

Abstract
Cell-based microarrays are being increasingly used as a tool for combinatorial and high throughput screening of cellular microenvironments. Analysis of microarrays requires several steps, including microarray imaging, identification of cell spots, quality control, and data exploration. While high content image analysis, cell counting, and cell pattern recognition methods are established, there is a need for new post-processing and quality control methods for cell-based microarrays used to investigate combinatorial microenvironments. Previously, microarrayed cell spot identification and quality control were performed manually, leading to excessive processing time and potentially resulting in human bias. This work introduces an automated approach to identify cell-based microarray spots and spot quality control. The approach was used to analyze the adhesion of murine cardiac side population cells on combinatorial arrays of extracellular matrix proteins. Microarrays were imaged by automated fluorescence microscopy and cells were identified using open-source image analysis software (CellProfiler™). From these images, clusters of cells making up single cell spots were reliably identified by analyzing the distances between cells using a density-based clustering algorithm (OPTICS). Naïve Bayesian classifiers trained on manually scored training sets identified good and poor quality spots using spot size, number of cells per spot, and cell location as quality control criteria. Combined, the approach identified 78% of high quality spots and 87% of poor quality spots. Full factorial analysis of the resulting microarray data revealed that collagen IV exhibited the highest positive effect on cell attachment. This data processing approach allows for fast and unbiased analysis of cell-based microarray data.
In vivo, cells receive and process tens of thousands of microenvironmental signals. Recapitulating these physical, chemical, and biological cues in vitro creates an enormous experimental design space that is exceedingly difficult to study in a parametric manner using traditional cell culture techniques. Cell-based1–6 and biomaterials microarrays7–10 can help address this problem. These technologies build on existing high throughput synthesis and screening techniques to create hundreds and thousands of combinatorial microenvironments to study cellular behaviors. The widespread use of nucleic acid microarrays has led to the standardization of analyzing fluorescence-based microarray data.11 While significant advances have been made in image acquisition and analysis of cell-based microarrays in terms of cell identification, high content analysis, and cell pattern recognition12–14, methods for automated identification of clusters of cells, or cell spots, and quality control of identified spots from high throughput data are needed to advance cell-based microarrays technologies.
Inherent to miniaturizing and multiplexing biological experiments is high variability in output data due to, among other things, high numbers of experimental repetitions and low numbers of biological events per repetition. Errors and variability in outputs from high throughput and multiplexed cell-based experiments can also originate in the generation of the combinatorial protein libraries, microarray fabrication, cell seeding, immunocytochemical analysis, and image acquisition.15
Obtaining high quality data from microarrays requires several steps including, imaging, spot identification, spot quality control, and data analysis. Each of these steps are well established in DNA based microarrays11, 16–18, but due to differences in experimental outcomes and microarray construction they cannot be directly applied to cell-based microarrays. For example, outputs of DNA microarray experiments are spots with homogeneous fluorescence, while immunocytochemical analysis and fluorescent staining of cell-based microarrays results in heterogeneous fluorescence within a cell spot.1, 6
Sophisticated commercial and open source software programs are available that readily identify cell nuclei, cytoskeletons, and immunohistochemical staining from fluorescence microscopy images12, 14 and techniques for confining cells within three-dimensional micropatterned arrays19 and two-dimensional geometries of cell adhesion proteins and biomaterials have been developed13. However, these techniques still require post-processing of image analysis data and quality control methods to successfully perform robust high throughput screening. For example, non-specific cell attachment to microarray backgrounds can distort spot size and interfere with spot detection, and uneven cell seeding can occur with cell types prone to forming aggregates. To address these and other potential sources of errors, cell-based microarrays are often evaluated manually1, 2, 6, 20: images of each spot are inspected, cells are counted, and spots that are clearly damaged, missing, abnormally sized, or otherwise visibly abnormal are excluded from further analyses. This method of quality control and spot identification limits the high throughput nature of the experiments and is prone to human error and bias.
Here, we implement an approach to microarray analysis that accounts for potential sources of variability and errors in cell-based microarrays (Figure 1). The approach is adapted from existing DNA microarray and cell-based imaging analyses and combines spot identification with quality control to extract high quality data from cell-based microarrays. Fluorescence images of stained microarrays were first acquired by automated microscopy, and all cells in a single image were identified by image analysis software (CellProfiler™; Broad Institute). For spot identification, we employed a density-based clustering algorithm (OPTICS)21 to analyze the distances between all cells within a single image. This algorithm was previously used for clustering protein sequences22, and was here used to identify cells confined to microarray spots from background fluorescence, non-specifically adhered cells, and debris and imaging artifacts erroneously identified as cells during image analysis. Two methods of quality control were tested and compared including a 3-step scoring algorithm and naïve Bayesian classifiers. Both methods applied quality control parameters of cell cluster size, number of cells per spot, and cell location. The methods were compared in terms of their sensitivity (identification of true positives), specificity (identification of true negatives), and correlation to a manually scored dataset. We used the best of these strategies (OPTICS followed by naïve Bayesian classifiers) to evaluate the main and interaction effects of murine cardiac side population (CSP) cell density on a combinatorial extracellular matrix (ECM) microarray. CSP cells have the potential for cardiomyogenic differentiation23 and study of these cells in combinatorial microenvironments will help in developing new cardiac therapies.
Figure 1.

Fabrication, image acquisition, and data processing of cell-based microarrays. (i) Cartoon representations of the fabrication and imaging of combinatorial extracellular matrix (ECM) microarrays with seeded cells. Microarrays are created by robotic contact printing and cell spots are imaged by automated microscopy after immunocytochemical staining. Fluorescence images and data are processed by different analysis approaches (ii and iii). The data processing approaches are general and can be applied to the evaluation of fluorescent markers and visible stains. The first step in each approach uses CellProfiler™ to identify all individual cells; the subsequent methods identify which cells belong to cell spots and which cell spots are of good quality.
To evaluate our data processing strategies we used an established cell-ECM microarray fabrication method that has previously been used to study mouse embryonic stem cell differentiation1 and hepatic stellate cell activation2. In this method, ECM protein microarrays are prepared on a standard glass slide modified with a polyacrylamide gel. ECM proteins were adsorbed on the ployacrylamid gel pad, which was hydrophobic when dried. In an aqueous environment, the hydrated polyacrylamide gel pad was resistant to the adsorption of serum proteins and cells preferentially adhered to the microarrayed ECM proteins.1 Herein, cell-ECM microarrays comprised of four randomized sub-arrays that each included all single, binary, and tertiary combinations of fibronectin, laminin, and collagens I, III, and IV were used. The microarrays also included intra-array negative controls of bovine serum albumin (of equal concentration to the ECM proteins) as well as negative controls with no added proteins.
Analysis of cell-based microarrays differs from nucleic acid based microarrays as the latter results in microarrayed spots with homogenously distributed fluorescence. In cell-based assays immunocytochemical analysis can provide useful information, but such analysis results in microarrayed spots with heterogeneous fluorescence. For example, nuclear staining results in discrete fluorescence for each attached cell. This information is useful in high content analysis but can present a challenge to reliable high throughput identification of cell spots. Previously established spot identification techniques are based on the homogeneous distribution of fluorescence seen in nucleic acid microarrays18 and therefore not applicable to the heterogeneities in cell-based microarrays. Existing image analysis software is capable of identifying individual cell bodies, cell nuclei, and specific fluorescent antibodies staining, but such analysis does not identify clustered cells confined to a microarray spot from other artifacts within an image.
A fluorescence microscope with automated staging was used to acquire two color images (DAPI stained nuclei and fluorescently-labeled phalloidin stained actin filaments) of each individual microarrayed spot after 12 hours of culture. While laser scanning, similar to that used in nucleic acid microarrays is feasible1, 7, 24, the low resolution of laser scanners limits this method to surrogate measurements of cell counts. A more direct measure of cell counts is possible by fluorescence microscopy and automatic cell detection software.25, 26 Nuclei and associated cell bodies on each image were identified using an image analysis protocol in CellProfiler™ software. Parameters used in the protocol and example images are shown in Supporting Information Figure 1 (SFigure 1). Spots within a microarray (Figure 2A) are variable and can result in high quality spots with minimal non-specific adhesion (Figure 2B and C), spots with high non-specific adhesion (Figure 2D), and damaged spots (Figure 2E). These images demonstrate the need to employ spot identification and subsequent quality control to attain high quality cell-based microarray data. This need is exemplified in Figure 2E, where the debris (circled in white) is classified as a number of connected cells by CellProfiler™ (SFigure 1).
Figure 2.
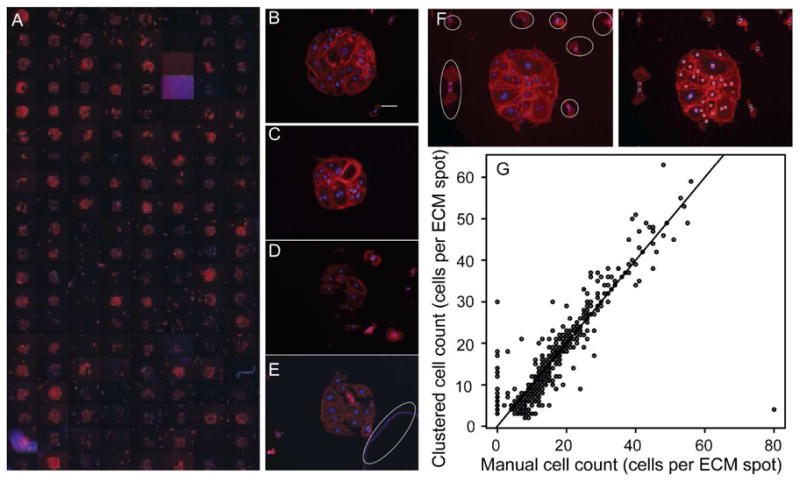
Combinatorial ECM-cardiac side population (CSP) cell microarrays. (A) A mosaic of fluorescent images of a combinatorial ECM microarray seeded with 10,000 CSP cells per cm2. Images were acquired at 10X magnification after 12 hours of culture and show phalloidin (red) and nuclear (blue) staining. The mosaic image shows one of four sub-arrays per glass slide. Each sub- array contains five replicates of 30 combinations of Fn, Lm, CI, CIII, and CIV (300 μg/mL total protein concentration) and five replicates of negative controls with no protein and five replicates containing 300 μg/mL of BSA. Sets of five replicates in each sub-array are randomly ordered. (B–E) Selected images of microarrayed spots that are of high quality (B,C), low quality (D), and damaged or contain debris (E; debris is circle in white). The brightness of image (E) has been increase so that the identified debris is clearly visible. Scale bar is 100 μm. (F) Fluorescent image of a single microarrayed spot with adhered CSP cells (left) with outliers from the microarrayed spot circled in white. Cells within a cluster are identified with a red spot, while cells outside of the cluster are identified with a black spot (right). Phalloidin stain is shown in red, while the nuclei are shown in blue. (G) Correlation of CSP cells per microarrayed spot as determined by counting each cell through visual inspection and by identification with the OPTICS clustering algorithm.
After initial processing of the single-spot images by CellProfiler™, the identified nuclei were analyzed and clustered using the OPTICS density based clustering algorithm.21 Cells were designated as belonging to a cluster if at least 4 cells were found within a specified distance or they were within the same distance of a cell already belonging to a cluster. Cells were reliably identified as belonging to one cluster and outlying cells were classified as noise (Figure 2F). To control for accurate cell counts per identified cluster, cells on spots were counted manually. Clustered cell counts shows good correlation (r = 0.88, p < 0.001) with the manually counted numbers (Figure 2G); however, there are many cell counts in the lower left quadrant of the correlation as well as in the upper right quadrant that correlate very poorly. We attribute the high number of false positives in the lower left quadrant that show positive cell counts when zero cells are present to a misidentification during image analysis of small indentations in the hydrogel substrate made by the contact printer as adhered cells. While the clustering algorithm does well in identifying many of the cell spots an additional level of quality control was required to account for damaged, missing, poorly imaged, or otherwise poor quality spots.
Two different approaches were used for quality control of the identified cell clusters: automatic scoring based on a predefined set of rules and naïve Bayesian classifiers. Both methods used cell cluster size, number of cells per spot, and cell location as parameters to evaluate each spot. These criteria were selected based on the most common errors and inconsistencies that we observed during microarray fabrication and analysis. For example, abnormally high cell counts resulted from imaging artifacts and debris (SFigure 1B) that were identified as cells, blank spots were identified as small cell clusters due to a small indentation on the substrate made by microarray pins, and moderately high levels of non-specific cell attachment resulted in the classification of two cell clusters. In the case of the automatic scoring method, spots were considered of good quality if the cluster size, the number of cells per spot, and the cell location were within two standard deviations of the mean. Cells and spots outside of two standard deviations were considered poor quality. Two manually scored arrays were used as training sets for the naïve Bayesian classifiers to identify the best values for the three quality control criteria.
The successes of the quality control methods were judged by evaluating the sensitivity (true positives) and specificity (true negatives) of the methods, and by correlation of processed data to a manually scored dataset. Sensitivity was defined as the percent of cell spots identified as good quality by a method (clustering, automatic scoring, or naïve Bayesian) and also by manual evaluation. Specificity was defined as the percentage of cell spots identified as poor quality by a method and also by manual evaluation. The manually scored dataset was considered a gold standard for comparison and was evaluated with the following four criteria: i) cells were present on an image, ii) cells formed a distinguishable cluster within the image, iii) there were no more than 5 cells outside of the cluster, and iv) there were no artifacts in the image.
Without quality control, the clustering algorithm results in datasets with 100% sensitivity and 0% specificity (Figure 3A, left). One hundred percent sensitivity and 0% specificity indicates that no quality control is applied and that all cell clusters are indiscriminately included (both true positives and false positives are classified as good quality). Application of the automatic scoring algorithm resulted in a high level of quality control with 94% specificity but only 44% sensitivity (Figure 3A, middle). In this case, a large number of poor quality spots were identified correctly but at the expense of incorrectly classifying many good quality spots. Application of the naïve Bayesian classifiers onto the same datasets improved sensitivity to 78% while maintaining high specificity of 87%, thus correctly classifying high percentages of poor and good quality spots, respectively.
Figure 3.

Comparison of quality control methods for cell attachment on combinatorial cell-ECM microarrays. (A) Graphical representations of the sensitivity and specificity of data from cluster analysis, automated scoring, and naïve Bayesian analysis. In a perfect dataset all high or low quality spots scored manually would also be considered high or low quality, respectively, by the quality control strategies (100% sensitive and 100% specific). (A, left) The clustered dataset was 100% sensitive (lower left quadrant) and 0% specific (upper right quadrant). (A, middle, right) The automated scoring quality control resulted in 44% sensitivity and 94% specificity, while the naïve Bayesian analysis quality control resulted in 78% sensitivity and 87% specificity. (B) Correlations of the average number of cells per ECM combination resultant from the cluster analysis (left), the automated scoring (middle), and naïve Bayesian analysis (right). The dashed line is x = y, and the solid line is a linear fit to the correlated data.
The quality control methods were also evaluated by comparing the mean cell counts per ECM combination. The mean cells per spot for a given ECM combination for the cluster analysis, automatically scored, and naïve Bayesian scored data were correlated with manually scored data to check for possible biasing by the quality control methods (Figure 3B). Clustered data showed strong correlation with manually scored data (r = 0.89) indicating good overall printing and seeding quality. Automatic scoring showed a weaker correlation (r = 0.74) indicating over correction of the dataset, while naïve Bayesian classifiers showed the highest correlation (r = 0.95) indicating successful removal of outliers and low quality spots. While automatic scoring performed well in identifying low quality spots (i.e. has high specificity), it overcorrected the data by excluding many good data points. Using naïve Bayesian classifiers trained on a manually scored subset to identify low quality spots resulted in both high sensitivity and specificity, thus successfully removing low quality spots without bias. Spot identification and quality control analysis leads to the exclusion of microarrayed spots from final analysis. Our analysis resulted in an exclusion rate of 65% after naïve Bayesian classification, which compares favorably with previous cell-based microarrays that ranged up to 50% exclusion.1
The power of cell-based microarrays is the ability to simultaneously evaluate combinatorial microenvironments from which the main and interaction effects of the independent variables can be extracted. Here, a full factorial analysis was used to determine the effects of ECM proteins on CSP cell attachment from the naïve Bayesian classifiers scored datasets (Figure 4). The main effects of all ECM components identified from the Bayesian analysis are positive and significant (p<0.05). This is also the case in the calculated effects of the manually scored subset (Figure 4). Microarrays were seeded with 10,000 cells per cm2, resulting in a theoretical initial density of 7 cells per spot (average spot diameter of 300 μm). The average number of cells per spot across the microarray was 18±10 cells, and each ECM exhibited a positive main effect (CIV, 4.3±0.4; Fn, 3.7±0.4; Lm, 3.4±0.4; CI, 2.5±0.4; CIII, 1.8±0.4; values are stated for the Bayesian classifiers scored dataset).
Figure 4.
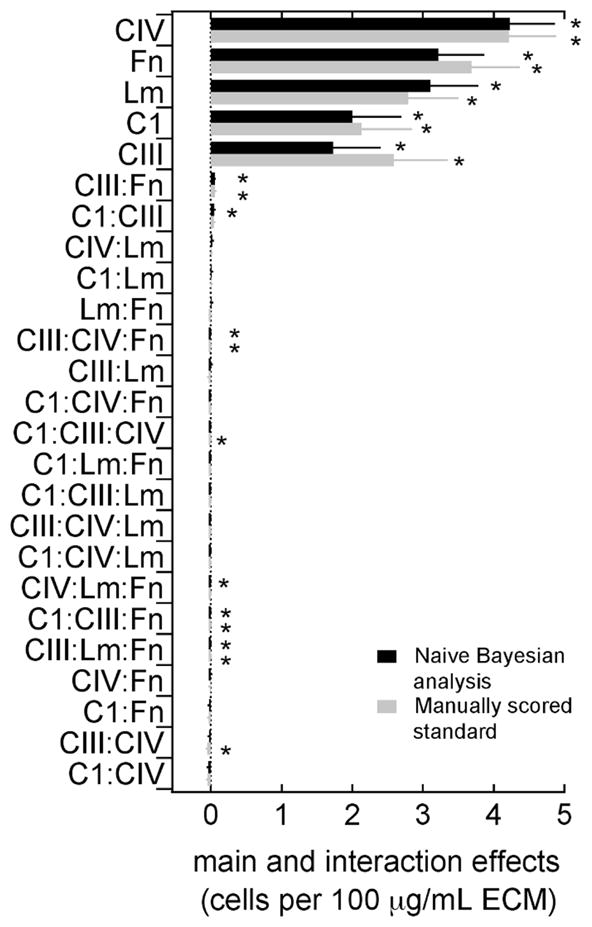
The main and interaction effects of extracellular matrix (ECM) proteins on adhesion of murine cardiac side population (CSP) cells for the Naïve Bayesian analysis quality control method (black) and for the manually scored standard (grey). Effects are ordered from top to bottom in descending order (*, p < 0.05).
Interaction effects between ECM components were at least 2 orders of magnitude smaller than the observed main effects, indicating that synergistic effects between ECM compounds were minimal. However, combinations of different ECM components were neither favored nor avoided in cell adhesion. Since cell density can alter individual cell biology, having similar attachment among all components can be beneficial in evaluating other cellular behaviors. It is expected that significant and large interaction effects will be observed in the analysis of more complex cell behaviors similar to what has previously been described.1, 2
Cell-based microarray experiments differ from macroscale experiments in several ways. Spots can be misprinted leading to a lack of ECM deposition, spots can be missed during cell seeding, and artifacts introduced during staining can render spots unusable. In previous experiments on cell-based microarrays such spots were excluded manually.1, 6, 20 Manual evaluation is a time consuming task especially when microarrays become large and consist of several thousand spots. Spot identification and single-spot quality control is well established in nucleic acid based assays.16, 27, 28 These techniques rely on homogenously distributed fluorescence values and therefore are difficult to directly translate to cell-based data as obtained from cell-based microarrays. The use of naïve Bayesian classifiers on nucleic acid microarrays has been described,29 and as the parameters used to construct naïve Bayesian classifiers could be defined, this strategy was successfully adapted to cell-based microarrays here. The methods described here are intended to augment image analysis software as the methods rely on the image analysis capabilities of CellProfiler™ (or similar software) to identify cells in acquired images. The combined application of image analysis and the methods described here will help advance new combinatorial and high throughput investigations of cellular microenvironment by automating post-processing (in this case spot identification) and quality control.
Cell-based microarrays are a promising tool for the exploration of the vast experimental space created by the physical, chemical, and biological parameters of the cellular microenvironment. Investigating this space will help advance cell therapies and tissue engineering technologies, and the automated spot detection and quality control tools developed here will help do so in a high throughput manner.
Materials and Methods
Fabrication of combinatorial extracellular matrix (ECM) microarrays
Microarrays of single, binary, and tertiary combinations of fibronectin, laminin, collagen I, collagen III, and collagen IV were fabricated using a robotic contact microarrayer (SpotBot3, ArrayIt Corp.). The microarrays were produced on standard-size glass microscope slides (25 × 75 mm) modified with a polyacrylamide gel pad.1, 2 Glass microscope slides were cleaned with 10 %(wt/v) NaOH and modified with 3-(trimethoxysilyl) propyl methacrylate. After curing at 80 °C for 10–12 hours, the silanized glass were washed (3-times in 100 % ethanol) and a 100 μL gel pad of 9.5 %(wt/v) acrylamide, 0.5 %(wt/v) bis-acrylamide and 20 μg mL−1 photoinitiator 4-(2-hydroxyethoxy)phenyl-(2-hydroxy-2-propyl)ketone; Irgacure 2959) was photopolymerized to each slide. The robotic spotter, equipped with a 946MP9 pin (0.24 nL per spot, 300 μm spot diameter) was used to create microarrays of ECM spots with a pitch of 900 μm and a total of 640 spots per modified glass slide. Each ECM was printed at 100 μg mL−1 concentration and a 300 mg μL−1 total protein concentration was maintained with bovine serum albumin as needed. ECM protein solutions were prepared in 100 mM acetate, 5 mM EDTA, 20 %(v/v) glycerol, and 0.25 % Trinton-X100 adjusted to pH 5. Printing was done in a controlled environment with a constant humidity of 40 % and a substrate temperature of 15 °C. Printed microarrays were incubated at 4 °C for 16 hours prior to use.
Cardiac side population (CSP) cell isolation and culture
CSP cells were isolated from adult mouse hearts as described before.30 Briefly, hearts were digested and mononuclear cells were stained with Hoechst 33423 and for the surface markers Sca1 and CD31. Sca1+ CD31- Hoechst low cells were selected using FACS. The cells were cultured in alpha-MEM containing 20% FBS (CSP media) and used between passages 4–6.
After incubation at 4 °C for 16 hours printed microarrays were washed with sterile phosphate buffered saline and the gel pads were allowed to swell for 30 minutes. CSP cells were seeded at a density of 10,000 cells cm−2 in 5 mL of CSP media. After 12 hours cells were fixed with 4% paraformaldehyde for 20 minutes and permeabilized with a solution of 0.5% Triton-X100 and 1% BSA. Microarrays were washed with 5 mL PBS 3-times between each step. F-actin was visualized by staining with phalloidin (Alexa Fluor® 594 phalloidin, dilution 1:40, 30 minutes at room temperature) and nuclei were visualized with mounting media containing DAPI (Vectashield).
Fluorescence microscopy and image acquisition
Series of grayscale images of immunostained samples were acquired using the automated stage of a Zeiss Axio Observer fluorescent microscope and were analyzed using the cell image analysis software CellProfiler™ (Broad Institute). A CellProfiler™ analysis pipeline was created to identify cell nuclei and cell bodies (SFigure 1).
Cell-based microarray spot detection
Cell clusters were identified from CellProfiler™ outputs by the density based algorithm OPTICS.21 Two hundred pixels (120 μm) were set as the maximum radius and 5 cells were defined as the minimum cluster size. In case of multiple identified clusters, the cluster with the highest cell number was chosen to proceed. Clustered cell numbers, dimensions, and cell positions within the cluster were exported for further quality scoring.
Manual scoring and quality control
Images were manually scored based on the following rules: 1) Cells were present; 2) Cells formed a cluster and the cluster was clearly detected by the clustering algorithm; 3) No more than 5 cells were outside the cluster; and, 4) no artifacts were present in the image.
Quality control: automated scoring and naïve Bayesian analysis
Two different strategies were used to detect erroneous spots: automatic scoring using a pre-defined set of rules and naïve Bayesian classifiers. For automatic scoring, cluster dimensions were evaluated first. Subsequently, spots were scored based on their cell number, and variation in cell location. For naïve Bayesian classifiers manually scored arrays were used as training data. The same criteria as were used for automatic scoring were used to determine Bayesian distributions. The distribution model was then used to predict the class of spots. The program scripts used for clustering and scoring are available in the supporting information. The two manually scored arrays were used separately to create training sets. Sensitivity and specificity of the prediction were determined by using the model from array 1 to predict array 2 and vice versa. The software used for clustering and scoring is available online at https://gitorious.org/optics-cellprofiler
Data processing and Statistical analysis
Spot identification by cluster analysis and automated quality control scoring were performed using python, and statistical analysis and naïve Bayesian classification was performed using R 2.10.1 with added software package e1071. Sensitivity and specificity were defined as (true positives/(true positives+false negatives)) and (true negatives/(true negatives+false positives)) respectively. Correlations were performed using Pearson’s test. Full factorial analysis for effect sizes was performed by using multi-factorial analysis of variance (ANOVA).
Supplementary Material
Acknowledgments
This study was supported by the NIH (HL099073) M.B. received an AHA Post-doctoral fellowship for the duration of the study. K.K. received a Natural Sciences and Engineering Research Council of Canada Post-doctoral fellowship. The authors thank the mechanical Turks for helping on manually counting and scoring microarray images.
Footnotes
M.B. and I.W. conceived and designed the experiments. K.K. designed and fabricated the combinatorial ECM arrays. B.C. and Y.Q. conducted experiments and imaging. M.B. conceived and developed approaches for spot identification and quality control. M.B. and I.W. wrote the manuscript with comments and editing by A.K. and R.L.
CellProfiler™ pipeline input parameters and example CellProfiler™ output images, spot identification and quality control scripts.
References
- 1.Flaim CJ, Chien S, Bhatia SN. An extracellular matrix microarray for probing cellular differentiation. Nat Methods. 2005;2(2):119–125. doi: 10.1038/nmeth736. [DOI] [PubMed] [Google Scholar]
- 2.Brafman DA, de Minicis S, Seki E, Shah KD, Teng DY, Brenner D, Willert K, Chien S. Investigating the role of the extracellular environment in modulating hepatic stellate cell biology with arrayed combinatorial microenvironments. Integrative Biology. 2009;1(8–9):513–524. doi: 10.1039/b912926j. [DOI] [PubMed] [Google Scholar]
- 3.Tavana H, Jovic A, Mosadegh B, Lee QY, Liu X, Luker KE, Luker GD, Weiss SJ, Takayama S. Nanolitre liquid patterning in aqueous environments for spatially defined reagent delivery to mammalian cells. Nat Mater. 2009;8(9):736–41. doi: 10.1038/nmat2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flaim CJ, Teng D, Chien S, Bhatia SN. Combinatorial signaling microenvironments for studying stem cell fate. Stem Cells Dev. 2008;17(1):29–39. doi: 10.1089/scd.2007.0085. [DOI] [PubMed] [Google Scholar]
- 5.Yarmush ML, King KR. Living-Cell Microarrays. Annual Review of Biomedical Engineering. 2009;11:235–257. doi: 10.1146/annurev.bioeng.10.061807.160502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu JH, Wheeldon I, Guo YQ, Lu TL, Du YN, Wang B, He JK, Hu YQ, Khademhosseini A. A sandwiched microarray platform for benchtop cell-based high throughput screening. Biomaterials. 2011;32(3):841–848. doi: 10.1016/j.biomaterials.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson DG, Levenberg S, Langer R. Nanoliter-scale synthesis of arrayed biomaterials and application to human embryonic stem cells. Nature Biotechnology. 2004;22(7):863–866. doi: 10.1038/nbt981. [DOI] [PubMed] [Google Scholar]
- 8.Anderson DG, Putnam D, Lavik EB, Mahmood TA, Langer R. Biomaterial microarrays: rapid, microscale screening of polymer-cell interaction. Biomaterials. 2005;26(23):4892–4897. doi: 10.1016/j.biomaterials.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 9.Lee MY, Kumar RA, Sukumaran SM, Hogg MG, Clark DS, Dordick JS. Three-dimensional cellular microarray for high-throughput toxicology assays. Proc Natl Acad Sci U S A. 2008;105(1):59–63. doi: 10.1073/pnas.0708756105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernandes TG, Kwon SJ, Bale SS, Lee MY, Diogo MM, Clark DS, Cabral JMS, Dordick JS. Three-Dimensional Cell Culture Microarray for High-Throughput Studies of Stem Cell Fate. Biotechnology and Bioengineering. 106(1):106–118. doi: 10.1002/bit.22661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karakach TK, Flight RM, Douglas SE, Wentzell PD. An introduction to DNA microarrays for gene expression analysis. Chemometrics Intell Lab Syst. 104(1):28–52. [Google Scholar]
- 12.Stromberg S, Bjorklund MG, Asplund C, Skollermo A, Persson A, Wester K, Kampf C, Nilsson P, Andersson AC, Uhlen M, Kononen J, Ponten F, Asplund A. A high-throughput strategy for protein profiling in cell microarrays using automated image analysis. Proteomics. 2007;7(13):2142–2150. doi: 10.1002/pmic.200700199. [DOI] [PubMed] [Google Scholar]
- 13.Unadkat HV, Hulsman M, Cornelissen K, Papenburg BJ, Truckenmuller RK, Post GF, Uetz M, Reinders MJT, Stamatialis D, van Blitterswijk CA, de Boer J. An algorithm-based topographical biomaterials library to instruct cell fate. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(40):16565–16570. doi: 10.1073/pnas.1109861108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamprecht MR, Sabatini DM, Carpenter AE. CellProfiler(TM): free, versatile software for automated biological image analysis. Biotechniques. 2007;42(1):71–75. doi: 10.2144/000112257. [DOI] [PubMed] [Google Scholar]
- 15.Inglese J, Shamu CE, Guy RK. Reporting data from high-throughput screening of small-molecule libraries. Nat Chem Biol. 2007;3(8):438–441. doi: 10.1038/nchembio0807-438. [DOI] [PubMed] [Google Scholar]
- 16.Kauffmann A, Gentleman R, Huber W. arrayQualityMetrics–a bioconductor package for quality assessment of microarray data. Bioinformatics. 2009;25:415. doi: 10.1093/bioinformatics/btn647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kauffmann A, Huber W. Genomics Microarray data quality control improves the detection of differentially expressed genes. Genomics. 2010;95:138–142. doi: 10.1016/j.ygeno.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Yang YH, Buckley MJ, Speed TP. Analysis of cDNA microarray images. Briefings in bioinformatics. 2001;2:341–9. doi: 10.1093/bib/2.4.341. [DOI] [PubMed] [Google Scholar]
- 19.Mercey E, Obeid P, Glaise D, Calvo-Munoz ML, Guguen-Guillouzo C, Fouque B. The application of 3D micropatterning of agarose substrate for cell culture and in situ comet assays. Biomaterials. 2010;31(12):3156–3165. doi: 10.1016/j.biomaterials.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 20.Kwon CH, Wheeldon I, Kachouie NN, Lee SH, Bae H, Sant S, Fukuda J, Kang JW, Khademhosseini A. Drug-Eluting Microarrays for Cell-Based Screening of Chemical-Induced Apoptosis. Anal Chem. 2011;83(11):4118–4125. doi: 10.1021/ac200267t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ankerst M, Breunig MM, Kriegel HP, Sander J. ACM SIGMOD Record, ACM. 1999. OPTICS: ordering points to identify the clustering structure; pp. 49–60. [Google Scholar]
- 22.Chen Y, Reilly KD, Sprague AP, Guan Z. SEQOPTICS: a protein sequence clustering system. BMC bioinformatics. 2006;7(Suppl 4):S10. doi: 10.1186/1471-2105-7-S4-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfister O, Mouquet F, Jain M, Summer R, Helmes M, Fine A, Colucci WS, Liao R. CD31- but Not CD31+ Cardiac Side Population Cells Exhibit Functional Cardiomyogenic Differentiation. Circulation Research. 2005;97(1):52–61. doi: 10.1161/01.RES.0000173297.53793.fa. [DOI] [PubMed] [Google Scholar]
- 24.Fernandes TG, Diogo MM, Clark DS, Dordick JS, Cabral JMS. High-throughput cellular microarray platforms: applications in drug discovery, toxicology and stem cell research. Trends Biotechnol. 2009;27(6):342–349. doi: 10.1016/j.tibtech.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin Da, Chang JH, Lindquist Ra, Moffat J, Golland P, Sabatini DM. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome biology. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kachouie N, Kang LF, Khademhosseini A. Arraycount, an algorithm for automatic cell counting in microwell arrays. Biotechniques. 2009;47(3):x–xvi. doi: 10.2144/000113202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sauer U, Preininger C, Hany-Schmatzberger R. Quick and simple: quality control of microarray data. Bioinformatics(Oxford, England) 2005;21:1572–8. doi: 10.1093/bioinformatics/bti238. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Ghosh S, Guo S-w. Quantitative quality control in microarray image processing and data acquisition. Nucleic Acids Research. 2001;29:e75. doi: 10.1093/nar/29.15.e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hautaniemi S, Edgren H, Vesanen P, Wolf M, Jarvinen AK, Yli-Harja O, Astola J, Kallioniemi O, Monni O. A novel strategy for microarray quality control using Bayesian networks. Bioinformatics. 2003;19:2031. doi: 10.1093/bioinformatics/btg275. [DOI] [PubMed] [Google Scholar]
- 30.Pfister O, Oikonomopoulos A, Sereti K-I, Liao R. Isolation of Resident Cardiac Progenitor Cells by Hoechst 33342 Staining. Stem Cells for Myocardial Regeneration. 660:53–63. doi: 10.1007/978-1-60761-705-1_4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.