

Abstract
Correct codon-anticodon pairing promotes translational fidelity, with these interactions greatly facilitated by modified nucleosides found in tRNA. We hypothesized that wobble uridine modifications catalyzed by tRNA methyltransferase 9 (Trm9) are essential for translational fidelity. In support, we have used phenotypic, reporter and protein-based assays to demonstrate increased translational infidelity in trm9Δ Saccharomyces cerevisiae cells. Codon reengineering studies suggest that Trm9-catalyzed tRNA modifications promote fidelity during the translation of specific genes, those rich in arginine and glutamic acid codons from mixed boxes. Using quantitative tRNA modification analysis, we determined that trm9Δ cells are only deficient in 2 of 23 tRNA modifications, with those 2, 5-methoxycarbonylmethyluridine (mcm5U) and 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U), classified as key determinants of translational fidelity. We also show that in the absence of mcm5U and mcm5s2U, the resulting translational infidelity promotes protein errors and activation of unfolded protein and heat shock responses. These data support a model in which Trm9-catalyzed tRNA modifications promote fidelity during the translation of specific transcripts, with decreased wobble base modification leading to translational infidelity, protein errors and activation of protein stress response pathways.
Keywords: Trm9, codon, heat shock response, modification, protein stress, tRNA, translation infidelity, unfolded protein response, wobble base
Introduction
Enzyme-catalyzed modification of the wobble nucleosides in tRNA can affect anticodon positioning in the ribosome and has been implicated in the codon-dependent translation of specific transcripts.1-3 Modifications found at the wobble position may be a simple methyl group or a more complicated moiety. For example, in Saccharomyces cerevisiae, the elongator (ELP) complex (consisting of Elp1, Elp2, Elp3, Elp4, Elp5 and Elp6 along with Kti11, Kti12 and Kti13) catalyzes the formation of the 5-carboxymethyluridine (cm5U) side chain on some wobble uridines.4 cm5U serves as a substrate for the S-adenosyl-methionine-dependent tRNA methyltransferase 9 (Trm9) that catalyzes the formation of mcm5U.5,6 The mcm5U modification can also be found in a thiolated form, mcm5s2U, arising from the action of Ncs2.7 The mcm5U and mcm5s2U modifications are present in the wobble uridine of 4 tRNA species (tRNAUCUArg, tRNAUUULys, tRNAUUGGln and tRNAUUCGlu), specific for codons in mixed boxes.6,8 Our recent work using trm9Δ cells, as well as other published studies, supports the idea that anticodons containing mcm5U and mcm5s2U have enhanced affinity for AGA and GAA codons and that such modifications can affect the speed of translation elongation through these codons.9
Wobble base tRNA modifications can also play a role in translation fidelity. In E. coli, it has been shown that these modifications can alter the geometry of the ribosome-decoding center and promote the binding of the anticodon to its cognate codons.1,10 It is important to note that there are both distinct and similar modifications when comparing E. coli and S. cerevisiae tRNA. In theory, tRNA wobble base modifications found in anticodons interacting with codons found in mixed boxes can enhance binding to A- and G-ending codons and prevent binding to U and C.1,10 Un- or under-modified uridines should therefore promote translational infidelity by failing to prevent anticodon binding to U- and C-ending codons. Translational infidelity can also be induced by aminoglycoside antibiotics, which include paromomycin and G418. These antibiotics disturb the ribosome-decoding center to promote misincorporation of amino acids at some near-cognate codons.11 In clinical practice, the aminoglycoside antibiotics have been demonstrated to cause elongation across premature stop codons in mutated ATM, cystic fibrosis and dystropin genes.12-15 While misincorporation can be used to rescue improperly truncated proteins, it also has the potential to promote errors and lead to misfolded proteins.16-18
Misfolded proteins are toxic to cells and can be refolded in the endoplasmic reticulum.19,20 As the cell accumulates misfolded proteins, it triggers a cascade of pathways called the unfolded protein response (UPR).21 Proteins associated with the UPR include Kar2, Ire1 and Hac1, which form the basis of a sensor, activator and transducer system to detect unfolded proteins and to reprogram the cells to respond to these toxic entities.22 The UPR is involved in the quality control of proteins in the endoplasmic reticulum (ER) and targets unfolded proteins for degradation via an endoplasmic reticulum assisted degradation pathway (ERAD).23-25 A corrupted UPR in humans can promote diabetes, Alzheimer disease, Parkinson disease, multiple myeloma, myocardial infarction and stroke.26,27 The ER stress pathways are also activated in many cancers to promote tumor growth.28-30 The strong association between defects or hyper-activation of the UPR with many diseases points to its central role in cellular stress responses.
Unfolded proteins can also activate the heat shock response (HSR),31 which is involved in the quality control of proteins in the cytoplasm.32 The HSR is activated by various stress signals that give rise to unfolded proteins, including high temperature, denaturing agents, heavy metals, free radicals, nutritional stress and hypoxia.33,34 In budding yeast, the HSR is mediated by the phosphorylation of the transcription factor Hsf1, which binds to the heat shock element (HSE) present in the promoter region of several genes and activates transcripts corresponding to chaperones, UPR and ubiquitin proteasomal machinery.35-37 The heat shock pathway in humans is activated during stroke and myocardial infarction,38 and is associated with the neurodegenerative disorders Alzheimer, Parkinson and Huntington’s diseases.34,39,40 In general, heat shock proteins serve a protective role by helping refold or degrade unfolded proteins to prevent aggregate formation.34,41-43
In our current investigation, we performed reporter and protein-based studies to demonstrate that increased amino acid misincorporation and frameshifting occur during translation in trm9Δ cells. Further, codon-reengineering studies support the idea that the translation errors observed in trm9Δ cells occur in specific transcripts. Using molecular and reporter studies, we demonstrate increased unfolded protein and heat shock responses in trm9Δ cells, with this data supporting the idea that increased translation errors lead to unstable proteins. In further support, agents that promote protein synthesis errors were found to increase the translational infidelity and activation of protein stress response pathways in trm9Δ cells. Quantitative analysis of 23 tRNA modifications demonstrated that only mcm5U and mcm5s2 levels are lower in trm9Δ mutants, thus implicating these wobble base modifications as major determinants of translational fidelity. Our study supports a mechanism by which decreased Trm9 activity promotes error-prone translation of specific transcripts, leading to protein stress and the activation of unfolded protein and heat shock responses.
Results
Markers of translational infidelity observed in trm9Δ cells
Paromomycin has been shown to increase translational errors and promote decreased growth and cell death.44,45 Using plate-based assays, we determined that trm9Δ cells, constructed for this study using a URA3 deletion cassette, were sensitive to paromomycin relative to wild-type cells (Fig. 1A). We complemented the paromomycin-sensitive phenotype of trm9 mutants by re-expressing TRM9 under the control of its natural promoter. We also tested other aminoglycoside antibiotics, as they should have a similar effect on trm9 mutants. We performed assays using G418 (Fig. 1A), which will also promote amino acid misincorporation at near-cognate codons, and determined that trm9Δ cells display increased sensitivity to this agent. We note that trm9Δ cells from two different strain backgrounds, By4741 and Cen.PK2 (Fig. 1A; Fig. S1), were sensitive to aminoglycoside antibiotics, which supports a role for Trm9 in preventing translation errors.

Figure 1. Molecular and viability phenotypes associated with translational infidelity identified in trm9Δ cells. (A) Wild-type (By4741), trm9Δ and trm9Δ + TRM9 cells were grown overnight in YPD at 30°C. Cultures were serially (10-fold) diluted and plated on YPD, YPD + paromomycin (350 ug/ml) or YPD + G418 (20 ug/ml). Figure S1 contains similar data for cells from the CEN.PK2–1C background. (B) Wild-type and trm9Δ cells from the BY4741 background were grown to ~5x106 cells/ml and were either mock treated or treated with paromomycin (1000 ug/ml) for 1 h. The cells were spun down and RNA was extracted using an RNeasy mini kit and used for northern blot analysis. (C) Wild-type and trm9Δ cells, both containing an integrated Pnc1-Tap tagged construct, were grown to ~5 x 106 cells/ml and left untreated or treated with 20 μg/ml of G418 for 60 min. Proteins were extracted and western blot analysis of Pnc1-TAP and tubulin was performed as described in Materials and Methods.
Expression of the longevity protein Pnc1 is increased by translation errors and has the potential to be a biomarker of translational infidelity.17 Pnc1 is a nicotinamidase that is part of the NAD(+) salvage pathway. Pnc1 protein levels have been also been demonstrated to increase in response to calorie restriction, heat shock or osmotic stress, making it a biomarker of many different environmental stresses.46,47 We have used northern blots against native PNC1 and immunoblots against an endogenously expressed Pnc1-TAP protein to quantitate corresponding transcript and protein levels in wild-type and trm9Δ cells. Under basal conditions, we have observed a 2.2-fold increase in PNC1 transcript levels in trm9Δ cells relative to wild-type cells (Fig. 1B). Similarly, after paromomycin treatment, we found that PNC1 transcript levels were increased 4.1-fold in trm9Δ cells, relative to treated wild-type cells. The paromomycin-induced increase in PNC1 in trm9Δ cells suggests that this agent exacerbates the existing translational infidelity in tRNA modification-deficient cells. Next, we wanted to determine if our PNC1 transcriptional results were supported at the protein level. Under basal conditions, we observed increased Pnc1-TAP levels in trm9 mutants, relative to wild-type cells (Fig. 1C), thus supporting our transcriptional results. In addition, we observed that, in trm9 mutants, treatment with the aminoglycoside antibiotic G418 increased the expression of Pnc1-TAP relative to untreated trm9Δ cells (Fig. 1C). The PNC1 transcriptional and Pnc1-TAP protein data support our hypothesis that trm9 mutants have increased translational errors.
Wobble base tRNA modifications are the only ones deficient in trm9Δ cells
The levels of mcm5U and mcm5s2U in tRNA in the trm9Δ cells used in this study have not been previously characterized. Further, in theory, the absence of a specific tRNA modification can prevent the addition of or promote the removal of other modifications in specific tRNA species. To confirm loss of mcm5U and mcm5s2U and to investigate the possibility that there will be other changes in tRNA modifications in trm9Δ cells, we used a recently developed liquid chromatography-coupled mass spectrometry (LC-MS) approach to quantify the level of 23 tRNA modifications.48 In trm9Δ cells, there was a significant reduction in mcm5U and mcm5s2U modifications relative to wild-type cells (Table 1). This loss of wobble base modifications is similar to previous observations.6,9 Note that, in this study, we have analyzed 21 other modifications and used a trm9 mutant generated by a URA3 deletion construct. Significantly, we did not detect differences between wild-type and trm9Δ cells in the levels of 21 other tRNA modifications quantitated in our analysis. Thus, the trm9Δ mutants represent a clean system where only the levels of specific wobble base modifications are affected. These observations support the general hypothesis that decreased mcm5U and mcm5s2U levels is driving the translational infidelity phenotypes observed in trm9 mutants.
Table 1. tRNA modification levels in wild-type and trm9Δ cells.
| |
Wild-type |
trm9Δ |
|
||
|---|---|---|---|---|---|
| Average | SD | Average | SD | P-value | |
|
Y |
221.4 |
14.5 |
211.3 |
17.3 |
4.8E-01 |
|
Um |
296.0 |
9.7 |
300.9 |
8.9 |
5.6E-01 |
|
D |
1,302.1 |
72.1 |
1,289.5 |
132.2 |
8.9E-01 |
|
yW |
1,334.7 |
93.9 |
1,585.9 |
290.2 |
2.3E-01 |
|
mcm5U |
2,122.2 |
79.6 |
0.0 |
0.0 |
1.3E-06 |
|
ncm5U |
4,061.9 |
567.7 |
3,647.9 |
155.3 |
2.9E-01 |
|
mcm5s2U |
4,831.6 |
237.1 |
0.0 |
0.0 |
3.8E-06 |
|
m5U |
13,819.9 |
269.4 |
14,332.5 |
1,644.2 |
6.2E-01 |
|
m1A |
21,101.4 |
2,036.6 |
21,694.4 |
6,728.1 |
8.9E-01 |
|
I |
24,622.7 |
1,134.5 |
23,197.7 |
4,595.2 |
6.3E-01 |
|
m1I |
27,372.9 |
3,631.4 |
23,544.9 |
1,578.3 |
1.7E-01 |
|
Am |
29,687.2 |
2,253.9 |
30,380.3 |
1,801.9 |
7.0E-01 |
|
i6A |
49,391.3 |
16,353.1 |
42,417.6 |
2,172.4 |
5.0E-01 |
|
m3C |
62,004.7 |
3,980.5 |
67,676.5 |
7,261.8 |
3.0E-01 |
|
ac4C |
74,377.3 |
3,329.2 |
78,210.6 |
10,272.8 |
5.7E-01 |
|
Cm |
95,365.6 |
4,053.8 |
91,435.4 |
7,942.5 |
4.9E-01 |
|
Gm |
267,292.1 |
11,890.5 |
283,795.0 |
38,692.2 |
5.2E-01 |
|
m7G |
423,221.7 |
14,848.4 |
433,472.0 |
21,609.8 |
5.4E-01 |
|
m5C |
470,526.2 |
34,912.3 |
419,122.1 |
132,576.3 |
5.5E-01 |
|
t6A |
494,106.4 |
29,619.1 |
499,505.8 |
57,128.1 |
8.9E-01 |
|
m2G |
788,418.1 |
36,110.2 |
806,881.3 |
98,987.6 |
7.8E-01 |
|
m1G |
812,697.9 |
59,833.2 |
770,962.5 |
26,921.7 |
3.3E-01 |
| m22G | 1,041,823.0 | 38,024.7 | 1,060,593.3 | 118,738.9 | 8.1E-01 |
Amino acid misincorporation is increased in trm9Δ cells
Our phenotypic and molecular data support the idea that translational infidelity is occurring in trm9Δ cells due to decreased wobble base modifications. We propose that one of the ways that Trm9-catalyzed tRNA modifications encourage translation fidelity is by promoting the discrimination of cognate from near-cognate codons (i.e., AGA and AGG from AGU and AGC) in mRNA. To test this hypothesis, we used a dual-luciferase reporter construct to measure amino acid misincorporation levels in wild-type and trm9Δ cells.11 The reporter construct contains Renilla and Firefly luciferase coding sequences under the control of an ADH promoter. Note that Renilla and Firefly luciferase are transcribed as a single open reading frame separated by a short linker region. In the control plasmid, Firefly luciferase has arginine 218 in the active site coded by an AGA codon. In the test plasmid, Firefly luciferase is rendered inactive by mutating arginine 218 (AGA) to serine, coded by either AGU, AGC, UCC or UCG (Fig. 2A). We note that AGA and AGU/AGC are in a mixed codon box and classified as near-cognate codons, while AGA and UCC/UCG codons are not found in the same codon box and are classified as non-cognate codons. In the mutated Firefly luciferase construct harboring a serine codon in the active site, high-fidelity translation will promote low Firefly luciferase activity. In contrast, translational infidelity can promote misincorporation of arginine, which will then produce an active form of Firefly luciferase. Under basal conditions, we observed an ~1.6-fold (p < 0.02) increase in amino acid misincorporation in trm9Δ cells harboring the AGC/AGU versions of the reporter relative to wild type cells (Fig. 2B and C). This increased misincorporation in trm9Δ cells was exacerbated after treatment with paromomycin for both AGC (1.8-fold, p < 0.009) and AGU (2.0-fold, p < 0.008). To determine whether Trm9-catalyzed tRNA modifications prevent amino acid misincorporation at any serine codon, we measured arginine to serine misincorporation in wild-type and trm9Δ cells transformed with a reporter containing a UCG or UCC codon at position 218 (Fig. 2D and E). We determined that the levels of arginine misincorporation in the UCC or UCG associated reporter were very similar between wild type and trm9Δ cells. Overall, our data support that Trm9-catalyzed wobble base modifications promote translational fidelity and that under-modified tRNA can promote the incorrect translation of some near-cognate codons.

Figure 2. Increased translational infidelity observed in trm9Δ cells. (A) On the left, the misincorporation construct containing Renilla luciferase attached by a linker region (black arrow) in frame to firefly luciferase. Misincorporation of an arginine in place of serine will result in firefly luciferase activity. On the right are four mixed codon boxes, with each box specific to two different amino acids. Codons that have the potential to interact with Trm9-modified tRNA’s are shaded gray. (B-E) Wild-type (white bars) and trm9Δ cells (black bars) from the CEN.PK2 background were transformed with misincorporation reporter plasmids and grown to ~5 x 106 cells / ml. The population was divided into equal halves and either mock or paromomycin (200 μg / ml) treated for 60 min. Cells were then spun down and firefly and Renilla luciferase activity was determined. (F-G) The frameshifting reporter system was modified to contain four GAG codons in the linker region connecting Renilla and firefly constructs. The Firefly reporter is either in the + 1 or -1 orientation, with frameshifting putting it in frame with Renilla luciferase. Assays were preformed similar to described for B-E.
Increased -1 frameshifting at 4XGAG codon runs in trm9Δ cells
Frameshifting is another component of translational infidelity and we hypothesized that the absence of the mcm5s2U modification in tRNAUUCGlu would lead to protein synthesis errors in trm9 mutants. Our rationale for this hypothesis is that in E. coli, the deletion of GidA, the gene involved in mnm5s2U modification, has been shown to increase frame shifting at GA-based repeats.49 We used a dual-luciferase reporter construct to test this hypothesis. We measured +1 and -1 frame shifting levels when 4 GAG codons are encountered in the linker region separating Renilla from Firefly luciferase, with frameshifting required to obtain this later activity (Fig. 2F and G). We observed that there was no difference in + 1 frameshifting levels between wild-type and trm9Δ cells (P-value ≤ 0.41). In contrast, the trm9Δ cells demonstrated a 1.9-fold increase in -1 frameshifting when compared with their wild-type counterparts (P-value ≤ 0.0002). When taken together with the amino acid misincorporation, the frameshifting data further supports the hypothesis that there is translational infidelity in cells lacking mcm5U and mcm5s2U tRNA modifications.
Yef3 and Rnr1, known proteins whose levels are dependent on Trm9-catalyzed tRNA modifications, are prone to translational infidelity
The mcm5U and mcm5s2U modifications are specific to codons found in 4 mixed codon boxes, which suggests that translational fidelity is linked to specific genes containing corresponding codons. YEF3 and RNR1 have distinct codon usage patterns linked to Trm9, in that they are over-represented with codons from mixed boxes. Yef3 is a translation elongation factor that stimulates the binding of aminoacyl-tRNA. Rnr1 is the primary large subunit of the ribonucleotide reductase complex, which catalyzes the rate limiting step in dNTP production.47 Efficient translation of the YEF3 and RNR1 transcripts has previously been linked to Trm9 activity, with a deficiency leading to decreased Yef3 and Rnr1 protein levels, which we have noted in a previous study.9 We reasoned that if YEF3 and RNR1 transcripts were prone to errors in translation, this could lead to decreased protein levels in trm9Δ cells. Further, translation of YEF3 and RNR1 should be perturbed in wild-type cells after treatment with an agent that promotes errors. We tested this prediction and measured the protein levels of endogenously expressed Yef3-TAP, Rnr1-TAP and tubulin, both pre- and post-paromomycin treatment (Fig. 3A). We observed a 3.2- and 1.6-fold decrease in Yef3 and Rnr1 (n = 3), respectively, in wild-type cells after paromomycin treatment, while noting little difference in tubulin levels. We have also analyzed Yef3 and Rnr1 protein levels in trm9Δ cells and found them to be at similar or lower levels than what was found in paromomycin treated wild-type samples. Thus, paromomycin treatment or removal of TRM9, both associated with translational infidelity, leads to similar decreases in Rnr1 and Yef3 protein levels. In contrast, we observed little difference in tubulin levels when comparing wild-type and trm9Δ cells (± paromomycin). These findings support our contention that YEF3 and RNR1 are prone to translational errors with decreased fidelity occurring in mcm5U- and mcm5s2U-deficient cells. It is also possible that the absence of Trm9, and under modification of tRNA, leads to a reduction in the decoding rate of a subset of codons enriched in YEF3 and RNR1, causing slow translation of these proteins. This in turn could lead to decreased protein levels. In addition, the slow translation could cause protein folding errors that promote protein degradation.
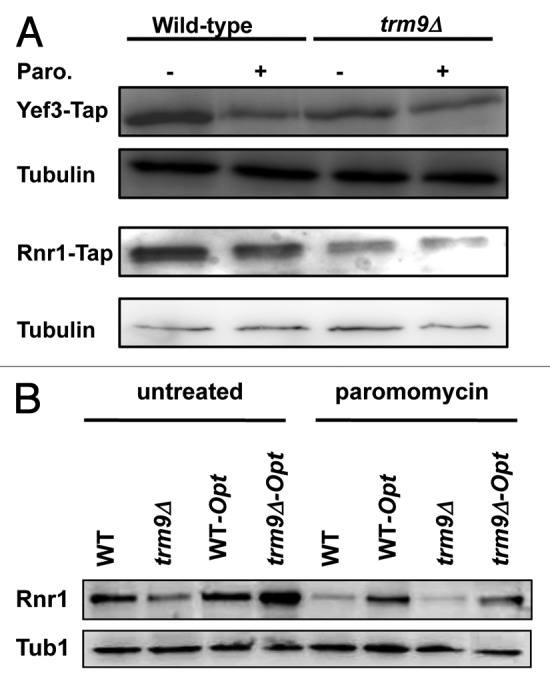
Figure 3. Some targets of Trm9-dependent translation are prone to errors. (A) Wild-type and trm9Δ cells in which either YEF3 or RNR1 genes were Tap tagged were grown to ~5 x 106 cells/ml and then treated with 1000 μg/ml of paromomycin for 1 h. Western blots performed using anti-Tap and anti-tubulin antibodies. (B) Wild-type and trm9Δ cells containing RNR1 or a codon-optimized RNR1 gene (Opt) were grown to ~5 x 106 cells/ml and then left untreated or treated with 1000 μg/ml of paromomycin for 1 h. Western blots performed using anti-Rnr1 and anti-tubulin antibodies. For both panels, we show representative blots (n = 3).
We propose that some codon usage patterns make the translation of specific transcripts error-prone, with YEF3 and RNR1 as potential examples. Further, we propose that paromomycin or decreased wobble base modification can increase these translation errors on specific transcripts. Should this prove true, then a change in codon usage should alleviate the translation errors and lead to a relative increase in protein levels after paromomycin treatment. A similar effect should also be observed in a trm9Δ background. To test these hypotheses, we used computational tools to design a new RNR1 gene, one that had an optimized codon usage pattern. The purpose was to make the most translatable gene by incorporating high-usage codons and removing codons, where possible, linked to Trm9 (i.e., the 18 AGA codons found in mixed boxes). We acknowledge the imperfect design of the optimized RNR1 sequence, which we attribute to constraints associated with the genetic code (only two codons for glutamic acid, glutamine and lysine) and the diversity of Trm9-target codons. This codon-optimized design for RNR1 generated the same amino acid sequence leading to a wild-type Rnr1 protein (Table S1). We replaced the native RNR1 gene with the codon-optimized RNR1 gene at the native locus in wild-type and trm9Δ cells. We assayed for Rnr1 protein levels in both cell types in the untreated and paromomycin treated samples (Fig. 3B). We demonstrate for the cases of paromomycin treatment, the presence of a trm9Δ allele, or paromomycin treatment of trm9Δ cells, that Rnr1 protein levels were higher in cells expressing the codon-optimized RNR1 gene relative to the native RNR1. These results provide support the conclusion that paromomycin promotes errors in codon-specific transcripts and that changing codon usage can restore translational fidelity. In addition, they support the notion that mcm5U and mcm5s2U modifications regulate codon-specific translation, in part, at the level of fidelity.
Markers of increased protein stress in trm9Δ cells
Increased translational infidelity in trm9Δ cells should lead to increased protein stress. To identify biomarkers of protein stress, we performed microarray analysis and compared basal transcript levels between wild-type and trm9Δ cells. Our microarray analysis results demonstrated that 99.5% of the transcripts were at similar levels in wild-type and trm9Δ cells. We previously demonstrated that transcript levels for YEF3, RNR1 and RNR3 were identical in wild-type and trm9Δ cells, a finding confirmed in our microarray analysis.9 One major difference between transcriptional profiles for each cell type was the expected absence of TRM9 transcripts in the trm9Δ cells. Detailed analysis of the transcripts differentially regulated (n = 60) between the two cell types demonstrated that many heat shock- and UPR-associated genes were upregulated in trm9Δ cells, when compared with wild-type cells (Table 2). The UPR associated genes HSP26, HSP78 and SSA4 were all upregulated in trm9Δ cells, relative to wild-type. Hsp26, Hsp78 and Ssa4 are all chaperones that work to prevent, re-solubilize or disassemble protein aggregates.47 The increased chaperone levels supports the idea that there is protein damage in trm9Δ cells. The absence of other transcriptionally regulated UPR-components in trm9Δ cells could be due to low levels of protein damage or perhaps some form of adaptation to the promoting stress. Twenty transcripts upregulated in trm9Δ cells have been reported to be upregulated by the ER stress inducers dithiothreitol (DTT) and tunicamycin (Fig. 4A).50 Gene Ontology analysis of transcripts upregulated in trm9Δ cells identified 11 functional processes significantly over-represented (p < 0.05). Those pertinent to protein stress included stress response, protein folding and stabilization, unfolded protein response/ER quality control and heat shock response (Table 3). Our microarray-based study not only supports the conclusion that there is increased protein stress in Trm9-deficient cells but also provides a putative link between deficiencies in tRNA modifications and activation of the unfolded protein and heat shock responses.
Table 2. List of transcripts upregulated more than 1.5 fold (p-value ≤ 0.06) in trm9Δ cells.
|
AAP1 |
FIT3 |
KDX1 |
REC114 |
YAL064W |
YKL096C-B |
|
AMS1 |
GRE2 |
MAM1 |
RNP1 |
YAL067W-A |
YLR162W |
|
ARN1 |
GSC2 |
MEI5 |
RTA1 |
YBL100W-C |
YLR307C-A |
|
ARO10 |
HSP12 |
NCA3 |
SAE3 |
YBR072C-A |
YLR412C-A |
|
ARO9 |
HSP26 |
NQM1 |
SPS100 |
YBR298C-A |
YMR317W |
|
DAN1 |
HSP78 |
OSW1 |
SRT1 |
YDR379C-A |
YNL067W-B |
|
ECM11 |
HXT10 |
PAU13 |
SSA4 |
YGL138C |
YOL155W-A |
|
ECM12 |
HXT12 |
PAU24 |
TSL1 |
YGR273C |
YOR214C |
|
FIT1 |
HXT5 |
PIR3 |
UBI4 |
YHR086W-A |
YOR268C |
| FIT2 | INO1 | PTR2 | XBP1 | YJR005C-A | YOR289W |
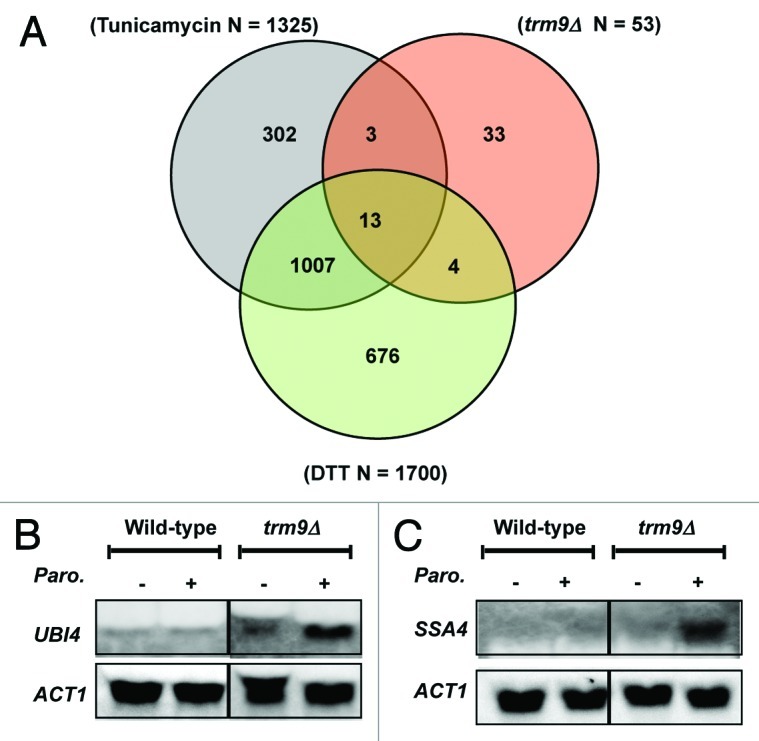
Figure 4. Protein stress transcripts are upregulated in trm9Δ cells. (A) A Venn diagram demonstrating that transcripts which are basally upregulated in trm9Δ cells are also regulated by agents that promote protein and ER stress. (B) northern blot analysis of UBI1 encoding ubiquitin and (C) the heat-shock marker SSA4, before and after paromomycin treatment. For both blots, ACT1 serves as a loading control. For both panels, we show representative blots (n = 3).
Table 3. Gene Ontology (GO) functional classifications over-represented in proteins corresponding to transcripts upregulated in trm9Δ cells.
| Category | p-value | Upregulated Out of Total | In Category from Cluster |
|---|---|---|---|
|
cell wall |
0.002701 |
7 out of 213 |
ECM11, GSC2, ECM12, PIR3, SRT1, YMR317W, OSW1 |
|
stress response |
0.003059 |
6 out of 162 |
PAU13, XBP1, KDX1, PIR3, UBI4, TSL1 |
|
protein folding and stabilization |
0.04994 |
3 out of 93 |
HSP26, HSP78, SSA4 |
|
unfolded protein response (e.g., ER quality control) |
0.02341 |
3 out of 69 |
HSP26, HSP78, SSA4 |
|
metabolism of energy reserves (e.g., glycogen, trehalose) |
0.01343 |
3 out of 56 |
GSC2, AAP1, TSL1 |
|
sugar, glucoside, polyol and carboxylate anabolism |
0.00362 |
3 out of 35 |
NQM1, INO1, TSL1 |
|
ion transport |
2.31E-05 |
3 out of 7 |
FIT1, FIT2, FIT3 |
|
meiotic recombination |
0.04493 |
2 out of 38 |
SAE3, REC114 |
|
osmosensing and response |
0.03868 |
2 out of 35 |
HSP12, KDX1 |
|
sugar transport |
0.03092 |
2 out of 31 |
HXT10, HXT5 |
| heat shock response | 0.01345 | 2 out of 20 | HSP12, GRE2 |
Unfolded proteins can be conjugated by ubiquitin and targeted for degradation via the ubiquitin-26S proteasome system.51-53 Using northern blots to quantitate UBI4 transcript levels corresponding to ubiquitin, we observed a 1.3-fold increase in trm9Δ cells relative to wild-type, a finding that is comparable to the 1.6-fold increase observed in the microarray studies described earlier. After treatment with paromomycin, an agent that promotes amino acid misincorporation, we observed a further increase of 3.2-fold in UBI4 transcripts in trm9Δ cells (Fig. 4B) relative to treated wild-type cells. This observation supports our contention that an already high basal level of translation errors in trm9Δ cells is exacerbated by treatment with an aminoglycoside antibiotic. We reasoned that, if trm9Δ cells have increased translational errors, they might express markers of protein aggregation such as Ssa4. The heat shock protein Ssa4 is a component of the cytosolic Hsp70 chaperone and its increased expression counteracts polyglutamine aggregation and toxicity in yeast cells.54,55 Using northern blots, we have demonstrated that there are increased basal levels of SSA4 (1.7-fold) in trm9Δ cells, a finding that is supported by the microarray analysis described earlier. We also observed increased expression of SSA4 (4.7-fold) in trm9Δ cells after paromomycin treatment when compared with treated wild-type cells (Fig. 4C). Examination of our data supports the hypothesis that there is both an increased protein stress and heat shock response in cells lacking Trm9-catalyzed tRNA modifications.
Increased translational errors activate the UPR in trm9Δ cells
Since our above microarray studies indicated that UPR and ER stress response pathways were also upregulated in trm9Δ cells, we used targeted assays to verify this upregulation. In the ER of S. cerevisiae, Kar2 normally binds to the ER domain of Ire1, a transmembrane serine threonine kinase, rendering it inactive.56 In the presence of misfolded proteins, Kar2 dissociates from Ire1, which promotes phosphorylation of the cytoplasmic domain of Ire1, activation of its endoribonuclease activity, and splicing of HAC1 mRNA.57 Spliced HAC1 is translated into an active Hac1 transcription factor and it binds to the unfolded protein response elements (UPREs) of several genes to induce their transcription.58 Using northern blots, we demonstrated that KAR2 and HAC1-spliced levels are higher in trm9Δ cells, relative to wild-type cells (Fig. 5A). In addition, we observed that trm9Δ cells further increased KAR2 and HAC1-spliced transcript levels after paromomycin treatment, relative to untreated cells (Fig. 5A).

Figure 5. Ire1-dependent activation of the UPR identified in trm9Δ cells. (A) northern blots analyses of KAR2, HAC1 and ACT1 were performed as described in materials and methods, using cells left untreated or treated with paromomycin (200 μg/ml) for 60 min. We show a representative blot (n = 3). (B) β-galactosidase activity is a measure of the unfolded protein response. (C) Cells were grown to ~107 cells/ml, centrifuged to remove the media, proteins were extracted and β-galactosidase activity was determined. (D) Cells were grown to ~5 x 106 cells/ml, treated with 1000 μg/ml of paromomycin for 1 h and β-galactosidase activity was determined. For panels C-D, average values are shown with standard deviations (n = 3).
Normally, the transcription of KAR2 is induced by both the UPRE and HSE in its promoter region.59 UPRE and HSE can work synergistically but independently of each other in the transcription of KAR2.60 To measure just the UPR, we used an integrated UPRE-lacZ reporter [AWY14, UPRE-PCYC1(−178)-lacZ] where transcription of the LacZ gene is driven by a CYC1 promoter under the control of the KAR2 UPRE only (Fig. 5B).22 We observed that β-galactosidase activity was ~2.5- to 3.5-fold higher in trm9Δ cells relative to wild-type cells (Fig. 5C and D). By integrating the TRM9 gene along with its promoter at the HO locus, we were able to complement the increased UPR activation observed in trm9Δ cells alone (Fig. 5C) as demonstrated by decreased β-galactosidase activity. We also investigated the role of a central activator of the UPR, Ire1, in the increased UPR activity found in trm9Δ cells. We deleted IRE1 in trm9Δ cells and observed markedly decreased β-galacotsidase activity (Fig. 5C). Thus, we have demonstrated that the observed increase in the UPR in trm9Δ cells was IRE1 dependent.
We propose that the activation of the UPR in trm9Δ cells was in response to increased translational errors. If this were the case, conditions that exacerbate translational infidelity should also promote increased activation of the UPR in trm9Δ cells, relative to basal conditions. To test this proposal, we have used wild-type and trm9Δ cells containing the UPR reporter and treated with paromomycin. We observed that there was an increase in UPR activity upon treatment with the translational error inducer paromomycin in both wild-type and trm9Δ cells. The magnitude of this increase in β-galactosidase activity was higher in trm9∆ cells when compared with wild-type, with an increase of 125 and 50 in specific activity, respectively, when subtracting treated from untreated values (Fig. 5D). Both the higher UPR reporter activity and increased KAR2 and HAC1Spliced transcript levels in trm9Δ cells support the contention that the UPR is basally activated in these tRNA modification deficient cells. In addition, when compared with untreated controls, the paromomycin-induced activation of the UPR reporter in trm9Δ cells, as well as the higher KAR2 and HAC1Spliced transcript levels, supports our finding that increased translational errors further activate the UPR. Ultimately, our studies support the hypothesis that there are unfolded or damaged proteins in trm9Δ cells and that they are caused by translational infidelity.
Discussion
Previous studies of Trm9 have revealed important roles for wobble base tRNA modifications during stress responses and cell cycle.9,61 Our current study is unique because it demonstrates that Trm9-catalyzed tRNA modifications prevent translational infidelity under normal growth conditions. We propose the following model (Fig. 6) to account for the observed phenotypes: in the absence of mcm5U and mcm5s2U modifications, amino acid misincorporations and frameshifting errors will occur during the translation of specific codons (i.e., ones belonging to Arginine, Glutamic acid, Glutamine and Lysine mixed codon boxes). As an extension of this idea, we predict that translational infidelity will occur in specific transcripts. These hypotheses are supported by the observations that mcm5U and mcm5s2U modifications are used to decode a select group of codons and by our misincorporation, frameshifting and codon optimization data, as well as immunoblot data detailing protein levels (Figs. Two and 3). In this study we have used protein level measurements as an indirect readout of translational infidelity, and theoretically this measures both normal and some error filled proteins. Proteomic analysis of error filled proteins is desirable, but requires identification of a hard to find 1% fraction that is sometimes easily degraded. We note that there are alternative possibilities for the decreased Yef3 and Rnr1 protein levels we observed. These include changes in chaperone levels, slow translation of individual transcripts and abrogation of some other form of translational regulation in trm9Δ cells. Further, we propose that the resulting protein errors are extensive, leading to error-laden and misfolded proteins, which activate the UPR and heat shock responses. This conclusion is supported by our data demonstrating basal activation of the heat shock and unfolded protein responses. It is noteworthy that activated heat shock and unfolded protein responses are components of some neurodegenerative diseases and cancers; thus, it will be interesting to analyze tRNA modification levels and translational infidelity in corresponding cells to determine if they are a root cause or a byproduct of these ailments.
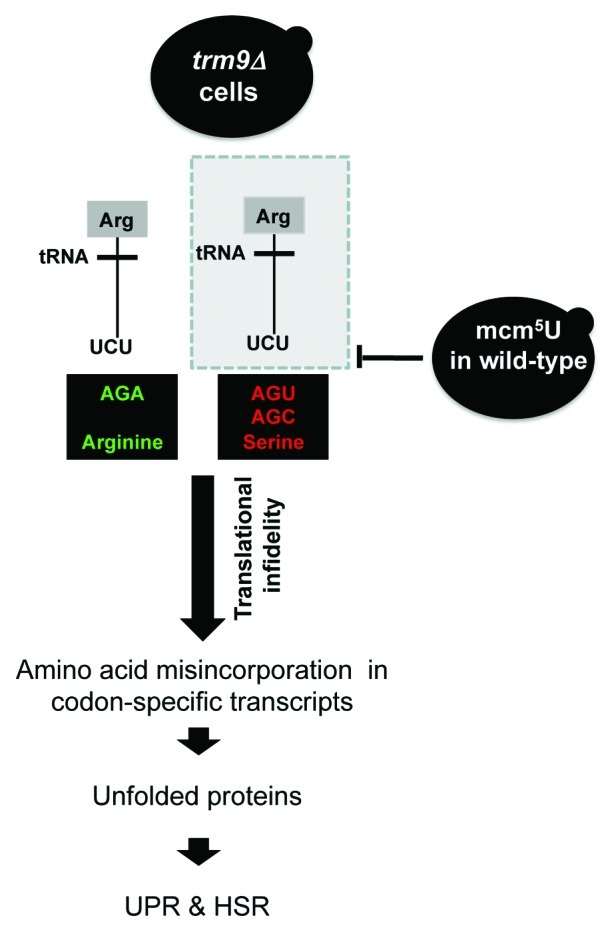
Figure 6. Model describing the link between Trm9-catalyzed tRNA modifications, translational infidelity and protein stress. In trm9Δ cells, the absence of the mcm5U modification causes the tRNAUCUArg to base pair with its near cognate AGC Serine or AGU Serine codons. This mispairing causes serine to arginine amino acid misincorporation. The higher level of amino acid misincorporation gives rise to increased unfolded proteins, thus activating the UPR and heat shock pathways in trm9Δ cells. Frameshifting (not shown) could also lead to protein errors.
Our misincorporation results are consistent with tRNAUCUArg being able to read, albeit at different efficiencies, all four codons in the arginine-serine mixed codon box containing AGA, AGG, AGC and AGU. While Ser to Arg is one type of misincorporation, a number of other amino acid misincorporations can be predicted based on their presence in a mixed codon box. These include glutamic to aspartic acid, lysine to asparagine and glutamine to histidine (Fig. 2A, right). Our misincorporation predictions are based on the assumption that all target codons are equal and the complete absence of mcm5U and mcm5s2U modifications, with both of these situations being a dramatic oversimplification of what might transpire inside the cell. Most likely, a portion of the codons predicted to have misincorporation in the absence of Trm9-catalyzed tRNA modifications will predominate. Misincorporation should depend on the number of copies of a tRNA species, other tRNA modifications outside the anticodon, and the 5′ and 3′ sequence context. Our results with Rnr1 and Yef3 support that specific codon sequences are affected by these translation errors. Defining the sequence rules leading to translation errors will help researchers identify susceptible transcripts.
Our data demonstrating -1 frameshifting in 4X GAG codon runs is an example of how a codon usage extreme can promote translation errors. In trm9Δ cells, the unmodified tRNA most likely binds weakly to its cognate codon, a molecular characteristic that promotes slippage during translation and, thus, protein synthesis errors. The local context of codons within a gene will likely play a major role in influencing infidelity, as long stretches of identical codons should be prime slippage sites. Using our recently reported Gene Specific Codon Counting (GSCC) database,62 we have identified 8 S. cerevisiae sequences (AVT3, CBF2, MPP10, MTC1, RLF2, TOD6, VPS13 and YOR223W) that have 4 GAG codons in a row and we suggest that frameshifting will occur during the translation of these proteins as well. Other then TOD6 and YOR223W, the 4X GAG is found in the beginning or middle of these above mentioned gene sequences and -1 frameshifting will lead to a drastically different protein sequence. The 4X GAG sequence is just one of several possible codon runs that could promote frameshifting in trm9Δ cells, with other amino acid runs currently under study.
We recently reported that tRNA modification levels change as a function of cellular stress,48 a change that predicts that misincorporation and frameshifting levels can be increased or decreased as part of the cell’s regulatory program. Increased fidelity could lead to more active enzymes and proteins at higher concentrations, both of which will promote more efficient responses to stress. This increased fidelity could also cause protein stress response systems to be repositioned to remove environmentally damaged proteins, instead of protein damage introduced by an endogenous process (i.e., error prone translation). Translational infidelity due to a decrease in a specific tRNA modification is also a possibility. It is interesting to note that we observed a ~2-fold decrease in mcm5U and mcm5s2U levels after sodium arsenite exposure,48 suggesting that this agent induces protein errors. It is also tempting to suggest that translational infidelity could be used as a cellular regulatory or survival strategy to introduce variation in the proteome or, in specific proteins, to provide a growth advantage to normal or disease associated cells. Mutator phenotypes at the DNA level have been commonly associated with colorectal cancers,63 and translational infidelity phenotypes could provide a similar growth advantage. While this last point is also speculative, our study has clearly shown that translational infidelity can influence protein stress response pathways and demonstrates a link between wobble base tRNA modifications and protein errors.
Materials and Methods
Yeast and growth conditions
Table S2 lists the oligonucleotides and S. cerevisiae strains used in these studies. Mutants were made using either a URA3-based strategy or a G418 knockout cassette from the S. cerevisiae Gene Deletion Project. Mutants were selected on either synthetic defined media lacking uracil (SD-URA) or yeast peptone dextrose (YPD) plates containing G418 (200 μg/ml). Mutants were confirmed by PCR. Media preparation and other yeast manipulations were performed using standard methods. Unless otherwise noted, all yeast cultures were grown at 30°C. All experiments using aminoglycoside antibiotics used mutants made with the URA3 deletion cassette.
Viability Analysis
Wild-type, trm9Δ and trm9Δ +TRM9 cells were grown overnight in YPD. Cultures were serially (10-fold) diluted and plated on agar plates containing YPD, YPD + paromomycin (350 ug/ml) or YPD + G418 (20 ugm/ml). The plates were incubated at 30°C for 3 d and imaged using an Alphaimager ™ (Alphainotech).
Northern and western blots
Wild-type and trm9Δ cells were grown to ~5 × 106 cells/ml in YPD and treated with 200 μg/ml paromomycin for 60 min. RNA was purified using an RNeasy Mini Kit and analyzed as described elsewhere.9 Detection of the RNA was facilitated using the Chemiluminescent Nucleic Acid Detection Module (Pierce). Protein extracts were prepared as described elsewhere,9 and protein concentrations were measured using a BCA Protein Assay Kit (Pierce). Western blots were performed as described elsewhere.9
Misincorporation reporter assay
Wild type and trm9Δ cells (CEN.PK2 background) were transformed with plasmids pJD375 (AGA as the active site at position 218 of firefly luciferase), pJD644 (AGA mutated to AGU at position 218 of firefly luciferase) and pJD643 (AGA mutated to AGC at position 218 of firefly luciferase). Transformed yeast cells were grown overnight in SD-Ura. Overnight cultures were diluted into fresh YPD to ~1 × 106 cells/ml and grown to ~5 x 106 cells/ml. Cells were either mock-treated or treated with 200 μg/ml of paromomycin for 60 min. Cells were pelleted by centrifugation, washed twice with 0.5 ml of cold lysis buffer containing 1 mM PMSF (Pierce), then resuspended in cold lysis buffer and broken by agitation with glass beads (0.5 mm Sigma-Aldrich). Lysates were clarified by centrifugation and supernatants were transferred to pre-chilled tubes. Luminescence reactions were initiated by the addition of 50 µl of Promega DLR (Promega) to 5 µl of clarified cell lysates and measured using a Victor Plate Reader (PerkinElmer).
Reporter assay for frameshifting
Wild type and trm9Δ cells (BY4741 background with TRM9 deletion using a HIS marker) were transformed with plasmids pJD375, pJDTB375+1 (pJD375 having 4XGAG +1 sequence in the linker region connecting Renilla and firefly luciferase) and pJDTB375–1 (pJD375 having 4XGAG +1 sequence between Renilla and Firefly luciferase). Transformed yeast cells were grown overnight in SD-Ura. The resulting cultures were then diluted into fresh YPD to ~1 × 106 cells/ml and grown to ~107 cells/ml. Cells were pelleted by centrifugation, washed twice with 0.5 ml of cold lysis buffer containing 1 mM PMSF (Pierce), then resuspended in cold lysis buffer and broken by agitation with glass beads (0.5 mm Sigma-Aldrich). Lysates were clarified by centrifugation and supernatants were transferred to pre-chilled tubes. Luminescence reactions were initiated by the addition of 50 µl of Promega DLR (Promega) to 5 µl of clarified cell lysates and measured using a Victor Plate Reader (PerkinElmer).
tRNA modification analysis
The LC/MS-based method for relative quantification of tRNA modifications in S. cerevisiae is reported in our previous study.48 Briefly, cells were lysed by mechanical disruption in Trizol reagent (Invitrogen) and all species of RNA were extracted by separating the lysate into two phases with chloroform, followed by the collection of the aqueous phase. tRNA in the aqueous phase was then enriched using the PureLink miRNA Isolation Kit (Invitrogen). RNA quantification was determined by UV-vis spectrophotometer and bioanalyzer analysis (Agilent Bioanalyzer Small RNA Kit). A known amount of tRNA-enriched sample was mixed with [15N]5-dA, as an internal standard, and tRNA was hydrolyzed enzymatically to ribonucleosides and the enzymes were removed by ultrafiltration with a 10 KDa membrane (Microcon YM10). Deaminase inhibitors (coformycin and tetrahydrouridine) and antioxidants (deferoxamine mesylate and butylated hydroxytoluene) were present throughout the processes to prevent damage artifacts.48 Digested ribonucleosides were resolved by reversed-phase HPLC with a gradient of aqueous ammonium acetate and acetonitrile as the mobile phase. The HPLC column was directly connected to an eletrospray ionization-triple quadrupole mass spectrometer (ESI-QQQ, Agilent). Modified ribonucleosides were identified by HPLC retention time and collision-induced dissociation fragmentation pattern, with quantification by comparison of signal intensities in wild type and trm9Δ cells or untreated and treated cells.
Microarray Analysis
Wild-type and trm9Δ cells (made using the G418 deletion cassette) were grown at 30°C to ~5 × 106 cells/ml in YPD media. The cells were pelleted by centrifugation and spheroblasts were generated by shaking the cells in 1 ml of sorbitol containing 250 U zymolase (Associates of Cape Cod), as described in the RNeasy Mini Handbook (Qiagen). RNA was extracted using RNeasy Mini Kit (Qiagen). The isolated RNA was examined spectroscopically by agarose gel electrophoresis and by Bioanalyzer analysis (Agilent). Affymetrix gene chip (Yeast 2.0) analysis was performed as previously described at the Center for Functional Genomics, University at Albany.64 Gene Chip data were analyzed using CyberT software and clustered into functional categories using Funspec.
β-galactosidase assays
Overnight cultures of wild-type and trm9Δ (AWY14 background) cells were diluted into fresh YPD to ~1 × 106 cells/ml and grown to ~107 cells/ml. Cells were harvested and then analyzed for β-galactosidase activity as previously described.9
Supplementary Material
Acknowledgments
Financial support was provided by the National Institute of Environmental Health Sciences (R01 ES015037, R01 ES017010 and P30 ES002109), the MIT Westaway Fund and the Singapore-MIT Alliance for Research and Technology. Special thanks to Dr. R.J. Kaufman (University of Michigan Medical School, Ann Arbor) for the UPR reporter strains, Dr. Jonathan Dinman (University of Maryland, MD) for the amino acid misincorporation reporter plasmids and Fraulin Joseph for help with immunoblots.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/20531
References
- 1.Agris PF. Decoding the genome: a modified view. Nucleic Acids Res. 2004;32:223–38. doi: 10.1093/nar/gkh185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agris PF. Bringing order to translation: the contributions of transfer RNA anticodon-domain modifications. EMBO Rep. 2008;9:629–35. doi: 10.1038/embor.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang B, Lu J, Byström AS. A genome-wide screen identifies genes required for formation of the wobble nucleoside 5-methoxycarbonylmethyl-2-thiouridine in Saccharomyces cerevisiae. RNA. 2008;14:2183–94. doi: 10.1261/rna.1184108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang BO, Johansson MJO, Byström AS. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA. 2005;11:424–36. doi: 10.1261/rna.7247705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leihne V, Kirpekar F, Vågbø CB, van den Born E, Krokan HE, Grini PE, et al. Roles of Trm9- and ALKBH8-like proteins in the formation of modified wobble uridines in Arabidopsis tRNA. Nucleic Acids Res. 2011;39:7688–701. doi: 10.1093/nar/gkr406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalhor HR, Clarke S. Novel methyltransferase for modified uridine residues at the wobble position of tRNA. Mol Cell Biol. 2003;23:9283–92. doi: 10.1128/MCB.23.24.9283-9292.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Björk GR, Huang B, Persson OP, Byström AS. A conserved modified wobble nucleoside (mcm5s2U) in lysyl-tRNA is required for viability in yeast. RNA. 2007;13:1245–55. doi: 10.1261/rna.558707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu J, Huang B, Esberg A, Johansson MJ, Byström AS. The Kluyveromyces lactis gamma-toxin targets tRNA anticodons. RNA. 2005;11:1648–54. doi: 10.1261/rna.2172105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Begley U, Dyavaiah M, Patil A, Rooney JP, DiRenzo D, Young CM, et al. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol Cell. 2007;28:860–70. doi: 10.1016/j.molcel.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durant PC, Bajji AC, Sundaram M, Kumar RK, Davis DR. Structural effects of hypermodified nucleosides in the Escherichia coli and human tRNALys anticodon loop: the effect of nucleosides s2U, mcm5U, mcm5s2U, mnm5s2U, t6A, and ms2t6A. Biochemistry. 2005;44:8078–89. doi: 10.1021/bi050343f. [DOI] [PubMed] [Google Scholar]
- 11.Plant EP, Nguyen P, Russ JR, Pittman YR, Nguyen T, Quesinberry JT, et al. Differentiating between near- and non-cognate codons in Saccharomyces cerevisiae. PLoS One. 2007;2:e517. doi: 10.1371/journal.pone.0000517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howard MT, Anderson CB, Fass U, Khatri S, Gesteland RF, Atkins JF, et al. Readthrough of dystrophin stop codon mutations induced by aminoglycosides. Ann Neurol. 2004;55:422–6. doi: 10.1002/ana.20052. [DOI] [PubMed] [Google Scholar]
- 13.Lai C-H, Chun HH, Nahas SA, Mitui M, Gamo KM, Du L, et al. Correction of ATM gene function by aminoglycoside-induced read-through of premature termination codons. Proc Natl Acad Sci U S A. 2004;101:15676–81. doi: 10.1073/pnas.0405155101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lukacs GL, Durie PR. Pharmacologic approaches to correcting the basic defect in cystic fibrosis. N Engl J Med. 2003;349:1401–4. doi: 10.1056/NEJMp038113. [DOI] [PubMed] [Google Scholar]
- 15.Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349:1433–41. doi: 10.1056/NEJMoa022170. [DOI] [PubMed] [Google Scholar]
- 16.Harger JW, Dinman JD. An in vivo dual-luciferase assay system for studying translational recoding in the yeast Saccharomyces cerevisiae. RNA. 2003;9:1019–24. doi: 10.1261/rna.5930803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silva RM, Duarte IC, Paredes JA, Lima-Costa T, Perrot M, Boucherie H, et al. The yeast PNC1 longevity gene is up-regulated by mRNA mistranslation. PLoS One. 2009;4:e5212. doi: 10.1371/journal.pone.0005212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–9. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 19.Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426:891–4. doi: 10.1038/nature02262. [DOI] [PubMed] [Google Scholar]
- 20.Herbst M, Wanker EE. Therapeutic approaches to polyglutamine diseases: combating protein misfolding and aggregation. Curr Pharm Des. 2006;12:2543–55. doi: 10.2174/138161206777698828. [DOI] [PubMed] [Google Scholar]
- 21.Hampton RY. ER stress response: getting the UPR hand on misfolded proteins. Curr Biol. 2000;10:R518–21. doi: 10.1016/S0960-9822(00)00583-2. [DOI] [PubMed] [Google Scholar]
- 22.Schröder M, Clark R, Kaufman RJ. IRE1- and HAC1-independent transcriptional regulation in the unfolded protein response of yeast. Mol Microbiol. 2003;49:591–606. doi: 10.1046/j.1365-2958.2003.03585.x. [DOI] [PubMed] [Google Scholar]
- 23.Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, et al. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Hum Mol Genet. 2007;16:618–29. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- 24.Stolz A, Wolf DH. Endoplasmic reticulum associated protein degradation: A chaperone assisted journey to hell. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research; 1803:694-705. [DOI] [PubMed] [Google Scholar]
- 25.Ishida Y, Yamamoto A, Kitamura A, Lamandé SR, Yoshimori T, Bateman JF, et al. Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol Biol Cell. 2009;20:2744–54. doi: 10.1091/mbc.E08-11-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–30. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 28.Carrasco DR, Sukhdeo K, Protopopova M, Sinha R, Enos M, Carrasco DE, et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell. 2007;11:349–60. doi: 10.1016/j.ccr.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boelens J, Lust S, Offner F, Bracke ME, Vanhoecke BW. Review. The endoplasmic reticulum: a target for new anticancer drugs. In Vivo. 2007;21:215–26. [PubMed] [Google Scholar]
- 30.Phillips-Jones MK, Hill LS, Atkinson J, Martin R. Context effects on misreading and suppression at UAG codons in human cells. Mol Cell Biol. 1995;15:6593–600. doi: 10.1128/mcb.15.12.6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Chang A. Heat shock response relieves ER stress. EMBO J. 2008;27:1049–59. doi: 10.1038/emboj.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mager WH, Ferreira PM. Stress response of yeast. Biochem J. 1993;290:1–13. doi: 10.1042/bj2900001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- 34.Turturici G, Sconzo G, Geraci F. Hsp70 and its molecular role in nervous system diseases. Biochem Res Int. 2011;2011:618127. doi: 10.1155/2011/618127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Estruch F. Stress-controlled transcription factors, stress-induced genes and stress tolerance in budding yeast. FEMS Microbiol Rev. 2000;24:469–86. doi: 10.1111/j.1574-6976.2000.tb00551.x. [DOI] [PubMed] [Google Scholar]
- 36.Hahn J-S, Hu Z, Thiele DJ, Iyer VR. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol Cell Biol. 2004;24:5249–56. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eastmond DL, Nelson HCM. Genome-wide analysis reveals new roles for the activation domains of the Saccharomyces cerevisiae heat shock transcription factor (Hsf1) during the transient heat shock response. J Biol Chem. 2006;281:32909–21. doi: 10.1074/jbc.M602454200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chi NC, Karliner JS. Molecular determinants of responses to myocardial ischemia/reperfusion injury: focus on hypoxia-inducible and heat shock factors. Cardiovasc Res. 2004;61:437–47. doi: 10.1016/j.cardiores.2003.11.033. [DOI] [PubMed] [Google Scholar]
- 39.Hoshino T, Murao N, Namba T, Takehara M, Adachi H, Katsuno M, et al. Suppression of Alzheimer’s disease-related phenotypes by expression of heat shock protein 70 in mice. J Neurosci. 2011;31:5225–34. doi: 10.1523/JNEUROSCI.5478-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Novo G, Cappello F, Rizzo M, Fazio G, Zambuto S, Tortorici E, et al. Hsp60 and heme oxygenase-1 (Hsp32) in acute myocardial infarction. Transl Res. 2011;157:285–92. doi: 10.1016/j.trsl.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 41.Yenari MA. Heat shock proteins and neuroprotection. Adv Exp Med Biol. 2002;513:281–99. doi: 10.1007/978-1-4615-0123-7_10. [DOI] [PubMed] [Google Scholar]
- 42.Kelly S, Yenari MA. Neuroprotection: heat shock proteins. Curr Med Res Opin. 2002;18(Suppl 2):s55–60. doi: 10.1185/030079902125000732. [DOI] [PubMed] [Google Scholar]
- 43.Trivedi S. Heat shock proteins and neuroprotection. Recent Pat DNA Gene Seq. 2007;1:134–7. doi: 10.2174/187221507780887054. [DOI] [PubMed] [Google Scholar]
- 44.Grant CM, Tuite MF. Mistranslation of human phosphoglycerate kinase in yeast in the presence of paromomycin. Curr Genet. 1994;26:95–9. doi: 10.1007/BF00313794. [DOI] [PubMed] [Google Scholar]
- 45.Stansfield I, Jones KM, Herbert P, Lewendon A, Shaw WV, Tuite MF. Missense translation errors in Saccharomyces cerevisiae. J Mol Biol. 1998;282:13–24. doi: 10.1006/jmbi.1998.1976. [DOI] [PubMed] [Google Scholar]
- 46.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–5. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cherry JM, Adler C, Ball C, Chervitz SA, Dwight SS, Hester ET, et al. SGD: Saccharomyces Genome Database. Nucleic Acids Res. 1998;26:73–9. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan CTY, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010;6:e1001247. doi: 10.1371/journal.pgen.1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brégeon D, Colot V, Radman M, Taddei F. Translational misreading: a tRNA modification counteracts a +2 ribosomal frameshift. Genes Dev. 2001;15:2295–306. doi: 10.1101/gad.207701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–58. doi: 10.1016/S0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 51.London MK, Keck BI, Ramos PC, Dohmen RJ. Regulatory mechanisms controlling biogenesis of ubiquitin and the proteasome. FEBS Lett. 2004;567:259–64. doi: 10.1016/j.febslet.2004.04.078. [DOI] [PubMed] [Google Scholar]
- 52.Goeckeler JL, Brodsky JL. Molecular chaperones and substrate ubiquitination control the efficiency of endoplasmic reticulum-associated degradation. Diabetes Obes Metab. 2010;12(Suppl 2):32–8. doi: 10.1111/j.1463-1326.2010.01273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riezman H. Why do cells require heat shock proteins to survive heat stress? Cell Cycle. 2004;3:61–3. doi: 10.4161/cc.3.1.625. [DOI] [PubMed] [Google Scholar]
- 54.Gokhale KC, Newnam GP, Sherman MY, Chernoff YO. Modulation of prion-dependent polyglutamine aggregation and toxicity by chaperone proteins in the yeast model. J Biol Chem. 2005;280:22809–18. doi: 10.1074/jbc.M500390200. [DOI] [PubMed] [Google Scholar]
- 55.Gong Y, Kakihara Y, Krogan N, Greenblatt J, Emili A, Zhang Z, et al. An atlas of chaperone-protein interactions in Saccharomyces cerevisiae: implications to protein folding pathways in the cell. Mol Syst Biol. 2009;5:275. doi: 10.1038/msb.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kimata Y, Kimata YI, Shimizu Y, Abe H, Farcasanu IC, Takeuchi M, et al. Genetic evidence for a role of BiP/Kar2 that regulates Ire1 in response to accumulation of unfolded proteins. Mol Biol Cell. 2003;14:2559–69. doi: 10.1091/mbc.E02-11-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okamura K, Kimata Y, Higashio H, Tsuru A, Kohno K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem Biophys Res Commun. 2000;279:445–50. doi: 10.1006/bbrc.2000.3987. [DOI] [PubMed] [Google Scholar]
- 58.Kawahara T, Yanagi H, Yura T, Mori K. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol Biol Cell. 1997;8:1845–62. doi: 10.1091/mbc.8.10.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mori K, Ogawa N, Kawahara T, Yanagi H, Yura T. Palindrome with spacer of one nucleotide is characteristic of the cis-acting unfolded protein response element in Saccharomyces cerevisiae. J Biol Chem. 1998;273:9912–20. doi: 10.1074/jbc.273.16.9912. [DOI] [PubMed] [Google Scholar]
- 60.Welihinda AA, Tirasophon W, Green SR, Kaufman RJ. Gene induction in response to unfolded protein in the endoplasmic reticulum is mediated through Ire1p kinase interaction with a transcriptional coactivator complex containing Ada5p. Proc Natl Acad Sci U S A. 1997;94:4289–94. doi: 10.1073/pnas.94.9.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bennett CB, Lewis LK, Karthikeyan G, Lobachev KS, Jin YH, Sterling JF, et al. Genes required for ionizing radiation resistance in yeast. Nat Genet. 2001;29:426–34. doi: 10.1038/ng778. [DOI] [PubMed] [Google Scholar]
- 62.Tumu S, Patil A, Towns WL, Dyavaiah M, Begley TJ. The gene-specific codon counting database: a genome-based catalog of one-, two-, three-, four- and five-codon combinations present in Saccharomyces cerevisiae genes. Database (Oxford) 2012;2012:bas002. doi: 10.1093/database/bas002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim JC, Lee KH, Ka IH, Koo KH, Roh SA, Kim HC, et al. Characterization of mutator phenotype in familial colorectal cancer patients not fulfilling amsterdam criteria. Clin Cancer Res. 2004;10:6159–68. doi: 10.1158/1078-0432.CCR-04-0651. [DOI] [PubMed] [Google Scholar]
- 64.Dyavaiah M, Rooney JP, Chittur SV, Lin Q, Begley TJ. Autophagy-dependent regulation of the DNA damage response protein ribonucleotide reductase 1. Mol Cancer Res. 2011;9:462–75. doi: 10.1158/1541-7786.MCR-10-0473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.