as previously reported11. Liposomes containing commercial cardiolipin were incubated with recombinant proteins, and PA was measured by thin layer chromatography. This did not reveal the production of measurable amounts of PA by mZuc, while a commercially available PLD from Streptomyces chromofuscus (scPLD) completely hydrolyzed the substrate (Fig. S2a). We also employed a more sensitive assay based upon mass spectrometry (MS). Selected reaction monitoring MS (SRM-MS) provides an extremely accurate and sensitive method for measuring reaction components. Using this approach, no significant changes in cardiolipin or PA were observed when comparing buffer controls to either wild-type or catalytically inactive mZuc proteins (Figs. 1bM; S2b,c); whereas scPLD again completely hydrolyzed the substrate. These results failed to provide support for Zucchini acting as a phospholipase to promote piRNA biogenesis and prompted us to examine alternative activities.
PLD family enzymes that have been demonstrated to act as nucleases, cleaving double-stranded DNA substrates15–17. For example, prototypic PLD nucleases, Nuc and BfiI, cleave dsDNA either non-specifically or at a defined recognition site10. We therefore incubated mZuc with a selection of end-labeled DNAs: single-stranded, double-stranded, or partially duplexed (Fig. 1c). The wild-type enzyme hydrolyzed single-stranded DNA or the single-stranded portions of partially duplexed substrates but did not cleave dsDNA. The mutant enzyme (H153N) was inactive against all substrates. As expected from studies of related PLD-family nucleases17, mZuc activity did not depend upon the presence of divalent cations for substrate cleavage, but it could be stimulated by the presence of specific cations, particularly Mn2+, Ca2+, and Zn2+ (Fig. S3a). The impact of Zn2+ may be explained by structural effects (see below), but as yet we do not understand how the other cations enhance activity.
The known catalytic mechanisms of PLD-family nucleases, including bacterial Nuc, suggest that cleavage proceeds via a two-step reaction scheme15. This includes the formation of a short-lived, covalent enzyme-substrate intermediate, joining a phosphate to the histidine in the HKD motif (Fig. S4a). A similar mechanism for mZuc is supported by our ability to label the enzyme simply by incubation with 32P-labeled inorganic phosphate in manner that depends on the presence of the HKD histidine (Fig. 1d). The specificity of this adduct was confirmed by performing a similar incubation of mZuc with PO43- followed by denaturation, proteolysis, and SRM-MS which detected phosphorylation of a His153-containing peptide. The fragmentation of this peptide was able to further resolve the location of the phosphorylation to residues 152–155 (Fig. S4b). Our phosphate-labeling data combined with the proposed catalytic mechanism for PLD family nucleases suggested that mZuc might be inhibited by phosphate analogs. Indeed, sodium orthovanadate completely abolished the ability of mZuc to cleave ssDNA substrates (Fig. 1e).
If mZuc functions as a nuclease in the piRNA pathway, it would most likely act on RNA substrates, and the biochemical mechanism used by PLD-family enzymes is consistent with their potentially cleaving either DNA or RNA. We therefore tested the ability of mZuc to cleave a variety of single-stranded or duplexed RNA substrates. mZuc cleaved only single stranded RNA, and this reaction was fully inhibited by orthovanadate (Fig. 2a). The mZuc mutant bearing an alteration in its catalytic site was inactive in these assays (Figs. 2b, S3b). These data indicate that mZuc can function as a backbone non-specific, single-strand specific nuclease, cleaving either RNA or DNA substrates. The binding affinity of mZuc for single stranded DNA or RNA substrates was nearly identical, measured at roughly 50 nM in each case (Fig. S5); however, in vivo, the physiologically relevant targets of mZuc are probably determined by its subcellular localization and perhaps with the help of additional proteins, which would focus its activity toward certain substrates.
Figure 2. mZuc acts as a single-strand specific endoribonuclease in vitro.

a) mZuc was incubated with ss, ds, and partially dsRNA substrates in the absence and presence of Na3VO4 as indicated. b) mZuc RNA cleavage products (from reactions shown in panel a, as indicated below) were tested for sensitivity to β-elimination and accessibility for polyadenylation.
HKD-family nucleases are predicted to leave 5' phosphate and 3' hydroxyl termini. These termini also correspond to the end polarities that would be expected of a nuclease that participated in the processing of primary piRNA transcripts. We tested whether the nucleolytic products of mZuc bore such termini in several ways. β-elimination shifted mZuc cleavage products by a single base, a reaction that is diagnostic of the presence of a 3' OH terminus (Fig. 2b). We also confirmed the presence of a 3' OH group by extending the cleavage products with poly A polymerase (Fig. 2b). DNA cleavage also produced 5' phosphate and 3' hydroxyl termini (Fig. S3c)
In order to gain further insight into mZuc activity and substrate specificity we determined its structure to 1.75 Å resolution. Like other members of the HKD family, the structure of mZuc consists of a conserved β-sheet core sandwiched between two α-helical layers (Fig. 3a). As seen in the crystal structure of the bacterial family member, Nuc18, the monomers of the mZuc dimer are related by a crystallographic two-fold axis. The dimer interface is extensive, with a buried surface area of 2600 Å2 as calculated by PISA19. The resulting arrangement of the active site residues and the distance between the catalytic histidines (His153) is consistent among both monomeric and dimeric PLD structures as evidenced by both the apo structure and the structure of the protein in complex with tungstate, a phosphate mimic that binds to the active site of the enzyme similar to Nuc18 (Fig. 3b, S6).
Figure 3. Crystal structure of mZuc.

a) The overall structure of the mZuc dimer is shown as a ribbon diagram. Helices are in green, strands in blue, and loops in beige. Each monomer binds one Zn2+ (yellow) in an extended zinc wing. The active site histidine residues (His153) are highlighted in red. b) A close-up of the zinc wing consisting of residues Cys49, Cys66, Cys68, and His72 is shown. c) A detailed view of mZuc co-crystallized with tungstate bound in the active site is presented.
Whilst mZuc is clearly an HKD family enzyme, a number of features unique to this structure support its biological activity as a nuclease and, more specifically, a single-stranded RNase. First, and most strikingly, a long, ordered loop is inserted between β1 and α2 (residues Pro44-Ser75). This loop extends away from the active site and contains three cysteines (Cys49, Cys66, and Cys68) as well as a histidine (His72) that form an unexpected CCCH “zinc wing” (Figs. 3c, S7). While zinc finger motifs are widely used for nucleic acid binding, this particular instance had escaped bioinformatic detection due to the atypical primary sequence of the CCCH (CX16CPCX3H as opposed to the canonical CX6–14CX4–5CX3H; see ref. 20). Interestingly, the CCCH class of zinc fingers, in particular, has been implicated in the binding of ssRNA molecules such as mRNA21–23 and viral RNAs24 in accord with ssRNA being the likely in vivo substrate for mZuc.
PLD family members that are responsible for lipid metabolism typically place the active site in a structural “pocket.” In contrast, we observed that those with nucleic acid substrates typically provide a larger substrate-binding “groove” (Figs. 4, S8). In the mZuc structure, the active site is flanked by a positively charged groove that extends to the zinc wings (Figs. 4a, S9), likely serving as a nucleic acid binding interface. When compared to the structure of Nuc, a PLD nuclease that acts on dsDNA (Fig. 4b), the width of the groove in mZuc is considerably narrower, consistent with the biochemically observed single-strand specificity of the enzyme.
Figure 4. Electrostatic surfaces of PLD family proteins indicate distinct binding surfaces for specific substrates.
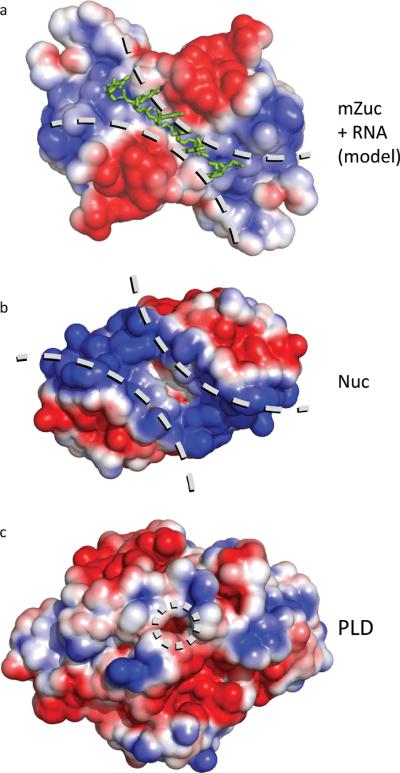
a) The electrostatic surface for mZuc displays a long, narrow, positively-charged grove laying across the zinc wings and active site. A short RNA molecule was manually built into the structure of mZuc, then subjected to energy minimization using GROMACS25. The minimized model shows the phosphates of the RNA backbone positioned in the most positively charged areas of the groove with the bases extending away from the dimer core. b) In Nuc (PDB ID 1BYR18), which acts on double-stranded DNA, the equivalent groove is significantly wider. c) A bone fide phospholipase, PLD (PDB ID 2ZE9), uses the same active site architecture in a strikingly different structural context. PLD has a small pocket for binding phospholipids, rather than an elongated groove. Each surface depicts the solvent-accessible surface contoured at ± 2 kB T/e using ABPS30.
We constructed a hypothetical model to examine the potential interaction between the proposed substrate-binding groove of mZuc and a single-stranded RNA. This was subjected to energy minimization using GROMACS25. The resulting model (Figs. 4a, S10) illustrates the shape and charge complementarity that a single-stranded nucleic acid substrate provides, including appropriate placement of the scissile phosphate in the active site. The surface complementarity is particularly striking; however, based on the projections of the bases away from the core of the protein, it seems unlikely that mZuc would display a strong sequence bias for binding or cleavage.
Our results strongly imply that Zucchini functions as a nuclease to promote primary piRNA biogenesis. Given the results of elegant biochemical analyses performed in silkworm extracts, it is highly probable that the mature 3' ends of piRNAs are formed by exonucleolytic trimming of precursor piRNAs that are already loaded into PIWI proteins3. This leaves open the possibility that Zucchini could generate the 5' ends of primary piRNAs. mZuc does produce products with the correct phosphate polarity. Yet, primary piRNAs show an overwhelming bias for a terminal U residue2,26,27, which does not seem to be a preference of mZuc, based either upon biochemical or structural studies. Therefore, if Zucchini does generate piRNA 5' ends, the prevalence of their characteristic terminal residue must result either from an unknown co-factor or from the selectivity of a 5' end-binding pocket within PIWI-family proteins. The latter is a reasonable possibility as there is ample biochemical and structural support within the Argonaute sub-family for strong binding preferences for 5' terminal nucleotides28,29. We must also consider the possibility that Zucchini proteins could contribute an additional, yet unanticipated activity, perhaps generating intermediate 3' ends of precursor piRNAs that are further resected by trimming to form mature termini. Attributing a definitive role to Zucchini will rest on further studies, perhaps ultimately requiring a full biochemical reconstitution of primary piRNA biogenesis to finally resolve its function.
In addition to their implications for piRNA production, our studies also highlight some general features of the phosphodiesterases, which use HKD motifs as their active sites. A comparison of the available structures of the PLD/nuclease family proteins indicates that these enzymes define their substrate specificity by their binding properties. While phospholipases within this family seem to harbor pocket-like substrate binding structures, nucleases display extended binding grooves for nucleic acid chains. These structural motifs are nearly diagnostic of the substrate specificity of these proteins. That having been said, based solely on our biochemical assays and taking into account existing literature11–13, we cannot exclude that mZuc can act in some circumstances as a phospholipase. We can simply argue that structural features are more consistent with its action as a nuclease. The ability to divide enzymes within this family into classes based upon their structural and biochemical features may also suggest that the PLD nomenclature for some of these enzymes, particularly for mZuc and Nuc, has become misleading and deserves reconsideration.
Online Methods
Cloning, expression, and purification of mZuc fragments
In order to screen for well-behaved targets, a panel of mZuc constructs was generated from M. musculus cDNA by SLIC cloning. These constructs presented various N- or C-terminal tags for enhanced expression and purification using either E. coli or baculoviral-induced insect cell culture systems. The sequence of each construct was verified by the Cold Spring Harbor Laboratory DNA Sequencing Facility.
The data presented herein resulted from a 30-residue N-terminal deletion of mZuc to create a protein that spans residues 31–221 fused to a Thrombin-cleavable Strep-tag at the C-terminus (mZuc△30-ThStrep2). This construct was cloned into the vector pFL for expression in Sf9 cells using the baculovirus expression system33. After expression, cells were harvested by centrifugation at 1000×g, resuspended in lysis buffer (0.1 MMES, pH 6.5, 0.15 M NaCl, 1 mM DTT) (~20 mL per liter culture), and lysed by sonication. The cell lysate was then clarified by ultracentrifugation at 125,000×g for 1 h and the supernatant applied to a Strep-Tactin (IBA) column equilibrated with lysis buffer. The bound mZuc△30-ThStrep2 was subsequently washed with lysis buffer, further washed with lysis buffer containing 2 mM ATP, and finally eluted in lysis buffer containing 5 mM D-desthiobiotin. To remove the C-terminal affinity purification tag, 15 units of thrombin protease were added per mg of purified protein and incubated overnight at 4 °C. The cleaved mZuc△30 (herein referred to as mZuc) was the further purified by gel filtration using a Superdex75 column equilibrated with lysis buffer. Once purified, the protein was concentrated to 5–10 mg/mL and stored at 4 °C for short periods or in 50% (v/v) glycerol at −20 °C for extended periods. Typical yields were 1–2 mg of purified protein (>98% purity as assessed by SDS-PAGE) per liter culture. Active site mutants were constructed using SLIC cloning methods. Purification of mutant proteins was identical to that for the wild type.
For SRM-MS experiments, mZuc wildtype + H153N heterodimer was purified by co-expressing mZucΔ30-ThStrep2 and mZucΔ30-ThHis6 H153N. After Strep-Tactin purification, a second round of affinity purification with Ni-NTA resin was performed to isolate heterodimers. The purified protein was immediately concentrated and desalted into 20 mM MES, pH 6.5, 0.15 M NaH2PO4, 1 mM DTT for subsequent SRM-MS processing.
Multiangle Light Scattering (MALS)
Multiangle light scattering was utilized to determine the oligomeric state of the purified proteins. Roughly 1 mg of purified protein (at 2 mg/mL) was taken for in-line gel filtration on a Superdex75 column followed by light scattering analysis. MALS was measured with a Wyatt Dawn Heleos-II and processed using the included software (ASTRA Version 5.3.4). Bovine Serum Albumin (BSA) was used as a control to ensure proper calibration.
Mass Spectrometry (MS) – Intact mass measurements
Each purified protein was diluted in water to 1 μM and applied to a Zorbax 300SB-C8 enrichment chip at 600 nL/min. In total, ~20 pmol of material was injected. A mobile phase gradient from 0.1% formic acid and 3% acetonitrile in water to 0.1% formic acid and 90% acetonitrile was used to chromatograph each sample, which was then taken for in-line electrospray ionization mass spectrometry. Mass spectra were obtained on an Agilent 6520 Accurate-Mass quadrupole time-of flight mass spectrometer with an accelerating voltage of 1850 V. Deconvolution was performed with the included software, Agilent MassHunter Qualitative Analysis Version B.04.00. Masses of all proteins were within 1.0 Da of those predicted.
Liposome preparation
In order to assess the phospholipase activity of the mZuc, cardiolipin-containing liposomes were prepared. Defined liposomes (PC:PE:PS:CL at 2:2:1:1) or extract-based liposomes (made with bovine heart lipid extract supplemented with CL) were made using standard methods. Briefly, lipids (Avanti Polar Lipids) in chloroform were mixed then dried using a stream of nitrogen followed by vacuum evaporation. Lipid films were then resuspended in 10 mM HEPES, pH 7.4, 0.1 M NaCl such that the final concentration of total lipids was 1 mg/mL. The resuspended lipids were vortexed vigorously and sonicated using a bath sonicator for ~15 minutes, until the suspension clarified. Absorbance measurements of the pre- and post-sonication liposomes were used to estimate the effective diameters34 of the resulting small unilamellar vesicles (SUVs) at less than 200 nm. Lipids and liposomes were stored under nitrogen at −20 °C and 4 °C, respectively, to limit oxidation when not in use.
Cardiolipase activity assay – Thin Layer Chromatography (TLC)
Cardiolipin cleavage assays were performed in a similar manner to those previously reported11. For each reaction, 20 μL of liposome solution (20 μg of total lipids) were mixed with 5 μg purfied protein in a reaction buffer of either 50 mM MES, pH 6.5, 75 mM NaCl, 1 mM DTT (nuclease buffer conditions) or 50 mM HEPES, pH 7.4, 80 mM KCl, 3 mM MgCl2, 2 mM CaCl2, 1 mM DTT (previously reported lipase conditions11). Commercially available PLD from S. chromofuscus (MP Biomedicals) was used as a positive control. Each 200 μL reaction was incubated at 37 °C for two hours, then quenched by the addition of 750 μL of methanol:chloroform (2:1).
Lipids were extracted by sequentially adding 250 μL chloroform and 250 μL 0.5 M NaCl in 0.1 M HCl (in water) with vigorous vortexing following each addition. The lower (organic) phase was removed, dried by vacuum evaporation, and resuspended in a small volume of methanol:chloroform for TLC analysis.
Each of the extracts was spotted onto 2.5 × 7.5 cm, glass-backed silica 60 F254 TLC plates (EMD). The extracts were chromatographed using a running solvent of chloroform:methanol:water:ammonium hydroxide (120:75:6:2), stained using permanganate (0.75% KMnO4, 5% KH2PO4, 0.06% NaOH), and developed by heating. Extract components appeared as yellow spots on a magenta background. Lipid identification was based on standards run in parallel.
Cardiolipase activity assay – Selected Reaction Monitoring (SRM) Mass Spectrometry (MS)
To confirm the findings of the TLC-based phospholipase activity assay and to obtain higher sensitivity for detecting the presumed phosphatidic acid product, an SRM-MS assay was employed. After performing the lipase reaction described above, 5 μ each (non-extracted) defined liposome lipase reaction were infused into a Thermo Scientific triple stage quadrupole (TSQ) Vantage mass spectrometer. Mass-to-charge ratio (m/z) peaks corresponding to cardiolipin [(18:2)4 species with a +1 charge state at 1448 Da; (18:2)4 species with a +2 charge state at 723 Da] and phosphatidic acid [(18:2)2 species with +1 charge state at 695 Da] were monitored. Injections were performed in triplicate.
Nuclease activity assays
DNA and RNA oligonucleotides ordered from Integrated DNA Technologies (IDT) or Dharmacon.
TGGCACTGTACCTCATCGACATTGAGAAGTGGCTCTCTGACTGGCGAATA (50 nt DNA)
TATTCGCCAGTCAGAGAGCCACTTCTCAATGTCGATGAGGTACAGTGCCA (50 nt DNA)
TATTCGCCAGTCAGAGAGCCACTTC (25 nt DNA)
TCAATGTCGATGAGGTACAGTGCCA (25 nt DNA)
TATTCGCCAGTCAGAGAGCC (20 nt DNA)
GTCGATGAGGTACAGTGCCA (20 nt DNA)
UGGCACUGUACCUCAUCGACAUUGAGAAGUGGCUCUCUGACUGGCGAAUA (50 nt RNA)
UAUUCGCCAGUCAGAGAGCCACUUCUCAAUGUCGAUGAGGUACAGUGCCA (50 nt RNA)
UAUUCGCCAGUCAGAGAGCC (20nt RNA)
GUCGAUGAGGUACAGUGCCA (20nt RNA)
UGGCACUGUACCUCAUCGAC (20nt RNA)
GGCUCUCUGACUGGCGAAUA (20nt RNA)
Oligonucleotides were 5'-labeled with 32P γ-ATP (PerkinElmer) or ATP (Sigma-Aldrich) using T4 polynucleotide kinase (NEB) according to the manufacturer's instructions. Free ATP was removed and the buffer exchanged using illustra MicroSpin™ G-25 Columns (GE Healthcare). Double-stranded substrates were annealed by mixing equimolar amounts of the complementary strands in water, denaturing at 95 °C for one minute, then cooled slowly from 65 °C to 25 °C in the presence of 30 mM NaCl, 10 mM MES, pH 6.5.
For enzymatic assays, mZuc was stored in 50% (v/v) glycerol at −20 °C. scPLD (EMD) was freshly prepared from the lyophilized stock according to the manufacturer's protocol. To assess nuclease activity, substrates were incubated with recombinant protein in 50 mM MES, pH 6.5, 75 mM NaCl, 2 mM CaCl2,1 mM DTT for DNase activity, or 5 mM MES, pH 6.5, 7.5 mM NaCl, 2 mM CaCl2,1 mM DTT for RNase activity at 37 °C for three hours (Fig. 1c and Supplementary Fig. 3c) or six hours (Figs 1e, 2 and Supplementary Fig. 2b). DNA and RNA substrates were used at a final concentration of 5 μM (Fig. 1 and Supplementary Fig. 3), or 50 nM (Fig. 2). Recombinant mZuc-dimer was used at a final concentration of 5 μM (Fig. 1) or 50 μM (Fig. 2 and Supplementary Fig. 2b). When RNA was used as a substrate, RNasin® Plus RNase Inhibitor (Promega) was added. Protein was removed from the cleavage reactions by proteinase K treatment (Roche) at a final concentration of 2 mg/mL (in 50 mM Tris-HCl, pH 7.5, 75 mM NaCl, 6 mM EDTA, 1% (w/v) SDS) at 50 °C for 30 minutes. Nucleic acids were extracted using phenol/chloroform (Ambion), precipitated with sodium acetate/ethanol and separated by 15% urea-polyacrylamide gel electophoresis (urea-PAGE). Low Molecular Weight Marker 10–100 nt (Affimetrix) was labeled with 32P γ-ATP and used as a size ladder. Visualization was accomplished using a Storm PhosphorImager.
To assess the chemical properties of mZuc cleavage products, nucleic acids were recovered from the cleavage reactions using proteinase K and phenol/chloroform extraction followed by an additional chloroform extraction and sodium acetate/ethanol precipitation. For DNA products, Terminal deoxynucleotidyl Transferase (TdT) (NEB) was incubated with the extracted oligonucleotides in the presence of ddATP or dATP according to the manufacturer's protocol. For RNA products, β-elimination was performed according to Vagin et al.35. Poly-A-tailing was performed using Poly(A) tailing kit (Ambion) at 37 °C for 15 minutes.
Detection of the phosphohistidine intermediate using 32P-disodium phosphate
mZuc wt and H135N (at a dimer concentration of 50 μM) were incubated with 0.4 mM 32P-disodium phosphate (PerkinElmer) in 50 mM MES, pH 6.5, 75 mM NaCl, 1 mM DTT at 37 °C for 3 hours. Proteins were separated by SDS-PAGE on a 10% NuPAGE gel (Invitrogen) and transferred to a nitrocellulose membrane. Proteins bound to the membrane were visualized by Ponceau S staining (0.1% Ponceau S, 5% acetic acid), and 32P was detected by phosphoimaging.
Detection of the phosphohistidine intermediate – Selected Reaction Monitoring (SRM) Mass Spectrometry (MS)
To confirm the findings of the 32PO43- labeling assay and further resolve the phosphorylation site, SRM-MS was employed. mZuc wildtype + H153N heterodimer was incubated in 50 mM MES, pH 6.5, 0.15 M NaH2PO4, 1 mM DTT for 1 hour at 4 °C. The protein (20 μg) was then denatured and reduced by adding 0.1% Protease-Max surfactant (Promega) and 5 mM TCEP in 200 mM triethylammonium bicarbonate, pH 10, and incubated at 55 °C for 20 min. The sample was then treated with 5 mM methyl methanethiosulfonate and proteolyzed with 2 μg Lys-C overnight at 37 °C. After digestion, 1 μg of the proteolytic fragments were applied to a home-packed C18 column. Peptides were chromatographed with a mobile phase gradient from 0.1% formic acid and 3% acetonitrile in water to 0.1% formic acid and 90% acetonitrile followed by in-line electrospray ionization mass spectrometry.
Mass spectra were collected on a Thermo Scientific triple stage quadrupole (TSQ) Vantage mass spectrometer. In the first round of MS, mass-to-charge ratio (m/z) peaks corresponding to a Lys-C generated, His153-containing peptide (AGIQVRHDQDLGYMHHK) were selected (+4 charge state masses of 522.0 Da and 502.0 Da for the phosphorylated and non-phosphorylated precursors, respectively). These precursors were then fragmented while monitoring for several y and b ion transitions.
Fluorescence Polarization
The affinity of mZuc for ssDNA and ssRNA was assessed using fluorescence polarization. Fluorescein-labeled 20-mer probes (5'–acagagtgccacttctcaac–Fluorescein–3' DNA, Sigma; 5'–acagagugccacuucucaac–Fluorescein–3' RNA, Dharmacon) at 10 nM were mixed with mZuc at various concentrations in a binding buffer of 0.1 M MES, pH 6.5, 75 mM NaCl, 1 mM DTT and incubated at room temperature for 10 minutes. Fluorescence polaration was then measured at ambient temperature (27 °C) on a BioTek Synergy4 microplate reader using excitation and emission wavelengths of 485 and 528 nm, respectively. Readings were performed in duplicate.
Crystallization
Crystals of mZuc were grown by hanging-drop vapor diffusion on siliconized glass coverslips (Hampton Research, Aliso Viejo, CA). Immediately prior to crystallization, the purified protein was mixed in a 1:100 (m/m) ratio with chymotrypsin. Two microliters of the protein solution at 3 mg/mL were then mixed with an equal volume of reservoir solution containing 50 mM Bis-Tris, pH 6.5, 18% PEG-3350, and 2% tascimate, pH 6.0, and suspended over 0.5 mL of reservoir solution. Rectangular prismatic crystals (~150 μm × 50 μm × 50 μm) grew at 4 °C within 24 h. Crystals were harvested and frozen in liquid nitrogen after serial transfer to 50 mM Bis-Tris, pH 6.5, 18% PEG-3350, and 2% tascimate, pH 6.0, and 20% ethylene glycol in increasing steps of 5% ethylene glycol. Tungstate-derivatized crystals were obtained by co-crystallization with 10 mM Na2WO4 added to the crystallization drop.
Data collection, crystal parameters, structure determination, and refinement
Data were collected at 100 K using synchrotron radiation at the ×29 beamline at the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory, Upton, NY. Data were processed with XDS36 and scaled with SCALA37. Additional processing was performed with programs from the CCP4 suite37.
The crystals belong to the space group P43212 with a=b=38.7 Å, c=213.1 Å, α=β=γ=90° with one molecule per asymmetric unit and a solvent content of ~35%. The structure was solved by molecular replacement with the program Phaser38 using the structure of Nuc, a bacterial PLD-family nuclease (PDB: 1BYR) as a search model18. Automatic model building was performed using ARP/wARP39, which correctly built approximately 140 residues of the 158 in the final structure. Subsequent model building and inspection were carried out using the program Coot40. Model refinement was performed using REFMAC541. TLS refinement was carried out using one TLS group. The final native model consists of residues 35–58, 66–126, and 130–209 in addition to one coordinated Zn2+ and 106 water molecules. The final models were validated by Molprobity42. All residues in the final structures were in the allowed regions of the Ramachandran plot with 152 / 158 residues being in favored regions. Data collection and refinement statistics are listed in Supplemental Table I.
In addition to native crystals, tungstate-derivatized crystals were also produced. These diffracted to similar resolution and had nearly identical unit cell parameters. Initial processing was performed as described above. The native structure was used as an isomorphous replacement search model then refined accordingly. To avoid over-fitting, refinement of the tungstate derivative utilized the same Rfree set as the native. To confirm the identity of the Zn2+, two data sets were collected from a single tungstate-derivatized crystal at ±100 eV from the Zinc K edge. Anomalous maps for each data set confirmed the identity of the metal. The r.m.s.d. for all protein atoms in the native and tungstate derivative was 0.49 Å. Crystallographic statistics for the tungstate-derivatized crystals are also presented in Supplemental Table I.
Figures
Figures of molecular models were generated using PyMOL43. Electrostatic surface calculations were performed with APBS30 with a solvent ion concentration of 0.15 M using the PARSE force field. Superpositioning of structural homologs was performed by the DALI server44.
Data deposition
Coordinates and structure factors have been deposited in the Protein Data Bank (PDB) with the accession numbers 4GGJ (native) and 4GGK (tungstate derivative).
RNA Modeling and Energy Minimization
The refined mZuc structure was used as a template for manual model building of a short, ssRNA using Coot40. The initial ssRNA model placed the phosphate backbone along the observed positively-charged active site groove (with the scissile phosphate positioned at the tungstate position in the derivative structure) while maintaining approximate two-fold symmetry of phosphates with respect to the homodimer (the RNA breaks the strict two-fold symmetry due to its polarity). Energy mimization was performed using GROMACS v. 4.5.525 with the AMBER-99SB-ILDN force field45 and Particle-mesh Ewald (PME) long-range electrostatic modeling46 with a conjugate gradient energy minimization algorithm, keeping the protein model (including Zn2+) fixed while allowing minimization of the RNA molecule and solvent model. A nearly indistinguishable model was also generated using the steepest descent algorithm.
Supplementary Material
Acknowledgements
We thank Witold Filipowicz, Robert MacDonald, and members of the Hannon and Joshua-Tor labs for fruitful discussions; Gyula Bencze, Keith Rivera, and Darryl Pappin of the CSHL proteomics facility, which is funded in part by an NCI Cancer Center Support Grant (CA045508), for support with mass spectrometry; and Howard Robinson for help at the National Synchrotron Light Source, which is supported by Department of Energy, Office of Basic Energy Sciences. J.J.I. was supported by the Harvey L. Karp award and by a Ruth L. Kirschstein National Research Service Awards NIH fellowship F32GM97888. This work was supported by US National Institutes of Health grant R01GM062534. G.J.H. and L.J. are Howard Hughes Medical Institute Investigators.
Footnotes
Author Contribution Statement LJ, GJH, ADH, and JII planned studies and wrote the paper. ADH and JII performed the experiments and SRK analyzed datasets.
References
- 1.Senti KA, Brennecke J. The piRNA pathway: a fly's perspective on the guardian of the genome. Trends Genet. 2010;26:499–509. doi: 10.1016/j.tig.2010.08.007. doi:10.1016/j.tig.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brennecke J, et al. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007;128:1089–1103. doi: 10.1016/j.cell.2007.01.043. doi:10.1016/j.cell.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 3.Kawaoka S, Izumi N, Katsuma S, Tomari Y. 3' end formation of PIWI-interacting RNAs in vitro. Mol Cell. 2011;43:1015–1022. doi: 10.1016/j.molcel.2011.07.029. doi:10.1016/j.molcel.2011.07.029. [DOI] [PubMed] [Google Scholar]
- 4.Gunawardane LS, et al. A slicer-mediated mechanism for repeat-associated siRNA 5' end formation in Drosophila. Science. 2007;315:1587–1590. doi: 10.1126/science.1140494. doi:10.1126/science.1140494. [DOI] [PubMed] [Google Scholar]
- 5.Haase AD, et al. Probing the initiation and effector phases of the somatic piRNA pathway in Drosophila. Genes Dev. 2010;24:2499–2504. doi: 10.1101/gad.1968110. doi:10.1101/gad.1968110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pane A, Wehr K, Schupbach T. zucchini and squash encode two putative nucleases required for rasiRNA production in the Drosophila germline. Dev Cell. 2007;12:851–862. doi: 10.1016/j.devcel.2007.03.022. doi:10.1016/j.devcel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schupbach T, Wieschaus E. Female sterile mutations on the second chromosome of Drosophila melanogaster. II. Mutations blocking oogenesis or altering egg morphology. Genetics. 1991;129:1119–1136. doi: 10.1093/genetics/129.4.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malone CD, et al. Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell. 2009;137:522–535. doi: 10.1016/j.cell.2009.03.040. doi:10.1016/j.cell.2009.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olivieri D, Sykora MM, Sachidanandam R, Mechtler K, Brennecke J. An in vivo RNAi assay identifies major genetic and cellular requirements for primary piRNA biogenesis in Drosophila. Embo J. 2010;29:3301–3317. doi: 10.1038/emboj.2010.212. doi:10.1038/emboj.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selvy PE, Lavieri RR, Lindsley CW, Brown HA. Phospholipase D: enzymology, functionality, and chemical modulation. Chem Rev. 2011;111:6064–6119. doi: 10.1021/cr200296t. doi:10.1021/cr200296t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi SY, et al. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol. 2006;8:1255–1262. doi: 10.1038/ncb1487. doi:10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- 12.Huang H, et al. piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Dev Cell. 2011;20:376–387. doi: 10.1016/j.devcel.2011.01.004. doi:10.1016/j.devcel.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watanabe T, et al. MITOPLD is a mitochondrial protein essential for nuage formation and piRNA biogenesis in the mouse germline. Dev Cell. 2011;20:364–375. doi: 10.1016/j.devcel.2011.01.005. doi:10.1016/j.devcel.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalvez F, Gottlieb E. Cardiolipin: setting the beat of apoptosis. Apoptosis. 2007;12:877–885. doi: 10.1007/s10495-007-0718-8. doi:10.1007/s10495-007-0718-8. [DOI] [PubMed] [Google Scholar]
- 15.Gottlin EB, Rudolph AE, Zhao Y, Matthews HR, Dixon JE. Catalytic mechanism of the phospholipase D superfamily proceeds via a covalent phosphohistidine intermediate. Proc Natl Acad Sci U S A. 1998;95:9202–9207. doi: 10.1073/pnas.95.16.9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lackey D, Walker GC, Keng T, Linn S. Characterization of an endonuclease associated with the drug resistance plasmid pKM101. J Bacteriol. 1977;131:583–588. doi: 10.1128/jb.131.2.583-588.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pohlman RF, Liu F, Wang L, More MI, Winans SC. Genetic and biochemical analysis of an endonuclease encoded by the IncN plasmid pKM101. Nucleic Acids Res. 1993;21:4867–4872. doi: 10.1093/nar/21.21.4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stuckey JA, Dixon JE. Crystal structure of a phospholipase D family member. Nat Struct Biol. 1999;6:278–284. doi: 10.1038/6716. doi:10.1038/6716. [DOI] [PubMed] [Google Scholar]
- 19.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. doi:10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 20.Berg JM, Shi Y. The galvanization of biology: a growing appreciation for the roles of zinc. Science. 1996;271:1081–1085. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- 21.Lai WS, Carballo E, Thorn JM, Kennington EA, Blackshear PJ. Interactions of CCCH zinc finger proteins with mRNA. Binding of tristetraprolin-related zinc finger proteins to Au-rich elements and destabilization of mRNA. J Biol Chem. 2000;275:17827–17837. doi: 10.1074/jbc.M001696200. doi:10.1074/jbc.M001696200. [DOI] [PubMed] [Google Scholar]
- 22.Kelly SM, et al. Recognition of polyadenosine RNA by zinc finger proteins. Proc Natl Acad Sci U S A. 2007;104:12306–12311. doi: 10.1073/pnas.0701244104. doi:10.1073/pnas.0701244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hurt JA, et al. A conserved CCCH-type zinc finger protein regulates mRNA nuclear adenylation and export. J Cell Biol. 2009;185:265–277. doi: 10.1083/jcb.200811072. doi:10.1083/jcb.200811072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao G, Guo X, Goff SP. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science. 2002;297:1703–1706. doi: 10.1126/science.1074276. doi:10.1126/science.1074276. [DOI] [PubMed] [Google Scholar]
- 25.Van Der Spoel D, et al. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. doi:10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 26.Aravin A, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–207. doi: 10.1038/nature04916. doi:10.1038/nature04916. [DOI] [PubMed] [Google Scholar]
- 27.Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199–202. doi: 10.1038/nature04917. doi:10.1038/nature04917. [DOI] [PubMed] [Google Scholar]
- 28.Frank F, Sonenberg N, Nagar B. Structural basis for 5'-nucleotide base-specific recognition of guide RNA by human AGO2. Nature. 2010;465:818–822. doi: 10.1038/nature09039. doi:10.1038/nature09039. [DOI] [PubMed] [Google Scholar]
- 29.Mi S, et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5' terminal nucleotide. Cell. 2008;133:116–127. doi: 10.1016/j.cell.2008.02.034. doi:10.1016/j.cell.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esnouf RM. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr D Biol Crystallogr. 1999;55:938–940. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.