

Abstract
The Mycobacterium tuberculosis enhanced intracellular survival (Eis_Mtb) protein is a clinically important aminoglycoside (AG) multi-acetylating enzyme. Eis homologues are found in a variety of mycobacterial and non-mycobacterial species. Variation of the residues lining the AG-binding pocket and positions of the loops bearing these residues in the Eis homologues dictates the substrate specificity and, thus, Eis homologues are Nature-made tools for elucidating principles of AG recognition by Eis. Here, we demonstrate that the Eis from Anabaena variabilis (Eis_Ava), the first non-mycobacterial Eis homologue reported, is a multi-acetylating AG-acetyltransferase. Eis_Ava, Eis from Mycobacterium tuberculosis (Eis_Mtb), and Eis from Mycobacterium smegmatis (Eis_Msm) have different structures of their AG-binding pockets. We perform comparative analysis of these differences and investigate how they dictate the substrate and cosubstrate recognition and acetylation of AGs by Eis.
Introduction
Upregulation of the enzyme Eis from Mycobacterium tuberculosis (Eis_Mtb) has been shown to be responsible for kanamycin A (KAN) resistance in a large fraction of KAN-resistant clinical isolates from tuberculosis patients around the globe.1,2 We recently reported the unique and unprecedented ability of Eis_Mtb3 and of its homologue from Mycobacterium smegmatis (Eis_Msm)4 to multi-acetylate a variety of aminoglycosides (AGs). Even though Eis_Mtb and Eis_Msm are structurally very similar,5 we identified differences in the substrate recognition by these two Eis homologues and in their inhibition.4 Di-acetylation of the rigid fused-ring AGapramycin (APR)was observed with Eis_Msm, but APR was found to not be a substrate of Eis_Mtb. Moreover, the potency of common inhibitors of these two Eis proteins varied between the two homologues. These data were consistent with structural differences between the AG-binding cavities of the two enzymes.
Due to the rising threat of mycobacterial infection from non-tuberculosis mycobacteria and the ever-growing emergence of resistance to AG antibiotics, it is important to extend the analysis of Eis homologues beyond Eis_Mtb and Eis_Msm to aid in the understanding of Eis substrate recognition principles across the bacterial kingdom.
As a focus of this study, we chose a putative GCN5-related-N-acetyltransferase (GNAT) from the cyanobacterium Anabaena variabilis ATCC 29413, (termed Eis_Ava herein) structurally similar to Eis_Mtb. A crystal structure of Eis_Ava was deposited into the Protein Data Bank (PDB) by the Joint Center for Structural Genomics (PDB code: 2OZG). In this work, to gain insight into the structural requirements dictating substrate and cosubstrate profile as well as the inhibition of Eis proteins, we performed structural and biochemical comparative studies of Eis_Ava with Eis_Mtb and Eis_Msm. Cyanobacteria do not cause any clinical infections that are treated with AGs. However, the A. variabilis ATCC 29413 strain was isolated from a sewage oxidation pond,6 and the development of resistance to AGs through water contamination has been previously documented in various bacterial species.7 Thus, exposure to AGs through sewage waste provides a potential explanation as to why a putative AG-acetylating enzyme, Eis_Ava, has arisen in this cyanobacterial strain.
Results and discussion
In silico analysis of Eis homologues across 29 bacterial species
To evaluate the evolutionary relationship among the 3 Eis homologues of interest (Eis_Ava, Eis_Mtb, and Eis_Msm), we first performed a phylogenetic tree analysis using 29 Eis homologue sequences from a variety of mycobacterial and non-mycobacterial species (Fig. 1). In general, the mycobacterial and non-mycobacterial Eis homologues can be divided into two distinct clades, with Eis_Ava and Eis_Mtb being two of the most evolutionary distinct proteins in this set. Interestingly, the Eis homologue from Mycobacterium abscessus (Eis_Mab) is clustered with the Eis homologues from the non-mycobacterial clade instead of the expected mycobacterial group. The genetic proximity of Eis_Ava to Eis_Mab further highlights the importance of studying non-mycobacterial enzymes such as Eis_Ava to understand the mycobacterial AG-acetylation resistance mechanisms, due to the current increase in drug-resistant Mab infections.8 In the future, the information learned about Eis_Ava could additionally be applied to problematic bacterial species closely related to Ava that also contain Eis, such as Enterococcus faecalis (Efa).
Fig. 1.

Phylogenetic tree analysis of 29 Eis homologues. The numbers are proportional to the evolutionary distance between the branches of the tree. The Eis proteins discussed in this manuscript are in bold. The abbreviated names of the bacterial species correspond to the following: Anabaena variabilis ATCC 29413 (Ava), Bacillus anthracis str. Sterne (Ban), Enterococcus faecalis V583 (Efa), Mycobacterium abscessus ATCC 19977 (Mab), Streptomyces venezuelae ATCC 10712 (Sve), Streptomyces albus J1074 (Sal), Streptomyces coelicolor A3(2) (Sco), Kocuria rhizophila DC2201 (Krh), Brevibacterium mcbrellneri ATCC 49030 (Bmc), Tsukamurella paurometabola ATCC 8368 (Tpa), Gordonia bronchialis DSM 43247 (Gbr), Mycobacterium parascrofulaceum ATCC BAA-614 (Mpa), Mycobacterium intracellulare ATCC 13950 (Min), Mycobacterium avium subsp. avium ATCC 25291 (Mav), Mycobacterium phlei RIVM601174 (Mph), Mycobacterium vanbaalenii PYR-1 (Mva), Mycobacterium gilvum Spyr1 (Mgi1), Mycobacterium gilvum PYR-GCK (Mgi2), Mycobacterium sp. JDM601 (Msp1), Mycobacterium smegmatis str. MC2 155 (Msm), Mycobacterium sp. JLS (Msp2), Mycobacterium sp. MCS (Msp3), Mycobacterium kansasii ATCC 12478 (Mka), Mycobacterium marinum M (Mma), Mycobacterium canettii CIPT 140010059 (Mca), Mycobacterium bovis BCG str. Pasteur 1173P2 (Mbo), Mycobacterium tuberculosis SUMu002 (Mtb1), Mycobacterium tuberculosis H37Rv (Mtb2), and Mycobacterium tuberculosis T17 (Mtb3).
Structural comparison of Eis homologues
Eis_Ava, Eis_Mtb, and Eis_Msm all share a tripartite monomer structure containing N-terminal and central GNAT regions and a similar homo-hexameric organization.3,5 By performing structure-based sequence alignment of these three Eis homologues, we observed that Eis_Ava greatly differs in amino acid composition when compared to Eis_Mtb (18% sequence identity) and Eis_Msm (19% sequence identity; Fig. 2). In contrast, Eis_Mtb and Eis_Msm are more similar (53% identical). In all 3 Eis proteins, the key catalytic residue involved in catalysis (Tyr125 in Eis_Ava) (Fig. 2, yellow circles/squares) is strictly conserved, and those involved in AcCoA binding (Fig. 2, orange circles/squares) are highly conserved. However, the amino acid residues predicted to bind AGs through site-directed mutagenesis studies of Eis_Mtb3 and structure analysis of Eis_Mtb and Eis_Msm4 are highly divergent between Eis_Ava and Eis_Mtb/Eis_Msm (Fig. 2, red squares). Taken together, these observations demonstrate the evolutionary divergence between the Eis proteins from mycobacterial and non-mycobacterial species observed in Fig. 1.
Fig. 2.

Structure-based sequence alignment of Eis_Ava from A. variabilis ATCC 29413, Eis_Msm from M. smegmatis str. MC2 155, and Eis_Mtb from M. tuberculosis H37Rv generated using Secondary-structure matching (SSM).9 Residues in bold red in blue boxes are conserved between the 3 Eis homologues. The circles above and the squares below the Eis_Ava and Eis_Mtb sequences, respectively, correspond to important residues in these sequences. Based on structural and mutagenesis studies of Eis_Mtb, the residues proposed to be involved in catalysis, in AcCoA binding, and in the formation of the AG-binding pocket are marked by yellow, orange, and red circles/squares, respectively.3 The AG-binding pocket of Eis_Mtb is divided into two channels. Residues lining these two channels are marked by green and turquoise circles/squares. The * and the + symbols indicate residues that structurally aligned when superimposing the crystal structures of Eis_Ava and Eis_Mtb.
Even though Eis_Ava greatly differs in amino acid composition from both Eis_Mtb and Eis_Msm, its structure is generally very similar to those of Eis_Mtb and Eis_Msm (Fig. 3A and B). To explore the structural similarities and differences among the 3 studied Eis proteins and their relationship to function, we compared the structures of their substrate-binding cavities (Fig. 3C–E) and observed striking differences. We previously reported that the AG-binding site of Eis_Mtb is divided into two distinct narrow channels (highlighted in green and blue in Fig. 3E), while the AG-binding pocket of Eis_Msm consists of one wide and open cavity (Fig. 3D), as a result of the small amino acid side-chains of the residues lining one of the two Eis_Msm channels (blue channel of Eis_Msm: Ala289, Ala287, Gly266, and Gly401; corresponding residues in Eis_Mtb: Gln291, Trp289, Ile268, and Glu401). As a result of the broadening of its AG-binding site relative to that in Eis_Mtb, Eis_Msm can accommodate the structurally rigid APR AG while Eis_Mtb cannot.4 Interestingly, the substrate-binding cavity of Eis_Ava is divided into two distinct channels, as in the case of Eis_Mtb, but it differs structurally. As in Eis_Mtb, the blue channel of Eis_Ava composed of Asp283, Ser281, Lys261, and Phe394 (corresponding residues in Eis_Mtb: Gln291, Trp289, Ile268, and Glu401) (Fig. 2, blue circles/squares and Fig. 3C and E) is narrow. However, the green channel of Eis_Ava is much larger than that of Eis_Mtb and could potentially accept APR as a substrate as does Eis_Msm. Different positions of the loop between the α1 and α2-helices and the β13-sheet in Eis_Ava and Eis_Msm (Fig. 3B) are also in part responsible for the broadening of the substrate-binding cavity of these two Eis homologues relative to that in Eis_Mtb.
Fig. 3.

Structural comparison of Eis homologues from Ava, Msm, and Mtb as well as molecular models of Eis–APR–AcCoA complexes. (A) Structural alignment of Eis_Ava (PDB: 2OZG) with Eis_Msm (PDB: 3SXN) (the alignment of one of the monomers of the hexameric structures is shown). (B) Structural alignment of Eis_Ava (PDB: 2OZG) with Eis_Mtb (PDB: 3R1K) (the alignment of one of the monomers of the hexameric structures is shown). The active site of (C) Eis_Ava, (D) Eis_Msm, and (E) Eis_Mtb with residues lining the two channels (green and blue) of the AG-binding pocket highlighted. Surface representation of the Eis monomer active sites of (F) Eis_Ava, (G) Eis_Msm, and (H) Eis_Mtb colored according to their electrostatic potential, positive in blue, negative in red, and hydrophobic in white. (I) A model of AcCoA and APR bound to Eis_Ava. (J) A model of AcCoA and APR bound to Eis_Msm. (K) Structure of APR.
Multi-acetylation, substrate, and cosubstrate specificity profiles of Eis_Ava
We recently reported the substrate and multi-acetylation profiles of Eis_Mtb and Eis_Msm, demonstrating the unique ability of the Eis enzymes to multi-acetylate many AGs.3,4 To investigate a potential effect of the broadening of the substrate-binding pocket of Eis_Ava on its substrate specificity profile, we monitored the acetylation by Eis_Ava of 11 AGs: amikacin (AMK), APR, KAN, neamine (NEA), neomycin B (NEO), netilmicin (NET), paromomycin (PAR), ribostamycin (RIB), sisomicin (SIS), spectinomycin (SPT), and streptomycin (STR) (Table 1; Fig. S1 and S3, ESI†), by UV-Vis and mass spectrometry assays. To ensure the validity of the comparisons made in this work, we cloned and overexpressed Eis_Ava in an identical fashion to Eis_Mtb and Eis_Msm, which we previously reported (Fig. S2, ESI†). As predicted, based on the structural homology with other Eis proteins, Eis_Ava is a functional AG-acetyltransferase. As we previously observed with Eis_Mtb and Eis_Msm, we found SPT and STR to not be substrates of Eis_Ava. By mass spectrometry, we observed that the number of acetylated positions on AMK, NEA, NEO, NET, and RIB by the 3 Eis homologues studied was the same for the 3 enzymes. With other AGs, such as KAN and SIS, we found the number of acetylations with Eis_Ava to be less than those observed with Eis_Mtb and Eis_Msm, which were equal for any given AG. Of particular note is the tri-acetylation of PAR by Eis_Ava and Eis_Msm, in contrast to only di-acetylation of PAR by Eis_Mtb. This result suggests that the broader substrate-binding cavities of Eis_Ava (Fig. 3C and F) and Eis_Msm (Fig. 3D and G) accommodate the large PAR molecule in more orientations than Eis_Mtb does. In addition, we found APR to be mono-acetylated by Eis_Ava, which corroborated the previously proposed role of the expanded substrate-binding cavity in binding this rigid extended AG. The presence of two channels (one narrow (blue) and one large (green) in Fig. 3C) in the substrate-binding site of Eis_Ava versus a single AG-binding pocket in Eis_Msm that is larger than the green channel of Eis_Ava (Fig. 3D) could explain the mono-acetylation and di-acetylation of APR observed with Eis_Ava and Eis_Msm, respectively.
Table 1.
Comparison of number of acetylations of various AGs by Eis homologues from A. variabilis, M. tuberculosis, and M. smegmatis
To explore how APR may be accommodated in the binding pocket of Eis_Ava for acetylation, we performed docking of APR to Eis_Ava and Eis_Msm in complex with AcCoA (Fig. 3I and J). The shorter loop between the α1 and α2-helices of Eis_Ava that is responsible for the expansion of the green channel of this enzyme when compared to its counterpart in Eis_Msm (Fig. 3B, I, and J) could account for the different conformations adopted by APR in the AG-binding site and the different number of acetylations of APR by these two Eis enzymes, mono- versus di-acetylation by Eis_Ava and Eis_Msm, respectively (Table 1). Taken together, the structural analysis of the substrate-binding cavities of Eis homologues and their multi-acetylation profiles revealed that Eis_Ava behaves more similarly to Eis_Msm than to Eis_Mtb.
In addition to studying the AG substrate promiscuity of Eis_Ava, we also investigated its cosubstrate profile. We recently showed that Eis_Mtb is limited in its cosubstrates, efficiently accepting only AcCoA and n-propionyl-CoA and moderately so crotonyl-CoA and malonyl-CoA for transfer onto a few AGs.10 We determined that among the 6 acyl-CoA cosubstrates tested with Eis_Ava (AcCoA, butyryl-CoA, crotonyl-CoA, malonyl-CoA, myristoyl-CoA, and n-propionyl-CoA), only the natural AcCoA cosubstrate and its close structural homologue n-propionyl-CoA were accepted by the cyanobacterial Eis.
Steady-state kinetic parameters for Eis_Ava
To further understand the action of Eis_Ava and the subtleties differentiating the 3 investigated Eis homologues, we performed steady-state kinetic measurements of AG acetylation by Eis_Ava and compared the Michaelis–Menten parameters, Km and kcat, to those of Eis_Mtb and Eis_Msm (Table 2 and Fig. S4, ESI†). In this study, we determined all kinetic parameters (for Eis_Ava, Eis_Mtb, and Eis_Msm) for 500 μM AcCoA, in a range of 0–2500 μM AG, and 0.25 μM Eis. In a previous report, we determined and compared the Km and kcat values for Eis_Mtb and Eis_Msm using 100 μM AcCoA, 0–2500 μM AG, and 0.25 μM Eis.4 AcCoA was used at a higher concentration in this study to ensure that all enzymes were bound to AcCoA thereby allowing a direct interpretation of kinetics as a property of this holo-Eis. For APR, a substrate of Eis_Ava and Eis_Msm, but not of Eis_Mtb, we found that Km and kcat values are very similar (337 ± 98 μM and 0.019 ± 0.002 s−1 for Eis_Ava and of 150 ± 43 μM and 0.019 ± 0.002 s−1 for Eis_Msm). For KAN and NEO, the Km and kcat values for acetylation by Eis_Ava were also more similar to those of Eis_Msm than to those of Eis_Mtb. Overall, the kinetic parameters of Eis_Ava appear to be most similar to those of Eis_Msm, which can be explained by the larger substrate-binding cavities of these two enzymes compared to the divided AG-binding site of Eis_Mtb. When determining kinetic parameters by varying AG concentrations at a constant concentration of AcCoA, apparent kcat and Km are, generally, functions of the concentration of AcCoA and, therefore, they are not intrinsic microscopic mechanistic parameters. To determine the true microscopic turnover rate constant for Eis_Ava, we performed three sets of experiments varying the concentration of KAN and keeping the concentration of AcCoA constant at 100, 200, or 500 μM. From these experiments, we obtained kcat of Eis_Ava to be 0.24 ± 0.04 s−1, similar to the apparent kcat value measured at [AcCoA] = 0.5 mM. This means that this concentration of AcCoA is saturating in the kinetic sense and, unless binding of an AG very strongly disfavours subsequent binding of AcCoA (an unlikely scenario), the apparent kcat values for the other AGs that are given in Table 2 to a good approximation should represent microscopic kcat values.
Table 2.
Apparent steady-state kinetic parameters for AG concentration-dependent acetylation by Eis homologues from A. variabilis, M. smegmatis, and M. tuberculosis
| Eis_Ava
| |||
|---|---|---|---|
| AG | Km/μM | kcat/s−1 | kcat/Km/M−1 s−1 |
| APR | 337 ± 98 | 0.019 ± 0.002 | 57 ± 18 |
| KAN | 1002 ± 43 | 0.205 ± 0.004 | 205 ± 9 |
| NEA | 42 ± 9 | 0.352 ± 0.021 | 7413 ± 1938 |
| NEO | 101 ± 16 | 0.106 ± 0.006 | 1052 ± 184 |
| PAR | 237 ± 36 | 0.183 ± 0.013 | 773 ± 132 |
| Eis_Msm | |||
| APR | 150 ± 43 | 0.019 ± 0.002 | 127 ± 39 |
| KAN | 665 ± 42 | 0.360 ± 0.010 | 541 ± 37 |
| NEA | 1302 ± 312 | 0.687 ± 0.083 | 528 ± 142 |
| NEO | 110 ± 14 | 0.148 ± 0.006 | 1345 ± 180 |
| PAR | 738 ± 158 | 0.235 ± 0.033 | 318 ± 82 |
| Eis_Mtb | |||
| APR | —a | — | — |
| KAN | 330 ± 40 | 0.526 ± 0.027 | 1594 ± 250 |
| NEA | 1315 ± 492 | 0.699 ± 0.136 | 532 ± 224 |
| NEO | 122 ± 23 | 0.610 ± 0.029 | 5000 ± 972 |
| PAR | 110 ± 21 | 0.136 ± 0.012 | 1236 ± 260 |
—Indicates that APR is not a substrate for Eis_Mtb.
Regio-specificity of NEA di-acetylation by Eis_Ava
We previously demonstrated that Eis_Mtb3 and Eis_Msm4 sequentially tri-acetylate NEA at the 2′-, 6′-, and 1-position. Seeking a deeper understanding of the differences and similarities between the mycobacterial and non-mycobacterial Eis proteins, we sought to compare the regio-specificity and order of multi-acetylation of NEA by Eis_Ava to those by Eis_Mtb/Eis_Msm. Interestingly, by thin-layer chromatography (TLC) and NMR spectroscopy (1H, gCOSY, zTOCSY, and gHSQC), we could only observe di-acetylation of NEA by Eis_Ava (Fig. 4; Fig. S5–S12 and Table S1, ESI†). Remarkably, we also established that the first position of the NEA scaffold acetylated was the 2′-position, as previously observed with Eis_Mtb and Eis_Msm, but that the second site modified was the 1-position instead of the expected 6′-position. It is important to note that, as established by mass spectrometry, a third position can be acetylated by Eis_Ava on the NEA scaffold. The change in order of the position acetylated by Eis_Ava could potentially explain why for this enzyme we did not observe by UV-Vis and mass spectrometry assays a third acetylation, which could proceed at the much slower rate. These data suggest that the order and number of acetylations may vary for each AG based on the Eis homologue utilized. Further studies aimed at establishing the number and positions of acetylation on multiple AGs by a variety of Eis homologues are currently underway in our laboratory.
Fig. 4.

Formation of 1,2′-di-acetyl-NEA by Eis_Ava monitored by a TLC assay and confirmed by NMR spectroscopy. Lanes 1–6: a time course showing the mono- and di-acetyl-NEA products of the Eis_Ava reaction. Lanes 7–12: controls for mono- and di-acetylation of NEA by AAC(2′)-Ic, AAC(3)-IV, and AAC(6′)-Ie used individually or sequentially.
Inhibition of Eis_Ava, Eis_Msm, and Eis_Mtb
We recently reported the discovery and characterization of Eis_Mtb inhibitors,11 which we also showed in some cases to inhibit Eis_Msm.4 As Eis homologues are found in a variety of bacterial strains (Fig. 1), including emerging resistant species such as Mab, the importance of finding Eis inhibitors that efficiently work across various species is increasing with the number of resistant bacterial species that harbour Eis homologues continuously rising. To perform preliminary exploration of the potential of the identified Eis_Mtb inhibitors against Eis homologues from various bacterial species, we tested 5 Eis_Mtb inhibitors against Eis_Ava and Eis_Msm (Fig. 5 and Table 3). For chlorhexidine (1), the IC50 value of 20 ± 7 μM observed for Eis_Ava was 107-fold higher than that for Eis_Mtb (188 ± 30 nM) and 11-fold higher than that against Eis_Msm (1.85 ± 0.39 μM). For compounds 2 and 3, the IC50 values were comparable for both mycobacterial strains. Interestingly, for compound 4, the IC50 value of 455 ± 92 nM observed for Eis_Msm was 4.4-fold lower than that for Eis_Mtb (2.01 ± 0.12 μM). The poor inhibition of Eis_Ava by all 5 inhibitors (IC50 values of 20 ± 7 μM for compound 1 and >200 μM for compounds 2–5) tested could be attributed to the electrostatics of its substrate-binding cavity, which is more hydrophobic (Fig. 3F) than in Eis_Mtb (Fig. 3H) and in Eis_Msm (Fig. 3G). For example, the hydrophobicMet32 and Pro185 residues of the substrate-binding pocket in Eis_Ava replace the structurally homologous negatively charged Asp28 and Asp195 in Eis_Msm. Therefore, to assess the utility of discovered Eis_Mtb inhibitors on new potential Eis homologues, a thorough analysis of the substrate-binding pocket residues is needed.
Fig. 5.

Structures of inhibitors tested against Eis_Ava, Eis_Msm, and Eis_Mtb.
Table 3.
Inhibition of Eis homologues from A. variabilis, M. smegmatis, and M. tuberculosis by compounds 1–5 for NEO acetylation
| Compound | IC50/μM
|
||
|---|---|---|---|
| Eis_Ava | Eis_Msm | Eis_Mtba | |
| 1 | 20 ± 7 | 1.85 ± 0.39 | 0.188 ± 0.030 |
| 2 | >200 | 0.925 ± 0.201 | 1.09 ± 0.14 |
| 3 | >200 | 1.69 ± 0.32 | 1.24 ± 0.16 |
| 4 | >200 | 0.455 ± 0.092 | 2.01 ± 0.12 |
| 5 | >200 | >200 | 2.29 ± 0.52 |
These data were previously reported.11
Reduced activity of Eis homologues towards 6′-glycinyl-KAN
The development of AGs capable of avoiding acetylation by Eis enzymes could present an alternative strategy to the discovery of Eis inhibitors to combat the Eis resistance problem. We recently reported a simple chemical approach for the production of biologically active 6′-N-acylated AG derivatives.12 To explore the potential of these acylated AGs to evade Eis multi-acetylation, we tested 6′-glycinyl-KAN against Eis_Ava, Eis_Msm, and Eis_Mtb (Fig. 6). We observed that in contrast to KAN that efficiently gets multi-acetylated by all Eis homologues tested (blue, red, and green circles in Fig. 6), 6′-glycinyl-KAN is not a substrate for these enzymes (blue, red, and green triangles in Fig. 6). These data demonstrate the potential of developing novel AG derivatives towards combating AG acetyltransferase enzymes.
Fig. 6.
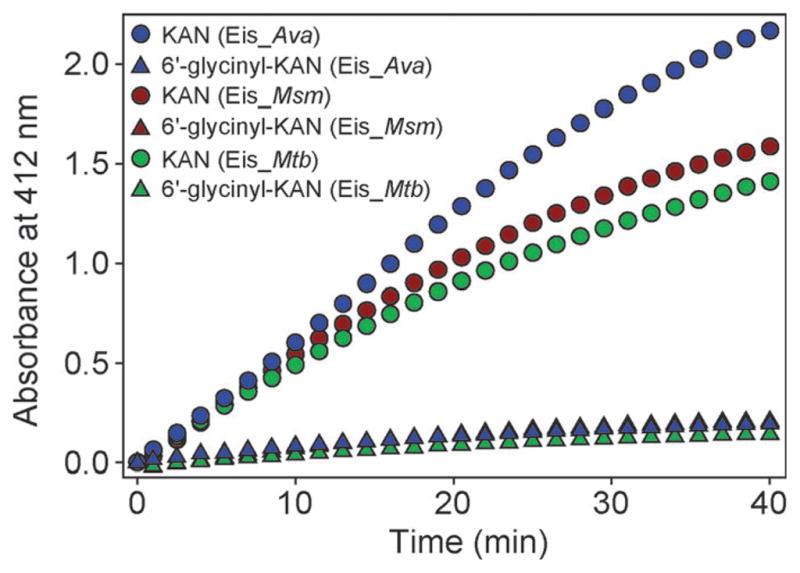
UV-Visible spectrum showing the activity of Eis_Ava (blue), Eis_Msm (red), and Eis_Mtb (green) against KAN (circles) and 6′-glycinyl-KAN (triangles). All Eis homologues are almost inactive against 6′-glycinyl-KAN (as a result the red triangles are completely hidden behind the blue triangles).
Conclusions
In this work, we refined our understanding of the details of the substrate recognition for multi-acetylation of AGs by Eis homologues from mycobacterial and non-mycobacterial species. We also explored the applicability of Eis_Mtb inhibitors to the inactivation of Eis homologues from other bacterial species. From these studies we concluded that (i) the substrate-binding cavities of Eis homologues vary greatly even though the overall structures of the enzymes are highly comparable, (ii) Eis_Ava behaves more similarly to Eis_Msm than to Eis_Mtb, (iii) the order of acetylation for a given AG can vary for different Eis homologues, (iv) inhibitors of Eis_Mtb could be applied with varying degrees of efficiency against bacterial strains containing Eis homologues, with higher potency against mycobacteria, and (v) glycinylation of the 6′-amine of KAN reduces the activity of Eis homologues. Further studies are underway in our laboratory aimed at establishing the positions acetylated by Eis homologues on a number of AG scaffolds. Structure activity relationship studies towards the development of Eis inhibitors with high potency and broad application are also ongoing.
Experimental
Bacterial strains, plasmids, materials, and instrumentation
Chemically competent Escherichia coli TOP10 and BL21 (DE3) strains were purchased from Invitrogen (Carlsbad, CA). A. variabilis ATCC 29413 genomic DNA was purchased from the American Type Culture Collection (ATCC). The pET28a plasmid used in this study was purchased from Novagen (Gibbstown, NJ). Primers used for PCR were purchased from Integrated DNA Technologies (Coralville, IA). All reagents and enzymes used for cloning, including restriction enzymes, Phusion DNA polymerase, and T4 DNA ligase, were purchased from New England Biolabs (Ipswich, MA). DNA sequencing was performed at the University ofMichigan DNA Sequencing Core. Dithionitrobenzoic acid (DTNB), AcCoA, acyl-CoAs (butyryl-CoA, crotonyl-CoA, malonyl-CoA, myristoyl-CoA, and n-propionyl-CoA), and aminoglycosides (AGs) (AMK, APR, KAN, NEO, RIB, SIS, SPT, and STR; Fig. S1, ESI†) were purchased from Sigma-Aldrich (Milwaukee, WI) and used without further purification. The remaining AGs (NEA, NET, and PAR; Fig. S1, ESI†) were purchased from AK Scientific (Mountain View, CA). Eis_Mtb and Eis_Msm were purified as previously described using the pEis_Mtb–pET28a3 and pEis_Msm–pET28a4 constructs, respectively. All UV-Vis spectrophotometric assays were performed in 96-well plates (Fisher Scientific; Pittsburg, PA) using a multimode Spectra-Max M5 plate reader. A Shimadzu LCMS-2019EV equipped with a SPD-20AV UV-Vis detector and a LC-20AD liquid chromatograph was used for all liquid chromatography mass spectrometry (LCMS) measurements. The PDB structures 3R1K (Eis_Mtb), 3SXN (Eis_Msm), and 2OZG (Eis_Aa) were analyzed using Coot13 and PyMOL (The PyMOL Molecular Graphics System, Version 1.4.1, Schrödinger, LLC).
Phylogenetic tree creation for Eis homologues
The amino acid sequences of 29 Eis homologues were retrieved from NCBI through the Geneious Pro 4.8.5 program. The phylogenetic tree (Fig. 1) was built by neighbor-joining using the Geneious Pro 4.8.5 program with the following parameters: Jukes–Cantor genetic distance and the cost matrix Blosum45. The NCBI accession numbers for the Eis homologues were YP_325469 (Ava), YP_029001 (Ban), NP_814755 (Efa), YP_001705255 (Mab), CCA55694 (Sve), ZP_06590713 (Sal), NP_628362 (Sco), YP_001855041 (Krh), ZP_06804610 (Bmc), YP_003645809 (Tpa), YP_003272441 (Gbr), ZP_06852004 (Mpa), ZP_05228133 (Min), ZP_05216001 (Mav), ZP_09975446 (Mph), YP_956800 (Mva), YP_004079667 (Mgi1), YP_001132138 (Mgi2), YP_004522750 (Msp1), YP_887817 (Msm), YP_001071002 (Msp2), YP_639864 (Msp3), ZP_04749484 (Mka), YP_001852011 (Mma), YP_004745879 (Mca), YP_978521 (Mbo), ZP_07418795 (Mtb1), CAB03742 (Mtb2), and ZP_06450792 (Mtb3).
Preparation of pEis_Ava–pET28a overexpression construct
PCR for the amplification of the homologous eis gene from A. variabilis was performed using Phusion DNA polymerase according to instructions provided by NEB. A. variabilis ATCC 29413 (Ava) genomic DNA was used as the template for PCR with forward primer 5′-GTGCTTCATATGGTAGAACCAATGAC-3′ and reverse primer 5′-CTATGCCTCGAGTTAAAAGAAATCAATC-3′. The amplified PCR product, eis_Ava, was inserted into a linearized pET28a vector between NdeI and XhoI restriction sites. The plasmid containing eis_Ava was transformed into chemically competent E. coli TOP10 cell and used to confirm the sequence of the Eis_Ava gene (gene locus Ava_4977) (University of Michigan DNA Sequencing Core).
Overproduction and purification of Eis_Ava protein
The Eis_Ava protein, containing a N-terminal His6-tag, was overexpressed and purified using the pEis_Ava–pET28a overexpression construct as previously reported for its homologue in Mycobacterium tuberculosis (Eis_Mtb).3 The Eis_Ava protein was dialyzed in Tris–HCl buffer (50 mM, pH 7.5 adjusted at rt), flash frozen in the dialysis buffer containing 10% glycerol, and stored at −80 °C. The protein yield after purification was 1.5 mg L−1 of culture (Fig. S2, ESI†).
UV-Vis spectrophotometric assay determination of AG selectivity profile of Eis_Ava
The Eis_Ava acetyltransferase activity was monitored by using Ellman’s method in which the CoA thiol, freed by the Eis_Ava catalyzed reaction, is reacted with DTNB to produce an increase in absorbance that can be monitored at 412 nm (ε412 = 13 600 M−1 cm−1).14 Reactions (200 μL) containing AG (0.1 mM, 1 eq.), acyl-CoA (0.5 mM, 5 eq.), DTNB (2 mM), and Tris–HCl (50 mM, pH 7.5 adjusted at rt) were initiated by the addition of Eis_Ava (0.5 μM) at 25 °C. The reactions were monitored by taking readings every 30 s for 1 h in 96-well plate format.
Determination by mass spectrometry of the number of AG amine functionalities acetylated by Eis_Ava
To determine the number of acetylations performed by Eis_Ava on each AG substrate, we used LCMS. Reactions (30 μL) containing AG (0.67 mM, 1 eq.), AcCoA (6.7 mM, 10 eq.), Tris–HCl (50 mM, pH 8.0 adjusted at rt), and Eis_Ava (10 μM) were run overnight at rt. To prepare samples for the LCMS, ice-cold MeOH (30 μL) was added to the reaction mixture, followed by incubation for 20 min at −20 °C to precipitate the protein. Centrifugation (13 000 rpm, rt, 10 min) was then used to pellet the protein and the supernatant was used for LCMS injection after its dilution in H2O (10 μL of supernatant into 20 μL of H2O). The whole sample (30 μL) was injected onto the LCMS, used in positive mode with H2O (0.1% formic acid). Mass spectra showing the number acetylations on AMK, APR, KAN, NEA, NEO, NET, PAR, RIB, and SIS are presented in Fig. S3 (ESI†). These data are also summarized in Table 1.
Molecular modeling of APR with Eis_Ava and Eis_Msm
The APR structure was built using the Sybyl-X software and minimized to 0.01 kcal mol−1 by the Powell method, using Gasteiger–Hückel charges and the Tripos force field. The coordinates of the Eis_Ava and Eis_Msm (PDB codes: 2OZG and 3SXN, respectively) were downloaded from the Protein Data Bank website. Except for CoA, the H2O molecules and all other substructures were removed from the two proteins. In the structure of Eis_Ava (PDB code: 2OZG), the selenium atoms were replaced manually by sulfur. An acetyl group was added to CoA in both Eis_Ava and Eis_Msm. The surrounding protein residues were then kept frozen and the AcCoA was subjected to energy minimization using the Steepest Descent method. Hydrogen atoms were added and the energy of the Eis_Ava–AcCoA and Eis_Msm–AcCoA complexes were minimized using the Amber force fields with Amber charges. The energy-optimized APR was docked into the AG-binding site in both Eis_Ava–AcCoA and Eis_Msm–AcCoA minimized complexes using GOLD.15 The parameters were set as the default values forGOLD. Themaximum distance between hydrogen bond donors and acceptors for hydrogen bonding was set to 3.5Å. After docking, the first-ranked conformation of APR in both cases was merged into the corresponding APR-free Eis_Ava–AcCoA and Eis_Msm–AcCoA complexes. The new Eis_Ava–APR–AcCoA and Eis_Msm–APR–AcCoA complexes were subsequently subjected to energy minimization using the Amber force fields with Amber charges. During the energy minimization, the structure of APR and residues within an 8Å radius was allowed to move. The remaining residues were kept frozen in order to save calculation time. The energy minimization, in both cases, was performed using the Powell method with a 0.05 kcal mol−1 energy gradient convergence criterion and a distance dependent dielectric function. The Eis_Ava–APR–AcCoA and Eis_Msm–APR–AcCoA complexes are depicted in Fig. 3I and J.
Characterization of Eis_Ava steady-state kinetic parameters
For several AGs tested in this study (APR, KAN, NEA, NEO, and PAR) the kinetic parameters (Km and kcat) were determined for acetylation by Eis_Ava in reactions (200 μL) with a fixed AcCoA (0.5 mM) concentration. For NEA, NEO, and PAR the AG concentration range used was 0, 20, 50, 100, 250, and 500 μM. Due to their higher Km values, APR and KAN were tested in a higher concentration range of 0, 50, 250, 500, 1000, and 2000 μM. The reaction mixture containing DTNB (2 mM), Tris–HCl (50 mM, pH 7.5 adjusted at rt), and Eis_Ava (0.25 μM) was initiated by the addition of AG. Reactions were monitored by taking readings every 20 s for 20 min at 25 °C. For determination of the Km and kcat parameters, a non-linear regression fit to the Michaelis–Menten dependence was carried out by using Sigma Plot 11.0 software (Systat Software Inc.; San Jose, CA) (Fig. S4, ESI†; Table 2). The same procedure was used to determine the kinetic parameters (Km and kcat) reported in Table 2 for Eis_Mtb and Eis_Msm. To determine the microscopic turnover rate constant for Eis_Ava, two additional sets of experiments were performed by keeping the concentration of AcCoA at 0.1 and 0.2 mM while varying the concentration of KAN (0, 50, 250, 500, 1000, and 2000 μM).
Monitoring of NEA acetylation by TLC
The eluent system utilized for all TLCs of NEA reactions was 92.5 : 7.5/MeOH : NH4OH. The TLCs showing various mono-, and di-acetylated NEA along with their respective Rf values are depicted in Fig. 4. The mono-acetylated standards 2′-, 3-, and 6′-acetyl-NEA were obtained by using AAC(2′)-Ic, AAC(3)-IV, and AAC(6′)-Ie/APH(2″)-Ia, respectively. These enzymes were overexpressed and purified as previously reported.3,16 The acetylation reactions (30 μL) by these enzymes were run at rt in MES buffer (50 mM, pH 6.0 adjusted at rt) (AAC(3)-IV and AAC(6′)-Ie) or in potassium phosphate buffer (100 mM, pH 7.0 adjusted at rt) (AAC(2′)-Ic) with NEA (5 mM, 1 eq.), AcCoA (25 mM, 5 eq.) and AAC enzyme (10 μM). After overnight incubation at rt, the protein was precipitated by addition of MeOH (30 μL; 20 min, −20 °C). The protein was pelleted by centrifugation (13 000 rpm, rt, 10 min). An aliquot (5 μL) of the supernatant was diluted with MeOH (5 μL) before loading onto SiO2 TLC plates. The di-acetylation control reactions (30 μL) were carried out in a similar fashion with the addition of the second AAC enzyme (10 μM) and an additional portion of AcCoA (25 mM, 5 eq.) after a 24 h incubation period.
The reactions with Eis_Ava (30 μL) were run in Tris–HCl (50 mM, pH 7.5 adjusted at rt) with AcCoA (25 mM, 5 eq.), NEA (5 mM, 1 eq.), and Eis_Ava (10 μM). At different time points: 0 (before Eis_Ava addition), 5, 10, 30, 120 min, and overnight, aliquots (5 μL) were removed and processed as described above before loading onto SiO2 TLC plates.
Determination by NMR of NEA amines acetylated by Eis_Ava
To determine which two positions of NEA are acetylated by Eis_Ava, a reaction was designed for characterization in solution by NMR without the need for purification. A reaction (200 μL) containing NEA (10 mM, 1 eq.), AcCoA (50 mM, 5 eq.), and Eis (10 μM) in Tris–HCl (50 mM, pH 7.5 adjusted at rt) containing 12% D2O was prepared and allowed to proceed to completion as monitored by TLC. After 24 h, 1H, gCOSY, zTOCSY, and gHSQC NMR experiments were performed on the reaction solution, applying PURGE solvent suppression to establish the positions of acetylation. LCMS was used to confirm the structure identification. Proton connectivity was assigned using zTOCSY, gCOSY, and gHSQC spectra. Representative spectra for 1,2′-di-acetyl-NEA are provided in Fig. S9–S12 (ESI†).
To establish unambiguously the two acetylated positions on the NEA scaffold, the NMR spectra of 1,2′-di-acetyl-NEA were compared to those of a standard of pure NEA [(10 mM in Tris–HCl (50 mM, pH 7.5 adjusted at rt) containing 12% D2O], which was prepared identically to the reaction mixture, but omitted AcCoA and Eis_Ava (Table S1, ESI†). Representative spectra for the standard reaction solution of NEA in the absence of AcCoA and Eis_Ava are provided in Fig. S5–S8 (ESI†).
Inhibition of Eis_Ava and Eis_Msm by compounds 1–5
IC50 values were measured by the UV-Vis assay described above. The reactions (200 μL total) were initiated in several steps. The inhibitors 1–5 (Fig. 5) were dissolved in Tris–HCl (50 mM, pH 8.0 adjusted at rt with 10% DMSO) (100 μL) with a fivefold serial dilution with the highest concentration of inhibitor being 250 μM. A second mixture (50 μL) containing Eis_Ava or Eis_Msm (2 μM), NEO (400 μM), and Tris–HCl (50 mM, pH 8.0 adjusted at rt) was added to the inhibitor solutions and incubated for 10 min at rt. Reactions were initiated by the addition of a third mixture (50 μL) containing AcCoA (8 mM), DTNB (8 mM), and Tris–HCl (50 mM, pH 8.0 adjusted at rt). The final concentrations of compound 1 ranged from 250 μM to 650 pM. The final concentrations of compounds 2–5 ranged from 200 μM to 102 pM. The assay was run in triplicate and the data were normalized to DMSO control reactions. A Hill plot analysis performed by using Kaleidagraph 4.1 software yielded the IC50 values.
UV-Vis spectrophotometric assay determination of Eis_Ava, Eis_Msm, and Eis_Mtb activity against KAN and 6′-glycinyl-KAN
The activity of Eis homologues against KAN and 6′-glycinyl-KAN was tested as described for the determination of the AG selectivity profile of Eis_Ava described above.
Supplementary Material
Acknowledgments
This work was supported by a National Institutes of Health Grant AI090048 (SGT), by Rackham Merit Fellowships from the University of Michigan (REP and JLH), by the NIH Cellular Biotechnology Training Program (CBTP) at the University of Michigan (JLH), by the American Foundation of Pharmaceutical Education Fellowship (AFPE) (JLH). We thank Dr Oleg V. Tsodikov for critical reading of the manuscript. We also thank Ahmed S. A. Mady for help with generating the phylogenetic tree of Eis homologues.
Footnotes
Electronic supplementary information (ESI) available: Figures showing the structures of the AGs used in this study, coomassie blue-stained SDS-PAGE of purified Eis_Ava, mass spectra of AGs multi-acetylated by Eis_Ava, Michealis–Menten plots for the kinetic parameters for the Eis_Ava catalyzed acetylation of various AGs, table of proton chemical shift determined for NEA and 1,2′-di-acetyl-NEA along with NMR spectra. See DOI: 10.1039/c2mb25341k
Notes and references
- 1.Zaunbrecher MA, Sikes RD, Jr, Metchock B, Shinnick TM, Posey JE. Proc Natl Acad Sci U S A. 2009;106:20004–20009. doi: 10.1073/pnas.0907925106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell PJ, Morlock GP, Sikes RD, Dalton TL, Metchock B, Starks AM, Hooks DP, Cowan LS, Plikaytis BB, Posey JE. Antimicrob Agents Chemother. 2011;55:2032–2041. doi: 10.1128/AAC.01550-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen W, Biswas T, Porter VR, Tsodikov OV, Garneau-Tsodikova S. Proc Natl Acad Sci U S A. 2011;108:9804–9808. doi: 10.1073/pnas.1105379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen W, Green KD, Tsodikov OV, Garneau-Tsodikova S. Biochemistry. 2012;51:4959–4967. doi: 10.1021/bi3004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim KH, An DR, Song J, Yoon JY, Kim HS, Yoon HJ, Im HN, Kim J, Kim do J, Lee SJ, Lee HM, Kim HJ, Jo EK, Lee JY, Suh SW. Proc Natl Acad Sci U S A. 2012;109:7729–7734. doi: 10.1073/pnas.1120251109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tischer RG. Nature. 1965;205:419–420. doi: 10.1038/205419a0. [DOI] [PubMed] [Google Scholar]
- 7.Rice EW, Messer JW, Johnson CH, Reasoner DJ. Appl Environ Microbiol. 1995;61:374–376. doi: 10.1128/aem.61.1.374-376.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. J Antimicrob Chemother. 2012;67:810–818. doi: 10.1093/jac/dkr578. [DOI] [PubMed] [Google Scholar]
- 9.Krissinel E, Henrick K. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 10.Chen W, Green KD, Garneau-Tsodikova S. Antimicrob Agents Chemother. 2012;56:5831–5838. doi: 10.1128/AAC.00932-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green KD, Chen W, Garneau-Tsodikova S. ChemMedChem. 2012;7:73–77. doi: 10.1002/cmdc.201100332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaul P, Green KD, Rutenberg R, Kramer M, Berkov-Zrihen Y, Breiner-Goldstein E, Garneau-Tsodikova S, Fridman M. Org Biomol Chem. 2011;9:4057–4063. doi: 10.1039/c0ob01133a. [DOI] [PubMed] [Google Scholar]
- 13.Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellman GL. Arch Biochem Biophys. 1958;74:443–450. doi: 10.1016/0003-9861(58)90014-6. [DOI] [PubMed] [Google Scholar]
- 15.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 16.Green KD, Chen W, Houghton JL, Fridman M, Garneau-Tsodikova S. ChemBioChem. 2010;11:119–126. doi: 10.1002/cbic.200900584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.