

Background: The molecular basis of variable cell fate after mitotic arrest is poorly understood.
Results: The robustness of Cdk1 signaling to antiapoptotic Bcl-2 proteins dictates mitotic death versus mitotic slippage.
Conclusion: Sustained Cdk1 activity coordinately promotes mitotic arrest and mitotic death.
Significance: Defining the molecular basis of antimitotic drug action is important for their rational use clinically.
Keywords: Anticancer Drug, Apoptosis, Bcl-2 Proteins, Cancer Biology, CDK (Cyclin-dependent Kinase), Cyclin B1, Mitotic Death, Mitotic Slippage, Taxol
Abstract
The prevailing model suggests that cell fate after mitotic arrest depends on two independent and competing networks that control cyclin B1 degradation and the generation of death signals. However, recent evidence for Cdk1/cyclin B1-mediated phosphorylation and inactivation of antiapoptotic Bcl-2 proteins suggests the existence of significant cross-talk and interdependence between these pathways. Further, the nature of the mitotic death signals has remained elusive. In this study, we sought to test the hypothesis that fate after mitotic arrest is dictated by the robustness of Cdk1/cyclin B1 signaling to Bcl-2 proteins and to identify signals that may represent a mitotic death signature. We show that when treated with Taxol, slippage-resistant HT29 colon carcinoma cells display robust Cdk1 activity and extensive Mcl-1/Bcl-xL phosphorylation and die in mitosis, whereas slippage-prone DLD-1 colon carcinoma cells display weak Cdk1 activity and partial and transient Mcl-1/Bcl-xL phosphorylation and die in subsequent interphase or survive. Furthermore, modulation of this signaling axis, either by inhibition of Cdk1 in slippage-resistant HT29 or by enforcing mitotic arrest in slippage-prone DLD-1 cells, evokes a switch in fate, indicating that the strength of Cdk1 signaling to Bcl-2 proteins is a key determinant of outcome. These findings provide novel insight into the pathways that regulate mitotic death, suggest that the robustness of these signaling events may be useful as a marker to define susceptibility to antimitotic drugs, and encourage a revision in the current model describing fate after mitotic arrest.
Introduction
The spindle-assembly checkpoint is a key regulatory mechanism that prevents advance to anaphase until all the chromosomes are properly attached to kinetochores (reviewed in Refs. 1 and 2). Antimitotic agents that inhibit microtubule function and dynamics, including the taxanes and vinca alkaloids, as well as newer drugs that target proteins with defined roles in mitosis, cause prolonged activation of this checkpoint, resulting in mitotic arrest (reviewed in Refs. 3–5). Failure to satisfy the spindle-assembly checkpoint leads to mitotic arrest by preventing activation of the anaphase-promoting complex through sequestration of its activator Cdc20. This in turn leads to elevated levels of cyclin B1, an anaphase-promoting complex substrate, and a subsequent increase in Cdk12 activity. Detailed cellular studies have indicated that mitotically arrested cells exhibit significant variation in their response, with several outcomes described (3–5). These include death during mitosis, mitotic slippage and death in the subsequent interphases, mitotic slippage and subsequent survival, or no mitotic entry. Elucidation of the factors that dictate these different outcomes is critically important for predicting which tumors may be responsive to these drugs and thus for the rational design of clinical trials testing efficacy of the newer drugs.
Although the pathways that regulate outcome after mitotic arrest are still unclear, recent work has suggested that fate may depend on two independent and competing networks that control cyclin B1 degradation on the one hand and the generation of death signals that activate caspases on the other (6, 7). If cyclin B1 levels fall below the mitotic exit threshold before sufficient death signals are generated, slippage occurs, whereas if the death signals accumulate before cyclin B1 is degraded sufficiently, death in mitosis occurs. However, this model does not account for recent evidence that the levels of cyclin B1 and the generation of death signals are not independent but, rather, are interdependent, via the actions of Cdk1/cyclin B1 on proteins that directly regulate apoptosis. Most notably, Cdk1/cyclin B1 has been shown to phosphorylate and inactivate antiapoptotic members of the Bcl-2 protein family (Bcl-2, Bcl-xL, and Mcl-1) (8–11), which in turn is necessary for mitochondrial apoptosis and caspase activation. The existence of significant cross-talk between these pathways raises the possibility that the choice between mitotic death and mitotic slippage may depend on the net effect of one pathway on the other, that is, on the robustness of Cdk1/cyclin B1 signaling to Bcl-2 proteins. However, direct approaches to test this hypothesis are limited because it is difficult to interrogate Cdk1 function via elimination or long term inhibition without perturbing mitotic entry or exit, processes that depend on Cdk1 activation and inactivation, respectively (12, 13).
In this study, we sought to test the hypothesis that fate after mitotic arrest is dictated by the robustness of Cdk1 signaling to Bcl-2 proteins by examining these pathways in two cell lines that differ markedly in fate. Earlier studies showed that upon Taxol treatment, HT29 colon carcinoma cells die predominately in mitosis, whereas DLD-1 colon carcinoma cells exhibit extensive slippage and then either die in subsequent interphase or survive (6). By comparing and modulating this signaling axis in these slippage-resistant or slippage-prone cell lines, we present results here that strongly support the hypothesis and suggest a revision in the current model describing fate after mitotic arrest. Our findings also show that death in mitosis is tightly associated with phosphorylation of Bcl-xL and phosphorylation/degradation of Mcl-1, suggesting that these events may represent elements of the elusive mitotic death signature.
EXPERIMENTAL PROCEDURES
Materials
Antibody against cyclin B1 (sc-245) and protein A/G PLUS-agarose beads were purchased from Santa Cruz (Santa Cruz, CA); antibodies against Bcl-xL (2762) and GAPDH (2118) were purchased from Cell Signaling Technology (Beverly, MA); antibody against poly(ADP-ribose) polymerase (PARP) (556362) and annexin V conjugated to phycoerythrin were purchased from Pharmingen; antibodies against Cdc20 (26483) and phospho-Ser-62-Bcl-xL (30655) were purchased from Abcam (Cambridge, MA); antibody against mitotic protein monoclonal 2 (MPM-2) (05-368) was purchased from Millipore (Temecula, CA); and antibody against Mcl-1 (ADI-AAP-240) was purchased from Enzo Life Sciences (Farmingdale, NY). Cy3-conjugated donkey anti-rabbit IgG secondary antibody (711-165-152) for immunofluorescence was purchased from Jackson ImmunoResearch (West Grover, PA). Taxol (paclitaxel) was purchased from Sigma, thymidine was purchased from EMD Biosciences (Gibbstown, NJ), and aminopurvalanol A and RO3306 were purchased from Axxora (San Diego, CA).
Cell Culture
Colon carcinoma cell lines HT29 and DLD-1 and HeLa sublines, KB3 and KBV1, were maintained in Dulbecco's modified Eagle's medium supplemented with 10% bovine growth serum, 2 mm l-glutamine, 50 units/ml penicillin, and 50 units/ml streptomycin at 37 °C under 5% CO2. Selection for GFP-tagged histone H2B in DLD-1 and HT29 cells was maintained with puromycin at 0.5 μg/ml (HT29) or 2 μg/ml (DLD-1). Selection for DsRed-tagged histone H2B in DLD-1 cells was maintained with G418 (0.75 mg/ml) and expression of GFP-cyclin B1 was induced with tetracycline (1 μg/ml). KBV1 cells, selected for resistance to vinblastine and exhibiting cross-resistance to other microtubule inhibitors (14), were maintained in 1 μm vinblastine and transferred to drug-free medium 5 days prior to conducting experiments.
Time-lapse Microscopy
Cells were seeded in Bioptechs Delta TC3 open dishes, synchronized by the addition of 2 mm thymidine for 16 h, released by washing with PBS, and then treated with 0.01 μm–1 μm Taxol 4.5 h after release. Changes in nuclear morphology were monitored by visual inspection of images collected by a Zeiss LSM410 confocal microscope every 15 min with 0.1-s exposure over 48–72 h. 50 cells from a representative field in each experiment were analyzed using the National Institutes of Health ImageJ software with individual cell fate plotted over time as a single line of different colors each denoting a particular fate (supplemental Fig. S1) (6).
Immunoblotting
Whole cell extracts were prepared by washing harvested cells with ice-cold PBS and suspending pellets in lysis buffer (40 mm HEPES, pH 7.5, 120 mm NaCl, 1% Triton X-100, 1 mm EDTA, 20 μg/ml aprotinin, 50 μg/ml leupeptin, 10 μm pepstatin, 1 mm phenylmethylsulfonyl fluoride, 20 mm β-glycerophosphate, 50 mm NaF, 1 mm Na3VO4, and 1 μm okadaic acid) for 1 h with occasional mixing. Insoluble material was removed by centrifugation (15 min at 15,000 × g), and the supernatant was retained as the whole cell extract. Protein concentration was determined using the Bio-Rad protein assay, and 50 μg was analyzed by SDS-PAGE and immunoblotting as described (15).
Immunofluorescence
Cells were grown on glass cover slips and after treatment nonadherent cells were collected and centrifuged onto a glass slide using a cytospin. Untreated cells were made nonadherent by trypsinization. Cells were fixed with 4% formaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 1% BSA. Cells were probed for expression of Mcl-1 or phospho-Bcl-xL using specific primary antibody (1:100) and Cy3-conjugated secondary antibody (1:100). Immunofluorescent protein (red) and nuclei (green) were visualized using a Zeiss LSM410 confocal microscope.
Cdk1/Cyclin B1 Assay
Cdk1 was assayed in cell extracts via cyclin B1 immunoprecipitation using as substrate FL62, a small peptide derived from the major Cdk1 phosphorylation site, Ser-62, in Bcl-xL as described (8). Briefly, whole cell extracts were precleared with protein A/G beads for 30 min at 4 °C and then incubated with mouse anti-cyclin B1 antibody for 1 h at 4 °C and incubated with protein A/G beads for 2 h at 4 °C. The pellets were washed twice with lysis buffer and incubated for 20 min at 37 °C in a reaction mixture containing 25 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 5 mm DTT, 1 μm ATP, 0.1–1 μCi of [γ32P]ATP, and 10 μg of FL62. Background kinase activity was determined with no substrate added to the reaction. Reaction mixtures were applied to P81 phosphocellulose filter discs (Whatman) and washed with 75 mm phosphoric acid, and 32P incorporation was determined by scintillation counting.
Cell Viability and Death Assays
Cell viability was determined using (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT) reagent as described.3 Cells (2000/well) were seeded in 96-well plates, and the following day, Taxol was added at concentrations up to 10 μm in a fixed final concentration of 0.1% DMSO. Controls received vehicle (0.1% DMSO) alone. Data are expressed relative to vehicle-treated controls, and IC50 value was defined as the drug concentration reducing absorbance, or relative cell survival, to 50% of the control. Cell death was determined by an ELISA assay (Roche Applied Science), which quantitatively detects apoptotic cell death by photometric measurement of cytoplasmic histone-associated DNA fragments. Phycoerythrin-annexin V staining for early apoptotic cells was performed according to the manufacturer's instructions (BD Biosciences). Analysis was performed by flow cytometry on a FACSCalibur using the CellQuest Pro software.
Cdc20 Knockdown
Depletion of Cdc20 was performed using Ambion Silencer Select siRNA against Cdc20 (s2748) at a final concentration of 100 nm with Silencer Select negative control siRNA (4390844) (100 nm) as control. Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
RESULTS
Fate Profiles for Colon Carcinoma Cell Lines
To demonstrate the nuclear morphological characteristics used to define and quantitate individual cell fate, DLD-1 and HT29 cells stably expressing GFP-tagged H2B were synchronized with a single thymidine block, treated with 0.1 μm Taxol, and monitored by time-lapse fluorescence microscopy. Representative images of individual cells tracked over 72 h illustrating the different types of fate exhibited are shown in supplemental Fig. S1. Cells that stayed in interphase or underwent division were readily apparent; cells that exhibited discrete chromosomal condensation and then chromosomal fragmentation were scored as death in mitosis; and death in interphase was apparent with nuclear fragmentation occurring from a cell with initially diffuse chromatin and lack of discrete chromosomal structures and exit or slippage indicated by nuclear condensation and decondensation without fragmentation or division. Fate profiles derived from examination of 50 individual HT29 or DLD-1 cells synchronized and treated with 0.1 μm Taxol are shown in Fig. 1A. As described previously (6), fate was displayed by horizontal lines, each representing a single cell, with the color indicating the particular fate and the length indicating its duration. Consistent with earlier findings (6), HT29 cells died predominately in mitosis, whereas DLD-1 cells underwent mitotic slippage and then either died in subsequent interphase or survived. Because fate is influenced by antimitotic drug concentration and individual cell lines differ in drug sensitivity, a more rigorous comparison was conducted with drug concentrations normalized based on cell viability assays. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell viability assays indicated a 10-fold difference in Taxol sensitivity, with IC50 values of 5 nm for HT29 and 50 nm for DLD-1 cell lines (data not shown), consistent with reported values.3 At a Taxol concentration of 2× [IC50], 68% of HT29 cells died in mitosis (Fig. 1B) versus 16% of DLD-1 cells (Fig. 1A). At a Taxol concentration of 20× [IC50], 92% of HT29 cells died in mitosis (Fig. 1A) versus 48% of DLD-1 cells (Fig. 1C). These results, graphically represented in Fig. 1D, demonstrate that although the percentage of cells dying in mitosis increased with increasing Taxol concentration for both cell lines, highly significant differences in fate intrinsic to the cell line are evident, validating their utility for the present studies.
FIGURE 1.

Fate profiles of HT29 and DLD-1 cells treated with Taxol. A–C, HT29 or DLD-1 cells as indicated were synchronized by single thymidine block, treated with the indicated concentration of Taxol 4.5 h after release, and monitored by time-lapse microscopy for 72 h. Data for 50 cells per condition are presented with each horizontal line representing a single cell, the color of the line indicating the fate according to the key at the right, and the length of the line indicating the duration of the exhibited fate. Time 0 represents the time of the addition of Taxol. D, the proportions of cells undergoing mitotic death, derived from data presented in panels A–C, are shown. Other fates include cells that exited mitotic arrest and died in interphase, cells that exited mitotic arrest and survived, and cells that did not enter mitosis. *** = p < 0.0001 (χ-squared test).
Kinetics of Phosphorylation of Antiapoptotic Bcl-2 Proteins
After verifying opposing cell fates in response to Taxol, we sought to compare antiapoptotic Bcl-2 protein phosphorylation to determine whether a relationship to fate existed. HT29 or DLD-1 cells were synchronized and treated with Taxol for up to 48 h, and immunoblotting was performed for markers of mitosis (MPM-2, cyclin B1), apoptosis (PARP cleavage), and antiapoptotic Bcl-2 proteins Mcl-1 and Bcl-xL. (Bcl-2 was undetectable in both cell lines.) When HT29 cells were treated with 0.1 μm Taxol (Fig. 2A, left), cyclin B1 levels were markedly increased at 4–8 h and sustained through 28–32 h, with strong MPM-2 immunoreactivity also evident at 12–20 h. Mcl-1 underwent a phosphorylation-dependent mobility shift and degradation, becoming undetectable by 20 h, and Bcl-xL underwent complete phosphorylation, observed both by mobility shift and by immunoreactivity with a phospho-Ser-62-dependent antibody (Fig. 2A). PARP cleavage was initiated at 16–20 h and was essentially complete by 28–32 h when cells still expressed mitotic markers. In contrast, when DLD-1 cells were treated with 0.1 μm Taxol (Fig. 2A, right), cyclin B1 levels and MPM-2 immunoreactivity were increased at early time points, but more weakly than observed in HT29 cells, and PARP cleavage was only partial, beginning at 20 h. In addition, transient phosphorylation of Mcl-1 and Bcl-xL was observed at early time points, and Mcl-1 remained detectable throughout. In DLD-1 cells treated with 1 μm Taxol (Fig. 2B), each of the signals was more pronounced than at 0.1 μm Taxol, but the degree of alteration in each protein was much less pronounced than in HT29 cells treated with 0.1 μm Taxol (Fig. 2A, left). Overall, the dynamic changes in protein expression and phosphorylation viewed in the context of the corresponding fate profiles indicate that mitotic death is associated with degradation of Mcl-1 and robust Bcl-xL phosphorylation, the latter resulting in loss of its antiapoptotic function (15). In contrast, in cells undergoing slippage, these events are transient and incomplete. In synchronized cells treated with vehicle instead of Taxol, pronounced changes in Mcl-1 phosphorylation/expression or Bcl-xL phosphorylation were not observed, and there was no evidence of PARP cleavage, as expected (data not shown). To quantify the extent of cell death in Taxol-treated cells, a cell death ELISA assay was performed, as described under “Experimental Procedures.” In HT29 cells treated with 0.1 μm Taxol, cell death increased at 24 h and further increased at 48 h, whereas in DLD-1 cells under the same condition, cell death was increased at 24 h but did not increase further, with a lower magnitude versus HT29 cells (Fig. 2C). These results are consistent with the fate profiles and immunoblot analysis, which showed little survival in HT29 cells when compared with significant slippage and survival in DLD-1 cells after 48 h treatment.
FIGURE 2.

Death in mitosis is accompanied by phosphorylation of Bcl-xL and degradation of Mcl-1. A, HT29 (left panel) or DLD-1 (right panel) cells were released from single thymidine block, and after 4.5 h, treated with 0.1 μm Taxol (Tax) and harvested at the indicated time intervals. Immunoblots were performed with MPM-2 antibody or for the proteins indicated. GAPDH was used as a loading control. Phosphorylated Bcl-xL was detected by mobility shift (p-Bcl-xL) or reactivity with the phospho-Ser-62-specific antibody (phospho-Bcl-xL). Intact (116-kDa) and cleaved (85-kDa) species of PARP are shown. B, DLD-1 cells were released from single thymidine block, and after 4.5 h, treated with 1 μm Taxol and harvested at the indicated time intervals after treatment. C, HT29 or DLD-1 cells were untreated or treated with 0.1 μm Taxol for 24 or 48 h, and apoptosis was measured by cell death ELISA, as described under “Experimental Procedures.” Values represent mean ± S.D. (n = 3). *** = p < 0.001 (Student's t test). D, synchronized HT29 or DLD-1 cells were untreated or treated with either 0.1 μm or 1 μm Taxol for the times indicated (Harvest time (h)), and cell extracts were prepared and subjected to immunoblotting for BubR1 expression. BubR1 phosphorylation was detected by mobility shift (p-BubR1). GAPDH was used as a loading control.
The relatively modest and transient increase in cyclin B1 expression in DLD-1 cells in response to Taxol is consistent with their tendency to undergo slippage and contrasts with the sustained cyclin B1 expression observed in HT29 cells, consistent with their propensity to undergo mitotic arrest and mitotic death. The spindle-assembly checkpoint acts to sequester Cdc20, preventing activation of the anaphase-promoting complex resulting in elevated levels of cyclin B1 and sustained Cdk1 activity (1–3). To determine whether the activation status of the spindle-assembly checkpoint was a factor underlying the differential responses observed in the two cell lines, HT29 and DLD-1 cells were untreated or treated with Taxol, and BubR1 phosphorylation was examined by immunoblotting. It has previously been shown that BubR1 is essential for spindle checkpoint activation and that BubR1 function is dependent on its phosphorylation status (2). As shown in Fig. 2D, 0.1 μm Taxol induced sustained expression and phosphorylation of BubR1 in slippage-resistant HT29 cells, whereas BubR1 phosphorylation was transient in slippage-prone DLD-1 cells treated with either 0.1 μm or 1 μm Taxol. Thus, the robustness of spindle-assembly checkpoint activation is likely an important factor in the sustainability of mitotic arrest and the different fate profiles observed.
To further demonstrate a link between Mcl-1/Bcl-xL phosphorylation and mitotic death, we used a pair of HeLa cell sublines that displayed either sensitivity (KB3) or resistance (KBV1) to Taxol (14). Treatment of KB3 cells with 0.1 μm Taxol resulted in mitotic death, as indicated by high levels of MPM-2 immunoreactivity and increased cyclin B1 expression in concert with PARP cleavage, and this occurred in association with phosphorylation of Bcl-xL and phosphorylation and degradation of Mcl-1 (supplemental Fig. S2). In contrast, none of these events were observed after similar treatment of KBV1 cells, which are resistant to 0.1 μm Taxol (supplemental Fig. S2). These results strengthen the link and indicate that phosphorylation of Bcl-xL and degradation of Mcl-1 are events not restricted to specific cell types but instead may represent elements of a conserved mitotic death pathway.
Analysis of Mcl-1 Expression and Bcl-xL Phosphorylation by Immunofluorescence Microscopy
Immunofluorescence microscopy was performed to evaluate changes in Mcl-1 expression and Bcl-xL phosphorylation in relation to nuclear morphology in single cells after Taxol treatment. To account for cells that became detached, adherent and nonadherent populations were collected and examined separately. Untreated HT29 cells maintained expression of Mcl-1 and exhibited intact nuclear morphology as expected (Fig. 3A). When treated with 0.1 μm Taxol, the nonadherent population of HT29 cells showed loss of Mcl-1 expression in concert with nuclear fragmentation (Fig. 3A). Cells that remained adherent at 24 h had abundant expression of Mcl-1 and intact nuclear morphology, whereas at 48 h, there was a mixed population of adherent cells, with reduced Mcl-1 expression in cells exhibiting nuclear fragmentation and more prominent Mcl-1 expression in cells with intact nuclei (Fig. 3A, bottom right, merged image). These data suggest that Taxol-induced death in HT29 cells is closely associated with degradation of Mcl-1. When phospho-Ser-62-Bcl-xL expression was examined in Taxol-treated HT29 cells, expression was absent in control cells, evident at 24 h, and diminished by 48 h (Fig. 3B), corresponding with the immunoblot data (Fig. 2A). In the population remaining adherent at 24 h, it was evident that phospho-Ser-62-Bcl-xL was more pronounced in cells with intact nuclei and less pronounced in cells with highly fragmented nuclei, suggesting that Bcl-xL phosphorylation preceded death, as also indicated by the immunoblots. In DLD-1 cells treated with 0.1 μm Taxol, both adherent and nonadherent populations expressed Mcl-1 at levels that remained largely unchanged at 0, 24, or 48 h (Fig. 3C), and phospho-Bcl-xL was undetectable (Fig. 3D). These results are consistent with the immunoblot data of Fig. 2A and confirm that conditions that largely promote slippage and nonmitotic death or survival are associated with maintenance of Mcl-1 expression and lack of robust Bcl-xL phosphorylation.
FIGURE 3.

Taxol-induced mitotic death is closely associated with phosphorylation of Bcl-xL and degradation of Mcl-1. A–D, HT29 (A and B) or DLD-1 (C and D) cells were untreated or treated for 24 or 48 h with 0.1 μm Taxol, and adherent and nonadherent cells were collected. Cells were fixed, permeabilized, and probed for expression of Mcl-1 (A and C) or phospho-Bcl-xL (p-Bcl-xL) (B and D) using fluorescent Cy3 secondary antibody (red signal). Nuclear fluorescence derived from GFP-histone H2B is indicated by the green signal.
Cdk1/Cyclin B1 Kinase Activity Is Greater in Slippage-resistant HT29 versus Slippage-prone DLD-1 Cell Lines
We and others have previously demonstrated that Cdk1 phosphorylates antiapoptotic Bcl-2 proteins, including Bcl-2, Bcl-xL, and Mcl-1, during mitotic arrest (8–11), with phosphorylation of Mcl-1 priming it for action by other kinases and subsequent degradation (10) and phosphorylation of Bcl-xL negatively affecting Bax binding (15). Because cyclin B1 levels increased to a greater extent and were more sustained, and Mcl-1 phosphorylation/degradation and Bcl-xL phosphorylation were much more robust in Taxol-treated HT29 cells versus DLD-1 cells, we sought to determine whether Cdk1 activity differed. Cells were synchronized and treated with Taxol for 16 h, and extracts were subjected to Cdk1/cyclin B1 assay, as described under “Experimental Procedures.” At 0.1 μm Taxol, Cdk1 activity was 14-fold higher in extracts from HT29 versus DLD-1 cells (Fig. 4). In DLD-1 cells treated with 1 μm Taxol, Cdk1 activity was higher than at 0.1 μm Taxol, but still only 25% of that found in HT-29 cells at the equally effective concentration of 0.1 μm Taxol (Fig. 4). Thus, slippage-resistant HT29 cells display much more robust Cdk1 activity in response to Taxol than slippage-prone DLD-1 cells, paralleling the high and sustained levels of Bcl-2 protein phosphorylation evident in the immunoblots (Fig. 2) and fluorescence micrographs (Fig. 3).
FIGURE 4.
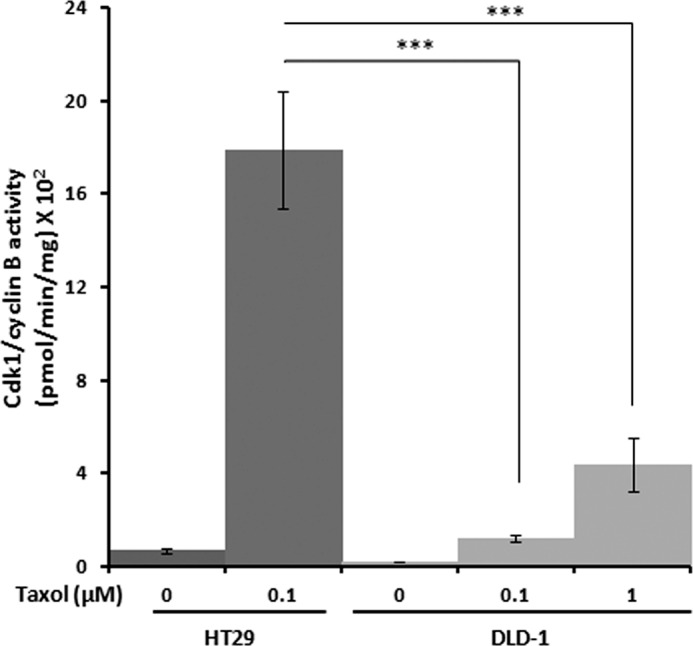
Increased Cdk1/cyclin B1 activity in Taxol-treated HT29 versus DLD-1 cells. HT29 or DLD-1 cells were synchronized by single thymidine block and treated after 4.5 h with the indicated concentration of Taxol for 16 h. Cell extracts were subjected to Cdk1/cyclin B1 assay, as described under “Experimental Procedures.” Values have been corrected for background kinase activity (mean ± S.D.; n = 3). *** = p < 0.001 (Student's t test).
Modulation of Cdk1/Bcl-2 Signaling Promotes a Switch in Cell Fate
Inducing Arrest in DLD-1 Cells
The results presented above show that mitotic death is strongly associated with robust Cdk1 activity and extensive Mcl-1 and Bcl-xL phosphorylation, and conversely, that mitotic slippage is associated with relatively low Cdk1 activity and incomplete phosphorylation of Mcl-1 and Bcl-xL. To strengthen the evidence supporting this hypothesis, we sought approaches to modulate Cdk1/Bcl-2 signaling oppositely in the two cell lines to determine whether a corresponding switch in fate occurred. First, we investigated ways to promote persistent Cdk1 activation via sustained mitotic arrest in DLD-1 cells. It has been reported that knockdown of the anaphase-promoting complex/cyclosome activator Cdc20 induces mitotic arrest, even in cell lines that characteristically fail to arrest in response to conventional mitotic inhibitors (17). To test this approach, knockdown of Cdc20 using siRNA transfection was performed in both HT29 and DLD-1 cells. In HT29 cells, Cdc20 knockdown caused significant cell rounding and loss of adherence, and nonadherent and adherent cells were collected and examined separately. Cdc20 protein expression in the nonadherent population was below the level of detection after 48 h of transfection and caused profound mitotic arrest and cell death, as indicated by strong MPM-2 immunoreactivity, elevated cyclin B1, phosphorylation and highly diminished expression of Mcl-1, complete Bcl-xL phosphorylation, and extensive PARP cleavage (Fig. 5, left). Notably, all these events were far less pronounced or absent in the adherent HT29 population, which retained detectable Cdc20 expression, further reinforcing Mcl-1/Bcl-xL phosphorylation as a key feature of mitotic arrest-induced death, independent of the stimulus. In contrast, even after 72 h of transfection with Cdc20 siRNA and despite efficient Cdc20 knockdown, DLD-1 cells remained largely adherent, there was no change in MPM-2 immunoreactivity, cyclin B1, PARP, Mcl-1, or Bcl-xL (Fig. 5, right), and cell viability was unaffected (data not shown). These results indicated that DLD-1 cells are markedly slippage-prone and failed to arrest in response to either Cdc20 knockdown or Taxol. Therefore, we sought to test whether a combination of the two would promote mitotic arrest and the signaling events characteristic of mitotic death. Cells were transfected with Cdc20 siRNA for 24 h and then treated with either 0.1 μm or 1 μm Taxol for a further 24 h. Immunoblot analysis (Fig. 6A) showed little change in the examined proteins with Cdc20 knockdown alone (lane 2) or with 0.1 μm Taxol alone (lane 3) and modest changes with the combination of these (lane 4) or with 1 μm Taxol (lane 5). However, the combination of 1 μm Taxol plus knockdown of Cdc20 (lane 6) gave a much stronger response, with increased MPM-2 staining and cyclin B1 expression, degradation of Mcl-1, robust phosphorylation of Bcl-xL, and a greater degree of PARP cleavage. Indeed, this signature was comparable with that found in mitotically arrested HT29 cells (Figs. 2 and 5). Fate profiles were next conducted, and a greater percentage of DLD-1 cells died in mitosis in response to the combined treatment versus Taxol alone (Fig. 6, B and C). Thus, by combining treatments that alone were ineffective, mitotic arrest could be attained in slippage-prone DLD-1 cells, with characteristic degradation of Mcl-1 and robust Bcl-xL phosphorylation, associated with a switch to predominately mitotic death.
FIGURE 5.
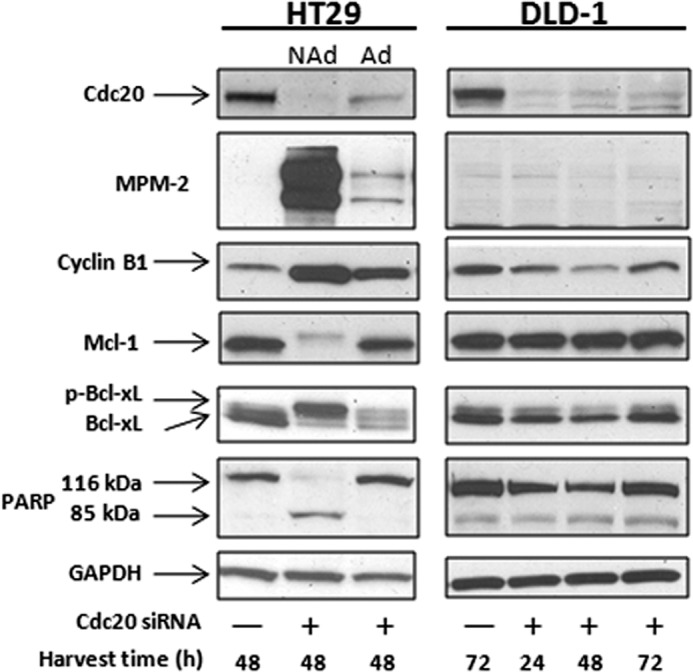
Knockdown of Cdc20 induces mitotic death in HT29 cells but not in slippage-prone DLD-1 cells. HT29 or DLD-1 cells were transfected with 100 nm control siRNA (−) or Cdc20 siRNA (+), and cell extracts were prepared at the times indicated after transfection and subjected to immunoblot analysis. Nonadherent (NAd) HT29 cells were collected by shake-off and pooled separately from the remaining adherent (Ad) cells.
FIGURE 6.

Combining Taxol and Cdc20 knockdown blocks slippage and induces mitotic death in DLD-1 cells. A, DLD-1 cells were transfected with 100 nm control siRNA (−) or Cdc20 siRNA (+) for 24 h and then treated with either 0.1 μm or 1 μm Taxol for an additional 24 h, and cell extracts were prepared and subjected to immunoblot analysis. B, DLD-1 cells were transfected with Cdc20 siRNA for 24 h and subsequently treated with 1 μm Taxol for an additional 48 h, during which time-lapse microscopy was performed. The fate profiles of 50 cells are shown in the right panel and compared with DLD-1 cells treated with 1 μm Taxol alone and monitored for 72 h (left panel). Time 0 represents the time of the addition of Taxol. C, tabulation of data from panel B.
Cyclin B1 overexpression was utilized as an additional strategy to promote sustained mitotic arrest in DLD-1 cells. For this purpose, we used cells transfected with GFP-tagged cyclin B1 under the control of a tetracycline-inducible promoter; these cells also stably expressed DsRed histone H2B to allow cell fate to be monitored by changes in nuclear morphology (6). After the addition of tetracycline, GFP-cyclin B1 expression was induced by 7 h and was sustained for up to 36 h (data not shown). When DLD-1 cells were treated with tetracycline in the absence of Taxol, GFP-cyclin B1 expression had little effect on the signals monitored; Bcl-xL remained unphosphorylated, Mcl-1 levels were maintained, and PARP was uncleaved, and further, MPM-2 immunoreactivity remained undetectable (Fig. 7A, lanes 1 and 2). Thus, a modest increase in cyclin B1 expression was insufficient to cause mitotic death in these cells, consistent with their tendency to undergo mitotic slippage, as also indicated by Cdc20 knockdown (Fig. 5). We therefore treated DLD-1 cells with Taxol in the absence or presence of tetracycline. As shown in Fig. 7A (lane 3), Taxol alone induced partial Bcl-xL phosphorylation and degradation of Mcl-1. Expression of GFP-cyclin B1 clearly enhanced the effects of Taxol on phosphorylation of Bcl-xL and MPM-2 immunoreactivity (lane 4). Fate profiles were next conducted, and expression of GFP-cyclin B1 greatly increased the percentage of Taxol-treated cells dying in mitosis, from 50 to 86%, largely at the expense of cells that died in interphase or survived (Fig. 7, B and C). These results further strengthen the conclusion that Cdk1/cyclin B1 drives mitotic death in concert with increased Bcl-xL phosphorylation and degradation of Mcl-1.
FIGURE 7.

Combining Taxol and cyclin B1 overexpression blocks slippage and induces mitotic death in DLD-1 cells. A, DLD-1 cells stably expressing DsRed histone H2B and GFP-cyclin B1, the latter under tetracycline control, were synchronized and treated with 1 μm Taxol either alone or in combination with tetracycline (1 μg/μl) for 24 h, and cell extracts were prepared and subjected to immunoblot analysis. GFP-cyclin B1 (80 kDa) was detected using an antibody against cyclin B1. B, DLD-1 cells were synchronized and released into medium containing 1 μg/μl tetracycline to induce GFP-cyclin B1 expression. 1 μm Taxol was added 4.5 h after release, and cells were monitored for 48 h using time-lapse microscopy. The fate profiles of 50 cells are shown in the right panel and compared with DLD-1 cells treated with 1 μm Taxol alone (left panel). Time 0 represents the time of the addition of Taxol. C, tabulation of data from panel B.
Inducing Slippage in HT29 Cells
As a complement to the studies described above, we sought next to block Cdk1 in mitotically arrested HT29 cells and examine the effects on Bcl-2 proteins, cell survival, and cell fate. Two Cdk inhibitors, RO3306 (18) and aminopurvalanol A (19), were employed. Cells were synchronized and treated with 0.1 μm Taxol, and 8 h later, 10 μm RO3306 or 10 μm aminopurvalanol A was added for an additional 16 h. This strategy allowed cells to progress to M phase in the absence of Cdk inhibition, whereas effectively inhibiting Cdk1/cyclin B1 during mitotic arrest. Immunoblot analysis indicated that the inhibitors caused mitotic slippage in Taxol-treated cells, with partial reversal in MPM-2 staining, together with partial protection of Mcl-1 degradation and partial reversal of Bcl-xL phosphorylation (Fig. 8A). The inhibitors had no effect in the absence of Taxol under these conditions (Fig. 8A). Furthermore, the Cdk inhibitors protected cells from Taxol-induced death at 24 h, as indicated both by reduced PARP cleavage (Fig. 8A) and by highly significantly reduced annexin V staining, which is an appropriate quantitative method for the early stages of apoptosis (Fig. 8B). Fate profiles were then conducted, with cells treated with Taxol followed by 10 μm RO3306 and tracked for 48 h (Fig. 8C). The Cdk inhibitor caused a marked change from predominately mitotic death with Taxol alone, to a combination of mitotic and interphase death, with data summarized in Fig. 8D. The data of Fig. 8, C and D, also indicate a greater degree of cell survival with co-treatment, consistent with reduced annexin V staining shown in Fig. 8B. Thus, inhibiting Cdk1 during mitotic arrest in HT29 cells partially reverses Cdk1-mediated the effects on Mcl-1 and Bcl-xL in concert with decreased mitotic death and increased survival.
FIGURE 8.

Inhibition of Cdk1 during mitotic arrest in HT29 cells reverses effects on Mcl-1 and Bcl-xL in concert with decreased mitotic death. A, HT29 cells were synchronized by single thymidine block for 16 h, released for 4.5 h, untreated or treated with 0.1 μm Taxol (Tax) for 8 h as indicated, and then co-treated with vehicle or 10 μm RO3306 (RO) or 10 μm aminopurvalanol A (PA) for an additional 16 h, as indicated. Cells were harvested and subjected to immunoblotting with MPM-2 antibody or for the proteins indicated. B, HT29 cells were synchronized, untreated or treated with 0.1 μm Taxol for 8 h, and then co-treated with vehicle or 10 μm RO3306 or 10 μm aminopurvalanol A for an additional 16 h (24 h after Taxol), and cell death was analyzed by phycoerythrin-annexin V staining, as described under “Experimental Procedures.” *** = p < 0.001 (Student's t test). C, HT29 cells were synchronized, treated with 0.1 μm Taxol for 8 h, then co-treated with 10 μm RO3306 for an additional 40 h, and monitored by time-lapse microscopy. Fate profiles of 50 cells are shown on the right and compared with fate profiles for synchronized HT29 cells treated with 0.1 μm Taxol 4.5 h after release and monitored for 72 h (Fig. 1A). D, graphical representation of data from panel C, showing proportions of cells undergoing different fates in response to Taxol alone versus Taxol plus RO3306 co-treatment, which was highly significantly different (p < 0.0001, Fisher's exact test).
DISCUSSION
In addition to acting as a master regulator of mitosis, recent evidence has indicated that Cdk1/cyclin B1 plays a key role in regulation of cell death during mitotic arrest and directly phosphorylates several apoptotic regulatory proteins including antiapoptotic Bcl-2 proteins (8–11), caspase-9 (20), caspase-2 (21), and survivin (22). Work from our laboratory has shown that antiapoptotic Bcl-2 proteins (Bcl-2, Bcl-xL and Mcl-1) act as sensors for Cdk1 signal duration, with partial and transient phosphorylation during normal mitosis but sustained phosphorylation during mitotic arrest leading to their functional or actual elimination (8). Indeed, degradation of Mcl-1, initiated via the action of Cdk1/cyclin B1 and several other kinases, is a common response to mitotic arrest (8–11, 23). These findings illustrate Cdk1 proapoptotic signaling and the existence of considerable cross-talk between pathways regulating cyclin B1 degradation and the generation of death signals. Based on these observations, we speculated that fate after mitotic arrest may depend on the robustness of Cdk1 signaling to antiapoptotic Bcl-2 proteins and sought here to test this hypothesis. Although the functions of many proteins can be explored by knockdown approaches, such a strategy is not feasible for Cdk1 because of its essential function in cell cycling and mitotic progression (12, 13, 24, 25).
The results presented here show that mitotic death is closely associated with high Cdk1 activity and extensive Mcl-1 phosphorylation/degradation and Bcl-xL phosphorylation, whereas slippage is closely associated with lower Cdk1 activity and partial and transient Mcl-1/Bcl-xL phosphorylation. Enforcing mitotic arrest in slippage-prone DLD-1 cells led to considerably more mitotic death in association with robust Bcl-xL phosphorylation and degradation of Mcl-1, whereas inhibiting Cdk1 during mitotic arrest in slippage-resistant HT29 cells led to reduced Bcl-xL phosphorylation, protection of Mcl-1 expression, increased slippage, and decreased mitotic death. The ability to modulate fate by manipulating Cdk1 signaling indicates that the relationship is not merely correlative but that a mechanistic link exists. As emphasized in several recent reviews (3–5), one of the major gaps in our current knowledge concerns the nature of the death signals that accumulate during mitotic arrest. Under each experimental condition examined, death in mitosis was always associated with complete Bcl-xL phosphorylation and Mcl-1 degradation, and mitotic slippage was always associated with incomplete Bcl-xL phosphorylation and maintenance of Mcl-1 expression. We propose that loss of antiapoptotic Bcl-2 protein function is the major driver of mitotic death and that phosphorylation and degradation of Mcl-1 and phosphorylation of Bcl-xL (and Bcl-2 when present) represent key elements of a mitotic death signature. KB3 cells also die predominately in mitosis when exposed to microtubule inhibitors and exhibit a highly similar signature (supplemental Fig. S2) (8, 11, 15), strengthening the link and indicating that Bcl-xL phosphorylation and degradation of Mcl-1 are key events in a conserved mitotic death pathway. Thus, our findings represent an important advance in our understanding of mitotic death regulation. Additional studies in our laboratory have indicated that knockdown of Mcl-1 or Bcl-xL individually does not affect the viability of HT29 or DLD-1 cell lines (data not shown), but simultaneous knockdown induces cell death, suggesting that each can compensate for loss of the other. Thus, functional elimination of both proteins may be important for mitotic death in this system. In addition to playing a proapoptotic role through Bcl-2 protein phosphorylation, Cdk1/cyclin B1 also plays a protective role through phosphorylation and inactivation of caspase-9 (20, 26) and caspase-2 (21) and stabilization of survivin (22). Understanding how the proapoptotic and prosurvival functions of sustained Cdk1 signaling are coupled and integrated is an important challenge for future studies of fate control after mitotic arrest.
Studies examining fate after mitotic arrest using single cell analysis have indicated that the pathways regulating cyclin B1 destruction and caspase activation are kinetically and mechanistically independent (6, 7). Accelerating or inhibiting one pathway failed to influence the kinetics of the other pathway, and thus, evidence for kinetic independence appears firm. However, based on the observations made here and elsewhere, we would argue that the pathways are in fact mechanistically linked and interdependent, with significant cross-talk evident. Indeed, our findings suggest that sustained expression of cyclin B1 maintains Cdk1 in an active state, and this has a dual role, maintaining mitotic arrest on the one hand and inducing apoptosis on the other. Conversely, low or declining cyclin B1 expression causes Cdk1 inactivation, which has a dual and reciprocal consequence, mitotic slippage on the one hand and protection from apoptosis on the other. Thus, persistent cyclin B1 expression drives both mitotic arrest and mitotic death, as illustrated in the model shown in Fig. 9. Because sustained mitotic arrest is an abnormal state ideally necessitating elimination of the cell, having the same protein kinase maintain arrest and actively promote death appears an efficient solution. In addition, mitotic death occurs in the context of highly condensed chromatin where transcription is repressed and up-regulation of proapoptotic Bcl-2 proteins is not achievable. Functional elimination of pre-existing antiapoptotic Bcl-2 proteins, using the processes of phosphorylation and proteolysis, which are highly active in mitotic cells (16), represents a logical and economical alternate mechanism. The findings presented here represent a conceptual advance in our understanding of the regulation of fate after mitotic arrest and also have clinical implications as the robustness of these signaling events may be useful as a marker to define susceptibility to antimitotic drugs.
FIGURE 9.

Model for a dual role of Cdk1/cyclin B1 in maintaining mitotic arrest and promoting mitotic death.
Acknowledgments
We are very grateful to Stephen Taylor and Karen Gascoigne for providing the HT29 and DLD-1 cell lines and for advice and detailed protocols for analysis of cell fate. We thank Michael Gottesman for providing KB3 and KBV1 cell lines, Richard Kurten for assistance with microscopy, and Horace Spencer for assistance with statistical analysis.
This work was supported, in whole or in part, by National Institutes of Health Grant CA-109821 from the NCI (to T. C. C.). This work was also supported in part by pilot funds from Translational Research Institute Grant UL1TR000039.

This article contains supplemental Figs. S1 and S2.
NCI-60 DTP Human Tumor Cell Line Screen, National Cancer Institute Developmental Therapeutics Program, dtp.nci.nih.gov.
- Cdk1
- cyclin-dependent kinase-1
- PARP
- poly(ADP-ribose) polymerase
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Taylor S. S., Scott M. I., Holland A. J. (2004) The spindle checkpoint: a quality control mechanism which ensures accurate chromosome segregation. Chromosome Res. 12, 599–616 [DOI] [PubMed] [Google Scholar]
- 2. Musacchio A., Salmon E. D. (2007) The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8, 379–393 [DOI] [PubMed] [Google Scholar]
- 3. Rieder C. L., Maiato H. (2004) Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell 7, 637–651 [DOI] [PubMed] [Google Scholar]
- 4. Gascoigne K. E., Taylor S. S. (2009) How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 122, 2579–2585 [DOI] [PubMed] [Google Scholar]
- 5. Manchado E., Guillamot M., Malumbres M. (2012) Killing cells by targeting mitosis. Cell Death Differ. 19, 369–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gascoigne K. E., Taylor S. S. (2008) Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14, 111–122 [DOI] [PubMed] [Google Scholar]
- 7. Huang H. C., Mitchison T. J., Shi J. (2010) Stochastic competition between mechanistically independent slippage and death pathways determines cell fate during mitotic arrest. PLoS One 5, e15724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Terrano D. T., Upreti M., Chambers T. C. (2010) Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol. Cell. Biol. 30, 640–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harley M. E., Allan L. A., Sanderson H. S., Clarke P. R. (2010) Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 29, 2407–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wertz I. E., Kusam S., Lam C., Okamoto T., Sandoval W., Anderson D. J., Helgason E., Ernst J. A., Eby M., Liu J., Belmont L. D., Kaminker J. S., O'Rourke K. M., Pujara K., Kohli P. B., Johnson A. R., Chiu M. L., Lill J. R., Jackson P. K., Fairbrother W. J., Seshagiri S., Ludlam M. J., Leong K. G., Dueber E. C., Maecker H., Huang D. C., Dixit V. M. (2011) Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471, 110–114 [DOI] [PubMed] [Google Scholar]
- 11. Chu R., Terrano D. T., Chambers T. C. (2012) Cdk1/cyclin B plays a key role in mitotic arrest-induced apoptosis by phosphorylation of Mcl-1, promoting its degradation and freeing Bak from sequestration. Biochem. Pharmacol. 83, 199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lindqvist A., Rodríguez-Bravo V., Medema R. H. (2009) The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J. Cell Biol. 185, 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wolf F., Sigl R., Geley S. (2007)' … The end of the beginning': cdk1 thresholds and exit from mitosis. Cell Cycle 6, 1408–1411 [PubMed] [Google Scholar]
- 14. Shen D. W., Cardarelli C., Hwang J., Cornwell M., Richert N., Ishii S., Pastan I., Gottesman M. M. (1986) Multiple drug-resistant human KB carcinoma cells independently selected for high-level resistance to colchicine, adriamycin, or vinblastine show changes in expression of specific proteins. J. Biol. Chem. 261, 7762–7770 [PubMed] [Google Scholar]
- 15. Upreti M., Galitovskaya E. N., Chu R., Tackett A. J., Terrano D. T., Granell S., Chambers T. C. (2008) Identification of the major phosphorylation site in Bcl-xL induced by microtubule inhibitors and analysis of its functional significance. J. Biol. Chem. 283, 35517–35525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sullivan M., Morgan D. O. (2007) Finishing mitosis, one step at a time. Nat. Rev. Mol. Cell Biol. 8, 894–903 [DOI] [PubMed] [Google Scholar]
- 17. Huang H. C., Shi J., Orth J. D., Mitchison T. J. (2009) Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell 16, 347–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vassilev L. T., Tovar C., Chen S., Knezevic D., Zhao X., Sun H., Heimbrook D. C., Chen L. (2006) Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. U.S.A. 103, 10660–10665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rosania G. R., Merlie J., Jr., Gray N., Chang Y. T., Schultz P. G., Heald R. (1999) A cyclin-dependent kinase inhibitor inducing cancer cell differentiation: biochemical identification using Xenopus egg extracts. Proc. Natl. Acad. Sci. U.S.A. 96, 4797–4802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Allan L. A., Clarke P. R. (2007) Phosphorylation of caspase-9 by CDK1/cyclin B1 protects mitotic cells against apoptosis. Mol. Cell 26, 301–310 [DOI] [PubMed] [Google Scholar]
- 21. Andersen J. L., Johnson C. E., Freel C. D., Parrish A. B., Day J. L., Buchakjian M. R., Nutt L. K., Thompson J. W., Moseley M. A., Kornbluth S. (2009) Restraint of apoptosis during mitosis through interdomain phosphorylation of caspase-2. EMBO J. 28, 3216–3227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Connor D. S., Wall N. R., Porter A. C., Altieri D. C. (2002) A p34cdc2 survival checkpoint in cancer. Cancer Cell 2, 43–54 [DOI] [PubMed] [Google Scholar]
- 23. Tunquist B. J., Woessner R. D., Walker D. H. (2010) Mcl-1 stability determines mitotic cell fate of human multiple myeloma tumor cells treated with kinesin spindle protein inhibitor ARRY-520. Mol. Cancer Ther. 9, 2046–2056 [DOI] [PubMed] [Google Scholar]
- 24. Santamaría D., Barrière C., Cerqueira A., Hunt S., Tardy C., Newton K., Cáceres J. F., Dubus P., Malumbres M., Barbacid M. (2007) Cdk1 is sufficient to drive mammalian cell cycle. Nature 448, 811–815 [DOI] [PubMed] [Google Scholar]
- 25. Malumbres M., Barbacid M. (2009) CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 [DOI] [PubMed] [Google Scholar]
- 26. Clarke P. R., Allan L. A. (2010) Destruction's our delight: controlling apoptosis during mitotic arrest. Cell Cycle 9, 4035–4036 [DOI] [PubMed] [Google Scholar]