

Abstract
The causative agent of leishmaniasis is the protozoan parasite Leishmania major. Part of the host protective mechanism is the production of reactive oxygen species including hydrogen peroxide. In response, L. major produces a peroxidase, L. major peroxidase (LmP), that helps to protect the parasite from oxidative stress. LmP is a heme peroxidase that catalyzes the peroxidation of mitochondrial cytochrome c. We have determined the crystal structure of LmP in a complex with its substrate, L. major cytochrome c (LmCytc) to 1.84 Å, and compared the structure to its close homolog, the yeast cytochrome c peroxidase–cytochrome c complex. The binding interface between LmP and LmCytc has one strong and one weak ionic interaction that the yeast system lacks. The differences between the steady-state kinetics correlate well with the Lm redox pair being more dependent on ionic interactions, whereas the yeast redox pair depends more on nonpolar interactions. Mutagenesis studies confirm that the ion pairs at the intermolecular interface are important to both kcat and KM. Despite these differences, the electron transfer path, with respect to the distance between hemes, along the polypeptide chain is exactly the same in both redox systems. A potentially important difference, however, is the side chains involved. LmP has more polar groups (Asp and His) along the pathway compared with the nonpolar groups (Leu and Ala) in the yeast system, and as a result, the electrostatic environment along the presumed electron transfer path is substantially different.
Certain Leishmania species are parasitic protozoa that cause the tropical disease visceral and cutaneous leishmaniasis. Upon infection, Leishmania survive the oxidative environment of host macrophages by scavenging toxic oxidative species, which include H2O2 (1). Leishmania major produces a mitochondrial peroxidase [L. major peroxidase (LmP)] (1), and studies with knockout mutants illustrate that LmP is a key player in protecting the parasite from macrophage-generated peroxide (2). LmP is very similar to the well-studied yeast cytochrome c peroxidase (CCP) (1). While CCP has served as a paradigm for enzyme structure and function studies for many years, there are very few studies on its biological function (3). Owing to the importance of L. major as a disease-causing parasite, there has been a greater focus on its biological function and role in pathogenesis (2). Thus, LmP offers the opportunity to better bridge the relationship between structure and biological function. We therefore have undertaken structure function studies on LmP, which include the recent structure determination of both LmP (4) and its substrate, L. major cytochrome c (LmCytc), which established LmP as a cytochrome c peroxidase (5).
Of particular interest is the complex formed between LmP and LmCytc because interprotein biological electron transfer (ET) is a challenging and complex problem. To ensure specificity, the binding surfaces of redox partners must complement each other electrostatically and/or by nonpolar interactions. Specificity, however, is not the only problem in interprotein ET because there must also be a balance between on and off rates to maintain rapid and efficient ET. If the electron donor binds too tightly, then dissociation of the donor after ET will be too slow to maintain high turnover rates. Such opposing requirements of specificity and relatively fast dissociation present challenges in trapping redox complexes to study the ET event without the complication of on/off rates. Another problem is defining the path of electron transfer. Extensive work with ruthenium-modified redox proteins indicates that ET proteins are wired for specific and efficient ET through σ and hydrogen bonds (6). Although precisely defining such pathways in single proteins covalently modified with a redox active ruthenium is fairly straightforward, defining natural pathways in protein complexes is far more difficult. Unlike specific and tight protein–protein complexes, redox pairs can be viewed as complementary surfaces where a nonproductive complex is nearly isoenergetic with an ET active complex (7), so trapping the lowest energy active complex can be challenging. These are the underlying reasons why there are so few crystal structures of redox complexes. Our search of the Protein Data Bank (PDB) gives nine unique crystal structures of noncovalent endogenous binary redox complexes: 2PCC (8), 2ZON (9), 2IAA (9), 1T9G (10), 2V3B (11), 2DE5 (12), 2H3X (13), 1L9B (14), 1KYO (15), and 2YVJ (16). Of these, one of the most extensively studied redox complexes is between yeast CCP and cytochrome c (Cytc) (17), which includes the crystal of the noncovalent (8) and a disulfide engineered covalent complex (18). CCP is a traditional heme peroxidase and reacts with H2O2 to form compound I. The heme iron and an active site Trp residue, Trp191 (19), are oxidized by H2O2 to give compound I, Fe(IV)=O Trp.+. Then in two successive one-electron transfer steps, both the iron and Trp.+ are reduced back to the resting state. LmP is very similar to CCP, and recently we showed that the biological function of LmP is to oxidize cytochrome c (5). Moreover, like yeast CCP, LmP has a redox active Trp (4). Using recombinant LmP and LmCytc, we found that the steady-state kinetics of the Lm system is far simpler than the yeast system (5). First, the Lm system obeys straightforward Michaelis–Menten kinetics, whereas yeast CCP does not (20) possibly because yeast CCP has a second weak site for Cytc (21, 22). Second, LmP exhibits a steady decrease in activity with increasing ionic strength (5), whereas yeast CCP activity increases with ionic strength up to ∼100 mM and then decreases (20). These clear contrasts suggest that the structure of LmP–LmCytc complex might be substantially different from its yeast counterpart. To understand the structural basis for these differences, we have solved the 1.84-Å structure of the LmP–LmCytc noncovalent complex.
Results and Discussion
Crystal Structure of the LmP–LmCytc Complex.
The crystal structures of each protein in the complex resembles the individual protein structures with the only significant difference being in the N terminus of LmP, which is disordered in the complex. The final refined model exhibits excellent stereochemical parameters with top scores in MolProbity (23). The average thermal displacement (B) factors for LmCytc are significantly higher than LmP: 66.9 versus 55.3, indicating higher relative mobility for LmCytc.
Based on our previous experience with yeast CCP (24), it is likely that LmP–LmCytc complex is a mix of redox states. Given that Cytc exhibits a higher redox potential, the LmCytc heme iron should be more susceptible to photoreduction, although the reported differences between oxidized and reduced Cytc (25) are subtle so we cannot conclude which redox state dominates based on such parameters as ligand–iron distances. In the complex, the presence of a water 2.2 Å above the peroxidase heme iron is consistent with predominantly high-spin ferric heme because ferrous heme should have no water close to the iron.
As expected, the complementary electrostatic surfaces that interact in the yeast and Lm ET complexes are the same (Fig. 1). However, the details at the interface and the orientation of Cytc relative to the peroxidase are substantially different. In the following comparisons, the heme groups of LmP and CCP were aligned, which provides a comparison on how each Cytc is positioned relative to its peroxidase binding partner. Relative to yeast Cytc, the LmCytc heme translates ∼4 Å and rotates ∼40° (Fig. 1D), and as such the specific interactions at the interface are substantially different. First, the Lm complex has about 1,179 Å2 of surface area buried, whereas 547 Å2 is buried in the yeast complex. As a result, the Lm complex is tighter with less solvent at the interface. Second, the Lm system has direct ion pairing at the interface, whereas the yeast complex has no direct contacts (8, 18). The key interactions are between Arg24 in LmCytc and Asp211 in LmP and Lys98 in LmCytC and Glu49 in LmP (Figs. 1 and 2). The basic residues are conserved in Cytc so the differences in the complexes result from differences in the peroxidases. The residues corresponding to Asp211 and Asp49 in LmP are Ala and Leu in yeast CCP, respectively. This suggests that the driving force in forming the complexes might be quite different with the burial of nonpolar groups being more important in the yeast complex whereas ionic interactions are more important in the Lm system. These differences are reflected in the steady-state kinetics. LmP exhibits simple Michaelis–Menten kinetics (5), whereas yeast CCP does not. In addition, the effect of ionic strength is straightforward with LmP: the activity steadily decreases with increasing ionic strength. However, with yeast CCP, there is first an increase in activity with increasing ionic strength up to ∼100 mM and then the activity decreases (20). Such behavior is a hallmark for nonpolar interactions helping to drive complex formation. Thus, the differences in steady-state kinetics are readily understood based on the differences at the interface.
Fig. 1.

Comparison of the LmP–LmCytc and CCP–Cytc complexes. In A–C, the peroxidases are cyan and the Cytcs are yellow. The hemes of each peroxidase were superimposed, so in D the orientation of the two Cytcs is relative to the superimposed peroxidases. In D, LmCytc is magenta and yeast Cytc is green.
Fig. 2.

The 2Fo − Fc electron density map contoured at 1.5σ showing the key ion pair between Arg24 in Cytc and Asp211 in LmP. The position of Arg24 in the free Cytc molecule is in cyan. The small red spheres represent ordered water molecules.
The main reason why the LmP–LmCytc interface is more extensive is due to fewer bulky, less conformationally restricted groups at the interface. For example, Val197 in yeast CCP forms a tight nonpolar contact with Ala81 in Cytc. Val197 is a Gly214 in LmP (Fig. 1C). In addition Gln120 in yeast CCP, which is Gly in LmP, protrudes toward Cytc. The smaller side chains in LmP thus allow for a firmer packing at the interface. Furthermore, the LmCytc Arg24 undergoes a conformational change from the unbound to the bound structure facilitating this tight interaction, as illustrated in Fig. 2. Such tight packing from the surrounding residues in concert with this conformational change enables the Arg24–Asp211 ion pair to be shielded from bulk solvent, which undoubtedly makes this a fairly strong electrostatic interaction. In sharp contrast, the yeast system relies primarily on nonbonded contacts.
Despite these differences, the electron transfer path and distances are the same. To fully reduce compound I, there are two one-electron transfer events. The generally accepted mechanism for CCP is shown in the following scheme (26):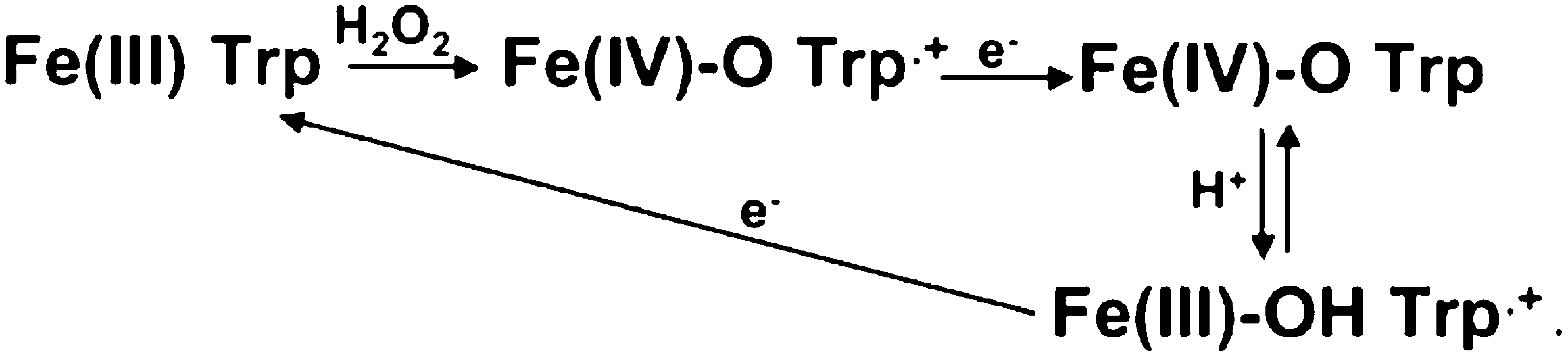
The first electron from Cytc reduces the Trp191.+ cationic radical. This is followed by an internal electron transfer event in which Trp191 reduces Fe(IV)=O to give Fe(III)-OH Trp.+. Therefore, there is only one functional docking site for both electron transfer events. In the CCP complex, the shortest through-space jump from the Cytc heme is about 4.3 Å to Ala193 (Fig. 3), and from there it is a short σ bond route to Trp191. In the Lm complex, it is the same 4.3 Å through-space jump to His210 in LmP, which corresponds to Ala193 in CCP, and again it is a short σ bond distance to the redox active Trp208 in LmP. Thus, despite the differences at the interface and orientation of the heme, the electron transfer distances and path are the same. What is substantially different is the type of amino acids that lie along the electron transfer route (Fig. 3). The route to the redox active Trp191 in CCP is composed of Ala194, Ala193, and Gly192, whereas in LmP the corresponding residues are Asp211, His210, and Thr209. Asp211 is especially interesting because in LmP Asp211 forms the key ionic interaction at the interface with the side chain of Cytc Arg24. Cβ of Asp211 also is only 4.1 Å from the LmCytc heme. In addition, the carbonyl backbone of Asp211 forms one of the K+ ligands (Fig. 1) in LmP, and this K+ is absent in yeast CCP. Therefore, the electrostatic environment right at the point where one expects the Cytc electron to exit is quite different in the two complexes. Whether or not this is important in controlling ET rates is not known, although the steady-state activities of both systems are about the same (4, 5).
Fig. 3.

Comparison of the electron transfer path between the LmP and yeast CCP complexes.
Steady-State Activity Assays.
Since there are two salt bridges between LmCytc and LmP, Arg24–Asp211 and Lys98–Glu49, we wanted to probe the importance of each interaction. We mutated LmCytc and not LmP because the backbone carbonyl of Asp211 is a ligand to the K+ ion and disrupting the K+ site might complicate interpretation of the kinetic data due to potential perturbation of the electrostatic environment, which is known to influence the stability of the Trp radical and hence enzyme activity (27, 28). Fig. 4 provides the enzymatic rate data, whereas Table 1 provides the kinetic information as determined from the equations obtained from the best fit line of the Lineweaver–Burk reciprocal plots (Fig. S1).
Fig. 4.

Michaelis–Menten plots of LmP activity using wild-type and mutant Cytcs as the substrate.
Table 1.
Kinetic information from Lineweaver–Burk plots (Fig. S1) of LmCytc and mutants
| Lineweaver–Burk | |||
| Substrate | KM, µM | kcat, s−1 | kcat/KM, M−1⋅s−1 |
| LmCytc | 8 ± 2 | 1,700 ± 200 | 2 × 108 |
| Δ10LmCytc | 10 ± 1 | 1,900 ± 10 | 2 × 108 |
| K98ALmCytc | 40 ± 11 | 1,100 ± 300 | 3 × 107 |
| R24ALmCytc | 20 ± 10 | 60 ± 20 | 3 × 106 |
| R24A/K98ALmCytc | ND | 400 ± 100* | ND |
*Rate, not kcat because saturation not reached, determined at 75 µM.
Δ10LmCytc (the LmCytc used for crystallization and missing the first 10 residues) exhibits almost identical Michaelis–Menten behavior to the wild-type LmCytc so the use of Δ10LmCytc for the cocrystal structure does not compromise the functional relevance of the crystal structure. Both single mutants, K98A and R24A, exhibit decreased kcat and increased KM. K98A retains about ∼74% of wild-type activity, so clearly this interaction is not of critical importance. Note that this interaction is near the edge of the complex exposed to solvent (Fig. 1), so this is unlikely to be a very strong interaction. R24A, however, exhibits significantly decreased activity at ∼5% of wild type and two orders of magnitude decreased efficiency (kcat/KM) from the wild type (Fig. 4). Although this ion pair is right at the interface, it is somewhat surprising that a single mutant has such a large effect because this interaction represents only a small part of the total buried surface area. For example, six different yeast CCP mutants at the interface exhibit between 40–105% of the wild-type control (29) and some of these mutants introduce substantial bulk at the interface.
The double mutant, R24A/K98A, does not follow Michaelis–Menten kinetics and cannot be saturated with increasing LmCytc concentration, so it is not possible to directly compare kcat (Fig. 4). At low LmCytc concentrations, the activity of the double mutant is similar to R24A, but the rate does continue to increase with increasing LmCytc (Fig. 4). Because this mutant binds so poorly, it is likely to exhibit a fast off-rate, which translates into faster turnover at very high concentrations of LmCytc. This same trend was observed in yeast CCP, in which some mutants at the interface actually increased kcat by increasing the off-rate (30).
We further probed the potential intermolecular ion pairs using the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) computational method for estimating the free energy of binding. Because this approach takes into account solvation energies, it should be particularly sensitive to estimating the differences in ionic interactions in wild type and mutants. We carried out the calculation for the wild-type complex and the R24A and K98A mutants, which were generated in silico by simply replacing the Arg or Lys residue with Ala. We compared the calculated free energies with the experimental free energies by assuming that KM is a true measure of KD and then converted KD to free energies followed by plotting the calculated values vs. the experimental values (Fig. S2). The double mutant, R24A/K98A, was not included in these calculations because KM could not be determined. The agreement in Fig. S2 is quite good, which suggests that the KM is indeed a good approximation of KD and that the mutations have a straightforward effect on binding. The larger effect of the R24A mutant is very likely because the Arg24–Asp211 ion pair is buried at the interface and sequestered from bulk solvent whereas the Lys98–Glu49 pair is solvent exposed.
If binding were the only effect of the R24A mutant, then we might expect kcat to be similar to wild type, but kcat is only 5% of wild type. The R24A mutant could have multiple effects owing to its close proximity to the heme. Arg24 also forms an intramolecular ion pair with Glu201. Disruption of this ion pair in R24A will provide greater exposure of the heme, thereby increasing the negative charge near the heme. This should decrease the redox potential, which also should decrease the ET driving force and ET rate. However, the homologous Arg in yeast Cytc, Arg13, has been converted to Ile and the ET rate measured by flash photolysis methods in the CCP–Cytc complex increases (31). Moreover, the actual rate-limiting step is very likely associated with on/off-rates of the complex and not the electron transfer event, which, by analogy with the yeast system, should be very fast (26). By removing a key ion pair at the interface, the off-rate of R24A should be faster than wild type, so kcat might actually increase or at least not be as low as 5% of wild type. This must mean that rate-limiting state is tied to the on-rate. To better understand the effect of these mutations, we refer to the following scheme taken after Selzer and Schreiber (32).
In this mechanism, the two partners form the initial encounter complex at a near diffusion controlled rate owing to the complementary electrostatic surfaces. The probability of the initial encounter complex being the lowest energy complex required for electron transfer is low, so after the initial encounter LmCytc binds and then samples the surface of LmP via a “bind and crawl” process until it ultimately reaches the lowest energy productive orientation. It is this process, k2 in the above scheme, that is most likely limiting and ionic strength dependent.
Conclusions
The crystal structure of the LmP–LmCytc complex adds to a short list of redox protein complexes with known 3D structure. The LmP–LmCytc complex is particularly interesting owing to its close similarity to the yeast CCP–Cytc complex and illustrates what structural features must be evolutionarily conserved and which can diverge. Although the docking surfaces are similar in terms of electrostatic complementarity, the precise orientation of Cytc and the structural features at the interface are very different. Unique to LmP–LmCytc is a central ion pair sequestered from solvent that our mutagenesis studies show is quite important for enzyme activity. In sharp contrast, the yeast system has no strong ion pairs and relies primarily on nonbonded contacts. These differences are due primarily to amino acid differences in the peroxidases and not the Cytcs, so it appears that evolutionary divergence on the peroxidase docking site can be tolerated. Even so, the Cytc heme edge approaches the peroxidase exactly the same in both complexes such that the polypeptide route of electron transfer is the same, which argues for an evolutionarily conserved ET pathway. Although the ET distance and polypeptide path are similar, the types of amino acid side chains and local electrostatic environment sharply contrast with the LmP–LmCytc systems having a much more polar environment. It remains to be seen whether the local electrostatic properties along the σ-bonded ET route is relevant to the actual ET event.
Materials and Methods
Site-Directed Mutagenesis and N-terminal Truncation.
LmCytc has an N-terminal extension compared with other c-type cytochromes such as horse heart cytochrome c. Because we were unable to cocrystallize full-length LmCytc with LmP, we decided to remove 10 residues from the N terminus. Our structure of LmCytc (5) showed that the first 5 residues are not visible in electron density maps and thus are disordered, whereas the next four residues are not involved in regular secondary structure. Therefore, we correctly assumed that the removal of 10 residues should not affect either structure or function. The Δ10LmCytc N-terminal mutant and the R24ALmCytc, K98ALmCytc, and R24A/K98ALmCytc mutants were designed and prepared with the Stratagene Site-Directed Mutagenesis Kit.
Protein Expression and Purification.
Wild-type and mutant LmCytcs were purified as described earlier (5). The main difference is that, for R24A/K98ALmCytc and Δ10LmCytc, the pH for the CM Sepharose was at 5.8. Because Δ10LmCytc was to be used in crystallography, it was further purified over Superdex-75 in 50 mM potassium phosphate (pH 7.0), 100 mM NaCl, and 5% (vol/vol) glycerol after the CM Sepharose column. Mutants used for kinetics were only purified over CM Sepharose. Resulting Rz (A414/A280) for all oxidized samples was ∼4.2, and each mutant exhibited the expected spectra for oxidized and reduced LmCytc. Sample homogeneity was verified with 12–15% SDS/PAGE. LmP was expressed and purified as previously described (4, 5).
Steady-State Activity Assays.
Spectrophotometric steady-state activity assays were carried out as previously described (5). The concentration of each reduced sample was determined using the molar extinction coefficient, ε558red = 29 mM−1⋅cm−1, and the steady-state oxidation of reduced LmCytc was calculated using the previously determined Δε558 = 19.4 mM−1⋅cm−1 (5). LmP concentration was determined using the Soret molar extinction coefficient, ε408 = 113.6 mM−1⋅cm−1 (4). Hydrogen peroxide was standardized using permaganate.
Cocrystallization.
After extensive screening, the following aerobic procedure resulted in crystals of the complex. First, 340 μM LmP and 640 μM Δ10LmCytc were combined and diluted in 40 mM potassium phosphate, pH 6.5. The sample was then concentrated to the original volume to ensure the same concentration in a 3,000 MWCO Centricon at 4 °C, 3,500 × g. Crystals were obtained with 36% pentaerythritol ethoxylate (15/4 EO/OH), which is typically used as an additive and not as a precipitant, so it was quite surprising to see crystals. Unfortunately, these crystals did not diffract, but SDS/PAGE clearly showed that these crystals contained both proteins, so we were encouraged to continue screening with pentaerythritol ethoxylate. Decent crystals were obtained by setting up a hanging-drop tray with well solution of 32–33% pentaerythritol ethoxylate and 4% acetone. Crystals were cryoprotected by quick exposure (10–30 s) to the following solution: 15% pentaerythritol ethoxylate, 2% acetone, 40% ethylene glycol, 1.5% trehalose, 1.5% sucrose, and 1.25% glucose.
Structural Determination and Refinement.
One LmP:Δ10LmCytc crystal, out of 50, diffracted to 1.84-Å resolution at the Stanford Synchrotron Radiation Lightsource beamline 12.2. The data were collected in shutterless mode with a 0.2° thin sliced oscillation per frame for 140° on a Pilatus 6M detector. A separate crystal was used for the single-wavelength Fe anomalous dispersion (SAD) experiment, and this diffracted to 2.29 Å, with a very high redundancy. The crystals belong to space group P42212 with one LmP and one Δ10LmCytc molecule per asymmetric unit. The structure was solved by combining phase information from molecular replacement and SAD at the Fe absorption edge. Data for each were indexed, integrated using the March 15, 2012, version of XDS (33) with the CC1/2 criteria (34). Reflections were scaled with Scala and converted to structure factors with Truncate (35, 36). The molecular replacement calculations were carried out with Phaser (37) through the CCP4i graphic interface (38) using LmP (PDB ID code 3RIV) and LmCytc (PDB ID code 4DY9) as search models. Phaser was also used for the SAD phasing calculation. The anomalous signals at the metal positions confirmed the Fe and K sites. Lack of a strong anomalous signal and lower 2Fo−Fc peak suggested a magnesium rather than a calcium at the second metal site in LmP. The structure was then refined initially in phenix.refine using experimental phase restraints (39, 40). Later, the structures were refined with BUSTER, version 2.10.0 (41–43), allowing for the inclusion of hydrogen atoms during refinement. The quality of the model continuously improved as checked with MolProbity scores (23). The final model has a perfect MolProbity all-atoms clash score of 0 (100%), and the MolProbity score is 0.72 (100% for structures at similar resolution). All inspections and manual manipulation were completed with COOT (44, 45). Coordinate and structure factor files have been deposited in the Protein Data Bank (PDB ID code 4GED). Crystallographic data collection and refinement statistics are summarized in Table S1.
Computational Methods.
The MM-PBSA method as implemented in Amber 9.0 was used to compute binding free energies (46). In this method, the total free energy of the protein–protein complex or the individual proteins is taken as the sum of the following energy terms
where EMM is the total molecular mechanics energy computed with the Sander module in Amber 9.0, Gsolv is the solvation free energy estimated from the Poisson–Boltzman equation, Gnp is the nonpolar solvation energy estimated from the solvent accessible surface area, and TSsolute is the solute entropy. In this case, we ignore the entropy term because we are comparing mutant and wild-type protein complexes that differ in only a single amino acid, so the entropic differences are negligible especially because the native side chains in the individual proteins are well ordered. The computationally solvated LmP–LmCytc crystal structure was first energy minimized, and then stripped off all solvent and the free energy for the complex, and each of the separated proteins was calculated. The overall free energy of binding is computed from the following equation:
Mutants were generated by simply replacing the residues of interest with Ala and repeating the calculations.
Supplementary Material
Acknowledgments
Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of US Department of Energy, Office of Basic Energy Sciences. This work was supported in part by National Institutes of Health (NIH) Grant GM42614 (to T.L.P.). The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the NIH, National Cancer for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4GED).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1213295109/-/DCSupplemental.
References
- 1.Adak S, Datta AK. Leishmania major encodes an unusual peroxidase that is a close homologue of plant ascorbate peroxidase: A novel role of the transmembrane domain. Biochem J. 2005;390(Pt 2):465–474. doi: 10.1042/BJ20050311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pal S, Dolai S, Yadav RK, Adak S. Ascorbate peroxidase from Leishmania major controls the virulence of infective stage of promastigotes by regulating oxidative stress. PLoS One. 2010;5(6):e11271. doi: 10.1371/journal.pone.0011271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwon M, Chong S, Han S, Kim K. Oxidative stresses elevate the expression of cytochrome c peroxidase in Saccharomyces cerevisiae. Biochim Biophys Acta. 2003;1623(1):1–5. doi: 10.1016/s0304-4165(03)00151-x. [DOI] [PubMed] [Google Scholar]
- 4.Jasion VS, Polanco JA, Meharenna YT, Li H, Poulos TL. Crystal structure of Leishmania major peroxidase and characterization of the compound I tryptophan radical. J Biol Chem. 2011;286(28):24608–24615. doi: 10.1074/jbc.M111.230524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jasion VS, Poulos TL. Leishmania major peroxidase is a cytochrome c peroxidase. Biochemistry. 2012;51(12):2453–2460. doi: 10.1021/bi300169x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gray HB, Winkler JR. Electron flow through metalloproteins. Biochim Biophys Acta. 2010;1797(9):1563–1572. doi: 10.1016/j.bbabio.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Ubbink M. The courtship of proteins: Understanding the encounter complex. FEBS Lett. 2009;583(7):1060–1066. doi: 10.1016/j.febslet.2009.02.046. [DOI] [PubMed] [Google Scholar]
- 8.Pelletier H, Kraut J. Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c. Science. 1992;258(5089):1748–1755. doi: 10.1126/science.1334573. [DOI] [PubMed] [Google Scholar]
- 9.Nojiri M, et al. Structural basis of inter-protein electron transfer for nitrite reduction in denitrification. Nature. 2009;462(7269):117–120. doi: 10.1038/nature08507. [DOI] [PubMed] [Google Scholar]
- 10.Toogood HS, et al. Extensive domain motion and electron transfer in the human electron transferring flavoprotein.medium chain Acyl-CoA dehydrogenase complex. J Biol Chem. 2004;279(31):32904–32912. doi: 10.1074/jbc.M404884200. [DOI] [PubMed] [Google Scholar]
- 11.Hagelueken G, et al. Crystal structure of the electron transfer complex rubredoxin rubredoxin reductase of Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 2007;104(30):12276–12281. doi: 10.1073/pnas.0702919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashikawa Y, et al. Electron transfer complex formation between oxygenase and ferredoxin components in Rieske nonheme iron oxygenase system. Structure. 2006;14(12):1779–1789. doi: 10.1016/j.str.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 13.Sukumar N, et al. Crystal structure of an electron transfer complex between aromatic amine dehydrogenase and azurin from Alcaligenes faecalis. Biochemistry. 2006;45(45):13500–13510. doi: 10.1021/bi0612972. [DOI] [PubMed] [Google Scholar]
- 14.Axelrod HL, et al. X-ray structure determination of the cytochrome c2: Reaction center electron transfer complex from Rhodobacter sphaeroides. J Mol Biol. 2002;319(2):501–515. doi: 10.1016/S0022-2836(02)00168-7. [DOI] [PubMed] [Google Scholar]
- 15.Lange C, Hunte C. Crystal structure of the yeast cytochrome bc1 complex with its bound substrate cytochrome c. Proc Natl Acad Sci USA. 2002;99(5):2800–2805. doi: 10.1073/pnas.052704699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Senda M, et al. Molecular mechanism of the redox-dependent interaction between NADH-dependent ferredoxin reductase and Rieske-type [2Fe-2S] ferredoxin. J Mol Biol. 2007;373(2):382–400. doi: 10.1016/j.jmb.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Volkov AN, Nicholls P, Worrall JA. The complex of cytochrome c and cytochrome c peroxidase: The end of the road? Biochim Biophys Acta. 2011;1807(11):1482–1503. doi: 10.1016/j.bbabio.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Guo M, Bhaskar B, Li H, Barrows TP, Poulos TL. Crystal structure and characterization of a cytochrome c peroxidase-cytochrome c site-specific cross-link. Proc Natl Acad Sci USA. 2004;101(16):5940–5945. doi: 10.1073/pnas.0306708101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sivaraja M, Goodin DB, Smith M, Hoffman BM. Identification by ENDOR of Trp191 as the free-radical site in cytochrome c peroxidase compound ES. Science. 1989;245(4919):738–740. doi: 10.1126/science.2549632. [DOI] [PubMed] [Google Scholar]
- 20.Kang CH, Ferguson-Miller S, Margoliash E. Steady state kinetics and binding of eukaryotic cytochromes c with yeast cytochrome c peroxidase. J Biol Chem. 1977;252(3):919–926. [PubMed] [Google Scholar]
- 21.Leesch VW, Bujons J, Mauk AG, Hoffman BM. Cytochrome c peroxidase-cytochrome c complex: Locating the second binding domain on cytochrome c peroxidase with site-directed mutagenesis. Biochemistry. 2000;39(33):10132–10139. doi: 10.1021/bi000760m. [DOI] [PubMed] [Google Scholar]
- 22.Mauk MR, Ferrer JC, Mauk AG. Proton linkage in formation of the cytochrome c-cytochrome c peroxidase complex: Electrostatic properties of the high- and low-affinity cytochrome binding sites on the peroxidase. Biochemistry. 1994;33(42):12609–12614. doi: 10.1021/bi00208a011. [DOI] [PubMed] [Google Scholar]
- 23.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meharenna YT, Doukov T, Li H, Soltis SM, Poulos TL. Crystallographic and single-crystal spectral analysis of the peroxidase ferryl intermediate. Biochemistry. 2010;49(14):2984–2986. doi: 10.1021/bi100238r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berghuis AM, Brayer GD. Oxidation state-dependent conformational changes in cytochrome c. J Mol Biol. 1992;223(4):959–976. doi: 10.1016/0022-2836(92)90255-i. [DOI] [PubMed] [Google Scholar]
- 26.Millett F, Miller MA, Geren L, Durham B. Electron transfer between cytochrome c and cytochrome c peroxidase. J Bioenerg Biomembr. 1995;27(3):341–351. doi: 10.1007/BF02110103. [DOI] [PubMed] [Google Scholar]
- 27.Bonagura CA, Sundaramoorthy M, Bhaskar B, Poulos TL. The effects of an engineered cation site on the structure, activity, and EPR properties of cytochrome c peroxidase. Biochemistry. 1999;38(17):5538–5545. doi: 10.1021/bi982996k. [DOI] [PubMed] [Google Scholar]
- 28.Bonagura CA, Sundaramoorthy M, Pappa HS, Patterson WR, Poulos TL. An engineered cation site in cytochrome c peroxidase alters the reactivity of the redox active tryptophan. Biochemistry. 1996;35(19):6107–6115. doi: 10.1021/bi960122x. [DOI] [PubMed] [Google Scholar]
- 29.Miller MA, et al. Interaction domain for the reaction of cytochrome c with the radical and the oxyferryl heme in cytochrome c peroxidase compound I. Biochemistry. 1994;33(29):8686–8693. doi: 10.1021/bi00195a009. [DOI] [PubMed] [Google Scholar]
- 30.Miller MA. A complete mechanism for steady-state oxidation of yeast cytochrome c by yeast cytochrome c peroxidase. Biochemistry. 1996;35(49):15791–15799. doi: 10.1021/bi961488c. [DOI] [PubMed] [Google Scholar]
- 31.Hazzard JT, et al. Effects of amino acid replacements in yeast iso-1 cytochrome c on heme accessibility and intracomplex electron transfer in complexes with cytochrome c peroxidase. Biochemistry. 1988;27(12):4445–4451. doi: 10.1021/bi00412a035. [DOI] [PubMed] [Google Scholar]
- 32.Selzer T, Schreiber G. New insights into the mechanism of protein-protein association. Proteins. 2001;45(3):190–198. doi: 10.1002/prot.1139. [DOI] [PubMed] [Google Scholar]
- 33.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336(6084):1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 1):72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 36.French S, Wilson K. On the treatment of negative intensity observations. Acta Crystallogr A. 1978;34:517–525. [Google Scholar]
- 37.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 39.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Afonine PV, Grosse-Kunstleve RW, Adams PD. A robust bulk-solvent correction and anisotropic scaling procedure. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 7):850–855. doi: 10.1107/S0907444905007894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blanc E, et al. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2210–2221. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
- 42.Bricogne G, et al. BUSTER Version 2.10.0. Cambridge, UK: Global Phasing Ltd; 2011. [Google Scholar]
- 43.Smart OS, et al. Exploiting structure similarity in refinement: Automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt 4):368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chong LT, Duan Y, Wang L, Massova I, Kollman PA. Molecular dynamics and free-energy calculations applied to affinity maturation in antibody 48G7. Proc Natl Acad Sci USA. 1999;96(25):14330–14335. doi: 10.1073/pnas.96.25.14330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.