

Abstract
Big potassium (BK) ion channels have several spliced variants. One spliced variant initially described within human glioma cells is the glioma BK (gBK) channel. This isoform consists of 34 aa inserted into the intracellular region of the basic BK ion channel. PCR primers specific for this inserted region confirmed that human glioma cell lines and freshly resected surgical tissues from glioblastoma multiforme patients strongly expressed gBK mRNA. Normal human brain tissue very weakly expressed this transcript. An Ab specific for this gBK isoform confirmed that human glioma cells displayed this protein in the cell membrane, mitochondria, Golgi, and endoplasmic reticulum. Within the gBK region, two putative epitopes (gBK1 and gBK2) are predicted to bind to the HLA-A*0201 molecule. HLA-A*0201–restricted human CTLs were generated in vitro using gBK peptide-pulsed dendritic cells. Both gBK1 and gBK2 peptide-specific CTLs killed HLA-A2+/gBK+ gliomas, but they failed to kill non-HLA-A2–expressing but gBK+ target cells in cytolytic assays. T2 cells loaded with exogenous gBK peptides, but not with the influenza M1 control peptide, were only killed by their respective CTLs. The gBK-specific CTLs also killed a variety of other HLA-A*0201+ cancer cells that possess gBK, as well as HLA-A2+ HEK cells transfected with the gBK gene. Of clinical relevance, we found that T cells derived from glioblastoma multiforme patients that were sensitized to the gBK peptide could also kill target cells expressing gBK. This study shows that peptides derived from cancer-associated ion channels maybe useful targets for T cell-mediated immunotherapy.
Introduction
The prolonged elevation of internal Ca2+ levels or by disrupting Na+/H+ intracellular ratios results in a type of cell death called “paraptosis” (1–4). This programmed cell death pathway leads to necrosis (5, 6) and is characterized by swollen mitochondria and endoplasmic reticulum (ER). Human U251 (7) and rat T9 gliomas (8) immediately swelled upon activation of the big potassium (BK) channels using a BK ionophore; paraptosis ultimately occurred within 18–24 h. Functional membrane BK channels were detected on the human and rat glioma cells by using patch-clamping techniques. Additionally, BK channels were found on the ER and the mitochondria (7, 8), providing a plausible rationale for why these organelles are specifically targeted in paraptosis. These ion channels are also called Maxi-K, hSlo, mSlo, KCNMA1, calcium-dependent, large conductance-, or voltage-activated channels (9–17). The complex interaction between various ions and their respective ion channels at the invadopodia of the malignant gliomas is speculated to explain some of the invasive properties of gliomas (18–20). This infiltrative nature of high-grade gliomas is thought to be responsible for the lethality of this tumor, since surgery and local irradiation fail to eliminate these invasive cancer cells.
Four large BKα-chains associate with four smaller BKβ-chains to form the functional ion pore. Several variant BKα channels are produced via alternative splicing pathways (21–24). Liu et al. (25) described a novel BKα channel, which they named the glioma BK channel (gBK), because it was initially described and genetically cloned from malignant human D54 glioma cells. This variant gBK channel consists of 34 aa inserted into the intracellular region of the BKα ion channel. This variant form was only seen when an additional 29-aa insert called the hbr5 region was simultaneously coexpressed within the BKα channel. We developed PCR primers specific for this gBK/hbr5 region and confirmed that human glioma cell lines and freshly resected glioblastoma multiforme (GBM) surgical specimens expressed this alternatively spliced mRNA. These gBK transcripts were very weakly detected within the brains of autopsy patients who died of noncancer-related causes. This gBK marker should therefore be considered tumor-associated, instead of being classified as a novel tumor-specific biomarker. An Ab designed specifically for this gBK region confirmed that human gliomas contained this insert at the protein level, whereas normal brain was gBK−. Within the gBK-specific protein sequence two putative T cell epitopes, gBK1 and gBK2, are predicted to bind to HLA-A*0201 molecules. HLA-A*0201+ dendritic cells (DCs) were loaded with these gBK1 or gBK2 peptides and subsequently generated CTL responses in vitro. Both gBK1 and gBK2 peptide-specific CTL populations killed the HLA-A2+/gBK+ gliomas (LN18, U87, U251, and T98G) but failed to kill HLA-A2− glioma cells, D54 (HLA-A1+/HLA-A3+/gBK+), or LNZ308 (HLA-A24+/gBK+) glioma cells in [51Cr]-release cytolytic assays. Exogenously loaded gBK peptides added onto T2 cells, but not influenza M1 control peptide-loaded T2 cells, were also specifically killed by their respective CTLs. This provides further evidence that the gBK-specific CTLs killed gBK+ target cells by HLA-A2 restriction. gBK-specific CTLs also killed HLA-A2+ human embryonic kidney (HEK) cells that were transfected with a plasmid that contained the gBK gene. T cells derived from GBM patients could be restimulated with autologous DCs that were pulsed with the gBK2 peptides and induced cell lysis of gBK-transfected HEK cells and U251 glioma cells. To our knowledge, this is the first report to demonstrate successful targeting of HLA-A2+ tumors by CTLs directed toward ion channel peptides. We detected this variant BK channel within a variety of other human cancers, indicating the universal nature of this target molecule. The gBK-specific CTLs also killed AGS gastric, A549 lung adenocarcinoma, and MCF-7 breast cancer cell lines that were both HLA-A2+ and gBK+. Thus, this gBK ion channel provides novel epitopes for T cell immune-directed therapy for several types of cancers.
Materials and Methods
Cell lines and surgical specimens
The human gliomas U87, U251, D54, LN18, LN229, T98G, and LNZ308 were described in Schwartz et al. (26). The HLA typing provides evidence of the genetic differences of these cells. All cells were grown in DMEM media (Sigma-Aldrich, St. Louis, MO) supplemented with 5–10% FBS (Gemini Bioproducts, Calabasas, CA) and 0.1% antibiotics in a humidified atmosphere of 5% CO2 at 37°C. The human T2 cells were supplied to us by Dr. Hideho Okada (University of Pittsburgh, Pittsburgh, PA). These cells were grown in RPMI 1640 medium with 5–10% FBS.
Other human cells, including AGS (gastric carcinoma), HepG2 (hepatocellular carcinoma), A549 (lung adenocarcinoma), Caco-2 (colon cancer), HuTu80 (duodenal adenocarcinoma), NCI-H69 (HTB-119; small cell lung cancer), Panc-1 (pancreatic cancer), and 293T HEK cells were obtained from the American Type Culture Collection (Manassas, VA). MCF-7 breast cancer cells were obtained from Ali Pedgram (Long Beach, VA). All of these cells were grown in DMEM with 10% FBS.
Normal human brain neurosphere cultures
Neural stem cells were derived from the brain subventricular zone of premature neonates who died shortly after birth. The stem cells were cultured cells on Matrigel-coated dishes with DMEM/F12 (1:1 v/v) medium (Irvine Scientific, Irvine, CA) containing 10% BIT 9500 (BSA, insulin, transferrin; Stem Cell Technologies, Tukwila, WA), 292 μg/ml glutamine (Irvine Scientific), 40 ng/ml basic fibroblast growth factor, 20 ng/ml epidermal growth factor, and 20 ng/ml platelet-derived growth factor-AB, 100 U/ml penicillin (Irvine Scientific), 100 μg/ml streptomycin (Irvine Scientific), 50 μg/ml gentamicin (Sigma-Aldrich), 10 μg/ml ciprofloxacin (Bayer), and 2.5 μg/ml amphotericin (Life Technologies/Invitrogen, Carlsbad, CA) as previously described (26). For continuous expansion, half of this medium was replaced every other day, and the cultures were passaged every 7 d or when confluent using nonenzymatic cell dissociation solution (Sigma-Aldrich). These cells were supplied by Dr. Philip H. Schwartz (Children’s Hospital of Orange County, Orange, CA).
Human GBM and normal brain tissue
After informed consent forms were signed, remnant surgical tissue from GBM patients was provided for research purposes. The debulking surgery was performed by the Neurosurgery Department at the Chao Comprehensive Cancer Center (University of California, Irvine Medical Center, Orange, CA). Normal human brain tissue was collected from autopsies taken from patients who died of noncancer-related causes. Autopsies were only performed after the next of kin signed the release forms for performing autopsies. The nonglioma autopsies were performed at the Veterans Affairs Medical Center (Long Beach, CA).
Paraffin-embedded glioma samples were obtained from autopsies done at the Veterans Affairs Medical Center and University of California, Irvine. Dr. Ronald C. Kim supplied us with these specimens that were recut and mounted on glass slides. The sections were then deparaffined and treated using a target retrieval unmasking solution (Dako, Carpinteria, CA). The sections were then stained as described above. The specimens were viewed under epifluorescence using the Nikon microscope as described above.
Measuring image fluorescence/Gray intensity using ImageJ software
The fluorescence images were captured using a Nikon Eclipse E600 fluorescence microscope system with the RTKE SPOT camera and SPOT software (Diagnostic Instruments). The levels of fluorescence expression were measured with a set of measurements (area, integrated density, and mean Gray value) from ImageJ software, and the expressed average pixel intensity (104 pixels2) values were calculated using the formula: average pixel intensity = average integrated density − area of selected image mean fluorescence of background readings. In brief, 40 randomly selected areas (100 × 100 pixels) per image (n = 4 images) were used to analyze the fluorescence intensity expressed in various tissues samples.
RT-PCR analysis
Total RNA was isolated from the cells or tissues using an RNeasy Plus Mini kit (Qiagen, Valencia, CA). One microgram total RNA and random primers were used for cDNA synthesis using the AffinityScript qPCR cDNA synthesis kit (Agilent Technologies, Santa Clara, CA). The PCR was performed using the following primers to identify the gBK/hbr5 region within the gBK transcripts: forward, 5′-CGTTGGGAAGAACATTGTTC-3′, reverse, 5′-AACTGGCTCGGTCACAAG-3′. After the thermal cycling was complete, the PCR products were run on a 1.5% agarose gel and stained with the ethidium bromide. The gel image was then captured under the UV light using the Eagle Eye II imager (Stratagene, San Diego, CA). The predicted size of the amplicons was 186 bp. The image was scanned for its densitometric reading by using the Un-Scan-It program (Silk Scientific, Orem, UT).
The same gBK/hbr5 primers used above were also used for quantitative real-time PCRs. Real-time PCR reactions were performed on an iCycler iQ detection system using a Brilliant SYBR Green kit (Stratagene). We used identical techniques as described previously (27).
Cloning of the human gBK gene
We genetically cloned the BK into the plasmid pcDNA3.1 (Invitrogen) using the mRNA purified from U251 cells as the source of the cDNA. By DNA sequencing (DeWalch Technologies, Houston, TX) of this cDNA, this BK cDNA was determined to be the hbr5 isoform of the BK channel. For the sake of time, we used in vitro mutagenesis techniques to insert the gBK region into the hbr5 cDNA.
The following oligonucleotides were synthesized by Integrated DNA Technologies (San Diego, CA). The mutagenic oligonucleotide primer no. 1 sequence is 5′-GAATAAAAAAATGTGGCTGCAAACGGCGTTGGGAAGAACATTGTTCTTTGTGGAGACTGGAAAGCAAGGGAAATGTGAGAAGATTAAACTACTGCAGGGGTCAGCAAACTTTCTCTGTCAAAGTCAAGGTTGCAGCTAGATCACGCTATTC-3′. The mutagenic oligonucleotide primer no. 2 sequence is 5′-GAATAGCGTGATCTAGCTGCAACCTTGACTTTGACAGAGAAAGTTTGCTGACCCCTGCAGTAGTTTAATCTTCTCACATTTCCCTTGCTTTCCAGTCTCCACAAAGAACAATGTTCTTCCCAACGCCGTTTGCAGCCACATTTTTTTATTC-3′. Sequences in boldface type are nucleotides corresponding to the gBK.
In vitro mutagenesis was performed using a QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies). This plasmid was then transformed in XL10-Gold ultracompetent cells provided in the mutagenesis kit. The inserted gBK region was confirmed by sequencing (San Diego State University, San Diego, CA). The plasmid was then named pcDNA/gBK. The plasmids were isolated using the Qiagen Maxi Prep kits for further use.
Transfection of gBK into non-gBK–expressing HEK cells
HEK cells were seeded into 100-mm2 plates the day before transfection to achieve a 35–40% confluence in standard tissue culture medium. Qiagen Attractene transfection reagent was used to transfect the pcDNA/gBK plasmid into the HEK cells, according to the instructions provided by the manufacturer. The purified plasmid and Attractene transfection reagent were mixed together for 15 min and added to the cell cultures. After 48 h, the transfected cells were harvested as target cells for gBK-specific CTLs. Expression of the gBK was then confirmed within the transfected HEK cells using the purified anti-gBK Ab with intracellular flow cytometry.
Glioma BK channel peptides and polyclonal Abs against the gBK or BK ion channels
The gBK peptides were designed to have the best binding to HLA-A0201 via the University of Heidelburg (SYFPEITHI) database (http://www.syfpeithi.de/). The peptides toward gBK (SLWRLESKG gBK704–712 [gBK1 peptide] and GQQTFSVKV gBK722–730 [gBK2 peptide]) were synthesized by GenScript (Piscataway, NJ), whereas the influenza M1 peptide (M158–66, GILGFVFTL) was synthesized by Think Peptides (a division of ProImmune, Oxford, U.K.).
The goat polyclonal Ab directed toward gBK was made by GenScript. The gBK region that was deemed to be the best to generate an Ab toward the gBK insert was RLESKGNVRRLNYC. An Ab toward the extracellular region of the BKα channel (ENSGDPWENFQNNQC) was made in rabbits by GenScript. This latter Ab was designed to bind human, rat, and mouse BK channels. The C-terminal amino acids on both the gBK and BK peptides were added with a cysteine to facilitate a better conjugation with keyhole limpet hemocyanin as the carrier protein/immunogen.
Cell surface flow cytometry
The cells were stained with the mouse mAb specific for HLA-A2 allele purchased from Santa Cruz Biotechnology (Santa Cruz, CA). One half million cells were incubated with 5 μl of the Ab on ice for 1 h. The cells were washed three times and then the secondary anti-mouse IgG Ab (FITC-labeled; Vector Laboratories, Burlingame, CA) was added. These cells were incubated on ice for an additional hour. The cells were again washed three times and then immediately analyzed on a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA). Ten thousand cells were then analyzed. We used the Kolmogorov–Smirnov test to show significant differences in flow cytometry expressions (p < 0.05) between the cells stained with the specific Ab with that of cells stained with the secondary Ab alone.
To quantitate the CD4/CD8 ratio of the gBK-specific CTLs, one million cells were stained with the CD4-FITC and CD8-PE Abs on ice for 1 h. The cells were washed three times and 10,000 cells were analyzed with a FACSCalibur flow cytometer.
Intracellular flow cytometry
Exponentially growing glioma cells (1 × 107 cells) at 50–70% confluence were fixed in FCM Fixation buffer (Santa Cruz Biotechnology) on ice for 30 min. The cells were washed twice in ice-cold PBS. The cells were permeabilized with Santa Cruz Biotechnology permeabilization buffer for 15 min on ice. The cells were washed again with FCM Wash buffer twice. The resuspended cells were then divided into 106-cell aliquots and then incubated with the primary Ab (anti-BK channel or BK channel Ab) for 1 h. The cells were washed twice and the secondary Ab conjugated with FITC (Vector Laboratories) was incubated on ice for another hour. After washing the cells twice, 10,000 cells were analyzed with a FACSCalibur flow cytometer. We used the Kolmogorov–Smirnov test to show significant differences in flow cytometry expressions (p < 0.05) between the various cell lines.
Confocal microscopy
Glioma cells were incubated on cover slips that were collagen coated. The cells were fixed with 2% paraformaldehyde and permeabilized with ice-cold methanol/acetone. Tissues containing the glioma were stained with a 1:50 dilution of the Ab. The tissue was washed and then incubated 1 h with a 1:200 dilution of the FITC-conjugated secondary Ab (Vector Laboratories). Tissue samples were washed and mounted with ProLong Gold anti-fade reagent (Molecular Probes, Eugene, OR). Samples were imaged and analyzed using a Nikon two-laser (helium/neon and argon) PCM 2000 confocal system on the Eclipse E800 microscope. To view the glioma organelles, the cells were stained with either MitoTracker or Golgi tracker obtained from Molecular Probes/Invitrogen. To view the ER, we used a monoclonal mouse Ab directed toward calreticulin, which binds to the ER (MBL International, Woburn, MA). The two different fluorescent dyes in the labeled sample (FITC and Texas Red/Deep Red) were simultaneously visualized. As a result, it can be inferred that the red-emitting and green-emitting dyes are colocalized when areas are present in the images shown in yellow.
Induction and evaluation of CTLs against glioma Ag peptides
To generate DCs, elutriated monocytes were collected from healthy normal donors who were HLA-A0201+. The adherent monocytes derived from PBMCs were cultured in AIM-V medium (Invitrogen/Life Technologies) supplemented with 1000 U/ml recombinant human (rh)GM-CSF and 500 U/ml rhIL-4 (Cell Sciences, Canton, MA) at 37°C in a humidified CO2 (5%) incubator. Immature DCs were harvested on day 6, resuspended in AIM-V medium at 1 × 106 cells/ml with peptide (10 μg/ml), and incubated for 4 h at 37°C and then stimulated overnight with 500 IU/ml rhIFN-γ and 1 μg/ml LPS for maturation. Magnetic bead-purified (Miltenyi Biotec, Auburn, CA) CD8+ autologous T cells were cocultured with mature peptide-pulsed DCs at a ratio of 10:1 in AIM-V medium supplemented with 5% human AB serum with 10 U/ml each rhIL-2 (R&D Systems, Minneapolis, MN) and rhIL-7 (Cell Sciences). On day 7, lymphocytes were restimulated with peptide-loaded mature DCs in AIM-V medium supplemented with 5% human AB serum, 10 U/ml rhIL-2, and 10 ng/ml each rhIL-7 and rhIL-15 (R&D Systems). The CD8+ cultured cells were analyzed for CTL activity 5 d after the second stimulation by a standard 6-h [51Cr]-release assay.
The T2 cells were loaded with the exogenous peptides (1 μM) (same gBK peptides as described in gBK peptide section) in RPMI 1640 media without any serum for 1 h at 37°C as described previously (28–30). The cells were washed twice and then labeled with the [51Cr] (MP Biomedicals, Irvine, CA) for an additional hour. These cells were washed and then used as target cells (5 × 103 cells/well) in 6-h [51Cr]-release assays.
Glioma patient-derived material
Excess vitally frozen PBMCs, collected by leukapheresis of HLA-A*0201+ GBM patients for DC vaccine trials at the University of California, Los Angeles Medical Center, were used for these studies. GBM patient CTLs (pCTL-1, pCTL-2) and CTLs derived from a normal HLA-A2+ donor (nCTLs) were generated as described above with rIL-4 and GM-CSF. The DCs isolated from the PBMCs were pulsed with 10 ug/ml gBK2 peptide overnight. After DC maturation, the autologous PBMCs were then added and expanded as described above, with the exception of the CD8+ selection step with Miltenyi Biotec beads. The early passage (P2) autologous glioma cells derived from the patient from which pCTL-1 were generated (i.e., syngeneic) were available for testing. The UC-GBM1 glioma cells displayed moderate to strong positivity for gBK by intracellular flow cytometry. Cytotoxicity assays were performed with effector pCTL-1, pCTL-2, and nCTLs. The target cells included HEK, HEK/gBK, U251, and UC-GBM1 cells. The average percentage lysis from quadruplicate wells at various E:T ratios was determined from 6-h [51Cr]-release assays.
Results
Human glioma cells express the novel gBK channel mRNA
Liu et al. (25) genetically cloned a novel gBK channel from human D54 glioma cells, which was overexpressed by human high-grade (World Health Organization grades III and IV) gliomas by performing Western blotting. This novel BK channel contained a 63-aa insert that incorporated both an hbr5 sequence (29 aa) along with the gBK region (34 aa). We synthesized PCR primers designed to specifically detect most of the gBK/hbr5 region. RNA was isolated from four different freshly resected surgical World Health Organization grade IV GBMs (GBM-1, -2, -3, and -4) from the Neuro-Surgery Department at the Chao Comprehensive Cancer Center (University of California, Irvine, Orange, CA). Additionally, the mRNA derived from five malignant human glioma cell lines was also used (D54, LN18, LN229, U87 and U251). The cDNA was synthesized and standard PCR techniques were used to amplify this inserted region. The gBK/hbr5 PCR amplicons from all gliomas had the predicted 186 bp whether they originated from the surgical specimens or cell lines (Fig. 1A, top panel). These amplicons were sequenced and verified to contain the gBK insert. The densitometric readings of these RT-PCR products are displayed in Fig. 1A (lower panel). So far, every human glioma cell line tested (n = 10) has strongly expressed gBK/hbr5.
FIGURE 1.

RT-PCRs show the presence of gBK/hbr5 inserted sequence within clinical and human GBM cell lines. (A) RNA was isolated from four surgical specimens (GBM-1, -2, -3, and -4) and five human glioma cell lines (D54, LN18, LN229, U87, and U251). After the cDNA was synthesized, it was amplified for 40 cycles. The amplicons were loaded onto a 1.5% agarose gel and then stained with ethidium bromide and imaged (top panel). Bottom panel, Staining intensity was detected by using the UV transilluminator and then quantitated for its densitometric readings. (B) Top panel, PCR results of HEK cells, normal human neurosphere cells, and two human GBM surgical samples; bottom panel, densitometric readings.
Cells obtained from brain tissue collected from an autopsy patient who died of a noncancer-related cause was grown in neurosphere cultures, and tested for gBK transcripts. Additionally, HEK cells also showed minimal expression of gBK. The RT-PCR from the amplicons derived from neurosphere cells barely showed any of the expected gBK band (Fig. 1B), whereas two human GBM surgical specimens did. We used quantitative real-time RT-PCR to confirm that the gBK transcripts were only weakly present and represented only 3–5% of the amount of the mRNA made by the resected human GBM tissue.
gBK protein is expressed by malignant glioma cells
Fig. 2A shows that human U251 cells were positively stained with the Ab directed at the basic unmodified BK ion channel. Because the gBK contains an additional inserted region, all Abs directed toward BK recognize BK as well as the gBK isoforms with the hbr5 insert. A polyclonal Ab that specifically recognizes only the gBK insert was generated. Our anti-gBK Ab only recognizes the unique gBK insert and will not react toward the basic, unmodified BK ion channel. The U251 cells stained positively with our Ab toward gBK (Fig. 2B).
FIGURE 2.

Glioma cells express both BK and gBK channels as detected by intracellular flow cytometry. U251 cells were fixed, permeabilized, and stained with either the Ab toward the standard BK channel (A) or gBK-specific Ab (B). The shaded area represents the value of the isotypic controls. (C and D) Ten thousand D54, LN18, LNZ308, T98G, U87, and U251 glioma cells were analyzed by intracellular flow cytometry using either the BK or gBK Abs. The mean peak channel number and SEMs of these cell lines are presented on the y-axis. (C) BK profile. (D) gBK channel profile.
Screening of six different human glioma cell lines (D54, LN18, LNZ308, T98G, U87, and U251) using intracellular flow cytometry shows the presence of gBK channels within all glioma cells. Expression of gBK was somewhat variable and was dependent on which cell line was used. Fig. 2C contains data representing three different experiments with the BK-stained cells and shows mean peak channel numbers of these gBK+ cells. U251 cells demonstrated the best expression of gBK with a mean peak channel value of 300. The LNZ308 cell line possessed the least amount of gBK positivity with a mean peak channel value of 125, but it is still a good positive signal. We also simultaneously stained the glioma cells with the gBK channel Ab (Fig. 2D). An identical staining pattern was also seen here: U251 had the brightest staining intensity, wheras LNZ308 cells had the lowest intensity.
To prove the specificity of this goat polyclonal Ab, we performed intracellular flow cytometric analysis with U251 and HEK cells using the pre- and postimmunized antiserum. Fig. 3A shows some low binding of the preimmunized antisera with the U251 glioma cells, whereas the serum from the immunized goat displayed a much stronger staining of the U251 cells. However, when the purified Abs were used, the background staining of the gBK was dramatically reduced. Minimal staining was seen when the anti-gBK Abs using the gBK− cell line were derived from normal brain neurosphere cultures (Fig. 3B), although these cells strongly expressed the unmodified BK channels (Fig. 3C).
FIGURE 3.

Specificity of the goat anti-gBK antiserum and purified anti-gBK Ab. Fixed-and-permeabilized U251 and normal human brain neurosphere cells were stained with either the preimmunized or postimmunized goat sera or with the purified goat anti-gBK Ab. Results in (A) show that (preimmunized) serum has some elevated background staining, whereas the immunized antiserum had enhanced staining. When the isotypic control and purified anti-gBK Abs were used, a lowered left shift was observed in both groups, as compared with the samples stained using whole sera. Results in (B) show the normal brain neurosphere cells to have minimal expression of gBK staining using the purified Ab, but they are positive for BK channels (C). Results in (D) show the expression of gBK by HEK cells that were either unmodified or transfected with the pcDNA/gBK plasmid for 48 h using the purified anti-gBK Ab.
We genetically cloned the gBK/hbr5 gene from U251 cells and transfected the HEK cells with our gBK construct. Fig. 3D shows that upon transfection with the gBK plasmid for 48 h, gBK staining was observed within the HEK cells using the immunized anti-gBK Ab.
U251 cells were cultured as adherent cells overnight onto cover slips. The cells were first stained with the BK channel Ab since its epitope was on the extracellular region. The cells were fixed and permeabilized and then stained with the gBK Ab. Supplemental Fig. 1 shows the dual staining of the two different BK channels on the U251 cells.
gBK channels can be seen within clinical GBM, but not within normal human brain
We next tested whether clinical GBM specimens possessed gBK at the protein level by using the gBK Ab. Two autopsy cases of GBM patients were examined. Both cases showed strong gBK expression within the glioma. Fig. 4 (middle row) shows one representative GBM case. As a negative control, we used normal brain tissue from an autopsy of someone who died of a noncancer-related cause. BK channels were detected within the normal brain (Fig. 4, top middle panel), but no staining was seen within the normal brain when we used the gBK (Fig. 4, top right panel). Fluorescent densitometric readings are presented in Fig. 4 (bottom panel), which show that the normal brain largely expresses BK, whereas GBM tissue shows predominantly the gBK form.
FIGURE 4.

Human GBM, but not normal brain, displays gBK. Top panel, top row, Staining profile of normal human brain; bottom row, staining profile of human GBM tissue; left column, negative control staining in the presence of a nuclear Hoescht red stain; middle column, staining with the BK channel Ab (green); right column, staining of the brain tissue using the gBK Ab (green). Original magnification ×40. Bottom panel, Summary of the average and SDs of green fluorescent staining of 40 different fields using the ImageJ intensity staining detection profile (as described in Materials and Methods).
gBK channels are found at the same cellular locations as with BK channels
We stained the fixed-and-permeabilized U251 glioma cells with the anti-gBK channel Ab and viewed these cells using immunofluorescent microscopy. BK channels are found on the cell membrane, on the mitochondria, and on the ER (7, 8). Fig. 5 (left column) shows gBK+ staining (green). The yellow expressed in Fig. 5 (right column) shows the colocalization of the respective gBK expression with various red stains of the mitochondria, Golgi, and ER. No differences in staining were seen when gBK (green) and BK Abs (red) were simultaneously compared (Fig. 5A, right panel). Staining is particularly heavy at the edges of the cells, primarily at the lamellipodia, microvilli, and filopodia. A complete overlap was seen throughout the cells. Fig. 5B shows the colocalization of the gBK with the mitochondria. Fig. 5C shows the gBK colocalization with the Golgi. Finally, Fig. 5D shows the staining with the anti-calreticulin Ab that stains the ER, and the gBK also colocalizes there as well.
FIGURE 5.

gBK channels are found in the same cellular locations as the BK channels. U251 glioma cells were attached to glass cover slips overnight and fixed and stained with the anti-gBK channel Ab. Left column, gBK (green) staining of different U251 cells. (A) Middle panel, staining profile with a BK channel Ab (red); right panel, merged image. Yellow indicates that the proteins are located very close (100 nm) to each other. (B) Staining patterns when the cells were stained with the Mito-Tracker. (C) Golgi staining profile (Golgi tracker). (D) Cells stained with calreticulin, which specifically stained the ER (red). Original magnification ×100.
gBK peptide-pulsed DCs stimulate gBK-specific human CTLs in vitro
We analyzed the region within the gBK protein sequence using the SYFPEITHI database to identify potential HLA-A0201–restricted epitopes. Two potential epitopes were identified, SLWRLESKG (gBK704–712; gBK peptide 1) and GQQTFSVKV (gBK722–730; gBK peptide 2). Both peptides scored as 16, indicating a reasonable binding affinity to HLA-A2. These two gBK peptides were synthesized. Human HLA-A2+ peripheral blood monocytes were stimulated with IL-4 and GM-CSF for 1 wk. The immature DCs were pulsed with either the specific gBK1 or gBK2 peptides for 1 d. The immature DCs were induced to differentiate into mature DCs. We used these DCs as APCs to stimulate gBK-specific CTLs by adding naive peripheral blood HLA-A2+ T cells. Afterward, the T cells were expanded by stimulating them with rhIL-2, rhIL-7 and rhIL-15. We generated five different preparations of gBK1- and gBK2-specific CTLs. Most of the CTL preparations were CD8+ cells, with CD4+ cells comprising only 5–20% of the total population.
These human HLA-A2–restricted CTLs displayed good cytotoxicity toward the human HLA-A2+ glioma cells, U251 (Fig. 6A), and LN18, T98G, and U87 (Fig. 6B) that possessed gBK. These gBK1-specific CTLs effectively killed these HLA-A2+/gBK+ glioma cells at all E:T ratios within 6 h. These values were significantly different (p < 0.05) from the results of these same CTLs responding to those non-HLA-A2–expressing gliomas that are gBK+, D54, but are HLA-A1+ and HLA-A3+ (Fig. 6A), or LNZ308, which is HLA-A24+ (Fig. 6B). Identical data were also observed with the gBK2-specific CTLs that were specifically activated toward the GQQTFSVKV, gBK722–730 epitope. All HLA-A2+ U251, LN18, U87, and T98G glioma cells were killed (Fig. 6C, 6D). Concurrently, the HLA-A2−, but gBK+, glioma cells (D54 and LNZ308) were not lysed. These data illustrate that functional CTLs are generated against two predicted epitopes derived from the gBK and can kill the gBK+ gliomas. Thus, the gBK protein is being properly digested by the glioma’s proteasome and can be recognized by the CTLs in the context of their HLA-A2 molecules.
FIGURE 6.

gBK-specific CTLs will kill HLA-0201+ gliomas that also express gBK. gBK1- and gBK2-specific T cells were generated and tested against either LN18, T98G, U87, or U251 glioma (HLA-A0201+/gBK+) or D54 cells (HLA-A1+/A3+, gBK+) and LNZ308 (HLA-A24+, gBK+) in a 6-h assay. (A and B) Results of one representative study done three times that shows the results of the gBK1-specific CTLs. (C and D) Results generated with the gBK2-specific CTLs. *p < 0.05 from the non-HLA-A2− control values.
gBK peptide-loaded T2 cells are killed by their respective gBK-specific CTLs
The human T2 (B cell × T cell) hybridoma cell line is capable of being loaded with exogenous peptides that bind to the HLA-0201 molecule (29–31). The gBK-specific CTLs were tested against the T2 cells that were loaded with either influenza M1 control peptide or gBK1- or gBK2-specific peptides. Fig. 7 shows that gBK1-specific CTLs only killed the T2 cells loaded with the gBK1 peptide (Fig. 7A). Conversely, gBK2-specific CTLs only killed the T2 cells pulsed with the gBK2 peptide (Fig. 7B). No gBK-specific CTLs killed the T2 cells bearing the influenza M1 peptide. This work confirms the previous experiment that shows that functional CTLs can be specifically generated against gBK Ags via an HLA-A2 restriction.
FIGURE 7.
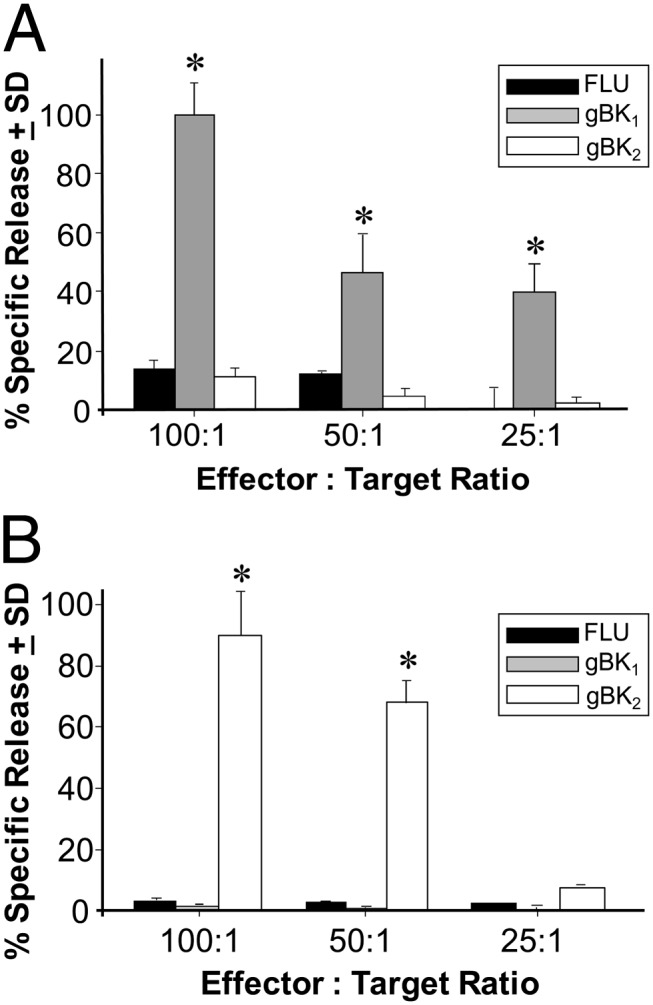
T2 cells loaded with exogenous gBK peptides are specifically killed by the gBK1- or gBK2-specific CTLs. gBK1- or gBK2-specific CTLs were generated in vitro and then tested against T2 cells that were separately loaded with three different exogenous peptides (1 μM) for 1 h at 37°C. The cells were washed and then labeled with [51Cr] for another hour at 37°C. The cells were then washed three times and used as target cells. (A) Data generated when gBK1-specific CTLs were used. (B) Data when gBK2-specific CTLs were tested against the various T2 cells. Representative data from three different experiments are shown. *p < 0.05 from the influenza M1 control peptide (FLU) cytotoxicity values.
gBK channels can be found on other human cancer cell lines
We tested a variety of other human cancer cell lines that were available to us and examined their gBK expression pattern. Fig. 8 shows the profile of gBK as detected by intracellular flow cytometry using the anti-gBK channel Ab. All of the tumor cells (Caco-2 colon cancer, HuTu-80 duodenal cancer, HepG2 hepatocellular carcinoma, and Panc-1 pancreatic cancers) showed good gBK expression that was comparable to the glioma cell lines. Only NCI-H69 small-cell lung cancer cells appeared to have low to moderate expression of gBK, possibly because the isotypic background staining was higher. This work demonstrates that gBK is not just specific for gliomas and can be found on other tumor types.
FIGURE 8.

gBK can be found on a variety of other types of human tumor cell lines. Caco-2 (colon cancer), HuTu80 (duodenal adenocarcinoma), HepG2 (hepatocellular carcinoma), NCI-H69 (small cell lung cancer) and Panc-1 (pancreatic cancer) cells were fixed, permeablized, and stained with the anti-gBK Ab. Afterward, the cells were washed and stained with the anti-goat FITC labeled Ab. Ten thousand stained cells were then analyzed. The U251 cells were supplied as a reference point for staining intensity.
gBK-specific CTLs can also kill other nonglioma cancer cells
The results obtained from Fig. 9 (right column) indicated that AGS gastric cancer cells, A549 adenocarcinoma lung cancer, and MCF-7 breast cancer cells all possess gBK as detected by intracellular flow cytometry. These three cell lines also tested positive for cell surface HLA-A2, as detected by flow cytometry. We tested whether the gBK-specific CTLs could also kill these cells, so cytotoxicity assays were initiated. Fig. 9 (left panels) demonstrates that both gBK1- and gBK2-specific CTLs killed these three cancer cells. The AGS and A549 cells were killed the best, whereas the MCF-7 cells were only weakly killed, even though MCF-7 cells possessed a higher level of gBK expression. We attribute this to the fact that the HLA-A2 expression of the MCF-7 cells was lower than that expressed on the other two cancer cell lines and hence limits the functional actions of the CTLs. Thus, the gBK Ag should therefore be considered a tumor-associated Ag and could be applicable for other types of immunotherapy toward those gBK+ types of tumors.
FIGURE 9.

gBK-specific CTLs will also kill AGS gastric cancer, A549 lung cancer and MCF-7 breast cancer cells. gBK1 and gBK2 CTLs were tested against these HLA-A2+/gBK+ cancer cells in a 6-h [51Cr]-release assay. Right column, Intracellular flow cytometry results using the gBK Ab and the cell-surface HLA-A2 flow cytometric profile showing the intensity of this Ag on nonpermeabilized cancer cells. The shaded area represents the value of the isotypic controls. Left column, Effects of gBK1- and gBK2-specific CTLs in 6-h assays. The cytolytic activities against the AGS (A) and MCF-7 (C) cells were performed on the same day, whereas the assay with the A549 adenocarcinoma cells (B) was done 1 wk after being stimulated with immobilized anti-CD3 and anti-CD28 beads.
gBK-specific CTLs can be generated from GBM-derived immune cells
We next tested whether gBK-specific CTLs could be generated from GBM patient PBMCs. Vitally frozen archival material from two HLA-A0201+ GBM patients and one normal donor (nCTL) was used. Their DCs were first pulsed with gBK2 peptide and then used to stimulate their respective autologous T cells. These T cells were then expanded and tested in [51Cr]-release assays. The data in Fig. 10 (top and middle panels) show that CTL populations derived from the GBM patients lysed the gBK+ U251 glioma cells and low-passage UC-GBM1 glioma cells (also gBK+), which were isolated from the same patient as UC-pCTL1. Identical results were obtained when nCTLs were derived from a normal HLA-0201+ donor (Fig. 10, bottom panel). All the gBK2-specific CTLs killed HLA-0201+ HEK cells that were transfected with the gBK gene, but they failed to kill the nontransfected HEK cells.
FIGURE 10.
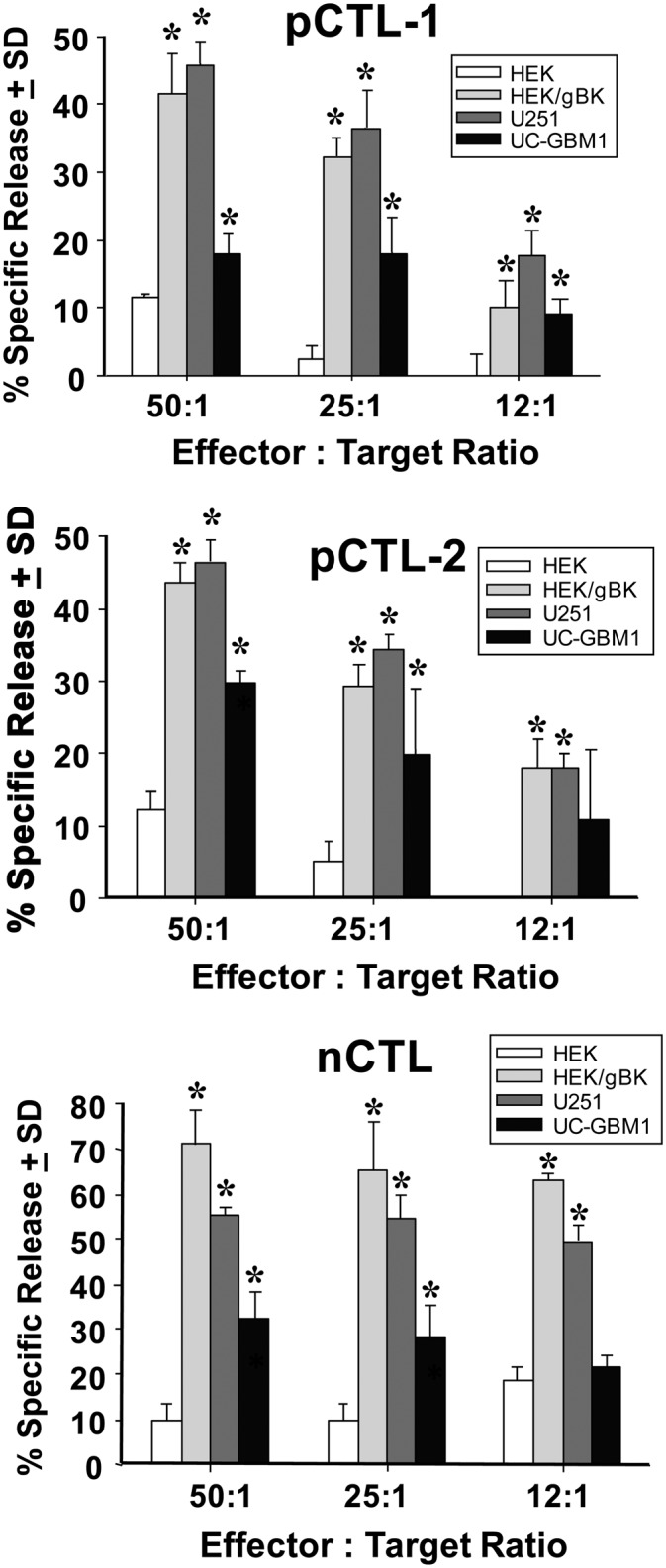
Cytotoxicity experiments with gBK-specific CTL effectors and gBK-expressing and nonexpressing target cells. pCTL-1 and pCTL-2 were generated from GBM patients and nCTLs were stimulated from a normal donor. All CTLs originated from HLA-A0201+ individuals. The DCs isolated from the PBMCs were pulsed with the gBK2 peptide prior to in vitro DC maturation. Autologous T cells were then stimulated and further expanded. The target cells included the U251 glioma cell line, early passage (P2) UC-GBM1 glioma cells (autologous to the pCTL-1), and HLA-0201+ HEK cells that were either nontransfected or transfected with the gBK plasmid 48 h prior to the assay. The gBK profile of the HEK cells is shown in Fig. 3D. All test symbols represent data that were done in quadruplicate cultures. Top panel, Results using the pCTL-1; middle panel, data generated with the pCTL-2; bottom panel, results using the nCTLs. *p < 0.05 from the nontransfected HEK control values.
Cold target inhibition studies were performed using either unlabeled HEK or U251 cells in the presence of [51Cr]-labeled U251 cells to show further specificity of gBK2-directed CTLs. A representative study is shown in Supplemental Fig. 2. The pCTL-1 lysed the 51Cr-labeled U251 cells in the absence of unlabeled U251 cells or in the presence of the HEK cells. Significant inhibition was observed when unlabeled U251 cells were added to the CTLs responding to the radiolabeled U251 cells.
Discussion
Ion channels (potassium, calcium, and chloride channels) are speculated to play an important role in glioma biology (18–20). BK channels are one member of this triumvirate and are overexpressed by malignant glioma cells (9–14). BKα channels have a complex splicing pattern with multiple forms known to be expressed (21–24). A novel BK isoform was genetically cloned by Liu and coworkers (25) in 2002 and was called the gBK. This gBK splicing variant is identified as BK isoform 7 (SwissProt accession no. Q12791-7; http://web.expasy.org/docs/swiss-prot_guideline.html). In the present study, we confirmed the previous work that shows that gBK mRNA exists genetically in surgically resected World Health Organization grade IV GBM and malignant glioma cell lines using specific PCR primers (Fig. 1). By using a specifically designed Ab toward gBK, we proved that this molecule is displayed as a protein (Figs. 2–5). Earlier studies (25) only used a BK channel Ab to deduce that gBK protein was present. That Ab used really could not distinguish whether either an hbr5 or gBK/hbr5 insert was actively expressed at the protein level within the gliomas. We show that gBK channels are specifically found in the same intracellular sites as nonmodified normal BK channels. gBK channels are found on the cell membrane and the edges of the cells (Fig. 5, Supplemental Fig. 1). The staining intensity with the gBK Ab was particularly high at the leading edges (lamellipodia) of the glioma. The gBK also colocalized with the mitochondria, Golgi, and ER. Thus, gBK resides in the identical places as do the BK channels. We found that normal human brain tissue only contains ∼3–5% the amount of gBK mRNA as that found within malignant human glioma tissue. Thus, gBK expression probably reflects an increased transcriptional rate within glial cancers. This Ag should be considered a tumor-associated Ag, as opposed to being truly a tumor-specific Ag.
Besides finding gBK on glioma cells, we also found gBK on different types of other human cancer cells (Figs. 8, 9). These cancers included breast, colon, pancreatic, stomach, duodenal, lung, and liver cancer cell lines. Others have shown differential effects of BK channel modulators on breast (32) and prostate cancers (33), but whether this reflects on the activity of gBK within these cancer cells is unknown. Thus, it could be appropriate to rename gBK to cancer BK (cBK) to reflect the more universal nature of this tumor-associated ion channel. At this time, we will continue to use the term gBK to promote a consistent terminology and minimize any confusion that could result as a new name to the literature. Besides, BK channels already have numerous other aliases, such as Maxi-K, hSlo, mSlo, KCNMA1, calcium-dependent, or large conductance- or voltage-activated channels (15–17).
Previous in vitro studies showed that disrupted BK channel functional activity inhibits glioma cell proliferation (33, 34) and migration (35, 36). Earlier, we showed that prolonged activation of BK channels of T9 or U251 gliomas in vitro causes cell swelling and eventually leads to paraptosis (7, 8). When paraptotic BK channel-activated/killed T9 cells were used as the immunogen, T9-specific tumor immunity toward the T9 glioma cells was generated within naive rats (8). BK channel activators were created and successfully prevented cardiac reperfusion injury in animals (37, 38). Clinical studies using these BK modulators failed to be effectively translated in humans (39). Severe side-effects and toxicities limited this approach. To directly kill glioma cells in situ by prolonged BK channel activation via paraptosis, one would have to continuously infuse large concentrations of BK channel activators either systemically or locally in the tumor mass with the risk of affecting the normal surrounding brain. Hence, either possible method would most probably lead to unwanted side-effects. Direct ways to specifically kill glioma in a clinical setting using the current BK channel activators are probably not feasible.
We also report in this study that the gBK inserted sequence contains protein sequences that can be processed into peptides that bind to the MHC and provoke human immune responses. Using the SYPEITHI database for searching epitopes that predicts binding to various MHC class I molecules, we found that two peptides, SLWRLESKG (gBK704–712; gBK1 peptide) and GQQTFSVKV (gBK722–730; gBK2 peptide), have reasonable scores (16) for binding to the HLA-A0201 molecules. We successfully generated human HLA-A2–restricted CTLs by loading DCs with gBK1 or gBK2 peptides and then stimulating the naive CD8+ cells. These CTLs killed all HLA-A2+ human glioma cells that coexpressed gBK. To our knowledge, this is the first study that immunologically targets peptides derived from ion channels. These same CTLs failed to kill gBK+ D54 (HLA-A1+ and HLA-3+ cells) or gBK+ LNZ308 CTLs that were HLA-A24+ (Fig. 6). The gBK1- and gBK2-specific CTLs also specifically killed human T2 hybridoma cells that were exogenously loaded with their respective gBK peptides. These CTLs failed to kill T2 cells loaded with the irrelevant influenza M1 peptide (Fig. 7). The gBK CTLs also killed the HLA-A2+ AGS gastric cancer, A549 adenocarcinoma lung cancer, and MCF-7 breast cancer cells, which are gBK+ (Fig. 9). Transfection of gBK− HEK cells with the gBK gene induced the expression of gBK (Fig. 3C), and more importantly now made the HEK cells susceptible to gBK2-specific CTLs (Fig. 10). Therefore, gBK represents a universal Ag that is expressed in many types of cancer cells, underscoring its broad therapeutic potential.
Using the gBK-specific sequence with the SYPEITHI analysis database, at least 17 other human MHC class I alleles have the predicted ability to bind gBK peptides with a score ≥16. Both gBK1or gBK2 peptides scored a 16 with HLA-A0201. The HLA-A03 allele had the ability to bind six gBK-specific peptides. The gBK region also is predicted to bind at least six different 15-mers to HLA-DR regions that can stimulate various CD4+ T cells. Thus, gBK peptides potentially can be used to treat many non–HLA-A2+ cancer patients as well. The gBK insert is always transcribed with another unique insert called hbr5. SYPEITHI analysis using the 29-aa hbr5 insert revealed another 13 possible human MHC class I and 5 MHC class II molecules theoretically capable of binding hbr5-specific peptides. Thus, the gBK/hbr5 sequence offers additional epitopes by which human T cell immune responses can be harnessed for immunotherapy against a variety of gBK+ tumor cell types.
If ion channels such as BK or other ion channels play a role in tumor cell invasiveness as they have been postulated (18–20), then targeting these membrane proteins might slow tumor cell invasion by interfering with a key mechanism by which the cells regulate their size/volume. It has been postulated that glioma cell size/volume allows these cells to squeeze through the brain parenchyma by shrinking its leading edge, perhaps at the filopodia region. Once the tips of the invadopodia shrink, they can penetrate between normal brain cells. Then these tips could swell and expand by gBK activation and help pull the remaining glioma cell through that narrow opening. BK channels have been described as being mechanosensitive; that is, when the membrane is stretched the BK channels become activated (40). Thus, this ionic regulation strategy can help explain some aspects of the motility of glioma cells. Even if only the high-expressing gBK glioma cells are killed by an immunotherapeutic approach, then the tumor cell population as a whole might be sufficiently reduced, thereby slowing down the tumor invasion. Targeting other ion channels, besides gBK channels, could include chloride channel-1 (CLIC1 or its other alias, CLCA1) or aquaporins. Both chloride channel-1 and aquaporins are also overexpressed in glioma (41–43). BK channel activation leads to cell swelling, whereas CLIC1 activation leads to cell shrinkage (7, 8, 44). Both of these functions, swelling and shrinking, are needed for cell volume regulation and motility. The BKβ-chain associates itself with the BKα-chain, but also heterodimerizes with this chloride ion channel and modulates CLIC1/CLCA1 activity (45). Therefore, attacking both ion channel regulatory pathways simultaneously could prove to produce synergistic effects and control the glioma cell infiltration within the brain. Thus, this immunotherapy may manage to slow down the glioma cell migration and invasion in the normal brain. Using the DCs and T cells from two human GBM patients, we generated CTLs that killed U251 cells as well as the allogeneic cells of low-passage glial tumor targets (UC-GBM1) (Fig. 10). Thus, this gBK Ag is expressed on freshly isolated and cultured GBM cells. We demonstrate that gBK-directed CTLs can be generated for GBM patients.
In summary, we find that gBK channels are genetically found within human malignant gliomas, either from surgical resections or from glioma cell lines. A gBK-specific Ab confirmed that the actual gBK sequence is produced as protein within human glioma cell lines and a number of other cancer cells. gBK-specific peptides provided sufficient stimulation to produce HLA-A2–restricted human CD8+ CTLs that killed gBK+ tumor cells. Thus, immunological targeting of tumor-associated ion channels maybe a novel way to target the infiltrative nature of malignant gliomas and perhaps other types of cancer.
Supplementary Material
Acknowledgments
We thank Dr. Michael Myers, National University (San Diego, CA), for previous useful discussions about BK ion channels. We also acknowledge the help of Dr. Ronald Kim (Veterans Affairs Medical Center–Long Beach and University of California, Irvine) for supplying the normal brain and GBM tissues that were used. We also acknowledge the help of Dr. Robert Prins (University of California at Los Angeles) who released the excess archived patient PBMCs and glioma cells for this study. Additionally, we thank Dr. Philip H. Schwartz for providing the human normal neurosphere cells, which were used as a negative control.
This work was supported by a Veterans Affairs Merit Review award (to M.R.J.) and by National Institutes of Health Grant R01 CA154256 (to C.A.K.).
The online version of this article contains supplemental material.
- BK
- big potassium
- DC
- dendritic cell
- ER
- endoplasmic reticulum
- gBK
- glioma big potassium
- GBM
- glioblastoma multiforme
- HEK
- human embryonic kidney
- nCTL
- normal donor CTL
- pCTL
- patient CTL
- rh
- recombinant human.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Rao R. V., Ellerby H. M., Bredesen D. E. 2004. Coupling endoplasmic reticulum stress to the cells death program. Cell Death Differ. 11: 372–380 [DOI] [PubMed] [Google Scholar]
- 2.Jambrina E., Alonso R., Alcalde M., del Carmen Rodríguez M., Serrano A., Martínez-A C., García-Sancho J., Izquierdo M. 2003. Calcium influx through receptor-operated channel induces mitochondria-triggered paraptotic cell death. J. Biol. Chem. 278: 14134–14145 [DOI] [PubMed] [Google Scholar]
- 3.Cassel D., Katz M., Rotman M. 1986. Depletion of cellular ATP inhibits Na+/H+ antiport in cultured human cells: modulation of the regulatory effect of intracellular protons on the antiporter activity. J. Biol. Chem. 261: 5460–5466 [PubMed] [Google Scholar]
- 4.Schneider D., Gerhardt E., Bock J., Müller M. M., Wolburg H., Lang F., Schulz J. B. 2004. Intracellular acidification by inhibition of the Na+/H+-exchanger leads to caspase-independent death of cerebellar granule neurons resembling paraptosis. Cell Death Differ. 11: 760–770 [DOI] [PubMed] [Google Scholar]
- 5.Sperandio S., de Belle I., Bredesen D. E. 2000. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 97: 14376–14381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bredesen D. E., Rao R. V., Mehlen P. 2006. Cell death in the nervous system. Nature 443: 796–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoa N. T., Zhang J. G., Delgado C. L., Myers M. P., Callahan L. L., Vandeusen G., Schiltz P. M., Wepsic H. T., Jadus M. R. 2007. Human monocytes kill M-CSF-expressing glioma cells by BK channel activation. Lab. Invest. 87: 115–129 [DOI] [PubMed] [Google Scholar]
- 8.Hoa N. T., Myers M. P., Douglass T. G., Zhang J. G., Delgado C., Driggers L., Callahan L. L., VanDeusen G., Pham J. T., Bhakta N., et al. 2009. Molecular mechanisms of paraptosis induction: implications for a non-genetically modified tumor vaccine. PLoS ONE 4: e4631 10.1371/journal.pone0004631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weaver A. K., Liu X., Sontheimer H. 2004. Role for calcium-activated potassium channels (BK) in growth control of human malignant glioma cells. J. Neurosci. Res. 78: 224–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ransom C. B., Liu X., Sontheimer H. 2002. BK channels in human glioma cells have enhanced calcium sensitivity. Glia 38: 281–291 [DOI] [PubMed] [Google Scholar]
- 11.Ransom C. B., Sontheimer H. 2001. BK channels in human glioma cells. J. Neurophysiol. 85: 790–803 [DOI] [PubMed] [Google Scholar]
- 12.Debska G., Kicinska A., Dobrucki J., Dworakowska B., Nurowska E., Skalska J., Dolowy K., Szewczyk A. 2003. Large-conductance K+ channel openers NS1619 and NS004 as inhibitors of mitochondrial function in glioma cells. Biochem. Pharmacol. 65: 1827–1834 [DOI] [PubMed] [Google Scholar]
- 13.Debska G., Kicińska A., Skalska J., Szewczyk A. 2001. Intracellular potassium and chloride channels: an update. Acta Biochim. Pol. 48: 137–144 [PubMed] [Google Scholar]
- 14.Abdullaev I. F., Rudkouskaya A., Mongin A. A., Kuo Y. H. 2010. Calcium-activated potassium channels BK and IK1 are functionally expressed in human gliomas but do not regulated cell proliferation. PLoS ONE 5: e12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korper S., Nolte F., Rojewski M. T., Thiel E., Schrezenmeier H. 2003. The K+ channel openers diazoxide and NS1619 induce depolarization of mitochondria and have differential effects on cell Ca2+ in CD34+ cell line KG-1a. Exp. Hematol. 31: 815–823 [DOI] [PubMed] [Google Scholar]
- 16.Spergel D. J., Catt K. J., Rojas E. 1996. Immortalized GnRH neurons express large-conductance calcium-activated potassium channels. Neuroendocrinology 63: 101–111 [DOI] [PubMed] [Google Scholar]
- 17.Grunnet M., Hay-Schmidt A., Klaerke D. A. 2005. Quantification and distribution of big conductance Ca2+-activated K+ channels in kidney epithelia. Biochim. Biophys. Acta 1714: 114–124 [DOI] [PubMed] [Google Scholar]
- 18.Sontheimer H. 2003. Malignant gliomas: perverting glutamate and ion homeostasis for selective advantage. Trends Neurosci. 26: 543–549 [DOI] [PubMed] [Google Scholar]
- 19.McFerrin M. B., Sontheimer H. 2006. A role for ion channels in glioma cell invasion. Neuron Glia Biol. 2: 39–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sontheimer H. 2008. An unexpected role for ion channels in brain tumor metastasis. Exp. Biol. Med. (Maywood) 233: 779–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adelman J. P., Shen K. Z., Kavanaugh M. P., Warren R. A., Wu Y. N., Lagrutta A., Bond C. T., North R. A. 1992. Calcium-activated potassium channels expressed from cloned complementary DNAs. Neuron 9: 209–216 [DOI] [PubMed] [Google Scholar]
- 22.Tseng-Crank J., Foster C. D., Krause J. D., Mertz R., Godinot N., DiChiara T. J., Reinhart P. H. 1994. Cloning, expression, and distribution of functionally distinct Ca2+-activated K+ channel isoforms from human brain. Neuron 13: 1315–1330 [DOI] [PubMed] [Google Scholar]
- 23.Shipston M. J. 2001. Alternative splicing of potassium channels: a dynamic switch of cellular excitability. Trends Cell Biol. 11: 353–358 [DOI] [PubMed] [Google Scholar]
- 24.Chen L., Tian L., MacDonald S. H. F., McClafferty H., Hammond M. S. L., Huibant J. M., Ruth P., Knaus H. G., Shipston M. J. 2005. Functionally diverse complement of large conductance calcium- and voltage-activated potassium channel (BK) α-subunits generated from a single site of splicing. J. Biol. Chem. 280: 33599–33609 [DOI] [PubMed] [Google Scholar]
- 25.Liu X., Chang Y., Reinhart P. H., Sontheimer H., Chang Y. 2002. Cloning and characterization of glioma BK, a novel BK channel isoform highly expressed in human glioma cells. J. Neurosci. 22: 1840–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwartz P. H., Bryant P. J., Fuja T. J., Su H., O’Dowd D. K., Klassen H. 2003. Isolation and characterization of neural progenitor cells from post-mortem human cortex. J. Neurosci. Res. 74: 838–851 [DOI] [PubMed] [Google Scholar]
- 27.Zhang J. G., Eguchi J., Kruse C. A., Gomez G. G., Fakhrai H., Schroter S., Ma W., Hoa N., Minev B., Delgado C., et al. 2007. Antigenic profiling of glioma cells to generate allogeneic vaccines or dendritic cell-based therapeutics. Clin. Cancer Res. 13: 566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ge L., Cornforth A. N., Hoa N. T., Delgado C., Zhou Y. H., Jadus M. R. Differential glioma-associated tumor antigen expression profiles of human glioma cells grown in hypoxia. PLoS One. 2011 doi: 10.1371/journal.pone.0042661. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parkhurst M. R., Salgaller M. L., Southwood S., Robbins P. F., Sette A., Rosenberg S. A., Kawakami Y. 1996. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J. Immunol. 157: 2539–2548 [PubMed] [Google Scholar]
- 30.Trojan A., Witzens M., Schultze J. L., Vonderheide R. H., Harig S., Krackhardt A. M., Stahel R. A., Gribben J. G. 2001. Generation of cytotoxic T lymphocytes against native and altered peptides of human leukocyte antigen-A*0201 restricted epitopes from the human epithelial cell adhesion molecule. Cancer Res. 61: 4761–4765 [PubMed] [Google Scholar]
- 31.Salter R. D., Cresswell P. 1986. Impaired assembly and transport of HLA-A and -B antigens in a mutant T×B cell hybrid. EMBO J. 5: 943–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coiret G., Borowiec A. S., Mariot P., Ouadid-Ahidouch H., Matifat F. 2007. The antiestrogen tamoxifen activates BK channels and stimulates proliferation of MCF-7 breast cancer cells. Mol. Pharmacol. 71: 843–851 [DOI] [PubMed] [Google Scholar]
- 33.Bloch M., Ousingsawat J., Simon R., Schraml P., Gasser T. C., Mihatsch M. J., Kunzelmann K., Bubendorf L. 2007. KCNMA1 gene amplification promotes tumor cell proliferation in human prostate cancer. Oncogene 26: 2525–2534 [DOI] [PubMed] [Google Scholar]
- 34.Basrai D., Kraft R., Bollensdorff C., Liebmann L., Benndorf K., Patt S. 2002. BK channel blockers inhibit potassium-induced proliferation of human astrocytoma cells. Neuroreport 13: 403–407 [DOI] [PubMed] [Google Scholar]
- 35.Bordey A., Sontheimer H., Trouslard J. 2000. Muscarinic activation of BK channels induces membrane oscillations in glioma cells and leads to inhibition of cell migration. J. Membr. Biol. 176: 31–40 [DOI] [PubMed] [Google Scholar]
- 36.Kraft R., Krause P., Jung S., Basrai D., Liebmann L., Bolz J., Patt S. 2003. BK channel openers inhibit migration of human glioma cells. Pflügers Arch. J. Physiol. 446: 248–255 [DOI] [PubMed] [Google Scholar]
- 37.Bentzen B. H., Osadchii O., Jespersen T., Hansen R. S., Olesen S. P., Grunnet M. 2009. Activation of big conductance Ca2+-activated K+ channels (BK) protects the heart against ischemia-reperfusion injury. Pflugers Arch. 457: 979–988 [DOI] [PubMed] [Google Scholar]
- 38.Hewawasam P., Ding M., Chen N., King D., Knipe J., Pajor L., Ortiz A., Gribkoff V. K., Starrett J. 2003. Synthesis of water-soluble prodrugs of BMS-191011: a maxi-K channel opener targeted for post-stroke neuroprotection. Bioorg. Med. Chem. Lett. 13: 1695–1698 [DOI] [PubMed] [Google Scholar]
- 39.Nardi A., Olesen S. P. 2008. BK channel modulators: a comprehensive overview. Curr. Med. Chem. 15: 1126–1146 [DOI] [PubMed] [Google Scholar]
- 40.Zhao H., Sokabe M. 2008. Tuning the mechanosensitivity of a BK channel by changing the linker length. Cell Res. 18: 871–878 [DOI] [PubMed] [Google Scholar]
- 41.Ransom C. B., O’Neal J. T., Sontheimer H. 2001. Volume-activated chloride currents contribute to the resting conductance and invasive migration of human glioma cells. J. Neurosci. 21: 7674–7683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayashi Y., Edwards N. A., Proescholdt M. A., Oldfield E. H., Merrill M. J. 2007. Regulation and function of aquaporin-1 in glioma cells. Neoplasia 9: 777–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ernest N. J., Weaver A. K., Van Duyn L. B., Sontheimer H. 2005. Relative contribution of chloride channels and transporters to regulatory volume decrease in human glioma cells. Am. J. Physiol. Cell. Physiol. 288: C1451–C1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colman H., Zhang L., Sulman E. P., McDonald J. M., Shooshtari N. L., Rivera A., Popoff S., Nutt C. L., Louis D. N., Cairncross J. G., et al. 2010. A multigene predictor of outcome in glioblastoma. Neuro-oncol. 12: 49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greenwood I. A., Miller L. J., Ohya S., Horowitz B. 2002. The large conductance potassium channel β-subunit can interact with and modulate the functional properties of a calcium-activated chloride channel, CLCA1. J. Biol. Chem. 277: 22119–22122 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.