

SUMMARY
Neuronal activity influences genes involved in circuit development and information processing. However, the molecular basis of this process remains poorly understood. We found that HDAC4, a histone deacetylase that shuttles between the nucleus and cytoplasm, controls a transcriptional program essential for synaptic plasticity and memory. The nuclear import of HDAC4 and its association with chromatin is negatively regulated by NMDA receptors. In the nucleus, HDAC4 represses genes encoding constituents of central synapses, thereby affecting synaptic architecture and strength. Furthermore, we show that a truncated form of HDAC4 encoded by an allele associated with mental retardation is a gain-of-function nuclear repressor that abolishes transcription and synaptic transmission despite the loss of the deacetylase domain. Accordingly, mice carrying a mutant that mimics this allele exhibit deficits in neurotransmission and spatial memory. These studies elucidate a mechanism of experience-dependent plasticity and define the biological role of HDAC4 in the brain.
INTRODUCTION
Neuronal activity guides the connectivity of developing circuits and regulates existing synapses in the adult brain (Kerschensteiner et al., 2009; Saneyoshi et al., 2010; Sin et al., 2002; Zito and Svoboda, 2002). Experience-dependent changes in synapse numbers and long-lasting modifications of functional synapses require induction and/or repression of specific genes. Numerous activity-regulated genes have been identified (Flavell and Greenberg, 2008; Leslie and Nedivi, 2011) yet, the molecular mechanisms that coordinate synaptic inputs with transcriptional programs essential for different aspects of neuronal differentiation, plasticity and information processing are incompletely understood. When neurons receive glutamatergic inputs, calcium influx through NMDA receptors and voltage-gated ion channels triggers signaling cascades that activate transcription factors (TFs) (Ch’ng and Martin, 2011; Deisseroth et al., 2003; Flavell et al., 2006; Flavell and Greenberg, 2008; Lai et al., 2008) In addition, these signals may disable nuclear repressor complexes that prevent gene expression in the absence of excitatory drive by binding to promoter or enhancer regions, altering the chromatin structure, and/or suppressing TFs (Chao and Zoghbi, 2009; Lai et al., 2008; Lunyak et al., 2002; McGraw et al., 2011; Qiu and Ghosh, 2008).
Class IIa histone deacetylases (HDACs) exhibit several features that make them attractive candidates for such a repressor mechanism. Unlike class I HDACs that reside in the nucleus and deacetylate histones, class IIa HDACs shuttle between the nucleus and cytoplasm (Haberland et al., 2009). The nuclear export of class IIa HDACs requires calcium-dependent phosphorylation, raising the possibility that in neurons, pathways regulated by these proteins may be affected by synaptic release of excitatory neurotransmitters (Chawla et al., 2003; McKinsey et al., 2000). In mice and flies, class IIa HDACs have been shown to play an essential role in skeletogenesis, muscle development, energy balance, and glucose homeostasis by interacting with TFs Runx2, MEF2, CAMTA, Dach2 and FOXO (McKinsey et al., 2000; Mihaylova et al., 2011; Vega et al., 2004; Wang et al., 2011; Zhang et al., 2002). Class IIa HDACs are expressed in the mammalian brain (Darcy et al., 2010; Haberland et al., 2009). However, their contribution to transcriptional control in the nervous system is poorly understood.
HDAC4 is a class IIa HDAC that has been implicated in neuroprotection. Although studies in animal models have demonstrated that loss of HDAC4 leads to neurodegeneration in the retina and cerebellum (Chen and Cepko, 2009; Majdzadeh et al., 2008), the underlying mechanisms remain controversial. In the retina, HDAC4 has been shown to promote the survival of photoreceptors and bipolar interneurons via an association with HIFα in the cytoplasm (Chen and Cepko, 2009). However, HDAC4 is also thought to accelerate the death of cerebellar granule and Purkinje neurons upon translocation to the nucleus, and through deacetylation of histones (Bolger and Yao, 2005; Li et al., 2012). The later conclusion is puzzling, considering that class IIa HDACs appear to have been evolutionarily inactivated as enzymes. Indeed, all vertebrate class IIa HDACs acquired a Histidine substitution of the Tyrosine residue in the active site of the deacetylase domain (H976 in humans). This Tyrosine is conserved in invertebrate class IIa HDACs and all class I HDACs, and plays a critical role in substrate deacetylation (Lahm et al., 2007).
Intriguingly, HDAC4 interacts with TFs that influence neuronal synapses (Benito and Barco, 2010; Flavell et al., 2008; Li et al., 2012), and studies in an ALS mouse model have shown that deletion of HDAC4 in the muscle enhances re-innervation through increased expression of FGFBP1 (Williams et al., 2009). Furthermore, heterozygous mutations in the human HDAC4 locus have been recently linked to a rare Brachydactyly mental retardation syndrome (Williams et al., 2010). The phenotypes of human subjects carrying mutant HDAC4 alleles are thought to be due to haploinsufficiency (Williams et al., 2010), but the exact causes of these deficiencies are unknown.
Here we report that HDAC4 regulates a transcriptional program essential for synaptic transmission and information processing in the brain. This pathway involves an activity-dependent association of HDAC4 with TFs and neuronal chromatin, and is dispensable for neuroprotection. In addition, we show that neither neuronal HDAC4 function requires deacetylation of substrates, suggesting that HDAC4 cannot be targeted with inhibitors that bind to catalytic sites of histone deacetylases.
RESULTS
Temporal and spatial pattern of HDAC4 expression in the mouse forebrain
The class IIa HDAC subfamily includes three highly homologous genes - HDACs 4, 5 and 7 (Haberland et al., 2009). To elucidate the roles of these HDACs in the nervous system, we examined their developmental expression profiles. HDAC7 was abundant in the embryonic forebrain, whereas HDAC5 was uniformly expressed throughout development. In contrast, HDAC4 was up-regulated during early postnatal stages, when massive synaptogenesis occurs (Figure 1C). Analysis of wildtype and mutant neuronal reporter Ai9/CamKIIα Cre mice revealed HDAC4 immunoreactivity in a broad spectrum of glutamatergic and GABAergic neurons (Figures 1A and 1B). Accordingly, immunoblotting of mixed neuronal cultures and astrocytes showed that HDAC4 is enriched in neurons (Figure 1D). Based on these observations and because mutations in the HDAC4 locus have been associated with neurological abnormalities (Williams et al., 2010), we hypothesized that HDAC4 may regulate the formation and/or function of central synapses.
Figure 1. Expression and activity-dependent shuttling of HDAC4 in the forebrain.

A and B, HDAC4 immunoreactivity overlaps with markers of glutamatergic (A) and GABAergic (B, marked by arrows) neurons in the postnatal cortex and hippocampus. In panel (A), asterisks mark the somas of differentiating granule cells.
C, Developmental profile of class IIa HDAC expression in the cortex. Protein extracts were probed by immunoblotting with antibodies to class IIa HDACs, synaptic proteins, and βTubulin (as a loading control).
D, Expression of HDAC4 in mixed cortical cultures and astrocytes.
E, NMDA receptors promote the export of native HDAC4 from the nucleus. Images of control neurons and cells that were treated for 12h with the NMDA receptor blocker, APV, are shown. Scale bar applies to all panels.
F, Quantitative analysis of the subcellular distribution of native and recombinant HDAC4 in wildtype neurons treated with various activity blockers, and Syb2- and NR1-deficient neurons. See Figure S1 for images.
G–J, Nuclear export of HDAC4 is regulated by vesicular release in vivo. G, Loss of Syb2 immunoreactivity in the cortex of R26floxstopTeNT/CamKIIα:Cre mutants (KI/Cre). H, KI/Cre mice have reduced excitatory drive from Entorhinal cortex to DG. Glutamatergic postsynaptic currents were monitored in acute slices from DG granule cells. I and J, KI/Cre mice exhibit accumulation of HDAC4 in the nucleus. Images of brain sections labeled for native HDAC4 (I) and quantifications of HDAC4 localization in the somatosensory cortex (J) are shown. Data from two littermate pairs of KI/Cre and control Cre-negative mice are plotted as Mean±S.E.M. (n=100 neurons per group)
The nuclear export of HDAC4 is induced by glutamatergic inputs
The subcellular distribution of class IIa HDACs is influenced by diverse signals including those elicited by neuronal activity (Chawla et al., 2003; McKinsey et al., 2000; Mihaylova et al., 2011; Wang et al., 2011). In the postnatal forebrain and cultured cortical neurons, HDAC4 is mainly cytoplasmic (Figures 1A, 1B and 1E). Supporting previous in vitro studies (Chawla et al., 2003), we found that treating mature neurons in culture with various activity blockers or a calcium/calmodulin-dependent kinase IIα inhibitor resulted in accumulation of native and recombinant HDAC4 in the nucleus. This effect was also induced by APV alone, a partial shRNA-mediated knock-down of NMDA receptor NR1 subunit, or in vivo injection of the competitive NMDA receptor inhibitor, MK801, suggesting that NMDA receptors play a major role in regulating the localization of HDAC4 neurons (Figures 1E, 1F and S1). Since NMDA receptors are expressed in neural progenitors and a fraction of these receptors are localized extrasynaptically in established circuits (Hardingham and Bading, 2010; Platel et al., 2010), we asked how nucleo-cytoplasmic shuttling of HDAC4 depends on vesicular release of glutamate. To this end, we examined dissociated cultures prepared from cortices of mouse embryos lacking Synaptobrevin/VAMP2 (Syb2), a SNARE protein essential for the exocytosis of neurotransmitter vesicles (Schoch et al., 2001). Similar to pharmacological treatments and NR1 knock-down, deletion of Syb2 resulted in nuclear HDAC4 accumulation (Figures 1F and S1B). To test how vesicular release affects the localization of HDAC4 in vivo, we generated a mouse strain carrying a Cre-inducible form of Tetanus toxin (Zhang et al., 2008) and a forebrain-specific transgene driving Cre recombinase in glutamatergic neurons (R26floxstopTeNT/CamKIIα:Cre). R26floxstopTeNT/CamKIIα:Cre mice exhibited a loss of Syb2-immunoreactive puncta and a decrease in excitatory synaptic strength (Figures 1G and 1H). When compared to control littermates, these mutants had a significant increase in native HDAC4 levels in neuronal nuclei (Figures 1I and 1J), suggesting that glutamatergic inputs trigger the nuclear export of HDAC4 in the brain.
HDAC4 represses genes essential for synaptic function
How do activity-dependent changes in HDAC4 localization affect neural circuits? Several studies have implicated HDAC4 in neuroprotection, raising the possibility that loss of synaptic excitation may lead to neurodegeneration due to depletion of HDAC4 from the cytoplasm and/or repression of genes that promote neuronal survival (Bolger and Yao, 2005; Chen and Cepko, 2009; Li et al., 2012; Majdzadeh et al., 2008). Contrary to this prediction, we found that even prolonged blockade of vesicular release or pharmacological silencing of NMDA receptors does not induce cell death in vitro and in vivo. Interestingly, chronic suppression of NMDA receptors in culture decreased glutamatergic neurotransmission (Figures S2A–S2D). These observations confirm findings published by other laboratories (Chubykin et al., 2007), and indirectly suggest that class IIa HDACs may participate in NMDA receptor-dependent modulation of synaptic strength.
To investigate the cellular consequences of nuclear HDAC4 signaling without affecting other NMDA receptor-dependent pathways, we performed genome-wide mRNA profiling of cultured neurons carrying a constitutively nuclear HDAC4 mutant containing Alanine substitutions of Serine residues 246, 467 and 633 whose phosphorylation is essential for nuclear export (3SA-OE) (McKinsey et al., 2001) (Figures 2A–2C). Because loss of HDAC4 has been shown to induce neurodegeneration (Chen and Cepko, 2009; Majdzadeh et al., 2008), we introduced this mutant via lentivirus-mediated gene transfer in a wildtype background. In parallel, we examined cultures expressing wildtype HDAC4 cDNA (WT-OE) to eliminate potential artifacts associated with viral integration. Control experiments showed no detectable effects of WT-OE on neuronal gene expression, morphology and physiology (Figures 5, S2G and data not shown).
Figure 2. HDAC4 represses genes essential for synaptic function.

A–E, Genome-wide mRNA profiling of cortical neurons expressing wildtype HDAC4 or the constitutively nuclear 3SA mutant. A, Schematic representation of HDAC4 domain structure. B and C, Expression, phosphorylation and localization of wildtype and 3SA HDAC4 was analyzed by immunoblotting with conventional and phospho-specific antibodies (B), and by immunostaining (C). D, DNA microarray heat maps of the top 100 differentially expressed transcripts. The magnitudes of fold change (FC) are indicated on the left. Genes encoding synaptic proteins are shown in green. Symbols mark hits whose regulation was confirmed by qPCR (#) and/or qPCR and immunoblotting (##). E, A diagram depicting HDAC4-dependent genes grouped into distinct categories based on localization and function of encoded proteins. SV=synaptic vesicle; AZ=active zone; PSD=postsynaptic density. See Figure S2 and Tables S1 and S2 for additional details and raw microarray data.
F, qPCR analysis of mRNA levels of genes identified by microarrays (shown in blue and red) and various controls.
G and H, Protein expression levels were examined by immunoblotting. In panels (F) and (H), averaged values from three independent experiments were plotted as 3SA/WT ratio. All data are represented as Mean±S.D. * = p<0.05.
Figure 5. HDAC4 regulates the strength and structural organization of excitatory synapses.

Cortical cultures were infected at 4–5 DIV with viruses encoding indicated HDAC4 cDNAs with or without shRNA, and analyzed at 14–16 DIV.
A and B, Effects of overexpression (OE) of nuclear 3SA, +C, ΔHDAC and ΔNES mutants or shRNA knock-down of native HDAC4 combined with rescue (KD+R) on magnitudes of evoked AMPA and NMDA-type excitatory postsynaptic currents (eEPSCs). Typical traces of eEPSCs recorded in whole-cell mode in the presence of picrotoxin, and averaged eEPSC amplitudes are shown. Data were normalized to the synaptic responses of control non-infected neurons.
C, Representative traces of quantal mEPSCs monitored in the presence of tetrodotoxin and picrotoxin.
D and E, Averaged mEPSC frequencies and amplitudes.
F, Synapses of WT-OE and 3SA-OE neurons were visualized by labeling with antibodies to native presynaptic proteins or with genetically encoded fluorescent tracers, CamKIIα GFP-SV2A and Synapsin:Homer-GFP. Top row: Immunostaining for native Vglut1. Second row: double labeling for Vglut1 and GAD67. Third row: neurons expressing CamKIIα GFP-SV2A were labeled with an antibody to MAP2. Fourth row: dendrites of live neurons expressing Synapsin:Homer-GFP.
G, Averaged densities of antigen and reporter-positive puncta per dendrite length in control, WT-OE and 3SA-OE neurons (n=20 neurons for each group).
H, Electron micrographs of excitatory synapses formed by WT-OE and 3SA-OE neurons. Top row: Examples of presynaptic boutons opposed to postsynaptic spines. The postsynaptic densities (PSD) are marked by arrows. Bottom row: Images of PSDs visualized by the PTA enhancement protocol.
I and J, Quantification of docked vesicle pools (I) and active zone length (J) in synapses of neurons expressing indicated HDAC4 constructs.
See Figure S6 and Tables S3A and S4 for additional results and statistics.
All data are represented as Mean±S.E.M. * = p<0.05.
Strikingly, nuclear HDAC4 repressed a group of genes highly enriched in those known to be essential for synaptic function. Using Affymetrix DNA microarrays, we identified 214 transcripts that were differentially expressed in WT-OE and 3SA-OE neurons. Most of these mRNAs were down-regulated by 3SA-OE, suggesting that HDAC4 predominantly acts as a transcriptional repressor (Figures 2D, S2E, S2F and Table S2). Approximately 40% of genes whose mRNA levels were reduced between 1.8 and 8-fold have been previously shown to be induced by neuronal activity in vitro and/or sensory experience in vivo (Flavell and Greenberg, 2008; Flavell et al., 2008; Majdan and Shatz, 2006; Tropea et al., 2006). Moreover, half of the genes in the entire pool fall into distinct functional classes related to synapses including constituents of neurotransmitter vesicles and presynaptic active zones, secreted proteins involved in synaptic differentiation and AMPA receptor trafficking, scaffolds, neurotransmitter receptors, and intracellular signaling molecules implicated in plasticity and memory (Figure 2E and Table S1). Intriguingly, Moesin (Msn), which restrains synaptic growth in flies and whose levels are elevated in the visual cortex of sensory-deprived mice (Seabrooke and Stewart, 2008; Tropea et al., 2006), was induced by 3SA-OE (Figure 2D). Nonetheless, the majority of neuronal and ubiquitously expressed mRNAs were unaffected, suggesting that these phenotypes were not due to non-specific changes in transcription and translation (Table S2). To validate these results, we further examined expression levels of genes identified as HDAC4 targets whose roles in synapses have been defined by gene knock-outs in mice and drosophila. Real-time qPCR and quantitative immunoblotting confirmed a 3SA-dependent repression of CamKIIa, Synapsins, Homers, Vglut1, Snap25, Dlg2, Rab3c and Lgi1, and up-regulation of Msn (Figures 2F–2H). Only a fraction of these genes were regulated by a nuclear form of HDAC7 (Figures S2H and S2I), indicating that different members of the class IIa HDAC family control largely non-overlapping transcriptional programs.
HDAC4 associates with neuronal chromatin and forms complexes with MEF2 in an NMDA receptor-dependent manner
Class IIa HDACs interact with tissue-specific TFs Runx2, CAMTA, Dach2, FOXO and MEF2 (McKinsey et al., 2000; Mihaylova et al., 2011; Vega et al., 2004; Wang et al., 2011; Zhang et al., 2002). An association between HDAC4 and MEF2 is intriguing, since members of the MEF2 family are expressed in the brain where they induce an activity-dependent transcriptional program that restricts excitatory synapse numbers (Flavell et al., 2006; Flavell et al., 2008). We compared our genome-wide mRNA profiling data to a recently described group of MEF2-dependent transcripts and found that HDAC4 and MEF2 both regulate at least 6 genes which include Homer1, Lgi1, Prkca, Syngap, Rgs2 and Mapk8 (Figure 2D). All of these genes were repressed by HDAC4 and activated by MEF2 in similar experimental settings.
Mechanistically, HDAC4 may abolish transcription through a cross-talk with MEF2 and/or other TFs upstream DNA binding, by associating with chromatin in a histone-like manner, or by coupling to promoters, enhancers or other regulatory sequences. To distinguish between these scenarios, we immunoprecipitated chromatin with epitope-tagged HDAC4 proteins, and analyzed genomic DNA by deep sequencing (ChIP-Seq). ChiP-Seq yielded ~1400 sites that were sparsely distributed across the genome and exhibited increased occupancy by the nuclear gain-of-function 3SA mutant relative to wildtype (Figures 3A and S3B). Moreover, we detected binding of HDAC4 to loci that are either immediately upstream of the first exons or in intronic regions of ~25% of genes identified in mRNA profiling studies, and confirmed the specificity of these interactions by qPCR (Figures 3B, 3C and data not shown). Because HDAC4 associates with MEF2 via its N-terminal domain (Backs et al., 2011) and none of the class IIa HDACs have canonical DNA-binding motifs, we asked if HDAC4 N-termini is also required for coupling to chromatin. Deletion of this domain (ΔN) did not affect the nuclear retention of the 3SA mutant, but completely disrupted its repressor activity and interaction with both MEF2 and genomic DNA (Figures S3D–S3F, S5E and data not shown).
Figure 3. HDAC4 associates with chromatin and MEF2 in an activity-dependent manner, and represses transcription without employing its catalytic domain.

A, Distribution of HDAC4-interacting loci (blue peaks) across chromosome 18. Green bars mark positions of individual genes.
B, Positions of HDAC4-binding sites within CamKIIa and Syn1 genes.
C, Chromatin was co-immunoprecipitated with wildtype or 3SA HDAC4 and analyzed by qPCR with primers specific for loci identified by deep sequencing (blue box) and various controls. Data are plotted as 3SA/WT RQ ratio.
D, qPCR analysis of wildtype HDAC4 interaction with its target sites in control neurons and neurons that were incubated for 12 hours with APV. Data are plotted as APV/Control RQ ratio.
E, NMDA receptors regulate the binding of HDAC4 to MEF2. HDAC4 was immunoprecipitated from control and APV-treated neurons. Protein complexes were analyzed by immunoblotting with antibodies to HDAC4 and MEF2.
F, Re-distribution of HDAC4 from the cytoplasm to nucleus does not affect the levels of acetylated histone H3. Subcellular fractions of control and APV-treated neurons were separated by centrifugation, and analyzed by immunoblotting with antibodies to native HDAC4, Acetyl-H3 and βTubulin.
G–J, A mutant HDAC4 allele associated with Brachydactyly mental retardation syndrome is a constitutively nuclear gain-of-function transcriptional repressor. G, Domain structures of wildtype HDAC4 and +C allele. Sequence alignment illustrates evolutional inactivation of vertebrate class IIa HDACs. H, A protein encoded by +C allele accumulates in neuronal nuclei. I, +C mutant constitutively binds to genomic loci that are occupied by wildtype HDAC4 in an activity-dependent manner (blue box). qPCR analysis was performed as described in Figure 3C. J, mRNA levels of HDAC4-regulated genes (shown in blue and red) and controls were measured by qPCR and plotted as +C/WT RQ ratio.
All qPCR data are represented as Mean±S.D. from RQ values obtained in three independent sets of experiments. * = p<0.05. See also Figures S3 and S4.
Next we asked whether coupling of HDAC4 to chromatin and transcription factors depends on glutamatergic neurotransmission. qPCR analysis of genomic DNA co-immunoprecipitated with wildtype HDAC4 demonstrated a robust increase in its binding to target sites in response to NMDA receptor blockade with APV (Figure 3D). The formation of HDAC4/MEF2 complexes was also strongly regulated by NMDA receptor activity, as evidenced by lack of detectable interaction between these proteins in extracts prepared from control neurons, and strong interaction in APV-treated neurons (Figure 3E). However, a fraction of HDAC4 was present in the nuclei in a chromatin-bound state even in neurons with normal levels of synaptic excitation (Figures 3F and S3C), raising the possibility that complete de-repression of HDAC4-dependent genes requires strong correlated stimulation. Notably, neither APV-induced nuclear translocation of native HDAC4 nor expression of 3SA mutant altered the levels of acetylated histone H3 (Figure 3F). In contrast, we found H3 acetylation to be increased upon treatments with a non-specific HDAC inhibitor, SAHA (Figures S4C and S4D).
A truncated form of HDAC4 encoded by a human +C allele associated with Brachydactyly mental retardation is a constitutively nuclear gain-of-function transcriptional repressor
Unlike their C.elegans and drosophila orthologs, vertebrate class IIa HDACs have a Histidine substitution of the Tyrosine residue in the catalytic pocket that plays a critical role in substrate deacetylation (H976 in humans)(Lahm et al., 2007). While our assessment of the effect of HDAC4 on histone H3 acetylation supports that vertebrate class IIa HDACs have been evolutionarily inactivated, HDAC4 may deacetylate other substrates or form complexes with effector proteins via its C-terminal domain. Interestingly, a recent human genetic study has linked a heterozygous mutation in the HDAC4 coding sequence with Brachydactyly mental retardation (Williams et al., 2010). This mutation is a single Cytosine (+C) insertion which leads to a frame shift 176 amino acids upstream H976, and results in a truncation of the deacetylase domain followed by the nuclear export signal (Figures 3G and S4A) (Williams et al., 2010). We expressed the human +C allele in cultured neurons and found that truncated protein was stable, migrated in SDS-page according to its estimated molecular weight, and acted as a constitutive transcriptional repressor. Specifically, +C HDAC4 was retained in the nucleus, bound to genomic loci that were occupied by wildtype HDAC4 in an activity-dependent manner, and abolished expression of HDAC4 target genes identified in our microarray screens (Figures 3H–3J and S4B). Taken together, these studies show that putative HDAC4 enzymatic function is dispensable for transcriptional regulation, and suggest that cognitive abnormalities associated with the +C mutation in humans were likely due to nuclear repression mediated by a gain-of-function allele.
HDAC4 promotes neuronal survival and regulates gene expression via distinct mechanisms and protein domains
Our experiments indicate that HDAC4 is an NMDA receptor-dependent transcriptional repressor that regulates a group of “synaptic” genes. To elucidate the interplay between this pathway and the known role of HDAC4 in neuroprotection, we designed a lentiviral RNAi/rescue system that enables the simultaneous shRNA-mediated knock-down of native HDAC4 and expression of shRNA-insensitive HDAC4 cDNAs (KD+Rescue). We introduced these viruses into mixed cortical cultures and examined the cells by immunostaining, immunoblotting, and by imaging a genetically encoded reporter, Synapsin:mCherry-H2B. Knock-down of HDAC4 resulted in a loss of all neuronal cell types without affecting astrocytes. This phenotype was rescued by wildtype cDNA, excluding off-target shRNA effects (Figures 4A and S5A–S5D). We then generated viruses encoding various HDAC4 mutants, and assessed their subcellular localization and capacity to promote neuronal survival in the absence of native protein. The 3SA, human +C allele and other constitutively nuclear HDAC4 forms lacking the C-terminal nuclear export signal (ΔHDAC and ΔNES) failed to fully rescue neuronal loss. Yet, these mutants did not induce detectable cell death in the wildtype background (OE) (Figures 4B–4D and S5E). Whereas the differences in the extent of rescue suggest that phosphorylation of HDAC4 serine residues is required for its neuroprotective activity, the death of neurons carrying mutants with a disrupted enzymatic domain was likely due to their nuclear retention rather than inability to deacetylase substrates. Indeed, full-length HDAC4 containing Alanine substitutions of five residues in the catalytic site (L-H/A) was cytoplasmic and rescued cell survival similar to wildtype. Finally, a mutant lacking the N-terminal domain essential for binding to MEF2 and chromatin (ΔN) was also cytoplasmic, failed to redistribute to the nucleus in response to NMDA receptor blockage as efficiently as wildtype protein, and completely rescued neuronal loss induced by RNAi (Figures 4B–4D and S5E). Remarkably, KD+ΔN neurons had increased levels of HDAC4-dependent genes, further suggesting that native HDAC4 is capable of supporting transcriptional regulation in the presence of synaptic input (Figures 4E–4G). Hence, HDAC4 acts in two non-overlapping pathways, and neither HDAC4 function involves deacetylation of substrates in the nucleus or cytoplasm.
Figure 4. HDAC4 represses transcription and promotes neuronal survival via distinct mechanisms.

A–D, Structure-function analysis of HDAC4-mediated neuroprotection. Cortical cultures were infected at 5 DIV with lentiviruses encoding shRNA against HDAC4 alone (KD) or shRNA together with shRNA-insensitive FLAG-tagged rescue HDAC4 cDNAs. Neuronal survival was analyzed as described in Figure S2A. A, Cell death induced by HDAC4 KD is fully rescued with wildtype HDAC4. B, Schematic representation of mutants used for rescue studies. C, Quantification of neuronal survival in cultures expressing either shRNA together with indicated cDNAs (KD+Rescue) or 3SA and +C mutants alone (OE). D, Localization of rescue mutants expressed without shRNA. See Figure S5 for images and additional data.
E–G, HDAC4 N-termini is essential for transcriptional repression. mRNA and protein expression was examined in neurons carrying shRNA and either the wildtype HDAC4 or the ΔN mutant. E, qPCR measurements of mRNA levels of HDAC4-dependent genes (shown in blue and red) and controls. Data are plotted as ΔN/WT RQ ratio. F and G, Representative immunoblots and quantifications of relative levels of indicated proteins.
All measurements were performed in three independent experiments and presented as Mean±S.D. * = p<0.05.
HDAC4 regulates the strength and structural organization of excitatory synapses
To directly test how nuclear HDAC4 signaling impacts the properties of central synapses, we monitored neurotransmission using electrophysiological methods. Mature cortical neurons carrying the gain-of-function 3SA mutant or +C allele in the wildtype background exhibited a drastic decrease in both the amplitudes of evoked AMPA- and NMDA-type excitatory postsynaptic currents (eEPSC) and frequencies of “spontaneous” quantal mEPSCs. Other nuclear mutants produced similar phenotypes. Conversely, KD+ΔN neurons had larger eEPSCs and higher rates of spontaneous events (Figures 5A–5D). The sizes of quantal AMPA currents were also significantly affected with ~30% smaller mEPSC amplitudes in 3SA-OE and +C-OE neurons, and ~20% larger currents in KD+ΔN neurons (Figure 5E). Surprisingly, inhibitory GABAergic neurotransmission was unaltered (Table S3A). The effects of HDAC4 gain- and loss-of-function on neuronal physiology appear to reflect changes in synaptic strength rather than synapse numbers. While confocal imaging of 3SA-OE neurons confirmed depletion of native synaptic proteins whose repression was detected by other assays, we found no significant difference in the density and distribution of nerve terminals and dendritic spines visualized with genetically encoded reporters, CamKIIα GFP-SV2A and Synapsin:Homer1-GFP (Figures 5F and 5G). However, electron microscopy revealed that 3SA-OE and +C-OE reduce the sizes of docked vesicle pools and the length of presynaptic active zones (AZ) and postsynaptic densities (PSDs), whereas knock-down of native HDAC4 combined with expression of ΔN rescue cDNA significantly increased the docked vesicle pool and AZ/PSD sizes (Figures 5H–5J and Table S4). Likewise, additional live imaging and electrophysiological tests showed that nuclear HDAC4 accumulation affects synaptic outputs and inputs of the same neuron (Figure S6).
Repression of the HDAC4-dependent transcriptional program reduces excitatory synaptic strength and impairs spatial memory in mice
To determine how nuclear HDAC4 activity impacts synaptic transmission and information processing in the brain, we generated mouse strains carrying either the full-length gain-of-function 3SA mutant or the truncated HDAC4 form lacking the C-terminal region (Figure 6A). The transgenes were expressed in glutamatergic neurons under the control of the forebrain-specific CamKIIα promoter that does not contain the HDAC4-binding site (Saura et al., 2004). We were unable to obtain offspring from 3SA-positive founders. However, mice harboring a truncated mutant (which we will refer to as TG) survived and had normal lifespan. Aside from its restricted expression pattern, TG functionally mimics the +C allele since both mutants had intact N-termini and phosphorylation sites, lacked the catalytic domains, and produced nearly identical phenotypes in culture (Figures 4B–4D and 5B). TG was stable in the brain, accumulated in neuronal nuclei, and acted as a transcriptional repressor, as evidenced from our quantitative assessments of its binding to chromatin and ability to down-regulate HDAC4-dependent genes (Figures 6A–6C and S7).
Figure 6. A truncated form of HDAC4 acts as a constitutively nuclear repressor in vivo.

A, Top: Schematic representation of wildtype HDAC4 and a truncated mutant used for the generation of transgenic mice (TG). Bottom: TG accumulates in nuclei and does not influence neuronal density and synapse numbers. Brain sections were labeled with antibodies to HDAC4, Prox1 and SyP. Enlarged images show neuronal somas in the DG granule cell layer (GCL) and synaptic puncta in the molecular layer (ML). Asterisks mark differentiating GCs.
B, TG binds to genomic DNA sites that are occupied by wildtype HDAC4 in an activity-dependent manner. Top: Immunoblot analysis of TG expression in the forebrain. Bottom: Chromatin was co-immunoprecipitated with anti-HDAC4 antibody and analyzed by qPCR with primers specific for HDAC4-interacting sites within CamKIIa, Syn1 and Syn2 genes. Data from three pairs of mice are plotted as TG/WT RQ ratio (Mean±S.D.)
C, TG mice have reduced expression of HDAC4-dependent genes. Proteins extracted from the DG were probed by immunoblotting with indicated antibodies. Note a selective reduction of CamKIIα, Synapsins 1/2 and Homer1. Data are from two pairs of mice. See Figure S7 for quantifications of protein levels.
D and E, TGs have reduced excitatory synaptic strength. Representative traces of evoked AMPA- and NMDA-type eEPSCs (D) and quantal AMPA-type mEPSCs monitored in acute slices from DG granule cells are shown. Recordings were performed in whole-cell mode in the presence of gabazine.
F, Summaries of synaptic properties of granule cell neurons in the DG of wildtype and TG mice. The densities of Prox1-positive neurons in the GCL and SyP-positive puncta in the ML were normalized to wildtype. All other values are raw. All experiments were performed at postnatal day 25.
Data are plotted as Mean±S.E.M. * = p<0.05. See Table S3B for statistics.
To evaluate the physiological consequences of increased repression of the HDAC4-dependent transcriptional program, we monitored postsynaptic currents from granule cells in the Dentate Gyrus (DG), which receive glutamatergic inputs from the Entorhinal cortex (Nakashiba et al., 2008). Recordings from acute slices isolated from TGs showed ~2-fold decrease in the amplitudes of evoked AMPA-type eEPSCs as well as frequencies of “spontaneous” mEPSCs. Again, we did not detect significant changes in the strength of GC inhibition by local interneurons (Figures 6D–6F and Table S3B). In addition, TGs had no detectable defects in global patterning of the hippocampal pathway, the numbers and membrane properties of Prox1-positive GCs in the DG, and densities of synapses visualized in dendritic fields of these cells by Synaptophysin staining, excluding the possibility of neuronal and synapse loss (Figures 6A and 6F).
We then interrogated TG mice and their wildtype littermates using behavioral tests designed to assess the functionality of various cortical centers, anxiety-like behavior and memory acquisition and retrieval. TGs exhibited decreased rearing, but had normal ambulation and total horizontal activity in the open field (Figure 7A). Despite the reduction of excitatory synaptic strength, these mice also had intact vision and anxiety (Figures 7E and 7F). Considering that HDAC4 regulates CamKIIα, which is known to be essential for spatial learning and memory (Bach et al., 1995), we subjected TGs to a series of tasks in the Barnes maze. Strikingly, TGs displayed significantly longer latencies to identify the correct target and escape from the maze, and lost the preference for the target quadrant in the probe test, indicating that their spatial memory was impaired. In contrast, wildtype mice showed evidence of spatial memory manifested as higher percentage of the trial spent in the target quadrant (Figures 7B and 7C). Furthermore, retention and reversal experiments performed in the maze two weeks following the acquisition phase revealed a loss of long-term memory in TGs (Figure 7D).
Figure 7. Repression of the HDAC4-dependent transcriptional program affects rearing behavior and impairs spatial memory.
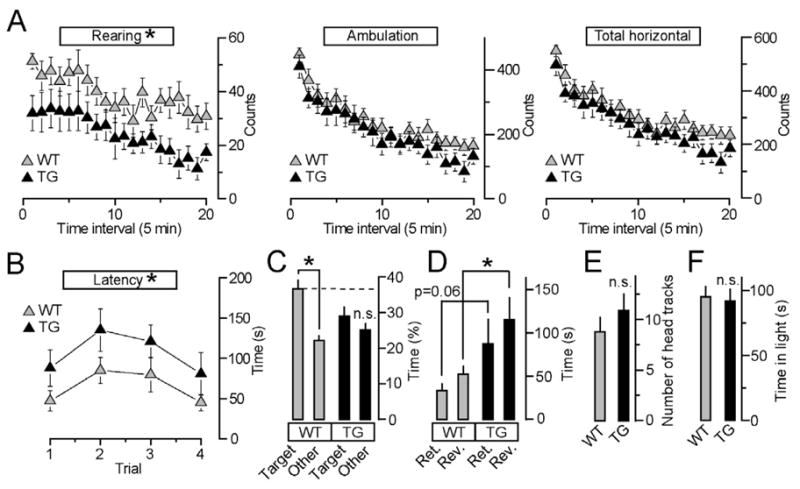
A, TG mice show decreased rearing (F(1,15)=4.9, p<0.05) but have normal ambulation and total horizontal activity.
B, TG mice exhibit significantly longer latencies to escape in the Barnes maze (F91,15)=5.2, p<0.05).
C, Wildtype mice use spatial cues and spend more time in the target quadrant in the Barnes maze probe test (F(1,8)=16.7, p<0.01), whereas HDAC4 TG mutants do not distinguish the target quadrant from the others.
D, TG mice exhibit longer latencies to escape relative to wildtype in the Barnes maze retention (Ret.) and reversal (Rev.) tests (t(15)=2.4, p <0.05).
E and F, TG mice have normal vision and anxiety-like behavior, as determined by the optomotor test (E) and quantification of time spent in the light compartment of the light/dark transfer box (F).
All studies were performed with 9 and 8 age-matched wildtype and TG males, respectively. Data are plotted as Mean±S.E.M.
DISCUSSION
Our study defines HDAC4 as a transcriptional repressor whose translocation from neuronal cytoplasm to the nucleus and coupling to chromatin and transcription factors is negatively regulated by glutamatergic inputs. HDAC4 represses genes encoding constituents of central synapses, thereby influencing synaptic structure, function, and information processing in the brain. In a general view, HDAC4 resembles a molecular substrate for Hebbian forms of plasticity expressed as a long-term increase in the synaptic strength in response to persistent neuronal activity (Turrigiano and Nelson, 2000). However, it is possible that HDAC4 is also involved in regulation of neural circuit development in the embryonic brain, prior to synapse formation.
The physiological effects associated with nuclear HDAC4 export/import may reflect altered transcriptional activity of multiple genes with diverse roles. These include secretory proteins essential for vesicular release of neurotransmitters (Vglut1, Snap25, Synapsins and Rabs) as well as postsynaptic scaffolds and signaling molecules (Homers, Lgi1, Syngap, Prkca, Prkcb and CamKIIα) (Table S1). The cognitive abnormalities of TG mice are highly reminiscent those observed in CamKIIa mutants (Bach et al., 1995), suggesting that HDAC4-dependent repression of CamKIIa may play an important role in memory acquisition and retrieval. Together with in vitro studies of the +C allele, our behavioral experiments provide mechanistic insight to neurological deficits associated with mutations in the HDAC4 locus in humans.
Consistent with reports from other laboratories (Chen and Cepko, 2009; Majdzadeh et al., 2008), we found that loss of HDAC4 causes neurodegeneration. Although HDAC4 promotes neuronal survival independently from regulating gene expression, both HDAC4 functions depend on Serine phosphorylation. Therefore, kinases activated by synaptic input may support neuronal survival and alter their synaptic properties through modification of one downstream target.
Previously characterized mechanisms of transcriptional repression in the brain involve class I HDACs, REST, which predominantly acts during embryogenesis (Ballas and Mandel, 2005; Lunyak et al., 2002; Mandel et al., 2011) and MeCP2, a methyl-CpG-binding protein implicated in Rett syndrome (McGraw et al., 2011). While we cannot exclude the possibility of a cross-talk between HDAC4 and these factors, the HDAC4-dependent pathway has several distinctive features. Recent studies have shown that class I HDACs affect synaptic development and function (Akhtar et al., 2009; Guan et al., 2009). However, the underlying mechanisms appear be constitutive and involve deacetylation of histones. In contrast, the putative catalytic activity of HDAC4 is dispensable, suggesting that HDAC4 cannot be manipulated with inhibitors that bind to active sites of histone deacetylases (Bantscheff et al., 2011).
Similar to HDAC4, the nuclear function of MeCP2 depends on site-specific phosphorylation (Chao and Zoghbi, 2009). Nonetheless, MeCP2 associates with genomic DNA in a histone-like fashion, globally alters chromatin structure, and impacts virtually thousands of genes triggering a genome-wide response of chromatin to neuronal activity (Cohen et al., 2011; Skene et al., 2010). Furthermore, MeCP2 acts as both a transcriptional repressor and activator (Chahrour et al., 2008), and MeCP2 loss-of-function studies revealed a variety of phenotypes including changes in neuronal branching, excitatory synapse numbers, and reduced inhibitory synaptic strength (Chao et al., 2010; Chao et al., 2007; Cohen et al., 2011). Unlike MeCP2, HDAC4 appears to interact with selective loci sparsely distributed across the neuronal genome, and influences a restricted pool of genes.
Whereas the interplay between HDAC4 and members of the MEF2 TF family accounts for control of a part of the HDAC4-dependent transcriptional program, HDAC4 also represses MEF2-independent genes and may therefore modulate synapses by interacting with distinct sets of effectors in the same neuron or in a cell-type-specific manner. It is important to note that we were unable to detect internal HDAC4-binding sites in a large fraction of genes identified in mRNA profiling screens. Potential reasons for this incomplete overlap include an association of HDAC4 with distal regulatory elements. In addition, HDAC4 may indirectly repress transcription through silencing of other factors, such as CAMTA (Song et al., 2006). Future studies elucidating the cross-talk between these proteins in the brain may uncover new mechanisms underlying neuronal differentiation, synapse formation, and plasticity.
EXPERIMENTAL PROCEDURES
Preparation of neuronal cultures, virus production and infection and all biochemical, imaging and electrophysiological studies were performed as described (Cao et al., 2011; Maximov et al., 2009).
Mice
Ai9 reporter, CamKIIα:Cre, Syb2 KO, and R26:floxstopTeNT mouse alleles were characterized previously (Madisen et al., 2010; Saura et al., 2004; Schoch et al., 2001; Zhang et al., 2008). New strains were established by crossing these alleles according to approved animal protocols. To generate HDAC4 transgenic mice, coding sequences were amplified by PCR and subcloned downstream of the ~8 kb CamKIIα promoter in a targeting vector that also included 5′ and 3′ introns flanking the cDNA and a 3′ SV40 polyA signal. The constructs were linearized and used for pronuclear microinjection at the TSRI Mouse Genetics Core. Positive founders were identified by PCR.
mRNA profiling
mRNA was extracted with the RNAeasy Kit (Qiagen) and processed at the TSRI DNA array facility using procedures recommended by Affymetrix. Data was normalized using RMA Express 1.0 (http://rmaexpress.bmbolstad.com) with quantile normalization, median polish and background adjustment. The sample clustering was performed using BRB-ArrayTools (http://linus.nci.nih.gov/BRB-ArrayTools.html). The ComBat function in the R software was used to adjust for batch effect. Heat maps were generated with dChip program (www.dChip.org).
Chromatin immunoprecipitation
Protein-DNA complexes were cross-linked in 1% formaldehyde. Chromatin was isolated from nuclear fractions, sheared by sonication and incubated with FLAG M2 mouse antibody (Sigma). Immune complexes were collected by incubating with Salmon Sperm DNA/Protein G sepharose and eluted in 1% SDS, 0.1M NaHCO3. Crosslinking was then reversed, and purified input and bound fractions were used for deep sequencing and qPCR.
Deep sequencing
Bar-coded genomic DNA was sequenced using Illumina HiSeq 2000 Analyzer. Image processing, base calling and alignments were performed with The Genome Analyzer Pipeline Software (Casava 1.8.1). Alignments were performed with ELAND2e (Efficient Large-Scale Alignment of Nucleotide Databases). Aligned reads were used as input to the Model-based Analysis for ChIP-Seq (MACS-1.4.1) program. Peaks were annotated within 50 kb of a refSeq transcript of the mouse version mm9 database (http://hgdownload.cse.ucsc.edu/goldenPath/mm9).
Electron microscopy
Neurons were fixed in 100 mM Na-cacodylate, 2% PFA, 2.5% glutaraldehyde and 1% sucrose (pH 7.4). For conventional labeling of synaptic boutons, fixed cells were incubated in 1% OsO4, 1.5% potassium hexacyanoferrate in 0.1 M cacodylate buffer, and then dehydrated in a graded series of ethanol solutions. For visualization of postsynaptic densities, cells were treated with 1% ethanolic phosphotungstic acid (PTA, MP biomedicals, USA). Subsequently, samples were contrasted in 2% uranyl acetate, washed and embedded in Epon. 70 nm sections were counterstained with lead citrate and examined under Philips CM 100 electron microscope.
Behavioral studies were conducted at the TSRI behavioral core according to approved animal protocols. All parameters were scored by an experimenter blind to the genotype. Spatial memory was assessed in the Barnes maze essentially as described (Bach et al., 1995; Barnes, 1979).
Full methods including a detailed description of all behavioral setups and data analysis are available in the supplementary materials accompanying the paper.
Supplementary Material
Acknowledgments
We thank Drs. Hollis Cline, Ulrich Mueller, Ardem Patapoutian, Franck Polleux, Mark Mayford and Kristin Baldwin for advice and critical comments; Drs. Thomas Südhof and Martyn Goulding for mouse strains and antibodies; members of the Cline, Mueller, Polleux, Baldwin, Feeney and McHeyzer-Williams labs for help with experiments; and Nina Torabi-Rander, TSRI DNA array, mouse transgenic and behavioral cores for expert technical assistance. This study was supported in part by the NIH grants MH085776 (A.M.), MH067880-09 (J.Y.), RR011823 (J.Y.), NS057096 (J.Y.), Novartis Advanced Discovery Institute (A.M.), The Baxter Foundation (A.M.), and Helen Dorris Postdoctoral Fellowship (S.P.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhtar MW, Raingo J, Nelson ED, Montgomery RL, Olson EN, Kavalali ET, Monteggia LM. Histone deacetylases 1 and 2 form a developmental switch that controls excitatory synapse maturation and function. J Neurosci. 2009;29:8288–8297. doi: 10.1523/JNEUROSCI.0097-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach ME, Hawkins RD, Osman M, Kandel ER, Mayford M. Impairment of spatial but not contextual memory in CaMKII mutant mice with a selective loss of hippocampal LTP in the range of the theta frequency. Cell. 1995;81:905–915. doi: 10.1016/0092-8674(95)90010-1. [DOI] [PubMed] [Google Scholar]
- Backs J, Worst BC, Lehmann LH, Patrick DM, Jebessa Z, Kreusser MM, Sun Q, Chen L, Heft C, Katus HA, et al. Selective repression of MEF2 activity by PKA-dependent proteolysis of HDAC4. J Cell Biol. 2011;195:403–415. doi: 10.1083/jcb.201105063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N, Mandel G. The many faces of REST oversee epigenetic programming of neuronal genes. Curr Opin Neurobiol. 2005;15:500–506. doi: 10.1016/j.conb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol. 2011;29:255–265. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- Benito E, Barco A. CREB’s control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33:230–240. doi: 10.1016/j.tins.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Bolger TA, Yao TP. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J Neurosci. 2005;25:9544–9553. doi: 10.1523/JNEUROSCI.1826-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Maximov A, Sudhof TC. Activity-dependent IGF-1 exocytosis is controlled by the Ca(2+)-sensor synaptotagmin-10. Cell. 2011;145:300–311. doi: 10.1016/j.cell.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch’ng TH, Martin KC. Synapse-to-nucleus signaling. Curr Opin Neurobiol. 2011;21:345–352. doi: 10.1016/j.conb.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY. The yin and yang of MeCP2 phosphorylation. Proc Natl Acad Sci U S A. 2009;106:4577–4578. doi: 10.1073/pnas.0901518106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla S, Vanhoutte P, Arnold FJ, Huang CL, Bading H. Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J Neurochem. 2003;85:151–159. doi: 10.1046/j.1471-4159.2003.01648.x. [DOI] [PubMed] [Google Scholar]
- Chen B, Cepko CL. HDAC4 regulates neuronal survival in normal and diseased retinas. Science. 2009;323:256–259. doi: 10.1126/science.1166226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, Sudhof TC. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. 2007;54:919–931. doi: 10.1016/j.neuron.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Gabel HW, Hemberg M, Hutchinson AN, Sadacca LA, Ebert DH, Harmin DA, Greenberg RS, Verdine VK, Zhou Z, et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron. 2011;72:72–85. doi: 10.1016/j.neuron.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darcy MJ, Calvin K, Cavnar K, Ouimet CC. Regional and subcellular distribution of HDAC4 in mouse brain. J Comp Neurol. 2010;518:722–740. doi: 10.1002/cne.22241. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, Tsien RW. Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol. 2003;13:354–365. doi: 10.1016/s0959-4388(03)00076-x. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Greenberg ME. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci. 2008;31:563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, Markenscoff-Papadimitriou E, Bear DM, Greenberg ME. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60:1022–1038. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner D, Morgan JL, Parker ED, Lewis RM, Wong RO. Neurotransmission selectively regulates synapse formation in parallel circuits in vivo. Nature. 2009;460:1016–1020. doi: 10.1038/nature08236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, Sambucini S, Bottomley MJ, Lo Surdo P, Carfi A, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A. 2007;104:17335–17340. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai KO, Zhao Y, Ch’ng TH, Martin KC. Importin-mediated retrograde transport of CREB2 from distal processes to the nucleus in neurons. Proc Natl Acad Sci U S A. 2008;105:17175–17180. doi: 10.1073/pnas.0803906105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie JH, Nedivi E. Activity-regulated genes as mediators of neural circuit plasticity. Prog Neurobiol. 2011;94:223–237. doi: 10.1016/j.pneurobio.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chen J, Ricupero CL, Hart RP, Schwartz MS, Kusnecov A, Herrup K. Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat Med. 2012 doi: 10.1038/nm.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunyak VV, Burgess R, Prefontaine GG, Nelson C, Sze SH, Chenoweth J, Schwartz P, Pevzner PA, Glass C, Mandel G, et al. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science. 2002;298:1747–1752. doi: 10.1126/science.1076469. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdan M, Shatz CJ. Effects of visual experience on activity-dependent gene regulation in cortex. Nat Neurosci. 2006;9:650–659. doi: 10.1038/nn1674. [DOI] [PubMed] [Google Scholar]
- Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D’Mello SR. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol. 2008;68:1076–1092. doi: 10.1002/dneu.20637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel G, Fiondella CG, Covey MV, Lu DD, Loturco JJ, Ballas N. Repressor element 1 silencing transcription factor (REST) controls radial migration and temporal neuronal specification during neocortical development. Proc Natl Acad Sci U S A. 2011;108:16789–16794. doi: 10.1073/pnas.1113486108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Sudhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw CM, Samaco RC, Zoghbi HY. Adult neural function requires MeCP2. Science. 2011;333:186. doi: 10.1126/science.1206593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol. 2001;21:6312–6321. doi: 10.1128/MCB.21.18.6312-6321.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, Downes M, Evans RM, Montminy M, Shaw RJ. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. 2011;145:607–621. doi: 10.1016/j.cell.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashiba T, Young JZ, McHugh TJ, Buhl DL, Tonegawa S. Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science. 2008;319:1260–1264. doi: 10.1126/science.1151120. [DOI] [PubMed] [Google Scholar]
- Platel JC, Dave KA, Gordon V, Lacar B, Rubio ME, Bordey A. NMDA receptors activated by subventricular zone astrocytic glutamate are critical for neuroblast survival prior to entering a synaptic network. Neuron. 2010;65:859–872. doi: 10.1016/j.neuron.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Ghosh A. A calcium-dependent switch in a CREST-BRG1 complex regulates activity-dependent gene expression. Neuron. 2008;60:775–787. doi: 10.1016/j.neuron.2008.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneyoshi T, Fortin DA, Soderling TR. Regulation of spine and synapse formation by activity-dependent intracellular signaling pathways. Curr Opin Neurobiol. 2010;20:108–115. doi: 10.1016/j.conb.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, 3rd, Kandel ER, Duff K, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Schoch S, Deak F, Konigstorfer A, Mozhayeva M, Sara Y, Sudhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- Seabrooke S, Stewart BA. Moesin helps to restrain synaptic growth at the Drosophila neuromuscular junction. Dev Neurobiol. 2008;68:379–391. doi: 10.1002/dneu.20595. [DOI] [PubMed] [Google Scholar]
- Sin WC, Haas K, Ruthazer ES, Cline HT. Dendrite growth increased by visual activity requires NMDA receptor and Rho GTPases. Nature. 2002;419:475–480. doi: 10.1038/nature00987. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K, Backs J, McAnally J, Qi X, Gerard RD, Richardson JA, Hill JA, Bassel-Duby R, Olson EN. The transcriptional coactivator CAMTA2 stimulates cardiac growth by opposing class II histone deacetylases. Cell. 2006;125:453–466. doi: 10.1016/j.cell.2006.02.048. [DOI] [PubMed] [Google Scholar]
- Tropea D, Kreiman G, Lyckman A, Mukherjee S, Yu H, Horng S, Sur M. Gene expression changes and molecular pathways mediating activity-dependent plasticity in visual cortex. Nat Neurosci. 2006;9:660–668. doi: 10.1038/nn1689. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol. 2000;10:358–364. doi: 10.1016/s0959-4388(00)00091-x. [DOI] [PubMed] [Google Scholar]
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, McAnally J, Pomajzl C, Shelton JM, Richardson JA, et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–566. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Wang B, Moya N, Niessen S, Hoover H, Mihaylova MM, Shaw RJ, Yates JR, 3rd, Fischer WH, Thomas JB, Montminy M. A hormone-dependent module regulating energy balance. Cell. 2011;145:596–606. doi: 10.1016/j.cell.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AH, Valdez G, Moresi V, Qi X, McAnally J, Elliott JL, Bassel-Duby R, Sanes JR, Olson EN. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science. 2009;326:1549–1554. doi: 10.1126/science.1181046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, McLeod DR, Zondag S, Toriello HV, Magenis RE, Elsea SH. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am J Hum Genet. 2010;87:219–228. doi: 10.1016/j.ajhg.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Narayan S, Geiman E, Lanuza GM, Velasquez T, Shanks B, Akay T, Dyck J, Pearson K, Gosgnach S, et al. V3 spinal neurons establish a robust and balanced locomotor rhythm during walking. Neuron. 2008;60:84–96. doi: 10.1016/j.neuron.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito K, Svoboda K. Activity-dependent synaptogenesis in the adult Mammalian cortex. Neuron. 2002;35:1015–1017. doi: 10.1016/s0896-6273(02)00903-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.