

Abstract
Objective
Deep vein thrombosis (DVT) and pulmonary embolism are frequent causes of morbidity and mortality. We wished to determine whether plasma high density lipoprotein (HDL), which inversely correlates with the risk of cardiovascular events, affects DVT.
Methods and Results
Using a murine DVT model of inferior vena cava stenosis, we demonstrate that deficiency of the HDL receptor, scavenger receptor class B type I (SR-BI), promotes venous thrombosis. As SR-BI−/− mice have increased plasma cholesterol levels and abnormal HDL particles, we tested SR-BI−/− mice with an SR-BI liver transgene that normalizes both parameters. These mice also exhibited increased susceptibility to DVT, indicating a protective role of extrahepatic SR-BI. Mice lacking the major HDL apolipoprotein apoA-I or eNOS (a downstream target of endothelial SR-BI signaling) also had a pro-thrombotic phenotype. Intravenous infusion of human apoA-I, an HDL component and SR-BI ligand, prevented DVT in wild-type but not SR-BI−/− or eNOS−/− mice, suggesting that its effect is mediated by SR-BI and eNOS. Intravenous apoA-I infusion abolished histamine-induced platelet-endothelial interactions, which are important for DVT initiation.
Conclusion
An apoA-I (HDL) - SR-BI - eNOS axis is highly protective in DVT and may provide new targets for prophylaxis and treatment of venous thrombosis.
Keywords: DVT, SR-BI, apoA-I, HDL, eNOS
Venous thromboembolism (VTE), which includes deep vein thrombosis (DVT) and its life-threatening complication pulmonary embolism (PE), is a major health problem in the Western world. In the United States alone, VTE affects ~900,000 people/year, and leads to hundreds of thousands of hospitalizations and deaths.1 Various factors predisposing to DVT, such as cancer, obesity or hereditary blood hypercoagulability, have been described.2 Distortion of blood flow resulting from immobilization, for example, due to post-surgery bed-ridden position, long airplane flights, and limb paralysis, constitutes the major provoking factor of idiopathic DVT. In contrast to arterial thrombosis that is initiated by vessel wall injury, for example when an atherosclerotic plaque ruptures, in venous thrombosis, the endothelial layer remains largely intact.3 As a consequence, the mechanisms initiating venous thrombosis are different from those underlying arterial thrombosis.
The metabolism of HDL has been the focus of much research, in part because the levels of plasma high density lipoprotein (HDL) cholesterol are inversely correlated with the risk of cardiovascular events4,5, i.e., diseases based on arterial thrombosis. In contrast, the relationship of plasma HDL levels and VTE in humans is unclear, with some studies demonstrating an inverse correlation6,7 and others reporting no such correlation8 or even a direct association of high HDL-cholesterol with increased risk of VTE in women.9 This discrepancy may reflect, in part, differences between the studied populations, as HDLs are heterogeneous populations of lipoprotein particles whose characteristics may vary depending of the physiologic state of an individual (for example, see reference 10).10
A major function proposed for HDL is reverse cholesterol transport, i.e., transport of cholesterol from peripheral tissues to the liver and subsequent excretion into the bile.11 One mechanism by which hepatocytes take up cholesterol from HDL is selective lipid uptake12,13 mediated by the HDL receptor called Scavenger Receptor, Class B, Type I (SR-BI) (reviewed in14). During selective lipid uptake, SR-BI binds to HDL particles via their apolipoproteins, particularly apoA-I, and the receptor mediates the transfer of HDL’s cholesteryl esters into the cells. The lipid depleted HDL particles are then released into the extracellular fluid.
SR-BI, a member of the CD36 family of proteins, is a multiligand receptor expressed on many cells, such as hepatocytes, macrophages, endothelial cells (including endothelium in veins15) and platelets.14,16,17,18 It plays an important role in protection against murine atherosclerosis.14,19 Some studies also implicated SR-BI in HDL-induced signal transduction in endothelial cells.20,21 For example, SR-BI mediates HDL-induced activation of endothelial nitric oxide synthase (eNOS)20, whose product, nitric oxide (NO), is anti-thrombotic, anti-atherogenic and plays a crucial role in vascular homeostasis.22 HDL binding to SR-BI activates eNOS20 via the following pathway: HDL-> SR-BI-> src -> PI3K -> (Akt or MAPK)->eNOS.21 In endothelial cells, HDL can also stimulate prostaglandin I2 synthesis23 and deliver sphingosine-1-phosphate to control endothelial functions.24
A cytoplasmic adaptor protein, PDZK1, binds to the intracellular C-terminus of SR-BI and influences SR-BI abundance and activity in a tissue-specific fashion.25,26 In cultured endothelial cells, PDZK1 is involved in SR-BI-dependent activation of eNOS,27 but is not required for normal SR-BI expression levels or binding to HDL.27,28 Similarly to SR-BI−/− mice,29 PDZK1−/− mice exhibit impaired post-injury re-endothelization.27 In PDZK1−/− mice, hepatic SR-BI levels fall to less than 5% of normal, resulting in abnormal plasma HDL (large particles, increased HDL cholesterol) that is reminiscent of, but not as severe as, that seen in SR-BI−/− mice.30 Mice lacking PDZK1 exhibit some,28,31 but not all19,30 of the abnormalities seen in SR-BI−/− mice, although with significantly less severity.
SR-BI expressed on platelets and hepatocytes modulates platelet function and arterial thrombosis. SR-BI−/− mice are thrombocytopenic, with ~38% of the platelet count seen in SR-BI+/+ mice, due to abnormally high platelet cholesterol levels and the consequent reduced platelet life-span in the circulation.32 Platelets lacking SR-BI exhibit moderate changes in aggregation response to some platelet agonists in vitro.32,33,34 It has recently been reported that deficiency of SR-BI on platelets protects them from hyperactivation mediated by increased platelet cholesterol content and also protects mice from arterial thrombosis induced by endothelial injury.34 Infusion of human apoA-I Milano (a mutant form of apoA-I identified in an Italian family) has been reported to delay FeCl3-induced thrombus formation in rat aorta.35 The question of how HDL and SR-BI affect venous thrombosis has not been addressed experimentally.
In the present work, we explored this question using an established murine model of DVT that employs flow restriction, which is one of the major factors triggering DVT in humans, to induce endothelial activation/dysfunction and consequent venous thrombosis.36 We demonstrate that an apparent HDL (apoA-I) - SR-BI - eNOS axis protects against flow restriction-induced venous thrombosis and that infusion of apoA-I protects mice from DVT.
Materials and Methods
Expanded materials and methods are available in the Supplemental Data, available online at http://atvb.ahajournals.org.
Mice
SR-BI+/−, SR-BI−/−37, and SR-BI−/−[Liver Tg]38 mice were generated and housed as previously described. The SR-BI−/−[Liver Tg] mice express a liver-specific, murine SR-BI transgene (~0.7 fold of the hepatic expression level in SR-BI+/+ mice38). For DVT and blood coagulation experiments not involving apoA-I infusion, these mice were on mixed 75:25 129-S4:C57BL/6 background. Littermates used in these experiments were obtained by crossing transgenic or nontransgenic SR-BI−/− males to nontransgenic SR-BI+/− or transgenic SR-BI−/− females, respectively. SR-BI−/− mice used in experiments involving apoA-I infusion followed by DVT, were on mixed 50:50 129-S4:C57BL/6 background. Littermates used in these experiments were obtained by crossing SR-BI−/− males to SR-BI+/− females (SR-BI−/− females are infertile38) Mice with homozygous null alleles for Nos3 (eNOS−/−) and APOA1 (apoA-I−/−) as well as their control mice were purchased from the Jackson Laboratory and were on a C57BL/6 background. PDZK1−/− mice30 and their PDZK1+/+ littermate controls were on 129SvEv background. All protocols for animal use were reviewed and approved by the Animal Care and Use Committee of the Immune Disease Institute.
Flow restriction surgery
Male 8–9 week old mice (weight 23–28 g) were anesthetized with isoflurane-oxygen mixture, placed in a supine position and the IVC was accessed through medial abdominal incision. For each mouse, the aorta was deliberately separated from the IVC just below the renal veins (in caudal direction) and the IVC was ligated with a polypropylene monofilament 7.0 over a 30G spacer as previously described.36 The spacer was placed externally in a way excluding any injury to the vessel, then the ligature was closed and the spacer was removed. This procedure decreases the IVC lumen by about 90%.36 All side branches of the IVC were completely closed as well. After surgery, peritoneum and skin were sutured separately. In 2, 3, 6 or 8 h, after initiating IVC stenosis, mice were anesthetized again, opened, and any thrombus present was taken and its weight and length determined. According to our experience, weight and length of thrombi may substantially vary between mice, which makes thrombi prevalence an important readout in this model. Our preliminary experiments showed that about 20–25% of wild-type mice on C57BL/6 background (WT) formed a thrombus within 2–3 h in this model, whereas the same percent of mice on mixed 129-S4:C57BL/6 background and about 40% of WT mice on the mixed or 129SvEv background developed a thrombus within 6 h.
Platelet-endothelial interactions in mice
Intravital microscopy was performed as described.39 In brief, mice were anesthetized with 2.5% tribromoethanol (150 μl per 10 g body weight) and apoA-I (3.5 mg/kg) or vehicle was intravenously infused through the retro-orbital plexus. Three min after apoA-I infusion, mesentery was exteriorized and veins (200 – 250 μm) were examined using an inverted microscope (Axiovert 135; Carl Zeiss, Inc., objective 10 x) connected to a video camera (C2400; Hamamatsu Photonics). Washed syngeneic platelets from C57BL/6 mice (2.5 × 109/kg) fluorescently labeled with CFSE (Molecular Probes) were infused. Observations from one vein per animal were recorded for 2 min and, then histamine (50 μL, 5 mmol/L, Sigma) was superfused and the recording from the same vessel continued for 15 min thereafter. Platelets interacting with the endothelium were counted by a ‘blinded’ observer for 60 s starting 1 min before histamine application and 5, 7 and 10 min after histamine. Platelet attachment to the vessel wall lasting more than 0.3 s was considered a platelet-endothelial interaction.
Statistics
The percentage of mice with thrombus in different experimental groups was compared using a contingency table and the Fisher’s exact test. Coagulation onset and coagulation rate were compared using one-way ANOVA followed by the Tukey’s multiple comparison test. Plasma D-dimer levels, platelet aggregation amplitude and numbers of platelets interacting with vascular wall with and without apoA-I were compared using Student’s t-test. Weight and length of thrombi were compared by the Mann-Whitney test except for the experiment with apoA-I infusion into C57BL/6J mice (Figure 4), in which the Mann-Whitney test was inapplicable due to uniformity of the results in all mice and therefore the Kruskal–Wallis test was used. Statistics was calculated using GraphPad Prism 5.0. Differences were considered significant for P < 0.05.
Figure 4. Infusion of apoA-I prevents DVT in WT but not SR-BI−/− or eNOS−/− mice.
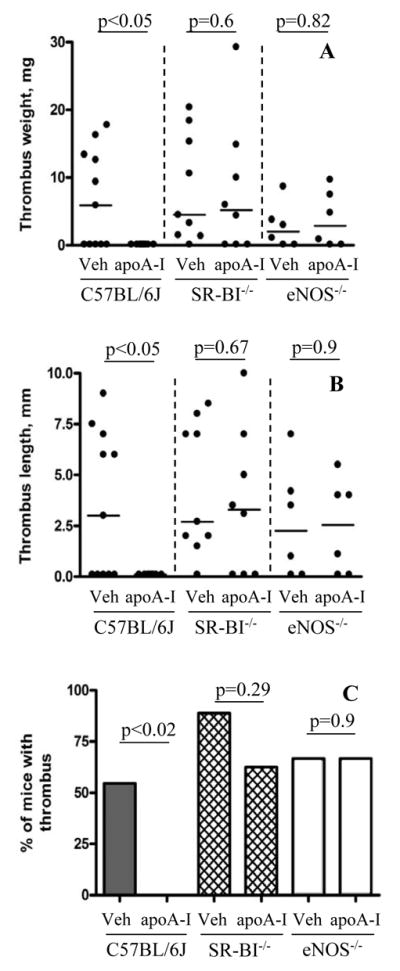
WT C57BL/6, SR-BI−/− or eNOS−/− mice underwent IVC stenosis. Immediately after surgery, native human apoA-I (3.5 mg/kg) or vehicle (Veh) was intravenously infused. The presence of thrombi was assessed at 6 h for WT and SR-BI−/− mice or at 2 h for eNOS−/− mice. (A) thrombus weight in mg; (B) thrombus length in mm; (C) thrombi prevalence. Number of animals (n): WT C57BL/6J 11 (vehicle or apoA-I infused); SR-BI−/− 9 (vehicle) or 8 (apoA-I); eNOS−/− 6 (vehicle or apoA-I). Horizontal bars in dot plots represent median values.
Results
Extrahepatic SR-BI deficiency enhances flow restriction-induced thrombosis
To explore the role of SR-BI in murine DVT we used a flow restriction (stenosis) model in the inferior vena cava (IVC). We chose a relatively short (2 – 8 h as compared to the usual 48 h 36) stenosis period to be able to detect a pro-thrombotic phenotype in the experimental mice. Stenosis of the IVC for 6 h resulted in thrombus development in only 17 % of the control SR-BI+/− mice (2 out of 12, Figure 1). In contrast, 100% of the SR-BI−/− littermates (9 out of 9) developed a thrombus under these conditions. Thus, expression of SR-BI protects mice against DVT.
Figure 1. SR-BI−/− mice with or without SR-BI liver transgene are prone to flow restriction-induced venous thrombosis.
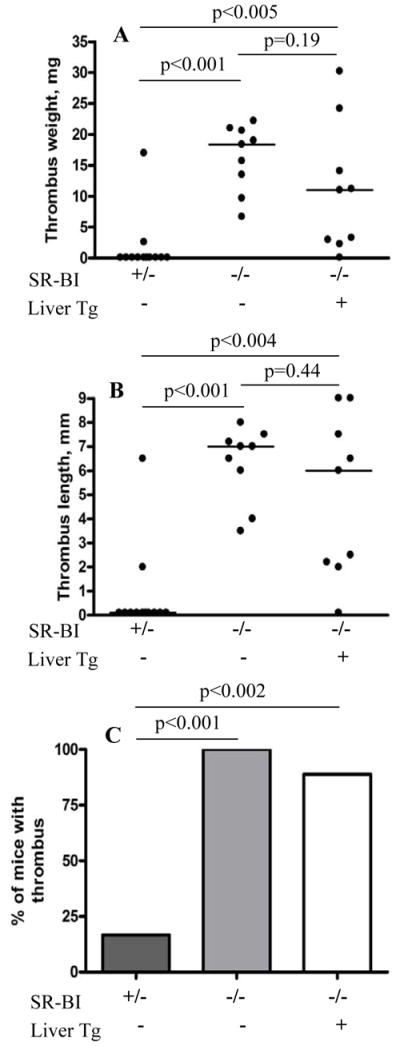
IVC stenosis surgery was performed in SR-BI+/− (n = 12), SR-BI−/−(n = 9), or SR-BI−/− [Liver Tg] littermates (n = 9). Mice were sacrificed after 6 h, thrombi excised and their weight and length measured. (A) thrombus weight in mg; (B) thrombus length in mm; (C) percent of mice that developed a thrombus (thrombi prevalence). Horizontal bars in dot plots represent median values.
Hepatic SR-BI expression influences the structure and abundance of plasma HDL and biliary cholesterol secretion.37,40 To determine if loss of hepatic SR-BI expression was responsible for the pro-thrombotic phenotype of SR-BI−/− mice, we studied DVT in transgenic SR-BI−/− mice (SR-BI−/−[Liver Tg])38 which express in the liver essentially WT levels of a murine SR-BI transgene driven by a primarily liver-specific promoter (‘Liver Tg’, see Methods). SR-BI−/−[Liver Tg] mice exhibit essentially WT type plasma cholesterol levels, HDL particle size distributions and plasma UC:TC ratios.38 We reported previously that the total plasma cholesterol (TC) for the SR-BI−/− [Liver Tg] mice is lower than that for the control nontransgenic SR-BI+/− mice (128±1 vs. 143± 4 mg/dL, respectively) or the SR-BI−/− mice (206±7 mg/dL).38 Figure 1 shows that 89% of the SR-BI−/−[Liver Tg] mice (8 out of 9) developed thrombi after 6 h of IVC flow restriction. No statistically significant differences in thrombus weight (P = 0.19) and length (P = 0.44) were observed between the non-transgenic and SR-BI−/−[Liver Tg] mice. Because WT levels of hepatic SR-BI did not ameliorate the pro-thrombotic phenotype in SR-BI−/− mice, we conclude that their common deficiency in extrahepatic SR-BI was the likely cause of their enhanced DVT.
It has been shown in vitro that the adaptor protein PDZK1 is implicated in SR-BI downstream signaling.21,27 We examined flow restriction-induced thrombosis (6–8 h periods of stenosis) in PDZK1−/− and PDZK1+/+ littermates. Surprisingly, thrombosis prevalence did not significantly differ between PDZK1−/− and PDZK1+/+ controls (Supplementary Figure 1).
SR-BI and blood coagulation
Procoagulant state of blood is frequently associated with venous thrombosis in humans,2 and HDL may influence coagulation as shown in vitro.41 We therefore tested ex vivo coagulation of recalcified citrated whole blood drawn from SR-BI+/−, SR-BI−/− and SR-BI−/−[Liver Tg] mice. SR-BI−/− mice had shorter coagulation onset times (Figure 2A) and higher coagulation rates (Figure 2B) than SR-BI+/− controls (Figure 2A–B, shaded bars). The SR-BI−/−[Liver Tg] mice had the longest onset time and lowest coagulation rate, (Figure 2A–B, open bars). Clot formation in recalcified blood from these mice was inversely correlated to their rank order of hepatic SR-BI protein expression [SR-BI−/−[Liver Tg]:SR-BI+/−:SR-BI−/−; ~0.7:0.5:0] and directly correlated with their rank order of plasma cholesterol levels (1.00:1.12:1.61, respectively)38 but no correlation with DVT susceptibility was observed. SR-BI−/− and SR-BI−/−[Liver Tg] mice had the fastest and slowest clot formation after recalcification, respectively, yet both exhibited dramatically higher flow restriction-induced DVT than the control SR-BI+/− mice. We measured plasma levels of D-dimer, a fibrin degradation product which reflects the extent of active fibrin formation and fibrinolysis, in untreated control and SR-BI−/− mice and the levels were similar (Figure 2C). We conclude that despite the propensity of blood from SR-BI−/− mice to clot faster in vitro, clotting in vivo is not constitutively hyper-activated and procoagulant state of SR-BI−/− mice is not the primary determinant of their increased DVT susceptibility.
Figure 2. In vivo expression of SR-BI can influence in vitro clot formation whereas in vivo plasma fibrin D-dimer levels remain unaltered.
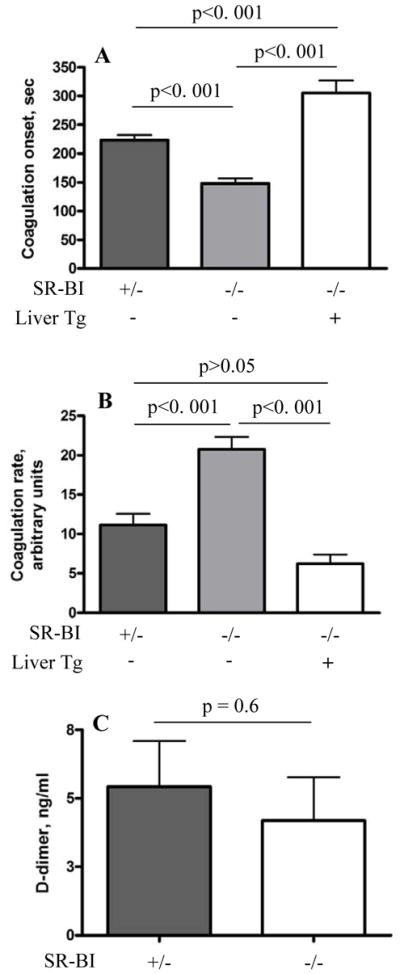
Whole blood was collected from SR-BI+/− (n = 16), SR-BI−/− (n = 12), or SR-BI−/−[Liver Tg] littermates (n = 8) and stabilized by sodium citrate. Calcium chloride was added to the blood within a minute after drawing and clotting monitored by a Sonoclot analyzer. (A) coagulation onset (time to first fibrin formation) and (B) coagulation rate. (C) D-dimer levels in plasma of SR-BI−/− mice (n=5) and SR-BI+/− controls (n=7). Error bars represent SEM.
Deficiencies in apoA-I or eNOS predispose mice to flow restriction-induced thrombosis
We next examined flow restriction-induced thrombosis in eNOS−/− and apoA-I−/− (reduced HDL42) mice. Because we anticipated pro-thrombotic phenotypes in the mutant mice, we performed the assays using 2 h rather than 6 h periods of IVC stenosis. While only 27% (3/11) of control C57BL/6J mice developed thrombi under these conditions, 88% (7/8) of apoA-I−/− mice (P < 0.02 vs. control) and 82% (9/11) of eNOS−/− mice (P < 0.04 vs. control) developed thrombi (Figure 3). These results do not establish directly that the SR-BI anti-thrombotic effect involves its main ligand apoA-I and eNOS, but they do show that both apoA-I and eNOS play protective roles in DVT.
Figure 3. ApoA-I and eNOS protect mice from flow restriction-induced thrombosis.
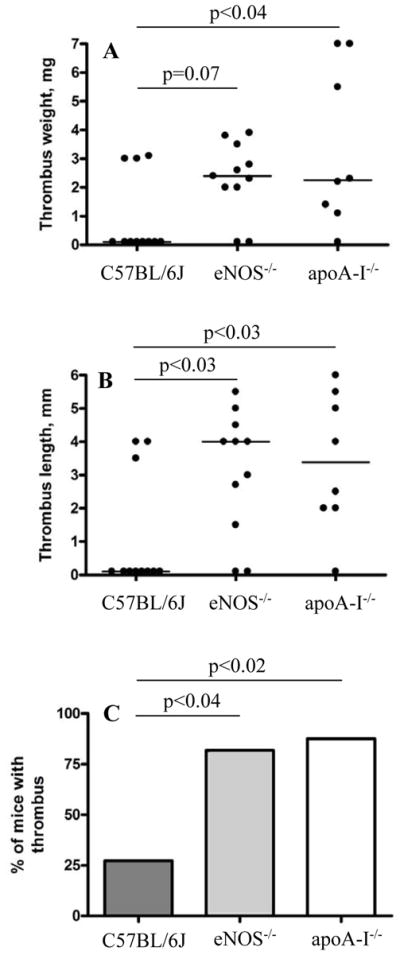
Stenosis of IVC was applied in WT (n = 11), eNOS−/− (n = 11) and apoA-I−/− (n = 8) mice and thrombus formation was examined after 2 h. (A) thrombus weight in mg; (B) thrombus length in mm; (C) thrombi prevalence. Horizontal bars in dot plots represent median values.
Infusion of apoA-I protects WT but not SR-BI−/− or eNOS−/− mice from venous thrombosis
Because apoA-I deficiency increased DVT susceptibility, we examined the effects on DVT of intravenous administration of exogenous human apoA-I. Indeed, Figure 4 (panels A-C) shows that one bolus infusion of apoA-I immediately after application of IVC stenosis for 6 h completely protected C57BL/6J mice from DVT, decreasing thrombi prevalence from 55% in vehicle-infused mice (n = 11) to zero (n = 11). This potent anti-thrombotic effect of apoA-I was SR-BI- and eNOS-dependent: apoA-I administration to SR-BI−/− or eNOS−/− mice did not significantly influence thrombosis (Figure 4). Thus, apoA-I is unable to exert its protective effect in the absence of either SR-BI or eNOS, raising the possibility that these molecules lay downstream of apoA-I in a signaling pathway that helps protect against venous thrombus development.
Because SR-BI is expressed on platelets, we asked if in vitro incubation of apoA-I (45 μg/mL) with platelets from C57BL/6J mice affects platelet aggregation induced by a set of agonists. The concentration used is similar to that expected in the blood of mice infused as described above with a bolus of 3.5 mg/kg of apoA-I. We observed no statistically significant effect of apoA-I on platelet aggregation induced by high or low concentrations of collagen, calcium ionophore A23187, or a high dose of thrombin, as determined by aggregation amplitude 10 min after the addition of agonist (Figure 5, A - D). There was a statistically insignificant trend of decreased aggregation of apoA-I-treated platelets with low dose thrombin (Figure 5, E, F). Therefore, infusion of apoA-I protected WT mice against flow restriction-induced thrombosis via a mechanism that apparently either directly or indirectly depends on SR-BI and may not involve direct alteration of platelet aggregation. These results raised the possibility that apoA-I infusion suppressed flow restriction-induced activation of venous endothelium.
Figure 5. ApoA-I does not affect platelet aggregation in vitro.

Washed platelets from WT C57BL/6J mice were incubated with apoA-I (45 μg/mL) or vehicle for 5 min before adding (A) calcium ionophore A23187 (10 μmol/L) (B, C) collagen (10 or 1 μg/mL), or (D–F) thrombin (1 U/mL (D) or 0.1 U/mL (E, F). Light transmission was recorded for 10 min. Each curve is a mean of 3 – 7 independent experiments. Arrows indicate time of agonist addition. Horizontal bars in (E) represent median values. Aggregation amplitudes of vehicle- and apoA-I-treated platelets at the end of 10 min period were compared statistically.
Infusion of apoA-I suppresses endothelial stimulation-induced platelet rolling
Platelet interactions with the venous endothelium are crucial for DVT in our stenosis model.36 Platelet recruitment to the endothelium is largely dependent on von Willebrand factor (VWF),39 a constituent of endothelial Weibel-Palade bodies (WPB), whose release is a key step in the initiation of DVT in the murine model employed in this study.36 Therefore, we utilized platelet-endothelial interactions as an experimental model entirely dependent on secretion of VWF from WPB and tested the effects of apoA-I on platelet-endothelial interactions in vivo using intravital microscopy. After intravenous administration of a bolus of apoA-I (3.5 mg/kg) or vehicle, we surgically exposed the mesenteric venules, infused fluorescently labeled platelets into the mice and then determined the number of platelets interacting with the endothelium before and after activation of the endothelium by histamine. Figure 6 shows that in vehicle-treated WT control mice, platelet/endothelium interactions increased with time from 19±6 at baseline to 84±39 platelets/view field/min at 10 min after histamine superfusion (black squares). In mice pre-treated with apoA-I (open circles), baseline platelet-vessel wall interactions were reduced compared to vehicle-treated controls, and there was no significant histamine-mediated increase with time. Thus, apoA-I inhibits platelet recruitment by the venous endothelium, presumably by suppressing WPB release.
Figure 6. Infusion of apoA-I suppresses platelet-endothelial interaction.
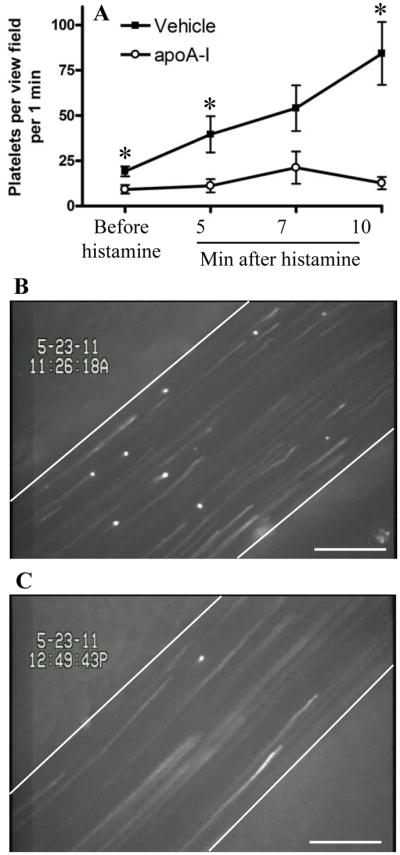
Mice were anesthetized and apoA-I (3.5 mg/kg, n = 4) or vehicle (n = 5) were administered intravenously. Mesenteric venules were surgically exposed and visualized using intravital microscopy. Fluorescently labeled platelets were then infused and baseline platelet-endothelial interactions were recorded for 2 min. Histamine (50 μL, 5 mmol/L) was then topically applied. One venule per mouse was recorded for 15 min. The number of platelets interacting with the vessel wall was counted for 60 s starting 1 min before and at 5, 7 and 10 min after histamine application. (A) number of interacting platelets per 1 min per view field. *, P < 0.05 between vehicle- and apoA-I-infused mice; (B–C) representative images demonstrating platelets interacting with activated endothelium at 11 min after histamine superfusion. More adherent platelets are seen in the vehicle-treated mouse (B) than in the apoA-I-treated animal (C). Error bars represent SEM. Bar, 100 μm.
Discussion
In this study, we showed that apoA-I, the major apolipoprotein in HDL, and extrahepatic expression of the HDL receptor SR-BI protected mice from flow-restriction-induced DVT. Mice with homozygous null mutations in the APOA1 (apoA-I−/−) or Scarb1 (SR-BI−/−) genes exhibited dramatically increased susceptibility to DVT relative to control WT or SR-BI+/− mice, respectively. SR-BI−/− mice expressing an essentially liver-specific murine SR-BI transgene (SR-BI−/−[Liver Tg]) exhibited susceptibility to DVT that was indistinguishable from that of non-transgenic SR-BI−/− mice. Thus, it appears that SR-BI deficiency in extrahepatic tissues was responsible for the increased susceptibility of SR-BI−/− mice to venous thrombosis.
Some6,7 but not all8,9 studies have shown that plasma HDL inversely correlates with the risk of DVT. Cholesterol enrichment of monocytes/macrophages can induce release of highly pro-coagulant microparticles from monocytes43 while intact HDL and apoA-I can activate fibrinolysis44 and therefore alter thrombus formation and stability. SR-BI−/− mice exhibit a distinctive dyslipidemia (elevated plasma levels of TC and unesterified cholesterol (UC), increased UC:TC ratio and abnormal HDL particles45,37), which is associated with increased UC accumulation in red blood cells46 and platelets,32 and hypercoagulability in an ex vivo assay (this study). Indeed, enhanced blood coagulation is known to predispose humans to venous thrombosis.2 However, hepatic expression of the SR-BI transgene in SR-BI−/−[Liver Tg] mice corrected both the dyslipidemia and abnormal clot formation in recalcified blood, but did not influence the increased susceptibility to DVT. PDZK1−/− mice exhibit a dyslipidemia similar to, but milder than, that of SR-BI−/− mice,30 yet do not show increased susceptibility to DVT. Also, the lack of SR-BI did not affect plasma D-dimer levels, which indicates that the dyslipidemia itself in SR-BI−/− mice does not cause a marked change in constitutive fibrin formation. Together these findings indicate that dyslipidemia and hypercoagulability were not major causes of the enhanced susceptibility of SR-BI−/− mice to DVT although SR-BI may regulate blood coagulation potential indirectly through controlling plasma lipids.
Two sites of extrahepatic SR-BI expression appeared to be particularly relevant with regard to the DVT susceptibility of SR-BI−/− mice: platelets and endothelial cells. Platelet SR-BI deficiency has been shown to protect mice from dyslipidemia-mediated arterial thrombosis in vivo.34 Also, SR-BI-deficient platelets have moderately decreased in vitro aggregation in response to some (but not all) agonists, such as ADP, collagen, PMA and convulxin.32,33,34 Thus, platelet dysfunction due to platelet SR-BI deficiency in SR-BI−/− mice might be expected to be anti-thrombotic and thus it is an unlikely mechanism underlying the observed pro-thrombotic phenotype.
In cultured endothelial cells, SR-BI can serve as a signaling receptor that regulates endothelial physiology by mediating HDL induction of eNOS activity20 via the following pathway: HDL-> SR-BI-> src -> PI3K -> (Akt or MAPK)->eNOS.21 The cytoplasmic SR-BI adaptor protein PDZK1 has been shown to influence this eNOS activation pathway in vitro and carotid artery reendothelialization following perivascular injury in vivo.27 We explored the potential role of this pathway in the enhanced susceptibility of SR-BI−/− mice to DVT by assessing flow-restriction-induced DVT in mice with homozygous null mutations in the APOA1, Nos3 or Pdzk1 genes. Both apoA-I−/− (which have decreased numbers of HDL particles and decreased total plasma cholesterol42) and eNOS−/− mice exhibited markedly increased venous thrombosis compared to WT controls, suggesting that this pathway may contribute to an anti-thrombotic phenotype. In contrast, PDZK1−/− mice were not prone to DVT. It is possible that there is a PDZK1-independent pathway that links SR-BI to eNOS and mediates protection from venous thrombosis in vivo. For example, it has been reported that lysophospholipids present in HDL induce eNOS activation through their receptor S1P3, a process in which SR-BI participate.47
Because both apoA-I−/− mice and SR-BI−/− mice were prone to venous thrombosis, we examined the effects of intravenous administration of exogenous apoA-I protein on flow-restriction-induced DVT. Infusion of apoA-I into mice has been reported to result in increased plasma concentrations of both relatively lipid-free apoA-I and apoA-I associated with HDL particles.48 We found that infusion of apoA-I into WT C57BL/6 animals virtually abolished flow-restriction-induced DVT. This dramatic protective effect of apoA-I was SR-BI-dependent, as there was no protection by apoA-I infusion in SR-BI−/− mice. Moreover, infusion of apoA-I did not protect eNOS-deficient mice from DVT. This result raises the possibility that the anti-thrombotic effect of apoA-I is eNOS-mediated. Taken together, these data suggest that a signal transduction pathway apoA-I(HDL) - SR-BI - eNOS, previously observed in vitro, may contribute to protection from venous thrombosis. It is possible that mechanisms not directly involving HDL signaling via SR-BI also contribute to this apoA-I effect in WT mice.49 However, our results support a pathway in which apoA-I-enriched HDL interacts directly with SR-BI on extrahepatic cells. Potential target cells include platelets and the venous endothelium. Indeed, using intravital microscopy we showed that infusion of apoA-I completely inhibited histamine-induced platelet-endothelial interactions in vivo. Treatment of isolated platelets with concentrations of apoA-I comparable to those expected in vivo after the apoA-I infusion did not affect platelet aggregation induced by a high dose of thrombin, A23187 or collagen. Thus, the anti-thrombotic effect of apoA-I infusion is unlikely to be a consequence of a direct apoA-I effect on platelets. However, we cannot completely rule out apoA-I effect on platelets in vivo.
HDL can activate eNOS in endothelial cells in vitro via an SR-BI-dependent signaling pathway.20,21 Activation of eNOS and release of NO, in turn, can inhibit the release of Weibel-Palade bodies,50 and thus interfere with the activation of the endothelium required for platelet/endothelium interactions and thrombosis.36 Thus, it appears that an apoA-I/HDL – SR-BI – eNOS pathway may operate in vivo to protect mice from venous thrombosis, at least in part via suppression of endothelial activation and subsequent platelet and leukocyte recruitment.51
In summary, our studies support the hypothesis that HDL has beneficial effects on endothelium52 and expand HDL’s range of benefits from arterial to venous circulation. The HDL receptor SR-BI appears to mediate, possibly via signaling through eNOS, some of these effects. Our observation that eNOS−/− mice exhibit increased DVT is consistent with this mechanism and suggests that eNOS can suppress endothelial activation, and thus the release of VWF and platelet adhesion, key features in the initiation of DVT.36 We found that apoA-I infusion prevents flow restriction-induced DVT in WT C57BL/6J mice. Thus, it may be possible that apoA-I mimetics, or other agonists of SR-BI, might provide a novel medical approach to prevent or treat DVT in humans.
Supplementary Material
Acknowledgments
We thank Lesley Cowan for help in preparing the manuscript.
Sources of Funding
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health grants P01 HL066105 (M.K. and D.D.W), HL52212 to M.K. and R01 HL041002 (D.D.W). S.F.D.M is a postdoctoral fellow of the ‘Fond voor Wetenschappelijk Onderzoek Vlaanderen’ (FWO).
Footnotes
Disclosures
None
References
- 1.Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol. 2008;28:370–372. doi: 10.1161/ATVBAHA.108.162545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldhaber SZ. Risk factors for venous thromboembolism. J Am Coll Cardiol. 2010;56:1–7. doi: 10.1016/j.jacc.2010.01.057. [DOI] [PubMed] [Google Scholar]
- 3.Sevitt S. The structure and growth of valve-pocket thrombi in femoral veins. J Clin Pathol. 1974;27:517–528. doi: 10.1136/jcp.27.7.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 5.Miller GJ, Miller NE. Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet. 1975;1:16–19. doi: 10.1016/s0140-6736(75)92376-4. [DOI] [PubMed] [Google Scholar]
- 6.Deguchi H, Pecheniuk NM, Elias DJ, Averell PM, Griffin JH. High-density lipoprotein deficiency and dyslipoproteinemia associated with venous thrombosis in men. Circulation. 2005;112:893–899. doi: 10.1161/CIRCULATIONAHA.104.521344. [DOI] [PubMed] [Google Scholar]
- 7.Ageno W, Becattini C, Brighton T, Selby R, Kamphuisen PW. Cardiovascular risk factors and venous thromboembolism: a meta-analysis. Circulation. 2008;117:93–102. doi: 10.1161/CIRCULATIONAHA.107.709204. [DOI] [PubMed] [Google Scholar]
- 8.Chamberlain AM, Folsom AR, Heckbert SR, Rosamond WD, Cushman M. High-density lipoprotein cholesterol and venous thromboembolism in the Longitudinal Investigation of Thromboembolism Etiology (LITE) Blood. 2008;112:2675–2680. doi: 10.1182/blood-2008-05-157412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braekkan SK, Borch KH, Mathiesen EB, Njolstad I, Hansen JB. HDL-cholesterol and future risk of venous thromboembolism: the Tromso Study. J Thromb Haemost. 2009;7:1428–1430. doi: 10.1111/j.1538-7836.2009.03481.x. [DOI] [PubMed] [Google Scholar]
- 10.Besler C, Heinrich K, Rohrer L, Doerries C, Riwanto M, Shih DM, Chroni A, Yonekawa K, Stein S, Schaefer N, Mueller M, Akhmedov A, Daniil G, Manes C, Templin C, Wyss C, Maier W, Tanner FC, Matter CM, Corti R, Furlong C, Lusis AJ, von Eckardstein A, Fogelman AM, Luscher TF, Landmesser U. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Invest. 2011;121:2693–2708. doi: 10.1172/JCI42946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glomset JA. The plasma lecithins:cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–167. [PubMed] [Google Scholar]
- 12.Glass C, Pittman RC, Weinstein DB, Steinberg D. Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-I of rat plasma high density lipoprotein: selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc Natl Acad Sci U S A. 1983;80:5435–5439. doi: 10.1073/pnas.80.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stein Y, Dabach Y, Hollander G, Halperin G, Stein O. Metabolism of HDL-cholesteryl ester in the rat, studied with a nonhydrolyzable analog, cholesteryl linoleyl ether. Biochim Biophys Acta. 1983;752:98–105. doi: 10.1016/0005-2760(83)90237-0. [DOI] [PubMed] [Google Scholar]
- 14.Rigotti A, Miettinen HE, Krieger M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr Rev. 2003;24:357–387. doi: 10.1210/er.2001-0037. [DOI] [PubMed] [Google Scholar]
- 15.Yu X, Murao K, Imachi H, Cao WM, Li J, Matsumoto K, Nishiuchi T, Ahmed RA, Wong NC, Kosaka H, Unterman TG, Ishida T. Regulation of scavenger receptor class BI gene expression by angiotensin II in vascular endothelial cells. Hypertension. 2007;49:1378–1384. doi: 10.1161/HYPERTENSIONAHA.106.082479. [DOI] [PubMed] [Google Scholar]
- 16.Buechler C, Ritter M, Quoc CD, Agildere A, Schmitz G. Lipopolysaccharide inhibits the expression of the scavenger receptor Cla-1 in human monocytes and macrophages. Biochem Biophys Res Commun. 1999;262:251–254. doi: 10.1006/bbrc.1999.1193. [DOI] [PubMed] [Google Scholar]
- 17.Imachi H, Murao K, Cao W, Tada S, Taminato T, Wong NC, Takahara J, Ishida T. Expression of human scavenger receptor B1 on and in human platelets. Arterioscler Thromb Vasc Biol. 2003;23:898–904. doi: 10.1161/01.ATV.0000067429.46333.7B. [DOI] [PubMed] [Google Scholar]
- 18.Valiyaveettil M, Kar N, Ashraf MZ, Byzova TV, Febbraio M, Podrez EA. Oxidized high-density lipoprotein inhibits platelet activation and aggregation via scavenger receptor BI. Blood. 2008;111:1962–1971. doi: 10.1182/blood-2007-08-107813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trigatti B, Rayburn H, Vinals M, Braun A, Miettinen H, Penman M, Hertz M, Schrenzel M, Amigo L, Rigotti A, Krieger M. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci U S A. 1999;96:9322–9327. doi: 10.1073/pnas.96.16.9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuhanna IS, Zhu Y, Cox BE, Hahner LD, Osborne-Lawrence S, Lu P, Marcel YL, Anderson RG, Mendelsohn ME, Hobbs HH, Shaul PW. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med. 2001;7:853–857. doi: 10.1038/89986. [DOI] [PubMed] [Google Scholar]
- 21.Saddar S, Mineo C, Shaul PW. Signaling by the high-affinity HDL receptor scavenger receptor B type I. Arterioscler Thromb Vasc Biol. 2010;30:144–150. doi: 10.1161/ATVBAHA.109.196170. [DOI] [PubMed] [Google Scholar]
- 22.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 23.Fleisher LN, Tall AR, Witte LD, Miller RW, Cannon PJ. Stimulation of arterial endothelial cell prostacyclin synthesis by high density lipoproteins. J Biol Chem. 1982;257:6653–6655. [PubMed] [Google Scholar]
- 24.Sattler K, Levkau B. Sphingosine-1-phosphate as a mediator of high-density lipoprotein effects in cardiovascular protection. Cardiovasc Res. 2009;82:201–211. doi: 10.1093/cvr/cvp070. [DOI] [PubMed] [Google Scholar]
- 25.Ikemoto M, Arai H, Feng D, Tanaka K, Aoki J, Dohmae N, Takio K, Adachi H, Tsujimoto M, Inoue K. Identification of a PDZ-domain-containing protein that interacts with the scavenger receptor class B type I. Proc Natl Acad Sci U S A. 2000;97:6538–6543. doi: 10.1073/pnas.100114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kocher O, Birrane G, Yesilaltay A, Shechter S, Pal R, Daniels K, Krieger M. Identification of the PDZ3 domain of the adaptor protein PDZK1 as a second, physiologically functional, binding site for the C-terminus of the HDL receptor SR-BI. J Biol Chem. 2011;286:25171–25186. doi: 10.1074/jbc.M111.242362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu W, Saddar S, Seetharam D, Chambliss KL, Longoria C, Silver DL, Yuhanna IS, Shaul PW, Mineo C. The scavenger receptor class B type I adaptor protein PDZK1 maintains endothelial monolayer integrity. Circ Res. 2008;102:480–487. doi: 10.1161/CIRCRESAHA.107.159079. [DOI] [PubMed] [Google Scholar]
- 28.Yesilaltay A, Daniels K, Pal R, Krieger M, Kocher O. Loss of PDZK1 causes coronary artery occlusion and myocardial infarction in Paigen diet-fed apolipoprotein E deficient mice. PLoS One. 2009;4:e8103. doi: 10.1371/journal.pone.0008103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seetharam D, Mineo C, Gormley AK, Gibson LL, Vongpatanasin W, Chambliss KL, Hahner LD, Cummings ML, Kitchens RL, Marcel YL, Rader DJ, Shaul PW. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ Res. 2006;98:63–72. doi: 10.1161/01.RES.0000199272.59432.5b. [DOI] [PubMed] [Google Scholar]
- 30.Kocher O, Yesilaltay A, Cirovic C, Pal R, Rigotti A, Krieger M. Targeted disruption of the PDZK1 gene in mice causes tissue-specific depletion of the high density lipoprotein receptor scavenger receptor class B type I and altered lipoprotein metabolism. J Biol Chem. 2003;278:52820–52825. doi: 10.1074/jbc.M310482200. [DOI] [PubMed] [Google Scholar]
- 31.Kocher O, Yesilaltay A, Shen CH, Zhang S, Daniels K, Pal R, Chen J, Krieger M. Influence of PDZK1 on lipoprotein metabolism and atherosclerosis. Biochim Biophys Acta. 2008;1782:310–316. doi: 10.1016/j.bbadis.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dole VS, Matuskova J, Vasile E, Yesilaltay A, Bergmeier W, Bernimoulin M, Wagner DD, Krieger M. Thrombocytopenia and platelet abnormalities in high-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:1111–1116. doi: 10.1161/ATVBAHA.108.162347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korporaal SJ, Meurs I, Hauer AD, Hildebrand RB, Hoekstra M, Cate HT, Pratico D, Akkerman JW, Van Berkel TJ, Kuiper J, Van Eck M. Deletion of the high-density lipoprotein receptor scavenger receptor BI in mice modulates thrombosis susceptibility and indirectly affects platelet function by elevation of plasma free cholesterol. Arterioscler Thromb Vasc Biol. 2011;31:34–42. doi: 10.1161/ATVBAHA.110.210252. [DOI] [PubMed] [Google Scholar]
- 34.Ma Y, Ashraf MZ, Podrez EA. Scavenger receptor BI modulates platelet reactivity and thrombosis in dyslipidemia. Blood. 2010;116:1932–1941. doi: 10.1182/blood-2010-02-268508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li D, Weng S, Yang B, Zander DS, Saldeen T, Nichols WW, Khan S, Mehta JL. Inhibition of arterial thrombus formation by ApoA1 Milano. Arterioscler Thromb Vasc Biol. 1999;19:378–383. doi: 10.1161/01.atv.19.2.378. [DOI] [PubMed] [Google Scholar]
- 36.Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Kollnberger M, Wakefield TW, Lammle B, Massberg S, Wagner DD. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117:1400–1407. doi: 10.1182/blood-2010-05-287623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci U S A. 1997;94:12610–12615. doi: 10.1073/pnas.94.23.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yesilaltay A, Morales MG, Amigo L, Zanlungo S, Rigotti A, Karackattu SL, Donahee MH, Kozarsky KF, Krieger M. Effects of hepatic expression of the high-density lipoprotein receptor SR-BI on lipoprotein metabolism and female fertility. Endocrinology. 2006;147:1577–1588. doi: 10.1210/en.2005-1286. [DOI] [PubMed] [Google Scholar]
- 39.Andre P, Denis CV, Ware J, Saffaripour S, Hynes RO, Ruggeri ZM, Wagner DD. Platelets adhere to and translocate on von Willebrand factor presented by endothelium in stimulated veins. Blood. 2000;96:3322–3328. [PubMed] [Google Scholar]
- 40.Kozarsky KF, Donahee MH, Rigotti A, Iqbal SN, Edelman ER, Krieger M. Overexpression of the HDL receptor SR-BI alters plasma HDL and bile cholesterol levels. Nature. 1997;387:414–417. doi: 10.1038/387414a0. [DOI] [PubMed] [Google Scholar]
- 41.Griffin JH, Kojima K, Banka CL, Curtiss LK, Fernandez JA. High-density lipoprotein enhancement of anticoagulant activities of plasma protein S and activated protein C. J Clin Invest. 1999;103:219–227. doi: 10.1172/JCI5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williamson R, Lee D, Hagaman J, Maeda N. Marked reduction of high density lipoprotein cholesterol in mice genetically modified to lack apolipoprotein A-I. Proc Natl Acad Sci U S A. 1992;89:7134–7138. doi: 10.1073/pnas.89.15.7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu ML, Reilly MP, Casasanto P, McKenzie SE, Williams KJ. Cholesterol enrichment of human monocyte/macrophages induces surface exposure of phosphatidylserine and the release of biologically-active tissue factor-positive microvesicles. Arterioscler Thromb Vasc Biol. 2007;27:430–435. doi: 10.1161/01.ATV.0000254674.47693.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saku K, Ahmad M, Glas-Greenwalt P, Kashyap ML. Activation of fibrinolysis by apolipoproteins of high density lipoproteins in man. Thromb Res. 1985;39:1–8. doi: 10.1016/0049-3848(85)90116-1. [DOI] [PubMed] [Google Scholar]
- 45.Braun A, Zhang S, Miettinen HE, Ebrahim S, Holm TM, Vasile E, Post MJ, Yoerger DM, Picard MH, Krieger JL, Andrews NC, Simons M, Krieger M. Probucol prevents early coronary heart disease and death in the high-density lipoprotein receptor SR-BI/apolipoprotein E double knockout mouse. Proc Natl Acad Sci U S A. 2003;100:7283–7288. doi: 10.1073/pnas.1237725100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holm TM, Braun A, Trigatti BL, Brugnara C, Sakamoto M, Krieger M, Andrews NC. Failure of red blood cell maturation in mice with defects in the high-density lipoprotein receptor SR-BI. Blood. 2002;99:1817–1824. doi: 10.1182/blood.v99.5.1817. [DOI] [PubMed] [Google Scholar]
- 47.Nofer JR, van der Giet M, Tolle M, Wolinska I, von Wnuck Lipinski K, Baba HA, Tietge UJ, Godecke A, Ishii I, Kleuser B, Schafers M, Fobker M, Zidek W, Assmann G, Chun J, Levkau B. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J Clin Invest. 2004;113:569–581. doi: 10.1172/JCI18004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murphy AJ, Woollard KJ, Suhartoyo A, Stirzaker RA, Shaw J, Sviridov D, Chin-Dusting JP. Neutrophil activation is attenuated by high-density lipoprotein and apolipoprotein a-I in in vitro and in vivo models of inflammation. Arterioscler Thromb Vasc Biol. 2011;31:1333–1341. doi: 10.1161/ATVBAHA.111.226258. [DOI] [PubMed] [Google Scholar]
- 49.Radojkovic C, Genoux A, Pons V, Combes G, de Jonge H, Champagne E, Rolland C, Perret B, Collet X, Terce F, Martinez LO. Stimulation of cell surface F1-ATPase activity by apolipoprotein A-I inhibits endothelial cell apoptosis and promotes proliferation. Arterioscler Thromb Vasc Biol. 2009;29:1125–1130. doi: 10.1161/ATVBAHA.109.187997. [DOI] [PubMed] [Google Scholar]
- 50.Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, Hara MR, Quick RA, Cao W, O’Rourke B, Lowenstein JM, Pevsner J, Wagner DD, Lowenstein CJ. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell. 2003;115:139–150. doi: 10.1016/s0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wagner DD, Frenette PS. The vessel wall and its interactions. Blood. 2008;111:5271– 5281. doi: 10.1182/blood-2008-01-078204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Calabresi L, Gomaraschi M, Franceschini G. Endothelial protection by high-density lipoproteins: from bench to bedside. Arterioscler Thromb Vasc Biol. 2003;23:1724–1731. doi: 10.1161/01.ATV.0000094961.74697.54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.