

Summary
γδ T cells contribute uniquely to host immune defense. However, how they function remains an enigma. Although it is unclear what most γδ T cells recognize, common dogma asserts that they recognize self-antigens. While they are the major initial Interleukin-17 (IL-17) producers in infections, it is unclear what is required to trigger these cells to act. Here, we report that a noted B cell antigen, the algae protein-phycoerythrin (PE) is an antigen for murine and human γδ T cells. PE also stained specific bovine γδ T cells. Employing this specificity, we demonstrated that antigen recognition, but not extensive clonal expansion, was required to activate naïve γδ T cells to make IL-17. In this activated state, γδ T cells gained the ability to respond to cytokine signals that perpetuated the IL-17 production. These results underscore the adaptability of lymphocyte antigen receptors and suggest a previously unrecognized antigen-driven rapid response in protective immunity prior to the maturation of classical adaptive immunity.
Introduction
γδ T cells, together with B cells and αβ T cells are the only cells that use somatic gene rearrangement to generate diverse antigen receptors. While αβ T cells perform most of the well-defined immune responses attributed to T cells, γδ T cells are present together with αβ T cells and B cells in all but the most primitive vertebrates. This conservation of γδ T cells during evolution suggests that these cells play a unique and important role in host immune defense. Nevertheless, the arrays of cytokines produced by most γδ T cells are similar to those of αβ T cells. γδ T cells also mount cytolytic responses upon activation that are much like those of cytotoxic αβ T cells. These effector similarities suggest that the principle difference between how γδ T cells and αβ T cells contribute to immune defense must lie in how they are triggered.
Indeed, γδ T cells differ from αβ T cells in antigen recognition, antigen specific repertoire development, and effector fate determination. While αβ T cells recognize proteins that are processed into peptides and presented on major histocompatibility complex (MHC) molecules on the cell surface, γδ T cells recognize antigens directly. There is no antigen processing and presentation requirement, and the MHC molecules are not an obligatory component of γδ T cell antigens (Chien and Konigshofer, 2007). Furthermore, based on lengths of key structural components for antigen binding, the complementarity determining region 3 (CDR3)s (the junctional regions formed by VJ, or VDJ recombination), γδ T cell receptors (TCRs) are more similar to immunoglobulins than to αβ TCRs (Rock, et al., 1994). While γδ T cells, like αβ T cells, require thymic maturation before entering the periphery, this process does not determine what peripheral γδ T cells can recognize. Instead, it determines how these cells function. In particular, γδ T cells that have developed without encountering cognate ligands in the thymus make IL-17 readily in response to TCR triggering in the periphery (Jensen, et al., 2008). These observations suggest that γδ T cells can recognize self and non-self antigens, and γδ TCR ligands could include pathogen-derived molecules, as well as infection- or injury-induced self-antigens, which may or may not be expressed in the thymus, and that γδ T cells specific for these antigens make IL-17.
IL-17 is a cytokine, which regulates the expansion and recruitment of neutrophils and monocytes to initiate the inflammatory response (Stark, et al., 2005). In acute inflammation, a swift IL-17 response must be elicited without prior antigen exposure. Therefore, γδ T cells may be uniquely suited to produce IL-17 at the onset of the inflammatory response. Indeed, γδ T cells are found to be the major initial IL-17 producers after immunization as well as in various infectious disease models, including Francisella tularensis (Henry, et al., 2010), Mycobacterium tuberculosis, Mycobacterium bovis, Escherichia coli and pulmonary aspergillosis in chronic granulomatous disease (Bonneville et al., 2010). However, it is unclear what most γδ T cells recognize in any of these infections, and how these cells are triggered to act.
To date, only a few molecules have been confirmed as γδ T cell antigens, and these are encoded by the host genome (Crowley, et al., 2000; Scotet, et al., 2005; Xu et al., 2011). It has been postulated that γδ TCRs have danger sensing molecular pattern associated receptor-like characteristics in that they focus on host molecules induced by cellular stress and infection (Bonneville, et al., 2010). Moreover, it has been argued that the IL-17 response mounted by γδ T cells is too rapid and too robust to be antigen-specific, and that this response is induced by the engagement of pathogen pattern recognition receptors and/or by inflammatory cytokine receptors (Hamada, et al., 2008; Kapsenberg, 2009). According to this line of reasoning, γδ T cells are triggered through receptors other than their TCRs, acting essentially as part of the innate immune system to provide an “innate” source of IL-17, and the ability of γδ T cells to recognize different antigens is irrelevant to their function. It’s clear that in order to understand how γδ T cells contribute to host immune defense and initiate an inflammatory response, we need to know what γδ T cells recognize, and what is required for γδ T cells to mount an effector response.
Results
Phycoerythrin (PE) is a murine γδ T cell antigen
Recently, we found that an algae protein, phycoerythrin (PE), stained a fraction of murine γδ T cells (Fig. 1). Although earlier reports indicated that PE can bind Fc receptors, these studies also showed that anti-Fc receptor antibodies and/or serum, or serum Ig completely block the interaction (Takizawa, et al., 1993; vanVugt, et al., 1996). Indeed, as Fc blockers, normal mouse serum and normal hamster serum were included in all our Fluorescence-activated Cell Sorting (FACS) analysis; we observed no above-background PE staining on myeloid cells or αβ T cells. PE is a noted B cell antigen and we routinely found ~0.1% of the B cells in naïve animals stained by PE (Fig. S1). This frequency is similar to what has been reported previously (Pape, et al., 2011; Wu, et al., 1991). Further analysis indicated that PE binding to γδ T cells is antigen receptor specificity dependent, as PE did not stain G8 γδ TCR (specific for the non-classical MHC class I molecules T10 and T22) transgenic T cells (Fig. 1A).
Fig. 1. PE recognition by murine γδ T cells.

FACS analysis of (A) PE staining of murine splenic γδ T cells and (B) PE binding and CD62L expression of splenic γδ T cells stimulated with immobilized PE for six hours; (C) PE staining of MA2 γδ TCR expressing 58α−β− cells (left) and in the presence of anti-PE Fab fragment (right); (D) IL-2 production of MA2-58α−β− cells activated with plate bound PE, anti-CD3 (1μg/ml) or PE in solution for 16 hours; (E) Surface plasmon resonance analysis of a soluble PE-specific γδ TCR (MA2) binding to immobilized PE (left) and the plot of steady state binding value (RU) vs. TCR concentration (right); (F) Kinetics of PE binding to MA2-58α−β− cells. The half-life (t1/2) was determined using real time flow cytometry in the presence of Fab of anti-PE antibody (left); KD was determined from Scatchard analysis, where the absolute number of PE bound per cell was determined using PE-quantum calibration beads (BD Biosciences) as reference (right).
PE stained ~0.02–0.4% of total γδ T cells in normal un-immunized mice, regardless of the tissue of origin (spleen, thymus and the intestine intraepithelial lymphocyte) or genetic background (including C57BL/6, BALB/c, B10.Br, 129, Lpr and NOD mice) (Fig. 1, 2, S1 and data not shown). Moreover, PE staining identified nearly all splenic γδ T cells that responded to PE stimulation (Fig. 1B). To further investigate this finding we identified PE-specific γδ TCR sequences at the single cell level (Table 1 and data not shown). Expression of these TCRs in 58α−β− cells or Jurkat β− (J.RT3-T3.5) cells enabled these cells to bind PE and to respond to PE stimulation (Fig. 1C, D). The observations that 1) the whole PE molecule stained PE-specific γδ TCR expressing cells, 2) the staining was completely inhibited by the Fab fragment of a PE specific monoclonal antibody, and 3) immobilized PE, but not PE in solution could activate PE-specific γδ T cells indicated that PE is recognized directly without being processed and presented.
Fig. 2. Antigen recognition determinants of PE specific γδ TCRs.

FACS analysis of (A) PE binding to Jurkat β− cells expressing γδ TCRs with different CDR3s; (B) PE binding and antibodies against Vγ1, Vγ4, and Vγ7 of splenic γδ T cells and IELs. The number within the gate indicates the percentage of PE-positive cells, or Vγ-subtype positive γδ T cells among total γδ T cells, representative of at least 3 independent experiments.
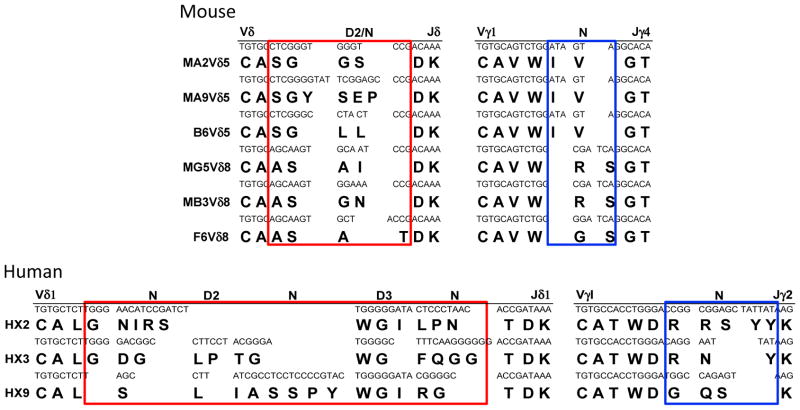
Table 1. Representative PE-specific γδ TCR sequences.
Each pair of the TCR sequences wereidentified from a single PE-specific γδ T cell (from mouse spleen or human PBL) and verified by their ability to confer PE binding to Jurkat β-cells expressing the TCR, and/or by establishing γδ T cell clones, which bind PE (by FACS) and respond to PE stimulation.
|
Indeed, direct binding between the soluble form of specific TCRs and PE could be demonstrated with Surface Plasmon Resonance (BIAcore). A KD (dissociation constant) of 2.7μM for a specific γδ TCR-MA2 binding to PE (Fig. 1E) indicated that this interaction was at the high affinity end of the spectrum for soluble αβ and γδ TCR-ligand interactions (~0.1 to >500 μM) (Crowley, et al., 2000; Newell, et al., 2011; van der Merwe and Davis, 2003; Xu, et al., 2011). It has been suggested that while highly specific, the affinity of the individual TCR-ligand interaction is necessarily low, as the interaction between antigen and the TCRs on T cell surface are of high avidity. PE is not a cell membrane associated protein, but is multimeric, composed of a γ subunit situated in the central channel of two alpha and beta chain trimers: ((αβ)3)2γ. Scatchard analysis of the apparent affinity of PE binding to MA2 expressed on 58α−β− cells showed a sub-nanomolar KD (0.3 nM) with a half-life of ~20 minutes as determined by real time FACS analysis (Fig. 1F). Taken together, these results indicate that PE is an antigen of γδ T cells, and is recognized directly by specific TCRs with an affinity that is high enough to be physiologically relevant.
The CDR3 regions of the TCR γ and δ chains confer antigen recognition specificity
The complementary determining region 3 (CDR3) generated by V, D, J recombination is essential in determining the antigen specificity of all immune receptors. One striking feature of murine PE-specific γδ TCRs was their short TCRδ CDR3 regions. Most of these were six amino acid residues long, with no discernable contribution from the Dδ1 or Dδ2 gene segments. In contrast, the TCRγ CDR3s were 9 or 10 residues in length, relatively long compared to the average CDR3 γ chain length (Table 1). However, not all TCRs with short CDR3δ recognized PE. Two pairs of γδ TCRs, MB3 and C2, utilized the same Vγ, Vδ, Jγ, Jδ gene segments and were of the same CDR3 γ and δ lengths, but differed in amino acid residues encoded by N nucleotides in the CDR3 γ and δ chain regions. MB3, but not C2, bound PE (Fig. 2A). Additional mutagenesis studies supported the supposition that both the CDR3γ and CDR3δ regions are essential for PE binding (Table S1).
The majority of the PE positive γδ T cells in the spleen express Vγ1 and Vγ4, those in the intraepithelial lymphocytes of the small intestine (IELs) express Vγ7 (Fig. 2B). This result, similar to what we observed for T10 and T22 specific γδ TCRs (Shin, et al., 2005), indicates that Vγ usage is more reflective of the tissue of origin than of the antigen specificity for the γδ TCRs. While the majority of the PE-specific γδ T cells we analyzed utilized Vδ5 and Vδ8, swapping the Vδ gene segment to Vδ10 weakened but did not abolish PE binding (Fig. S2). These observations indicate that the CDR3 regions of the TCR γ and δ chains encode PE-binding determinants, with contributions from the CDR1 and/or 2 encoded on the Vδ gene segment.
PE is recognized by human and bovine γδ T cells
The observation that PE, a microbial protein is a natural antigen of γδ T cells is consistent with previous studies showing that the antigen specificity of γδ T cells are not constrained by thymic selection (Jensen et al., 2008). Thus, γδ T cells can recognize microbial or foreign molecules that are not expressed in the thymus. In addition, as a group, γδ TCRs and immunoglobulins are similar in antigen recognition (Chien and Konigshofer, 2007). In fact, PE is a noted B cell antigen that has been used extensively to study memory B cell development (Hayakawa et al., 1987; Maruyama et al., 2000; Pape et al., 2011). It has been demonstrated that antibodies from multiple animal species can recognize the same molecule. In this context, we found that PE stained populations of bovine, sheep and human periphery γδ T cells (Fig. 3A and data not shown).
Fig. 3. PE recognition by human and bovine γδ T cells.

FACS analysis of PE staining of (A) human and bovine peripheral blood γδ T cells and (B) Jurkat β− cells expressing a PE-specific human γδ TCR-HX2, and a non-PE-specific γδ TCR, FQQ1; (C) human and mouse γδ TCRs expressing Jurkat cells stained with PE from Porphyra ternera, P1 or P5, representative of at least 3 independent experiments.
We have analyzed human PE-specific γδ T cells further by generating PE-specific human γδ T cell clones from individual PE-specific cells, determining their TCR sequences and demonstrating PE binding to Jurkat cells, which express PE-specific TCRγ and δ chain sequences (Table 1) (Fig. 3B). The CDR3δ lengths of human PE-specific γδ TCRs were ~15–17 amino acid residues long and consisted of a Vδ1 Jδ rearrangement with a clearly discernable Dδ3 region in a single open reading frame. The TCRγ chains were assembled from Vγ1 to Jγ1 rearrangement with different CDR3γ sequences and lengths. Two human γδ T cells clones, HX2 and FQQ1, express TCRs with the same Vγ, Vδ, Jγ, Jδ but differ in the CDR3 regions. HX2 but not FQQ1 binds PE (Fig. 3B), indicating that the CDR3 regions of human PE-specific γδ TCRs are also essential for antigen recognition.
Human and murine PE-specific γδ TCRs have different fine specificities
PE is a hexameric protein with tetrapyrrole chromophores covalently linked to cysteine residues. PE isolated from different algae has minor differences in amino acid sequences and the numbers, combinations and positions of chromophores (Apt, et al.,1995). Comparing the reactivity of a human γδ TCR with two prototypic PE-specific murine γδ TCRs indicated that each pair of the TCR bound R-PE from porphyra tenera (Cyanotech), PE from the red algae, P-1 (from Quantaphy) and P-5 (Prozyme, supplied by Quantaphy) differently. With respect to γδ T cells as a whole, we found that R-PE as well as P1 stained twice as many murine splenic γδ T cells as P5 did. In one human sample we tested, the percentage of P-1 and P-5 specific γδ T cells was similar (Fig. S1 and data not shown).
Taken together, these results indicate that γδ TCR’s binding to PE is highly specific and dependent on the sequences generated by somatic gene rearrangement. While there are common features shared among murine specific γδ TCRs, these features are not shared by human anti-PE specific γδ TCRs and there is no apparent sequence similarity between the human and the murine PE-specific γδ TCR sequences. Thus, the antigen specificity, but not the particular antigen-specific TCR sequences, is conserved through evolution. This aspect is a defining characteristic of adaptive immune recognition (Deng, et al.,2010).
PE can be used to track PE specific γδ T cells in immune response
Our past studies identified γδ T cells that are specific for the nonclassical MHC class I molecules T10 and T22. We also generated a tetrameric staining reagent that allows us to identify and analyze T10 and T22-specific γδ T cells (Crowley, et al., 2000; Jensen, et al., 2008; Shin, et al., 2005). However, this system is unsuitable for determining what is required to activate γδ T cells in an immune response. This is because the expression of the antigen themselves (i.e. T10 and T22) are induced in infection and after activation (Chang, et al., 2007; Crowley, et al., 2000 and data not shown). Thus, we could not distinguish antigen recognition from inflammatory cues in γδ T cell activation. In contrast, PE is a molecule of microbial origin. As a result, we can use PE to track PE-specific γδ T cells in an immune response to determine what is required in order for3 γδ T cells to mount an effector response.
To this end, we analyzed PE-specific and non-PE-specific γδ T cells in mice immunized with PE from P1. For comparison, we also analyzed PE-specific γδ T cells in mice immunized with ovalbumin (OVA). Alum was used as it is a non-antigenic adjuvant (Eisenbarth, et al., 2008), and we chose subcutaneous immunization as it focuses the immune response to the draining lymph nodes.
Antigen recognition, but not extensive clonal expansion is required to induce PE-specific γδ T cells to make IL-17
Prior to immunization, ~90% of PE-specific γδ T cells were CD44loCD62Lhi, a phenotype typical of naïve T cells. Within 24 hours after PE-alum immunization there was a ~3 fold increase of PE-specific γδ T cells and ~40% of these cells were CD44hiCD62Llo (Fig. 4A). Using BioMark (Warren, et al.,2006), which permits microfluidic quantitiative polymeriase chain reaction (qPCR) analysis on multiple genes from 1 to 100 cells, we found that PE-specific γδ T cells showed increased chemokine receptor CCR2 and decreased CCR7 expression. The transition from CD44loCD62LhiCCR2loCCR7hi to CD44hiCD62LloCCR2hiCCR7lo is commonly associated with the acquisition of a new cell migration pattern in antigen activated naïve αβ T cells (DeFranco A.L.,2007).
Fig. 4. γδ T cell responses after immunization.

(A) Mean and standard deviation of the number of total cells, total γδ T cells, PE+ γδ T cells, and activated (CD44hiCD62Llo) PE+ γδ T cells (per four mice), averaged from two or three independent experiments; and CD44, CD62L expression on PE+ (red) and PE− (blue) γδ T cells in the draining lymph nodes (dLN) of mice immunized with PE-alum or OVA-alum 24 hours prior and in the corresponding lymph nodes of naïve mice; (B) BioMark analysis of CD62LloPE+ and CD62LhiPE− γδ T cells isolated from the dLN of C57BL/6 mice immunized with PE-alum 48 and 60 hours prior (5 cells/sample). The heatmap, where rows are individual genes and the columns are individual samples, indicates the expression of a gene/sample pair (relative to the B2m expression) in false color scale. All non-varying genes were removed prior to performing two-class, unpaired analyses using the significant analysis of microarrays (SAM) (Tusher, et al.,2001). Only genes considered significantly different at a q-value < 0.01 were shown; (C) Intracellular IL-17 staining of γδ T cells of C57BL/6 mice (left) and Thy 1.1 expression on γδ T cells from Il17fthy1.1/thy1.1 mice (right) immunized with PE-alum 60 hours prior. The number indicated the percentage of total PE+ γδ T cells that are intracellular IL-17 positive. All results (A–C) are representative of at least 3 independent experiments. (D) The number of PE+ γδ T cells (left) and the number of activated (CD44hiCD62Llo) PE+ γδ T cells (right) (per four mice) in mice immunized with PE-alum or OVA-alum at the indicated time prior. Each square represents one independent experiment in each of the immunization schemes.
BioMark analysis showed that 48 hours after PE-alum immunization activated (CD62Llo) PE-specific γδ T cells expressed RORγt, a transcription factor necessary to induce IL-17 expression (Ivanov, et al.,2009), and another 12 hours later they expressed IL-17A and IL-17F (Fig. 4B). Indeed, intracellular staining and analysis of γδ T cells from IL-17F reporter mice (Il17f thy1.1/thy1.1) (Lee, et al., 2009) showed that 60 hours after PE immunization, activated PE specific γδ T cells expressed IL-17 protein or the reporter-Thy1.1 (Fig. 4C).
Surprisingly, during this time period (up to 60 hours) the numbers of either total PE-specific γδ T cells, or PE-specific γδ T cells that showed activated phenotypes (CD44hiCD62Llo) in the draining lymph nodes of PE-alum immunized mice did not change significantly after the initial increase (~3 fold for total PE-specific γδ T cells and ~10 fold for CD44hiCD62Llo PE-specific γδ T cells) at 24 hours after immunization (Fig. 4A, D). This differs from naïve αβ T cells, which acquire effector functions only after extensive proliferation for 5–7 days following initial antigenic challenge (DeFranco A.L., 2007). Importantly, OVA-alum immunization does not induce PE-specific γδ T cell activation, and immunization with either antigen does not induce general γδ T cell activation (Fig. 4).
Antigen activated γδ T cells gain the ability to respond to inflammatory cytokines
Notably, PE-alum but not OVA-alum immunization induced PE-specific γδ T cells to express IL-1 and IL-23 receptors (BioMark and FACS) (Fig. 5A, B), indicating that antigen recognition induces the expression of these receptors. This is consistent with a study showing that signaling through the TCR via the guanine nucleotide exchange factor VAV1 is essential for inducing IL-1R+ γδ T cells (Duan, et al., 2010). Along this line, rapid IL-17 responding γδ T cells are reported to be CCR6hi (chemokine receptor for MIP1a) (Haas, et al., 2009) or CD27lo (a TNF receptor) (Ribot, et al., 2009). We found that PE-specific γδ T cells from PE-alum, but not from OVA-alum immunized mice showed increased CCR6 and decreased CD27 expression (Fig. 5A, S3), suggesting that the expression of these molecules on γδ T cells can be regulated by antigen recognition in the context of an immune response. Indeed, CCR6hi or CD27lo γδ T cells were CD62Llo (Fig. 5C, S3).
Fig. 5. Regulation of IL-17 expression by TCR triggering and inflammatory cytokines.

(A) BioMark analysis of PE+ γδ T cells from the dLN of mice immunized with PE-alum or OVA-alum 60 hours prior, directly ex vivo (2 cells/sample); (B) IL-1 receptor expression on PE+ and PE− γδ T cells from dLN of mice immunized with PE-alum 60 hour prior; (C) CD44, CD62L expression of the total spleen γδ T cells from the naive C57BL/6 mouse, and the expression of IL-1R and CD27 expression of each subpopulation according to their CD44, CD62L expression. (D) BioMark analysis of splenic CD62Llo and CD62Lhi γδ T cells after in vitro stimulation with plate bound anti-CD3 (10 μg/ml), IL-1 and IL-23 (1ng/ml each), Pam3Csk4 (1μg/ml) and IL-23 (1ng/ml), or media alone. Mean and standard deviation of qPCR analysis of (E) IL-17 mRNA of total splenic CD62Llo γδ T cells (purified by FACS sorting, achieving >98% purity), stimulated with plate bound anti-CD3 and anti-CD3 together with IL-1 and IL-23 (1ng/ml each), in the absence or presence of 100 ng/ml Cyclosporine A and (F) IL-17 mRNA of total γδ T cells enriched from G8 TCR transgenic mice lymph nodes by negative depletion (achieving >98% purity as determined by FACS) stimulated with IL-1 and IL-23 (1ng/ml each), plate bound anti-CD3 and anti-CD3 together with IL-1 and IL-23, at indicated time points. All results are representative of at least 3 independent experiments.
In normal, un-immunized mice, only CD44hiCD62Llo γδ T cells express IL-1 and IL-23 receptors (Fig. 5C, S3). Accordingly, we found that CD62Llo, but not CD62Lhi γδ T cells responded to IL-1 and IL-23 stimulation to make IL-17 (Fig. 5D). While this response was initiated without explicit TCR triggering, it was inhibited by Cyclosporine A (CsA) (Fig. 5E) or by FK506 (Fig. S4). Both compounds reduce NFAT (nuclear factor of activated T-cells) activity and disrupt the calcineurin, NFAT signaling circuit activated by signaling through the antigen receptor (Flanagan, et al., 1991). Similarly, CD44hiCD62Llo PE+ cells isolated from naïve animals could make IL-17 in response to IL-1, IL-23 stimulation, but the response was also sensitive to CsA inhibition (Fig. S4). In addition, while Pam3Csk4 and IL-23 enhanced an anti-CD3 induced IL-17 response, this enhanced response was also inhibited by CsA or FK506 (Fig. S4). Thus, the observations that IL-1 and IL-23 or TLR agonists and IL-23 can induce IL-17 production from γδ T cells without explicit TCR ligation (Martin et al., 2009; Reynolds et al., 2010; Sutton, et al., 2009) may reflect the activation characteristics of the pre-activated γδ T cells (CD62LloCD44hi) in naïve animals that have been inadvertently included in the experiments.
While triggering through the TCR alone can activate γδ T cells to make IL-17, this response was limited in magnitude and duration. As shown in Fig. 5F, when IL-1 and IL-23 were included in the stimulation assay, the IL-17 response was much higher and lasted longer than stimulation with anti-CD3 alone. Thus, antigen induced IL-1 and IL-23 receptor expression could enhance and prolong the IL-17 response of antigen activated γδ T cells in inflammation. Taken together, our observations that signaling through the antigen receptors induces γδ T cells’ ability to respond to inflammatory cytokines suggest a way for γδ T cells to respond to environmental cues and perpetuate their response in inflammation, in a process that is initiated by antigen encounter.
Discussion
Despite intense effort, the process of identifying γδ TCR ligands has been long, challenging, and confusing. Few ‘agents’ have been shown to activate γδ T cells and even fewer are known to be both necessary and sufficient to trigger T-cell responses through the γδ TCR, which would make them bone fide γδ T-cell antigens. Nonetheless, it is clear that γδ T cell antigens include host molecules that are induced on activated, stressed or cancerous cells such as T10 and T22 in the mouse and ATP synthase, ApoA-1 complex and the major histocompatibility complex class I related chain MICA and MICB in the human. Moreover, our past observations that T10 and T22 specific γδ T cells are present and functional in C57BL/6 (express T10 and T22), BALB/c (express only the inducible T22) and B2m−/− mice (do not express either T10 or T22 on cell surface) further extend the γδ T cell antigen specific repertoire to include molecules that are either constitutively expressed, or not at all by the host. Here we have demonstrated that a molecule of microbial origin could bind specifically to γδ TCRs and could be recognized by γδ T cells from multiple animal species without prior immunization. This suggests that microbial and foreign antigens are also relevant γδ T cell targets.
Indeed, it appears that γδ T cells can respond to a variety of stimulations irrespective of the molecular or genetic nature of the stimuli. Immunizing mice with MHC mismatched spleen cells generates MHC reactive γδ T cell clones, including the I-E specific LBK5 and the T10 and T22 specific G8. Human γδ T cells that recognize MICA and B or CD1c were derived from culturing γδ T cells with MICA and B expressing CIR cells, or CD1c expressing dendritic cells, respectively. G115, a human γδ T cell clone that recognizes ATP synthase and ApoA-1 complex on tumor cells, was produced by culturing PBL γδ T cells with irradiated PBL and lymphoblastoid cells. In addition, stimulating lymph node cells from herples smplex virus (HSV) infected mice with herpes glycoprotein gI expressing L cells generates herpes glycoprotein gI reactive γδ T cell clones, and stimulating human γδ T cells with M. tuberculosis extract generates human γδ T cell clones that respond to small molecular weight phospho-molecules produced by microorganisms (Chien and Konigshofer, 2007). Although further experiments will be needed to ascertain that these microbial molecules are γδ T cell antigens, these observations suggest that the γδ T cell antigen repertoire is diverse.
While γδ T cells can recognize different kind of antigens; their repertoire may be smaller than that which is estimated for αβ T cells. We have noted previously that in non-immunized mice, ~0.1–1% of total γδ T cells are T10 and T22 specific (Crowley, et al., 2000). Here we found that ~0.02–0.4% of the γδ T cells recognize PE. These frequencies are much higher than the 0.0001% to 0.001% for antigen specific αβ T cells before clone expansion. If the frequency of other antigen specific γδ T cells are also in a similar range, then the numbers of distinct γδ T cell antigens would be ~103 to 104. In this context, it has been proposed that the immune repertoire can probably offer efficient protection against about 103 to 104 distinct infections relevant for the survival of a given species (Cohn and Langman, 1990; Mims, 1987; Zinkernagel, 1996). Based on this argument, Zinkernagel and Hangartner proposed, “--in mice --- immunity is generated from a starting number of about 100 to 1000 antigen-specific precursor (αβ) T and B cells.” (Zinkernagel and Hengartner, 2001). While this range turns out to be much higher than the frequencies of naïve antigen specific αβ T cells, it is surprisingly close to that of antigen specific γδ T cells in naïve animals. In fact, the high initial frequency of antigen specific γδ T cells is coupled with the lack of clonal expansion requirement to mount an effector function, features that allow γδ T cells to mount a rapid and substantial response in immune challenge.
However, the observation that γδ T cells mount a rapid and substantial IL-17 response after immune challenge has fuelled the supposition that γδ T cells provide an “innate” source of IL-17. Experiments showing that IL-17 can be elicited from γδ T cells by stimulation with IL-1, IL-23 or TLR agonists without explicitly triggering the TCR (Martin, et al., 2009; Reynolds, et al., 2010; Sutton, et al., 2009) seem to be consistent with this notion. Nonetheless, the amount of IL-17 induced by the inflammatory cytokines alone is much more reduced in magnitude than those induced by cytokines together with TCR stimulation, as was first demonstrated by Sutton and colleagues (Sutton, et al., 2009) and confirmed by our observation here; this suggests that robust IL-17 production requires combined signaling through the TCR and cytokine receptors. Moreover, we showed that only activated (CD44hiCD62Llo) γδ T cells express IL-1, IL-23 receptors and make IL-17 without explicit TCR triggering and, even then, the response is inhibited by CsA or FK506. These observations further underscore the importance of TCR signaling in γδ T cell IL-17 induction.
In fact, the very nature of γδ T cell antigen recognition makes it difficult to discount TCR triggering in an immune response. Our results here indicate that γδ T cells can recognize microbial antigens. γδ T cells can also recognize self-antigens that are induced on activated cells, such as T10 and/or T22. Increased T10 and/or T22 expression has been noted on lipospolysaccharide (LPS) stimulated B cells and on activated T cells (Crowley, et al., 2000 and data not shown). Relevant to the discussion here, it was reported that within two days after influenza virus infection, there is a significant increase in T22 expression on myeloid cells and T22 specific γδ T cells with activated phenotype (CD69hi and CD62Llo) in the regional lymph node (Chang, et al., 2007). Indeed, the increase of IL-17+ γδ T cells after immunization with LPS or malaria-infected red blood cells (Ribot, et al., 2010) could result from activation of γδ T cells that recognize microbial antigens and/or host antigens, that is amplified and sustained by inflammatory cytokines induced from myeloid cells in TLR and MyD88 dependent manner. In fact, the timing (assayed 3 days after immunization) and the magnitude of the response (~ 5 fold increase in the number of IL-17+ γδ T cells) are within the range of what we have observed for the development of PE-specific IL-17 responses after PE immunization.
Our past studies indicate that γδ T cells need not encounter cognate antigen in the thymus to signal through the TCR, mature and exit to the periphery. When triggered through the TCR, periphery γδ T cells derived from γδ thymocytes that have not encountered thymic ligands make IL-17 (Jensen, et al., 2008). Our results here confirm and extend these observations to provide a clear dissection of what is required to activate these cells in an immune response. In particular, we demonstrate that PE-specific γδ T cells differentiate toward an IL-17-producing phenotype upon antigen encounter: within 24 hours after PE immunization, PE specific γδ T cells in the draining lymph node increased in numbers and showed activated phenotypes, such as becoming CD44hi and CD62Llo. 48 hours after immunization, activated PE-specific γδ T cells express RORγt, and after another 12 hours, IL-17A and F. Importantly, encountering antigen in an immune response induces the expression of inflammatory cytokine receptors such as IL-1R and IL-23R on specific γδ T cells. The cytokine receptor signaling provides a “second signal” in addition to TCR engagement to perpetuate the response in inflammation. This synergistic effect between TCR and inflammatory cytokine signaling may also prevent the development of a prolonged and robust IL-17 response in the absence of inflammation.
Although diversity in antigen receptor specificities is the hallmark of adaptive immune system, not all antigenic responses produce results. In fact, effective adaptive immune responses are focused in antigen specificity. This coordinated antigen recognition is best illustrated in the T-dependent antibody response, where the participating αβ T cells and B cells must respond to the same antigen. Our observations that PE, a noted antigen for inducing (αβ) T cell dependent antibody response is also a γδ T cell antigen and that PE induces early IL-17 production from specific γδ T cells suggest that a focused adaptive immune response starts from an antigen specific γδ T cell response.
Experimental Procedures
Mice, Reagents and Immunization
C57BL/6 mice were purchased from the Jackson Laboratories and housed in the Stanford Animal Facility for at least a week before use. IL-17fThy1.1/Thy1.1 mice and G8 γδ TCR transgenic mice were bred and housed in the pathogen-free Stanford Animal Facility. All experiments were performed in accordance with the Institutional Biosafety Committee and the Institutional Animal Care and Use Committee. Human PBMCs were obtained from platelet apheresis donors through the Stanford Blood Bank in accordance with IRB protocol. 200 μg each of PE (from P1 strain, Quantaphy) or ovalbumine (OVA) (Sigma) in alum (Imject Alum; Thermo Scientific) per mouse and subcutaneous immunization was used in all studies.
Antibodies and FACS analysis
All antibodies are from eBioscience, unless stated otherwise. For PE staining, murine γδ T cells were first enriched (Jensen, et al.,2008), followed by staining with PE (0.5 μM, P1 strain) on ice for one hour and APC conjugated anti-γδ TCR (GL-3), Live/Dead Aqua (Invitrogen, Molecular probes), FITC conjugated anti-αβ TCR (H57-597), B220 (RA3-6B2), CD11b (M1/70), CD11c (N418), Gr-1 (RB6-8C5), F4/80 (BM8). Aqua and FITC positive cells were excluded from analysis.
For the analysis of CD62L and CD44 expression, enriched γδ T cells were stained with LIVE/DEAD Aqua, Pacific blue CD62L (MEL-14), FITC-CD44 (IM7), and PE. For the analysis of Thy1.1 expression on cells isolated from IL-17fThy1.1/Thy1.1 reporter mice, enriched γδ T cells were stained with LIVE/DEAD Aqua, Pacific blue-CD62L, FITC-Thy1.1 (OX-7; BioLegend), PE, APC-GL-3. For the analysis of IL-1R (CD121a) and CD27, enriched γδ T cells were stained with LIVE/DEAD Aqua, Pacific blue-CD62L, FITC-CD27 (LG.7F9), PE, APC-CD121a (JAMA-147; BioLegend), PerCP Cy5.5-GL-3, eFluor-605NC-CD44. All staining include the addition of APC-Cy7 conjugated anti-TCRβ, B220, CD11b, CD11c, Gr-1, F4/80. APC-Cy7 and Aqua positive cells were excluded from analysis.
For intracellular IL-17 staining, γδ T cells were enriched from pooled draining lymph node cells from 5-15 of immunized mice. Enriched γδ T cells were re-stimulated in vitro with PE (0.5 μM), IL-1 and IL-23 (1ng/ml each) for seven hours before analysis. Cells were then harvested, blocked with serum, FcBlocker and dump antibodies as described above, and fixed and permeabilized with BD Cytofix/Cytoperm solution (BD Biosciences) for 20 min on ice, followed by staining with APC conjugated IL-17A (eBio17B7) or Rat IgG2a isotype. Analysis of Vγ usage was as described (Shin, et al.,2005).
Human and bovine PBL γδ T cells were stained with PE (0.5 μM, P1 strain), without enrichment. Human cells were also stained APC conjugated anti-γδ TCR (5A6.E9; Invitrogen Molecular probes), Live/Dead Aqua, FITC conjugated anti-CD19 (HIB19), αβ TCR (IP-26), CD14 (HCD14), CD16 (3G8). Bovine cells were stained with APC conjugated anti-γδ TCR (GB21A) (all antibodies are from VMRD Inc.), LIVE/DEAD Aqua, FITC conjugated anti-CD4 (CC8), CD8 (CC63), CD14 (CC-G33; all from AbD Serotec), anti-B lymphocytes (LCT30A). Aqua and FITC positive cells were excluded from analysis.
Identification of PE-specific γδ TCR sequences
Identification of murine PE-specific γδ TCRs was as described (Shin, et al.,2005). Briefly, PE+ γδ T cells from naive spelnocytes were single cell sorted into 96 well U bottom plates pre-coated with 10μg/ml anti-TCRδ (GL-4) and cultured in the presence of 100U/ml rIL-2. After 7–9 days, γδ TCR genes were determined by amplifying the TCR chains from genomic DNA. PE-specific γδ TCRs were expressed on Jurkat β− cells or 58α− β− cell line. To identify human PE-specific γδ TCRs, individual PE+ γδ T cells were isolated from PBMC by FACS and expanded separately in wells pre-coated with 10μg/ml anti-CD3 antibody (OKT3), in the presence of 100U/ml rIL-2, 7.5×105 cells/ml irradiated fresh PBMC (4000 Curie) and 7.5×104 cells/ml irradiated JY cell line (12000 Curie) for 2~3 weeks with additional 100U/ml rIL-2 every week. Clones were re-analyzed by FACS with PE and APC conjugated anti-TCRδ. PE positive clones were isolated using FACSAria (BD Bioscience), and the γδ TCR genes were determined by amplifying the TCR chains from cDNA using primers as described in Supplemental Methods. PE-specific human γδ TCRs were expressed in Jurkat β− cells as described for the expression of the murine γδ TCRs (Shin, et al.,2005).
Production of soluble γδ TCRs
The extracellular domains of the γ and δ chains (residues 1-273 and 1-242, respectively) were cloned in frame with a gene encoding a Phinovirus protease site, followed by acidic (TCRδ) or basic (TCRγ) leucine zipper and a (histidine)6 tag in the pMSCV-P2 and Z4 retroviral expression vectors containing IRES puromycin resistance gene for γ chain or zeocin resistance gene for δ chain(Shin, et al.,2005), and expressed in BHK-21 cells (Baby Hamster Kidney, ATCC). The soluble TCRs were purified from the supernatant with lectin agarose (Sigma) and Ni-NTA beads (Qiagen). The acidic and basic zippers were removed with Precision Protease (GE lifescience).
BIAcore analysis
Approximately 230 RU of PE were immobilized on a CM3 BIAcore sensor chip using NHS and EDC reagents. A similar amount of allophycocyanin (APC) was immobilized on a reference flow-cell used for background signal subtraction. Soluble TCR were injected at various concentrations (40, 13, 4.4, 1.48, 0.49, 0.165, 0.05 and 0 mM) at a flow-rate of 50 ml/min. The decay rates of these curves were used to obtain the koff value of 0.058 s−1. The signal at the end of each injection was taken as the steady state binding value and plotted vs. TCR concentration. The fit to a 1:1 steady-state binding relationship was used to obtain the KD value of 2.69 μM. kon was calculated as 21,600 M−1s−1. From this same fit, the Rmax for this experiment was found to be 235 RU. Based on the molecular weights of PE (240 KD) and TCR (60 kD), an average of four TCR molecules can bind to each PE molecule at saturation.
Kinetics measurement of antigen binding to surface TCR by real time FACS analysis
MA2 (PE specific) γδ TCR expressing 58α−β− cells (1×106/ml) were incubated with 40 nM PE for 1 hour at 4°C in the presence of Aqua. Cells were spun down and re-suspended in 0.5 ml FACS buffer with 2400 nM anti-PE Fab prepared from an anti-PE monoclonal antibody (kindly provided by Dr. Richard Hardy) using the Pierce Fab Preparation Kit. The fluorescent intensities of 2000 cells were measured every 5 or 10 minutes (<10 s for each measurement) for the duration of 2.5 hours. The left-shift of the histogram toward lower fluorescent intensity was monitored in real time. The first-order decay kinetic model was used to fit the mean fluorescent intensities at different time points and obtain the off-rate koff and half-life t1/2.
Affinity Measurement of PE binding to surface TCR
1×105 MA2 -58α−β− cells or 58α−β− (control) cells were incubated with varying amount of PE (0.08–10.42 nM) at 4°C for one hour, followed by FACS analysis. The number of ligand bound on each cell was determined as described (Huang, et al., 2010). Briefly, the fluorescent intensity of the sample was compared with the calibration curve generated with quantum calibration beads (PE beads from BD) that were analyzed under the same setting after subtracting negative control fluorescent intensity. After multiplying the cell number (1×105/tube) with the bound ligands on each cell, it was divided by the Avogadro’s number and the volume to obtain the bound concentration [Bound]. The concentration of free ligands [Free] was calculated by subtracting the [Bound] from the concentration of added total ligands.
Stimulation of splenic γδ T cells with immobilized PE in vitro
PE-streptavidin (1mg/ml) was added to chamber pre-coated with biotinylated poly-lysine at room temperate for 1 hour. Enriched γδ T cells were isolated by FACS sorting (with > 98% purity), and incubated in PE coated chambers at 37°C for 6 hours, followed by analysis of PE binding and CD62L expression.
Immobilized PE or anti-CD3 for stimulation of MA2 TCR expressing 58α− β− cells in vitro
Various amount of PE-streptavidin (0.064–40μg/ml) was added to chamber pre-coated with biotinylated poly-lysine at room temperate for 1 hour. Control chamber was added 20 μg/ml streptavidin, following 1μg/ml biotinylated anti-CD3. 1×105 MA2 58α−β− cells (1×105 cells/ml) were incubated in PE-streptavidin or anti-CD3 coated chamber at 37°C for 16 hours. Supernatants were harvested and analyzed for the presence of IL-2 by ELISA (eBioscience).
BioMark analysis
To determine the gene expression pattern of PE-specific γδ T cells after PE-alum immunization, γδ T cells were enriched from the dLN of immunized mice, and incubated with PE (0.5 μM) for 6 hours in vitro. PE+ CD62Llo and PE− CD62Lhi γδ T cells were then FACS sorted into PCR plate with 5 cells/well for the analysis. To compare the gene expression pattern of PE+ γδ T cells from PE-alum or OVA-alum immunized mice directly ex vivo, γδ T cells were enriched from the dLN and stained with PE, CD62L, and CD44. PE+CD62LloCD44hi from PE-alum and PE+CD62LhiCD44lo from OVA-alum immunized mice were analyzed with 2 cells/well. To analyze the gene expression pattern of CD62Llo and CD62Lhi γδ T cells and their response to stimuli, γδ T cells were enriched from naive C57BL/6 mouse spleens, and CD62Llo and CD62Lhi γδ T cells were isolated by FACS. The sorted cells were stimulated with media alone, plate-coated anti-CD3 (10 μg/ml), IL-1 (1ng/ml) and IL-23 (1ng/ml), or Pam3Csk4 (1μg/ml) and IL-23 (1ng/ml) for 48 hours in vitro. γδ T cells were re-sorted with FACS, which deposits 100 live-cells to each well for BioMark analysis. To determine the gene expression pattern of PE+ CD62LloCD44hi and PE+CD62LhiCD44lo γδ T cells in naive mouse, γδ T cells were enriched from naive C57BL/6 mouse spleens and stimulated with IL-1 (1ng/ml) and IL-23 (1ng/ml), in the absence or presence of CsA (100ng/ml) for 4 hours, followed by surface marker staining. PE+CD62LloCD44hi and PE+ CD62LhiCD44lo cells were sorted with FACS, which deposits 2 live-cells to each well for the analysis. The primers for BioMark qPCR were purchased from Applied Biosystems.
Analyses of the expression data were performed with the R statistical package v. 2.12 (Team, RDCR: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria (2009). URL http://www.R-project.org. ISBN 3-900051-07-0). All non-varying genes were removed prior to performing two-class, unpaired analyses using the significance analysis of microarrays (SAM) package developed by Tusher, Tibshirani, and Chu (Tusher, et al.,2001). Genes were considered significantly different at a q-value < 0.01.
Supplementary Material
Acknowledgments
We thank Drs. P. Pereira, L. Puddington and L. Lefrancois for anti-Vγ antibodies; Dr. Richard Hardy for anti-PE monoclonal antibody; Dr. D. Cua for IL-23R-GFP.KI reporter mouse spleen; Dr. Fengqin Fang for the FQQ1 γδ TCR transfectant; Dr. Kirk Jensen for advice and encouragement at earlier part of this work; Sara Davis for manuscript editing, Burt and Marion Avery endowment and NIH for grant support (YC).
Footnotes
The authors declare no conflict of interest.
XZ and Y-h C design experiments, XZ, Y-l W, JH, EWN, HY, and BAK carried out experiments with JH, EWN, HY, and BAK contributing equivalently to this work. MSK, MMD, CTW provide unique protocol, reagent and/or equipment, Y-h C and BAK wrote the manuscript with inputs from all authors. MSK’s current address: Department of Immunobiology, University of Arizona College of Medicine, Tucson, AZ 85724
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Apt KE, Collier JL, Grossman AR. Evolution of the phycobiliproteins. J Mol Biol. 1995;248:79–96. doi: 10.1006/jmbi.1995.0203. [DOI] [PubMed] [Google Scholar]
- Awasthi A, Riol-Blanco L, Jager A, Korn T, Pot C, Galileos G, Bettelli E, Kuchroo VK, Oukka M. Cutting Edge: IL-23 Receptor GFP Reporter Mice Reveal Distinct Populations of IL-17-Producing Cells. J Immunol. 2009;182:5904–5908. doi: 10.4049/jimmunol.0900732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneville M, O’Brien RL, Born WK. Gammadelta T cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol. 2010;10:467–478. doi: 10.1038/nri2781. [DOI] [PubMed] [Google Scholar]
- Chang WLW, Coro ES, Rau FC, Xiao YY, Erle DJ, Baumgarth N. Influenza virus infection causes global respiratory tract B cell response modulation via innate immune signals. Journal of Immunology. 2007;178:1457–1467. doi: 10.4049/jimmunol.178.3.1457. [DOI] [PubMed] [Google Scholar]
- Chien YH, Konigshofer Y. Antigen recognition by gammadelta T cells. Immunol Rev. 2007;215:46–58. doi: 10.1111/j.1600-065X.2006.00470.x. [DOI] [PubMed] [Google Scholar]
- Cohn M, Langman RE. The Protecton - the Unit of Humoral Immunity Selected by Evolution. Immunological Reviews. 1990;115:7–147. doi: 10.1111/j.1600-065x.1990.tb00783.x. [DOI] [PubMed] [Google Scholar]
- Crowley MP, Fahrer AM, Baumgarth N, Hampl J, Gutgemann I, Teyton L, Chien Y. A population of murine gammadelta T cells that recognize an inducible MHC class Ib molecule. Science. 2000;287:314–316. doi: 10.1126/science.287.5451.314. [DOI] [PubMed] [Google Scholar]
- DeFranco AL, LRMaRMl . Immunity: the immune response in infectious and inflammatory disease. Sinauer Associates, Inc., Publishers; 2007. [Google Scholar]
- Deng L, Velikovsky CA, Xu G, Iyer LM, Tasumi S, Kerzic MC, Flajnik MF, Aravind L, Pancer Z, Mariuzza RA. A structural basis for antigen recognition by the T cell-like lymphocytes of sea lamprey. Proc Natl Acad Sci U S A. 2010;107:13408–13413. doi: 10.1073/pnas.1005475107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J, Chung H, Troy E, Kasper DL. Microbial colonization drives expansion of IL-1 receptor 1-expressing and IL-17-producing gamma/delta T cells. Cell Host Microbe. 2010;7:140–150. doi: 10.1016/j.chom.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan WM, Corthesy B, Bram RJ, Crabtree GR. Nuclear association of a T-cell transcription factor blocked by FK-506 and cyclosporin A. Nature. 1991;352:803–807. doi: 10.1038/352803a0. [DOI] [PubMed] [Google Scholar]
- Haas JD, Gonzalez FH, Schmitz S, Chennupati V, Fohse L, Kremmer E, Forster R, Prinz I. CCR6 and NK1.1 distinguish between IL-17A and IFN-gamma-producing gammadelta effector T cells. Eur J Immunol. 2009;39:3488–3497. doi: 10.1002/eji.200939922. [DOI] [PubMed] [Google Scholar]
- Hamada S, Umemura M, Shiono T, Tanaka K, Yahagi A, Begum MD, Oshiro K, Okamoto Y, Watanabe H, Kawakami K, Roark C, Born WK, O’Brien R, Ikuta K, Ishikawa H, Nakae S, Iwakura Y, Ohta T, Matsuzaki G. IL-17A produced by gammadelta T cells plays a critical role in innate immunity against listeria monocytogenes infection in the liver. J Immunol. 2008;181:3456–3463. doi: 10.4049/jimmunol.181.5.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Ishii R, Yamasaki K, Kishimoto T, Hardy RR. Isolation of high-affinity memory B cells: phycoerythrin as a probe for antigen-binding cells. Proc Natl Acad Sci U S A. 1987;84:1379–1383. doi: 10.1073/pnas.84.5.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry T, Kirimanjeswara GS, Ruby T, Jones JW, Peng K, Perret M, Ho L, Sauer JD, Iwakura Y, Metzger DW, Monack DM. Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J Immunol. 2010;184:3755–3767. doi: 10.4049/jimmunol.0902065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zarnitsyna VI, Liu B, Edwards LJ, Jiang N, Evavold BD, Zhu C. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature. 2010;464:932–936. doi: 10.1038/nature08944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KD, Su X, Shin S, Li L, Youssef S, Yamasaki S, Steinman L, Saito T, Locksley RM, Davis MM, Baumgarth N, Chien YH. Thymic selection determines gammadelta T cell effector fate: antigen-naive cells make interleukin-17 and antigen-experienced cells make interferon gamma. Immunity. 2008;29:90–100. doi: 10.1016/j.immuni.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapsenberg ML. Gammadelta T cell receptors without a job. Immunity. 2009;31:181–183. doi: 10.1016/j.immuni.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity. 2009;31:321–330. doi: 10.1016/j.immuni.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Maruyama M, Lam KP, Rajewsky K. Memory B-cell persistence is independent of persisting immunizing antigen. Nature. 2000;407:636–642. doi: 10.1038/35036600. [DOI] [PubMed] [Google Scholar]
- Mims CA. Pathogenesis of Infectious Disease. Academic Press; 1987. [Google Scholar]
- Newell EW, Ely LK, Kruse AC, Reay PA, Rodriguez SN, Lin AE, Kuhns MS, Garcia KC, Davis MM. Structural basis of specificity and cross-reactivity in T cell receptors specific for cytochrome c-I-E(k) J Immunol. 2011;186:5823–5832. doi: 10.4049/jimmunol.1100197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science. 2011;331:1203–1207. doi: 10.1126/science.1201730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JM, Angkasekwinai P, Dong C. IL-17 family member cytokines: Regulation and function in innate immunity. Cytokine Growth F R. 2010;21:413–423. doi: 10.1016/j.cytogfr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, Girardi M, Borst J, Hayday AC, Pennington DJ, Silva-Santos B. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nat Immunol. 2009;10:427–436. doi: 10.1038/ni.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribot JC, Chaves-Ferreira M, d’Orey F, Wencker M, Goncalves-Sousa N, Decalf J, Simas JP, Hayday AC, Silva-Santos B. Cutting Edge: Adaptive Versus Innate Receptor Signals Selectively Control the Pool Sizes of Murine IFN-gamma- or IL-17-Producing gamma delta T Cells upon Infection. J Immunol. 2010;185:6421–6425. doi: 10.4049/jimmunol.1002283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock EP, Sibbald PR, Davis MM, Chien YH. CDR3 length in antigen-specific immune receptors. J Exp Med. 1994;179:323–328. doi: 10.1084/jem.179.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotet E, Martinez LO, Grant E, Barbaras R, Jeno P, Guiraud M, Monsarrat B, Saulquin X, Maillet S, Esteve JP, Lopez F, Perret B, Collet X, Bonneville M, Champagne E. Tumor recognition following Vgamma9Vdelta2 T cell receptor interactions with a surface F1-ATPase-related structure and apolipoprotein A-I. Immunity. 2005;22:71–80. doi: 10.1016/j.immuni.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Shin S, El-Diwany R, Schaffert S, Adams EJ, Garcia KC, Pereira P, Chien YH. Antigen recognition determinants of gammadelta T cell receptors. Science. 2005;308:252–255. doi: 10.1126/science.1106480. [DOI] [PubMed] [Google Scholar]
- Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22:285–294. doi: 10.1016/j.immuni.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Takizawa F, Kinet JP, Adamczewski M. Binding of Phycoerythrin and Its Conjugates to Murine Low-Affinity Receptors for Immunoglobulin-G. J Immunol Methods. 1993;162:269–272. doi: 10.1016/0022-1759(93)90392-k. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Merwe PA, Davis SJ. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol. 2003;21:659–684. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- vanVugt MJ, vandenHerikOudijk IE, vandeWinkel JGJ. Binding of PE-CY5 conjugates to the human high-affinity receptor for IgG (CD64) Blood. 1996;88:2358–2360. [PubMed] [Google Scholar]
- Warren L, Bryder D, Weissman IL, Quake SR. Transcription factor profiling in individual hematopoietic progenitors by digital RT-PCR. Proc Natl Acad Sci U S A. 2006;103:17807–17812. doi: 10.1073/pnas.0608512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CJ, Karttunen JT, Chin DHL, Sen D, Gilbert W. Murine Memory B-Cells Are Multi-Isotype Expressors. Immunology. 1991;72:48–55. [PMC free article] [PubMed] [Google Scholar]
- Xu B, Pizarro JC, Holmes MA, McBeth C, Groh V, Spies T, Strong RK. Crystal structure of a gammadelta T-cell receptor specific for the human MHC class I homolog MICA. Proc Natl Acad Sci U S A. 2011;108:2414–2419. doi: 10.1073/pnas.1015433108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkernagel RM. Immunology taught by viruses. Science. 1996;271:173–178. doi: 10.1126/science.271.5246.173. [DOI] [PubMed] [Google Scholar]
- Zinkernagel RM, Hengartner H. Regulation of the immune response by antigen. Science. 2001;293:251–253. doi: 10.1126/science.1063005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.