

Abstract
Brucella spp. are facultative intracellular bacterial pathogens responsible for brucellosis, a worldwide zoonosis that causes abortion in domestic animals and chronic febrile disease associated with serious complications in humans. There is currently no approved vaccine against human brucellosis, and antibiotic therapy is long and costly. Development of a safe protective vaccine requires a better understanding of the roles played by components of adaptive immunity in the control of Brucella infection. The importance of lymphocyte subsets in the control of Brucella growth has been investigated separately by various research groups and remains unclear or controversial. Here, we used a large panel of genetically deficient mice to compare the importance of B cells, transporter associated with antigen processing (TAP-1), and major histocompatibility complex class II-dependent pathways of antigen presentation as well as T helper 1 (Th1), Th2, and Th17-mediated responses on the immune control of Brucella melitensis 16 M infection. We clearly confirmed the key function played by gamma interferon (IFN-γ)-producing Th1 CD4+ T cells in the control of B. melitensis infection, whereas IFN-γ-producing CD8+ T cells or B cell-mediated humoral immunity plays only a modest role in the clearance of bacteria during primary infection. In the presence of a Th1 response, Th2 or Th17 responses do not really develop or play a positive or negative role during the course of B. melitensis infection. On the whole, these results could improve our ability to develop protective vaccines or therapeutic treatments against brucellosis.
INTRODUCTION
Brucella (alphaproteobacteria) spp. are facultative intracellular Gram-negative coccobacilli that infect humans as well as domestic mammals (cattle, sheep, swine, camels, etc.) and wild mammals (deer, bison, etc.). Animal infection leads to abortion and infertility with huge economic costs. Human brucellosis is an ancient disease mainly transmitted through ingestion, inhalation, or contact with conjunctiva or skin lesions. It is characterized by undulant fever that if left untreated can result in chronic and sometimes lifelong disease, with serious clinical manifestations (12, 15, 23, 56). Human brucellosis remains a significant public health concern, with more than 500,000 new human cases reported annually (56). There is currently no available vaccine against human brucellosis, and all commercially available animal vaccines are based on live attenuated strains of Brucella (Brucella melitensis Rev.1, B. abortus S19, B. abortus RB51) (20, 54). Despite their effectiveness, these live vaccines have disadvantages, such as being infectious and causing disease in humans, interfering with diagnosis, resulting in abortions when administered to pregnant animals, and bearing antibiotic resistance (54). Thus, the ideal safe vaccine would be a nonreplicating subunit vaccine that specifically targets the critical aspects of the immune response necessary to induce immunity. The success of a subunit vaccine is strongly dependent on the choice of appropriate antigen-presenting pathways and a suitable adjuvant. Therefore, a better understanding of the role played by various lymphocyte subsets during infection and the identification of T helper cytokine profiles associated with protective and counterprotective immune response will be very useful for the development of a safe protective subunit vaccine.
Immune responses to Brucella spp. have been studied mainly in mouse models (4, 25). There has been much evidence demonstrating that the gamma interferon (IFN-γ)-mediated T helper 1 (Th1) immune response is crucial for the control of Brucella infection. Supplementing infected mice with recombinant IFN-γ (66) results in better clearance of B. abortus in the spleen, and neutralizing endogenous IFN-γ by administering monoclonal antibodies (MAbs) results in decreased control of bacterial growth (18, 19, 74). Brucella-infected MyD88-deficient (MyD88−/−) (10) and interleukin-12 p40 (IL-12p40)-deficient (IL-12p40−/−) (37) C57BL/6 mice display reduced production of IFN-γ and 10- to 100-fold higher bacterial counts in the spleen. Finally, IFN-γ-deficient (IFN-γ−/−) and interferon responsive factor 1 (IRF-1)-deficient (IRF-1−/−) C57BL/6 mice are the rare strains that succumb to B. abortus (6, 37, 52) or B. melitensis (2) infection. In contrast, Th2 responses are frequently considered detrimental during Brucella infection. Indeed, BALB/c mice, which present a well-known Th2 bias in numerous infectious models (27, 42), display a reduced ability to control Brucella infection (10, 19, 52). Moreover, neutralization of IL-4 by MAb can reduce bacterial counts in some models (19). Up to now, studies of the function of Th17 responses in immunity to Brucella organisms have been scarce. Nevertheless, Th17 responses have been shown to contribute to host defense against several intracellular microorganisms, such as Mycobacterium tuberculosis (14, 33, 68).
Adaptive immunity is clearly implicated in the control of chronic Brucella infection (29). However, the relative roles of B cells, CD4+ cells, and CD8+ T cells in protection remain controversial. B cells have been found to be involved in resistance to the attenuated B. abortus virB mutant (61) and susceptibility to virulent B. abortus (24). Depending on the studies, major histocompatibility complex class II (MHC-II)-deficient (MHC-II−/−) mice demonstrate that the presence of CD4+ T cells appears to be important (24) or dispensable (55) in the control of infection. CD8+ T cells have been shown to play a role in resistance to infection by depleting CD8+ T cells with anti-CD8α monoclonal antibodies (48, 51, 58) and using β2-microglobulin-deficient (β2-microglobulin−/−) mice (55). Both CD4 and CD8 T cells have been shown to be equally protective in adoptive transfer studies (1). These results are subject to multiple interpretations.
Using a large panel of gene-deficient mice, we evaluated the contributions of the humoral response and T cell subsets in protective immunity against B. melitensis, the most frequent cause of human brucellosis (12). To our knowledge, this study is the first to analyze the course of infection of virulent Brucella in transporter associated with antigen processing (TAP-1)-, IL-4-, IL-12p35-, IL-17α receptor (IL-17Rα)-, IL-21R-, and IL-22-deficient mice. Our results confirm the central role of MHC-II-dependent antigen presentation to CD4+ T cells and the IFN-γ-mediated Th1 response in the control of B. melitensis infection, whereas the absence of B cells, MHC-I-dependent antigen presentation, and Th2 and Th17 responses appears to have no important positive or negative impact.
MATERIALS AND METHODS
Ethics statement.
The animal handling and procedures of this study complied with current European legislation (directive 86/609/EEC) and the corresponding Belgian law, Arrêté Royal Relatif à la Protection des Animaux d'Expérience du 6 Avril 2010 Publié le 14 Mai 2010 (Royal Decree on the Protection of Experimental Animals of 6 April 2010, published on 14 May 2010). The complete protocol was reviewed and approved by the Animal Welfare Committee of the Facultés Universitaires Notre-Dame de la Paix (FUNDP; Namur, Belgium) (permit number 05-558).
Mice and reagents.
MyD88−/− C57BL/6 mice (31) were obtained from S. Akira (Osaka University, Osaka, Japan). TAP-1-deficient (TAP-1−/−) C57BL/6 mice (70) and MHC-II−/− C57BL/6 mice (13) were obtained from Jörg Reimann (University of Ulm, Ulm, Germany). IL-12p35−/− C57BL/6 mice (47) and IL-12p40−/− C57BL/6 mice (44) were obtained from B. Ryffel (University of Orleans, Orleans, France). IL-21R−/− C57BL/6 mice (39) were obtained from F. Andris (Université Libre de Bruxelles, Brussels, Belgium). IL-17Rα−/− C57BL/6 mice (53) and IL-22−/− C57BL/6 mice (38) were obtained from L. Dumoutier (Ludwig Institute for Cancer Research, Brussels Branch, Brussels, Belgium). Recombinant activating gene (RAG)-deficient (RAG1−/−) C57BL/6 mice (49) were obtained from S. Goriely (Université Libre de Bruxelles, Brussels, Belgium). RAG2γc−/− C57BL/6 mice (7) were obtained from Michel Y. Braun (Université Libre de Bruxelles, Brussels, Belgium). CD28−/− and CD40−/− C57BL/6 mice (30, 64) and B cell-deficient (MuMT−/−) C57BL/6 mice (36) were purchased from The Jackson Laboratory (Bar Harbor, ME). IL-12p40−/− BALB/c mice (44) were obtained from V. Flamand (Université Libre de Bruxelles, Brussels, Belgium). Wild-type C57BL/6 mice and BALB/c mice were purchased from Harlan (Bicester, United Kingdom) and were used as controls. All wild-type and deficient mice used in this study were bred in the animal facility of the Gosselies Campus of the Free University of Brussels (ULB; Brussels, Belgium).
B. melitensis strain 16 M (biotype 1, ATCC 23456) was initially isolated from an infected goat and grown in a biosafety level III laboratory facility. The culture was grown overnight with shaking at 37°C in 2YT medium (Luria-Bertani broth with a double quantity of yeast extract) to stationary phase and was washed twice in phosphate-buffered saline (PBS) by centrifugation at 3,500 × g for 10 min before use for mouse inoculation as previously described (10).
Mouse infection.
Mice were injected intraperitoneally (i.p.) with the indicated dose of B. melitensis in 500 μl of PBS. Control animals were injected with the same volume of PBS. The infectious doses were validated by plating serial dilutions of inocula. At selected time intervals, mice were sacrificed by cervical dislocation. Immediately after the mice were killed, the spleen was collected for bacterial count and flow cytometry analyses.
Bacterial count.
Spleens were recovered in PBS–0.1% Triton X-100 (Sigma). We performed successive serial dilutions in PBS to obtain the most accurate bacterial count and plated them onto 2YT medium plates. The numbers of CFU were counted after 4 days of culture at 37°C.
RNA purification and real-time RT-PCR.
RNA was extracted from total spleen tissue. Spleen samples were collected and immediately frozen and stored at −80°C until processing. RNA was then extracted from frozen tissue with TriPure isolation reagent (Roche) according to the instructions of the manufacturer. RNA samples were treated with DNase (Fermentas). Reverse transcription (RT) and real-time PCRs were carried out using LightCycler 480 RNA master hydrolysis probes (one-step procedure) on a LightCycler 480 real-time PCR system (Roche Diagnostics). Primer sequences are described in Table S1 in the supplemental material.
Cytofluorometric analysis.
As previously described (11), spleens were harvested, cut in very small pieces, and incubated with a cocktail of DNase I fraction IX (100 μg/ml; Sigma-Aldrich Chimie SARL, Lyon, France) and 1.6 mg/ml of collagenase (400 Mandl units/ml) at 37°C for 30 min. After washing, spleen cells were filtered and first incubated in saturating doses of purified 2.4G2 (anti-mouse Fc receptor; ATCC) in 200 μl PBS–0.2% bovine serum albumin–0.02% NaN3 (fluorescence-activated cell sorter [FACS] buffer) for 20 min on ice to prevent antibody binding to Fc receptor. A total of 3 × 106 to 5 × 106 cells were stained on ice with various fluorescent MAb combinations in FACS buffer and further collected on a FACSCalibur cytofluorometer (Becton Dickinson, BD). We purchased the following MAbs from BD Biosciences: fluorescein isothiocyanate (FITC)-coupled 145-2C11 (anti-CD3ε), FITC-coupled H57-597 (anti-TCRβ), phycoerythrin (PE)-coupled RM4-5 (anti-CD4), and PK136 (anti-NK1.1). The cells were analyzed on a FACSCalibur cytofluorometer. Cells were gated according to size and scatter to eliminate dead cells and debris from the analysis.
Intracellular cytokine staining.
Spleen cells were incubated for 4 h in RPMI 1640–5% fetal calf serum with 1 μl/ml Golgi Plug (BD Pharmingen) at 37°C in 5% CO2. The cells were washed with FACS buffer and stained for cell surface markers before fixation in PBS–1% paraformaldehyde (PFA) for 15 to 20 min on ice. These cells were then permeabilized for 30 min using a saponin-based buffer (10× Perm/Wash [BD Pharmingen] in FACS buffer) and stained with allophycocyanin (APC)-coupled XMG1.2 (anti-IFN-γ; BD Biosciences). After final fixation in PBS–1% PFA, cells were analyzed on a FACScalibur cytofluorometer. No signal was detectable with control isotypes.
Statistical analysis.
We used a Mann-Whitney or Wilcoxon-Mann-Whitney test provided by the GraphPad Prism program to statistically analyze our results. Each group of deficient mice was compared to wild-type mice. We also compared each group with each other and displayed the results when required. P values of <0.05 were considered to represent a significant difference.
RESULTS
Major role of CD4+ T cells in control of the plateau phase of chronic B. melitensis infection.
The precise role of different components of adaptive immunity in the control of Brucella growth is quite controversial (1, 3, 6, 24, 48, 51, 52, 55, 58, 60, 61). Here, we compared the course of B. melitensis infection in the spleen of various C57BL/6 mice rendered genetically deficient for key elements of adaptive immune response: RAG (T and B cells), MHC-II (CD4+ T cells), TAP-1 (CD8+ T cells), and MuMT (B cells). As an internal control, we used MyD88−/− mice that present well-established susceptibility to B. melitensis infection (10, 71). Wild-type and deficient mice were infected i.p. with 4 × 104 CFU of B. melitensis (Fig. 1A and B). As expected, RAG deficiency was correlated with an increased bacterial load in the spleen (Fig. 1A). Comparative analysis of TAP-1−/−, MHC-II−/−, and MuMT−/− mice showed a clear unique implication of CD4+ T cells in the late control of B. melitensis infection, as demonstrated by the fact that at 28 days postinfection (p.i.), only MHC-II−/− mice were unable to control the infection. In contrast, at 28 days p.i., TAP-1−/− mice displayed an unchanged CFU count in the spleen, and as previously reported by Goenka et al. (24), MuMT−/− mice exhibited a reduced bacterial load compared with wild-type mice.
Fig 1.
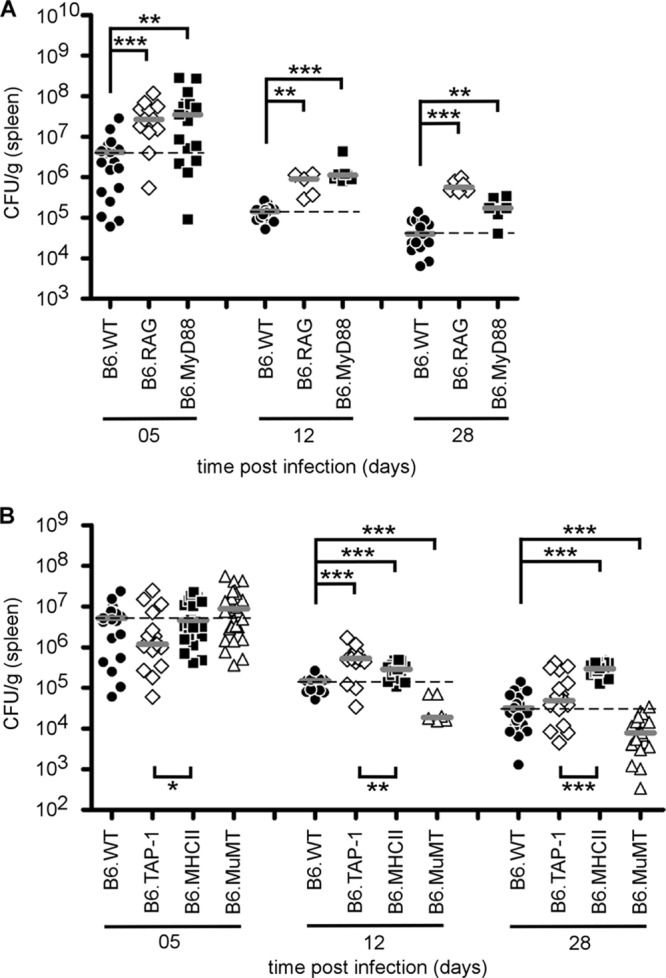
Course of B. melitensis infection in the spleen of C57BL/6 (B6) versus RAG−/−, MyD88−/−, TAP-1−/−, MHC-II−/−, and MuMT−/− mice. Wild-type (WT) and deficient mice were injected i.p. with 4 × 104 CFU of B. melitensis and sacrificed at the indicated times. The data represent the number of CFU per gram of spleen. Gray bars represent the medians. These results are representative of two independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
IFN-γ is well-known as a key cytokine that regulates resistance to Brucella infection (10, 19, 52), as mice deficient for IFN-γ do not survive infection (6, 52). In the spleen, early IFN-γ production (at 5 days) derives equally from NK cells, CD4+ T cells, and CD8+ T cells, while later sustained IFN-γ production (at 12 days) is mainly dependent on CD4+ T cells (10). Using flow cytometry analyses, we investigated the impact of different targeted genetic deficiencies on the frequency of IFN-γ-producing cells in the spleen. It is important to note that the cells were never stimulated in vitro before intracellular cytokine staining. Thus, the IFN-γ detected in these cells derived only from natural in vivo activation during the course of infection.
At 5 days p.i., a highly significant increase of IFN-γ-positive (IFN-γ+) cells was observed in RAG−/− and MuMT−/− mice compared with wild-type mice (Fig. 2A). In RAG−/− mice, this surprisingly elevated frequency of IFN-γ+ cells was mainly due to NK cells (Fig. 2B; see Fig. S1 in the supplemental material). This was confirmed by the complete absence of IFN-γ in the spleens of RAGγc−/− mice deficient for NK cells (see Fig. S1 in the supplemental material). As previously described by Goenka et al. (24), MuMT−/− mice displayed a higher frequency of IFN-γ-producing T cells, associated with better control of the infection (Fig. 2A). Despite a severe deficiency in CD4+ T or CD8+ T cells (see Fig. S2 in the supplemental material), the total frequency of IFN-γ-producing cells was not reduced either in MHC-II−/− mice or in TAP-1−/− mice. Similar results, due to transitory compensations, have been reported by Das (16) after injection of anti-CD3 antibodies.
Fig 2.
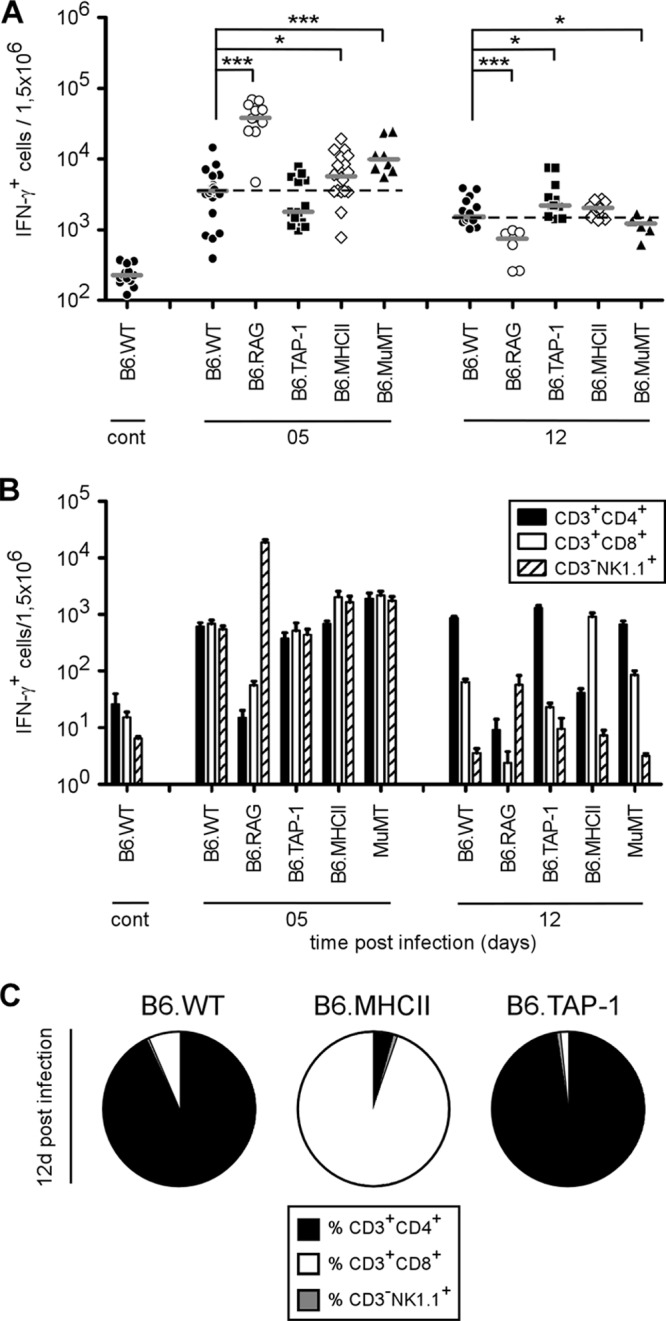
Flow cytometry analysis of IFN-γ-producing spleen cells following B. melitensis infection. Wild-type, RAG−/−, TAP-1−/−, MHC-II−/−, and MuMT−/− C57BL/6 mice were injected i.p. with PBS (as a control) or 4 × 104 CFU of B. melitensis and sacrificed at the indicated times. Spleen cells were harvested and analyzed by flow cytometry. Cells were gated according to size and scatter to exclude dead cells and debris from the analysis. Spleen cells from individual mice were first analyzed for forward size scatter (FSC) versus IFN-γ production and then for cell surface markers. The frequency of IFN-γ-positive cells in uninfected deficient mice was similar to the one observed in uninfected wild-type mice. (A) Number of IFN-γ-positive cells per 1.5 × 106 spleen cells acquired. Each piece of data represents the value obtained from an individual spleen, and the data are representative of two independent experiments. Gray bars represent the medians. cont, control; *, P < 0.05; ***, P < 0.001. (B) IFN-γ-positive cells in each group were then analyzed for CD3, CD4, CD8, and NK1.1 expression. The data represent the mean ± SEM of groups described in panel A. (C) The percentages of IFN-γ-positive cells 12 days p.i. are represented in a sectoral diagram according to cell surface markers.
At 12 days p.i., only RAG−/− mice displayed a strongly diminished frequency of IFN-γ-producing cells, since NK cells were no longer able to assume IFN-γ production (Fig. 2A). Interestingly, at 12 days p.i., MHC-II−/− and TAP-1−/− mice displayed a similar frequency of IFN-γ-producing cells, but at 28 days p.i., only MHC-II−/− mice failed to control Brucella infection (Fig. 1B). As expected, IFN-γ production was taken over by CD8+ T cells in MHC-II−/− mice (Fig. 2B and C). Note that splenomegaly and the number of spleen cells (data not shown) were similar in both strains of infected mice. This strongly suggests that resistance to Brucella not only is restricted to IFN-γ production but also requires functional CD4+ T cells.
BALB/c mice do not present a Th2 bias during the course of Brucella melitensis infection.
Since the discovery of the Th1 and Th2 paradigm, resistance to Brucella has been described to be associated with a Th1 response and susceptibility has been described to be associated with a Th2 response, notably, because BALB/c mice display reduced control of infection compared with that displayed by C57BL/6 mice (4, 19). BALB/c mice are well-known for their Th2 bias in several infection models, such as the Leishmania major infection model (41–43). The recent discovery of the Th17 response and its implication in the control of infection by several intracellular bacteria have sparked debate on the role of each subclass of Th response during the course of Brucella infection.
In order to identify the response profile dominating Brucella infection in C57BL/6 and BALB/c mice, we compared the expression of IFN-γ, IL-4, and IL-17A by quantitative real-time PCR in spleen at 5 and 12 days p.i. (Fig. 3). As for the flow cytometry analysis, this test was performed on the entire organ without in vitro stimulation or purification of the spleen cells. As expected, IFN-γ mRNA was indeed detected in C57BL/6 and BALB/c mice and peaked at 5 days p.i. Both IL-4 and IL-17A mRNA expression remained weakly detectable during infection, and only IL-17A expression displayed a small and short upregulation at 5 days p.i. No statistically significant differences were observed between C57BL/6 and BALB/c mice. Thus, as determined by quantitative RT-PCR, BALB/c mice do not seem to present a Th2 bias following Brucella infection.
Fig 3.
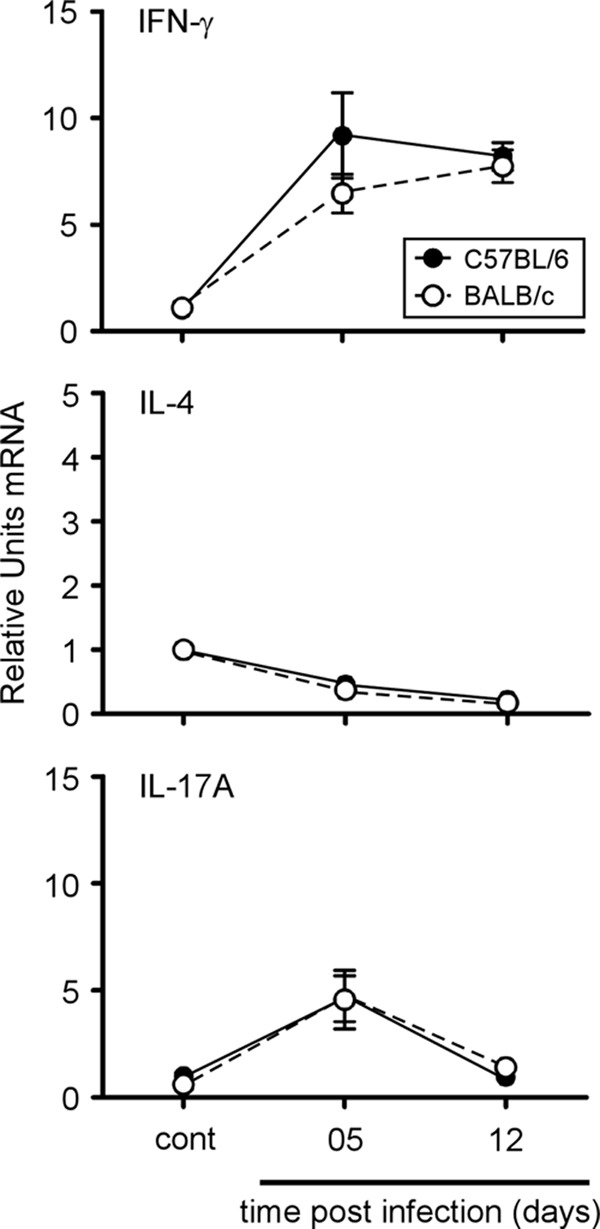
Kinetics of IFN-γ, IL-4, and IL-17A gene expression in spleen cells from C57BL/6 or BALB/c mice during the course of B. melitensis infection. C57BL/6 and BALB/c mice were injected i.p. with PBS (control) or 4 × 104 CFU B. melitensis. After the indicated times, spleen cells were collected and total RNA was extracted and analyzed by real-time PCR. mRNA levels were normalized using β-actin mRNA as the reference and compared with those obtained under uninfected conditions (control). The data are expressed as the mean ± SEM of 16 animals per time point pooled from two independent experiments.
Ablation of the Th2 or Th17 response does not affect the control of Brucella melitensis infection in C57BL/6 mice.
In order to functionally compare the impact of each subclass of Th response, we compared the susceptibility of C57BL/6 mice genetically deficient for IL-12p35 (Th1), IL-12p40 (Th1 and Th17), IL-4 (Th2), and IL-17Rα, IL-21R, and IL-22 (Th17). Mice were infected, and bacterial counts in the spleen were examined at 5, 12, and 28 days p.i. (Fig. 4A and B). Only IL-12p35−/− and IL-12p40−/− mice displayed significantly higher bacterial counts than wild-type mice at each time point after infection, confirming the crucial role of the Th1 response in the control of Brucella infection. Surprisingly, IL-4 deficiency only slightly but significantly improved the resistance of C57BL/6 mice at 28 days p.i. (Fig. 4A). Mice deficient for elements (IL-17Rα, IL-21R, IL-22) of the Th17 response controlled the infection like wild-type mice, suggesting no major positive role for the Th17 response (Fig. 4B).
Fig 4.
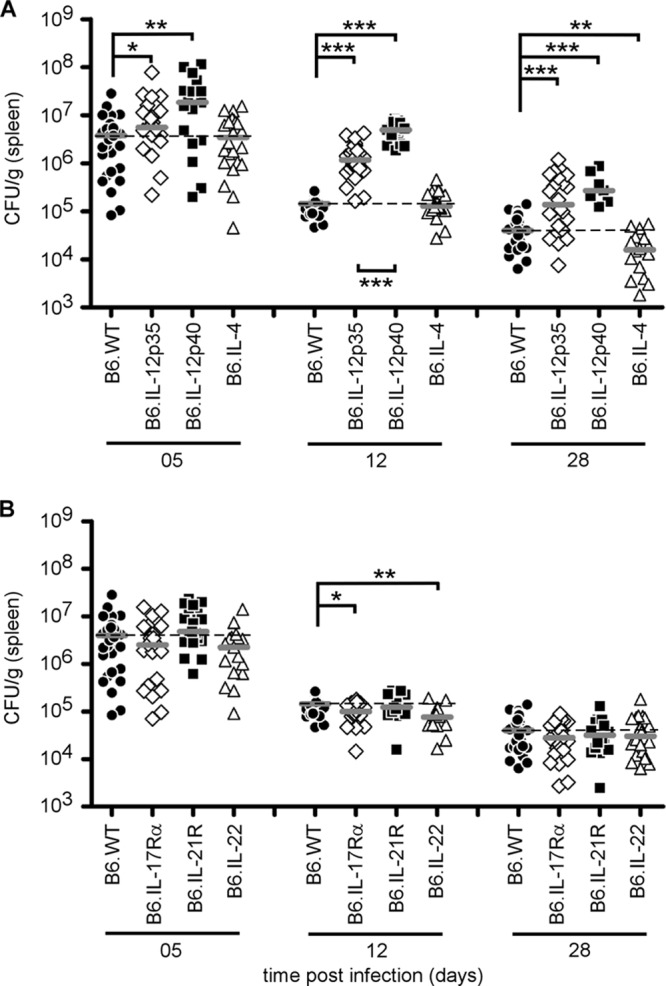
Course of B. melitensis infection in the spleen of wild-type and Th response-deficient C57BL/6 mice. Wild-type, IL-12p35−/− (Th1), IL-12p40−/− (Th1 and Th17), IL-4−/− (Th2), IL-17Rα−/−, IL-21R−/−, and IL-22−/− (Th17) C57BL/6 mice were injected i.p. with 4 × 104 CFU of B. melitensis and sacrificed at the indicated times. The data represent the number of CFU per gram of spleen. Gray bars represent the medians. These results are representative of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Flow cytometry analyses of the frequency of IFN-γ+ cells in the spleen during the course of B. melitensis infection confirm the close relationship existing between resistance to infection and IFN-γ production by CD4+ T cells (Fig. 5A to D). As expected, IL-12p35 and IL-12p40 deficiency strongly decreased the frequency of IFN-γ+ cells at 5 and 12 days p.i. compared with that in wild-type mice (Fig. 5A and B). IL-4, IL-17Rα, IL-21R, and IL-22 deficiency (Fig. 5C and D) affected this frequency very moderately, suggesting that the Th2 and Th17 responses have only minor effects on the Th1 immune response against B. melitensis infection in C57BL/6 mice.
Fig 5.

Flow cytometry analysis of IFN-γ-producing spleen cells following B. melitensis infection. Wild-type, IL-12p35−/−, IL-12p40−/−, IL-4−/−, IL-17Rα−/−, IL-21R−/−, and IL-22−/− C57BL/6 mice were injected i.p. with PBS (control) or 4 × 104 CFU of B. melitensis and sacrificed at the indicated times. Spleen cells were collected and analyzed by flow cytometry. Cells were gated according to size and scatter to exclude dead cells and debris from analysis. Spleen cells from individual mice were first analyzed for forward size scatter versus IFN-γ production and then for cell surface markers. The frequency of IFN-γ-positive cells in uninfected deficient mice was similar to the one observed in uninfected wild-type mice. (A, C) Number of IFN-γ-positive cells per 1.5 × 106 spleen cells acquired. (B, D) IFN-γ-positive cells were then analyzed for T cell receptor β (TCRβ) and CD4 expression. The data represent the number of IFN-γ+, T cell receptor β-positive, and CD4+ cells from the groups described in panels A and B. Each piece of data represents the value obtained from an individual spleen. These data are representative of two independent experiments. Gray bars represent the medians. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
In order to better identify the signal required to induce IFN-γ production during Brucella infection, we compared the frequency of IFN-γ-producing cells in the spleen of CD28−/−, CD40−/−, MyD88−/−, and IL-12p35−/− C57BL/6 infected mice. MyD88−/− and IL-12p35−/− mice were used as internal controls. The results (see Fig. S3 in the supplemental material) showed that deficiency of both CD28 and CD40 has only a minor impact on the frequency of IFN-γ-producing cells in the spleen, suggesting that the MyD88 and IL-12 pathways remain the main tested pathways regulating IFN-γ production during Brucella infection.
Ablation of the Th2 response does not improve the resistance of BALB/c mice to Brucella melitensis infection.
We then compared the susceptibility of wild-type, IL-12p40−/− (Th1 and Th17), and IL-4R−/− (Th2) BALB/c mice. As expected, IL-12p40 deficiency drastically reduced the resistance of BALB/c mice (Fig. 6), demonstrating that the Th1 response, even in this strain, is the main protective response. Surprisingly, IL-4R deficiency did not enhance the resistance of BALB/c mice at 5 and 12 days p.i. (Fig. 6). Only a very modest improvement of CFU counts in the spleen was observable at 28 days p.i.
Fig 6.
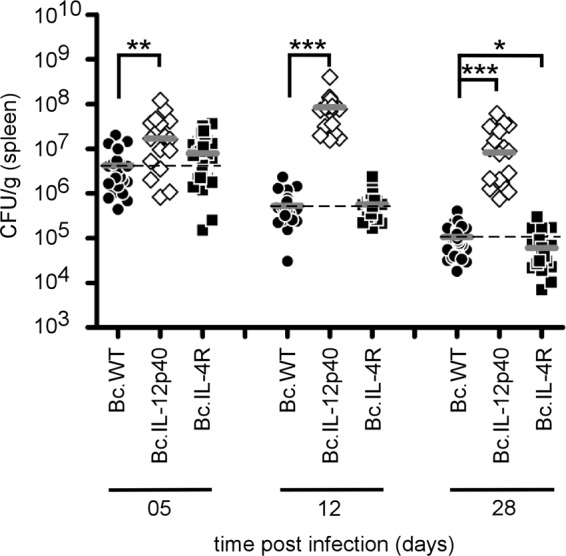
Course of B. melitensis infection in the spleen of wild-type and Th response-deficient BALB/c mice. Wild-type, IL-12p40−/− (Th1 and Th17), and IL-4R−/− (Th2) BALB/c mice were injected i.p. with 4 × 104 CFU of B. melitensis and euthanized at the indicated times. The data represent the number of CFU per gram of spleen. Gray bars represent the medians. These results are representative of two independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Analysis of IFN-γ-producing cell frequency by flow cytometry (Fig. 7A and B) showed a strong defect of IFN-γ production in IL-12p40−/− mice. In contrast to C57BL/6 mice, IL-4R deficiency in BALB/c mice was associated with an enhanced Th1 response, as shown by the 2-fold higher number of IFN-γ+ cells at 5 days p.i. However, this higher frequency was not maintained until 12 days (Fig. 7A and B) and was not associated with better control of the infection at the same time point (Fig. 6).
Fig 7.
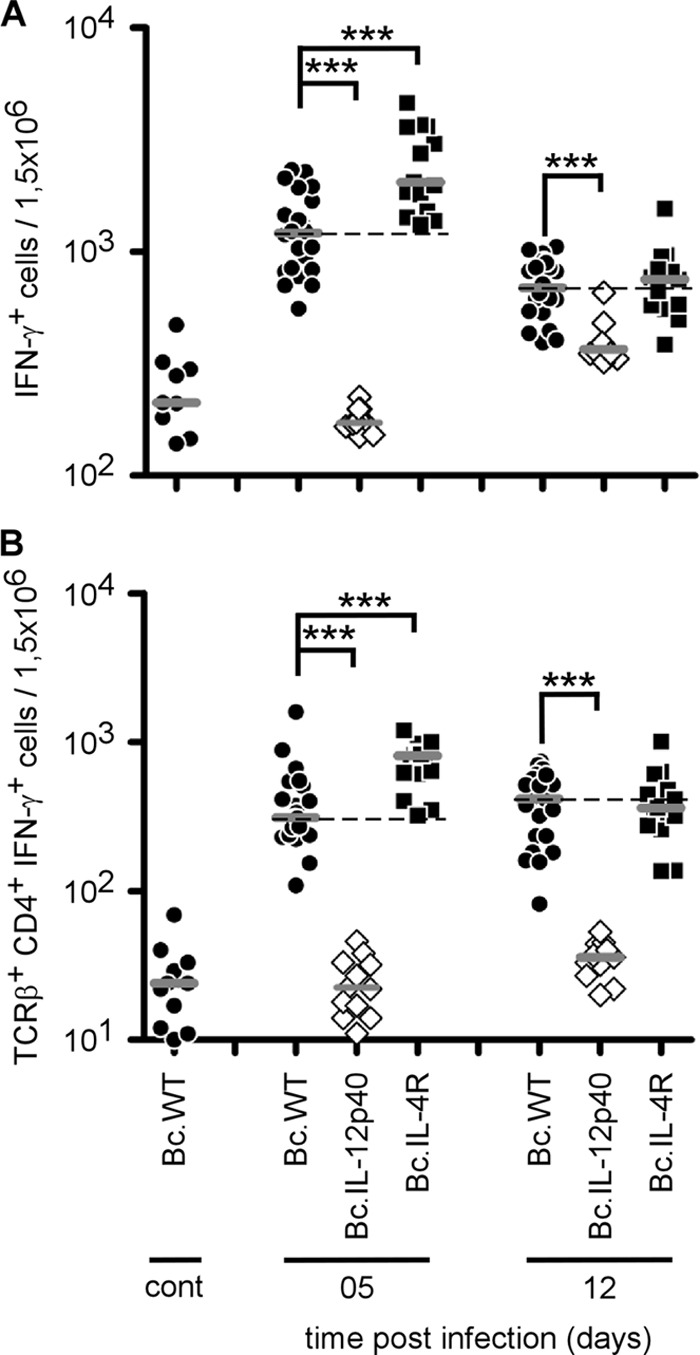
Flow cytometry analysis of IFN-γ-producing cells following B. melitensis infection. Wild-type, IL-12p40−/−, and IL-4R−/− BALB/c mice were injected i.p. with PBS (control) or 4 × 104 CFU of B. melitensis and sacrificed at the indicated times. Spleen cells were collected and analyzed by flow cytometry. Cells were gated according to size and scatter to exclude dead cells and debris from analysis. Spleen cells from individual mice were first analyzed for forward size scatter versus IFN-γ production and then for cell surface markers. The frequency of IFN-γ-positive cells in uninfected deficient mice was similar to the one observed in uninfected wild-type mice. (A) Number of IFN-γ-positive cells per 1.5 × 106 spleen cells acquired. (B) IFN-γ-positive cells were analyzed for T cell receptor β (TCRβ) and CD4 expression. The data represent the number of IFN-γ+, T cell receptor β-positive, and CD4+ cells from the groups described in panel A. Each piece of data represents the value obtained from an individual spleen, and the data are representative of two independent experiments. Gray bars represent the medians. ***, P < 0.001.
DISCUSSION
Brucellae are exquisitely well adapted to their mammalian hosts. They disseminate in all tissues and escape many immune system components through stealthy intracellular growth (46). However, this silent infection leads to a chronic febrile disease with potentially serious complications over the long term (12, 15, 23, 56). Empirical research has failed to develop a safe protective vaccine for humans (54, 59). Killed vaccines are not protective, and the live attenuated vaccines used in animals are too virulent for humans. One solution could be subunit vaccination candidates including Brucella antigens associated with adjuvants that mimic patterns of pathogenesis (POPs) in order to select appropriate immune effector mechanisms. POPs are defined as a combination of signals including the production of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) under specific infection conditions (69). Characterization of Brucella-specific POPs requires good knowledge of its in vivo infectious strategy, which, unfortunately, is still largely enigmatic. Using the same mouse model, we recently characterized (11) macrophages and dendritic cells as the major infected cells in the spleen and liver and identified granuloma formation as a main trait of resistance to Brucella infection in situ. Strikingly, the construction of granuloma structures during the early phase of chronic infection appeared to be largely independent of adaptive immunity, as the same structures are observed in RAG−/− mice. In contrast, they are strictly dependent on MyD88/IL-12p35/IFN-γ-mediated pathways (11). This observation could explain why IFN-γ (6, 52) or IRF-1 (37) deficiency, but not RAG deficiency (29), is fatal to Brucella-infected mice. Indeed, in RAG−/− mice, NK cells produce high counts of IFN-γ, as shown in this work. Though adaptive immunity components are not crucial to survival of Brucella infection, they do play a key role in successful vaccination. Depending on the studies considered, their roles appear to be very controversial (3).
We first attempted to determine if the control of B. melitensis infection involves a humoral immune response (circulating antibodies and B cells). Several groups have previously analyzed this question, with contradictory results. Natural antibodies have been reported to favor Brucella control by facilitating its elimination by phagocytosis (61). Many serum transfer experiments have demonstrated a positive role of antibodies in protecting mice against Brucella infection (1, 50, 72). However, as previously reported by Goenka et al. (24), we observed here that MuMT−/− mice cleared the infection from the spleen more rapidly than wild-type mice and displayed a higher frequency of IFN-γ-producing T cells at the peak of infection. Enhanced IFN-γ production could be due to a regulatory role of B cells, as hypothesized by Goenka et al. (24). On the whole, these results strongly suggest that natural or induced humoral immunity does not contribute positively to the control of primary Brucella infection, without excluding a possible protective role for specific antibodies in the secondary immune response.
The precise roles of CD4+ and CD8+ T cells during Brucella infection remain particularly unclear (reviewed in reference 3). Indeed, a positive role for CD8+ T cells in protection has been shown by adoptive transfer (1), depletion with anti-CD8α monoclonal antibodies (48, 51, 58), and infection of β2-microglobulin−/− mice (52, 55). In contrast, a negative role for CD8+ T cells has also been reported in studies (24, 52) showing that β2-microglobulin−/− mice cleared the infection better than their wild-type counterparts. On the other hand, the importance of CD4+ T cells has also been demonstrated by adoptive transfer (1) and, recently, by infecting MHC-II−/− mice (24). Finally, a study using mixed infection with B. abortus 2308 and the virB mutant showed no impact of β2-microglobulin deficiency on the control of wild-type bacterial infection and a greater ability to control this infection for CD4−/− mice (60). These contradictory results can be explained by multiple parameters, such as the mouse and Brucella strains and the experimental design used. For example, studies showing a central role for CD8+ T cells and no implication of CD4+ T cells in β2-microglobulin−/− and MHC-II−/− mice used Brucella abortus S19 (55), which is an attenuated strain of Brucella abortus known to be more susceptible to rapid intraphagocyte destruction after opsonization than a virulent strain (17). In depletion experiments, results must be interpreted cautiously, as inaccurate data can be generated by the administration procedure or a lack of specificity of the antibodies used. For example, the anti-CD8α antibodies used to deplete CD8+ T cells also target both CD8α-positive dendritic cells and a particular subset of γδ T cells (8). Discrepancies can also result from the choice of inadequately or poorly characterized deficient mice. Indeed β2-microglobulin−/− mice are now known to lack CD8+ T cells but also CD1d-restricted NK T cell responses and circulating IgG antibodies (35), findings which render interpretation of the results in the context of infection very hazardous. Moreover, comparison of studies using experimental animals performed in different laboratories has raised new concerns since environment and diet as well as immune deficiencies (22) have been demonstrated to shape the gut microbiota of mice and strongly modify the immune response. However, the impact of immune deficiencies appears to be strongly reduced under homeostatic conditions (67). In agreement, we initially worked with mice coming from different animal facilities, and we observed significant discrepancies following infection, even between wild-type mice that originated from the same supplier. As a consequence, the present study compared wild-type mice with a large panel of deficient mice that were bred in the same animal facilities for a minimum of 1 year and that thus displayed similar hygienic status. These factors and conditions therefore bring more consistent results on the role of multiple components of adaptive immunity.
Here, we chose to compare infection in TAP-1−/− and MHC-II−/− C57BL/6 mice to evaluate the contribution of CD8+ and CD4+ T cell functions, respectively. Both types of deficient mice were able to control bacterial growth at the peak of infection. Although transient susceptibility was observed in both groups of mice by 12 days p.i., TAP-1−/− mice were able to control the infection at 28 days p.i. In contrast, MHC-II−/− mice displayed a 10-fold increased bacterial load at 28 days p.i., suggesting a protective role for MHC-II-dependent CD4+ T cells during the chronic phase of infection. IFN-γ was produced by both CD4+ and CD8+ T cells at the peak of infection (5 days p.i.), but only CD4+ T cells retained the ability to produce high levels of IFN-γ during the plateau phase of infection in wild-type mice (10; this study). It is very interesting to note that the frequency of IFN-γ-producing cells was maintained at similar levels during the plateau phase of infection in both MHC-II−/− and TAP-1−/− mice. In MHC-II−/− mice, IFN-γ production was restricted to CD8+ T cells, showing that these cells were able to sustain IFN-γ production for more than 5 days. However, these cells did not provide protection since MHC-II−/− mice displayed enhanced CFU counts in the spleen compared with TAP-1−/− and wild-type mice. While several studies (4, 19, 52) support a correlation between IFN-γ production and protection, our data demonstrate that IFN-γ production must be associated with activated CD4+ T cells to be protective, suggesting that CD4+ T cells provide other undetermined additional protective cofactors.
The protective immune response against Brucella has historically been associated with the Th1 response, notably, because BALB/c mice, which display a well-known Th2 bias in several infection models (42, 43), show a reduced ability to control Brucella infection compared with C57BL/6 mice (4, 19). It has been hypothesized (19) that production of IL-4 in BALB/c mice reduces the production of IFN-γ and adversely affects the protective immune response to Brucella. Our data showed that infected C57BL/6 and BALB/c mice displayed similar weak expression of IL-4 mRNA in the spleen. Along the same lines, in BALB/c and C57BL/6 mice, the absence of IL-4 protein detectable by enzyme-linked immunosorbent assay following in vitro restimulation has been reported previously (19). Using IL-4−/− C57BL/6 and IL-4R−/− BALB/c mice, we demonstrate that IL-4 does not adversely affect host defense against Brucella infection. Indeed, IL-4 deficiency in C57BL/6 mice did not drastically enhance IFN-γ production. However, in IL-4R−/− BALB/c mice, a 2-fold increase in IFN-γ production was measurable at the peak of infection. This difference could be a consequence of the lack of responsiveness to both IL-4 and IL-13 in IL-4R−/− mice (32). Thus, we conclude that the low levels of IL-4 produced during Brucella infection in wild-type C57BL/6 and BALB/c mice do not affect the ability to mount a protective response. These observations support the idea, previously suggested by others (62), that the susceptibility of BALB/c mice to Brucella infection is not due to the development of a Th2 response but is due to an intrinsically lower ability to mount a Th1 response.
The recently characterized Th17 response has been shown to contribute to host defense against several extracellular pathogens, such as Klebsiella pneumoniae (26, 73), Citrobacter rodentium (45), and Candida albicans (28), as well as against intracellular microorganisms, such as Listeria monocytogenes, Salmonella enterica, and Mycobacterium tuberculosis (14, 33, 68). The role of Th17 during Brucella infection has not been extensively explored. Two studies have reported that IL-17A neutralization (57) or the absence of IL-17Rα (65) did not at all influence the subsistence and growth of Brucella at 1 month and 1 week postinfection, respectively. However, another recent study has demonstrated a role for IL-17-producing T cells in osteoclastogenesis during murine brucellosis (21). The Th17 response implicates the coordinated production of IL-17, IL-21, and IL-22 by CD4+ T cells following IL-23 secretion by antigen-presenting cells (for a review, see reference 5). In this study, we compared the ability of wild-type, IL-12p35−/−, IL-12p40−/− (IL-12/23−/−), IL-17Rα−/−, IL-21−/−, and IL-22−/− mice to control Brucella infection. Only IL-12p35−/− and IL-12p40−/− mice displayed susceptibility to Brucella infection, suggesting that components of the Th17 response are not essential to bacterial clearance. However, we observed significant transitory higher susceptibility in IL-12p40−/− mice than IL-12p35−/− mice. This difference suggests cooperation between the Th1 and Th17 responses during Brucella infection. A similar phenomenon during Mycobacterium tuberculosis infection has been reported previously (9, 34). When IL-12p70 is available, IL-23 is dispensable for protection. In the absence of IL-12p70, the lack of IL-23 enhanced susceptibility to M. tuberculosis. These compensations have also been described for Salmonella enterica (63) and Toxoplasma gondii (40).
In summary, our study demonstrates the key role played by IFN-γ-producing Th1 CD4+ T cells in the control of Brucella infection and the negligible importance of CD8+ T cells or humoral immunity in the clearance of bacteria during primary infection. We also showed that an IL-4-dependent Th2 response does not develop or play a negative role during the course of infection. However, the Th17 response could partially compensate for the lack of a Th1 response. These results could potentially improve our ability to develop a protective vaccine or therapeutic treatment against brucellosis. They suggest that favoring IL-12 production during Brucella vaccination, for example, by targeting CD8+ dendritic cells, should lead to a better induction and persistence of IFN-γ-producing CD4+ T cells and concomitantly allow better protection against Brucella infection.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Fonds National de la Recherche Scientifique (FNRS) (convention FRSM FNRS 3.4.600.06.F, Belgium), the Communauté Française de Belgique (Action de Recherches Concertées 08/13-015), the Fonds Emile Defay (Belgium), and the Fonds Van Buuren (Belgium). E.M. is a research associate from the FRS-FNRS (Belgium). B.R. is supported by the Agence Nationale pour la Recherche (ANR 2007 MIME-103-02), the Fondation pour la Recherche Médicale (FRM allergy DAL 2007 0822007), the Fonds Européen de Développement Régional” (FEDER Asthme 1575-32168), and Le Studium, Orleans, France. M.-A.V. holds a PhD grant from the FRS-FNRS (Belgium).
We thank S. Goriely (Université Libre de Bruxelles, Brussels, Belgium) and Michel Y. Braun (Université Libre de Bruxelles, Brussels, Belgium) for providing us with RAG1−/− and RAGγc−/− mice, respectively.
Footnotes
Published ahead of print 24 September 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Araya LN, Elzer PH, Rowe GE, Enright FM, Winter AJ. 1989. Temporal development of protective cell-mediated and humoral immunity in BALB/c mice infected with Brucella abortus. J. Immunol. 143:3330–3337 [PubMed] [Google Scholar]
- 2. Arenas-Gamboa AM, Rice-Ficht AC, Fan Y, Kahl-McDonagh MM, Ficht TA. 2012. Extended safety and efficacy studies of the attenuated Brucella vaccine candidates 16 M(Delta)vjbR and S19(Delta)vjbR in the immunocompromised IRF-1−/− mouse model. Clin. Vaccine Immunol. 19:249–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baldwin CL, Goenka R. 2006. Host immune responses to the intracellular bacteria Brucella: does the bacteria instruct the host to facilitate chronic infection? Crit. Rev. Immunol. 26:407–442 [DOI] [PubMed] [Google Scholar]
- 4. Baldwin CL, Parent M. 2002. Fundamentals of host immune response against Brucella abortus: what the mouse model has revealed about control of infection. Vet. Microbiol. 90:367–382 [DOI] [PubMed] [Google Scholar]
- 5. Bettelli E, Korn T, Kuchroo VK. 2007. Th17: the third member of the effector T cell trilogy. Curr. Opin. Immunol. 19:652–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brandao AP, et al. 2012. Host susceptibility to Brucella abortus infection is more pronounced in IFN-gamma knockout than IL-12/beta2-microglobulin double-deficient mice. Clin. Dev. Immunol. 2012:589494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Colucci F, et al. 1999. Dissecting NK cell development using a novel alymphoid mouse model: investigating the role of the c-abl proto-oncogene in murine NK cell differentiation. J. Immunol. 162:2761–2765 [PubMed] [Google Scholar]
- 8. Cook L, et al. 2008. Evidence that CD8+ dendritic cells enable the development of gammadelta T cells that modulate airway hyperresponsiveness. J. Immunol. 181:309–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cooper AM, et al. 2002. Mice lacking bioactive IL-12 can generate protective, antigen-specific cellular responses to mycobacterial infection only if the IL-12 p40 subunit is present. J. Immunol. 168:1322–1327 [DOI] [PubMed] [Google Scholar]
- 10. Copin R, De Baetselier P, Carlier Y, Letesson JJ, Muraille E. 2007. MyD88-dependent activation of B220-CD11b+LY-6C+ dendritic cells during Brucella melitensis infection. J. Immunol. 178:5182–5191 [DOI] [PubMed] [Google Scholar]
- 11. Copin R, et al. 2012. In situ microscopy analysis reveals local innate immune response developed around Brucella infected cells in resistant and susceptible mice. PLoS Pathog. 8:e1002575 doi:10.1371/journal.ppat.1002575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Corbel MJ. 1997. Brucellosis: an overview. Emerg. Infect. Dis. 3:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cosgrove D, et al. 1991. Mice lacking MHC class II molecules. Cell 66:1051–1066 [DOI] [PubMed] [Google Scholar]
- 14. Curtis MM, Way SS. 2009. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology 126:177–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cutler SJ, Whatmore AM, Commander NJ. 2005. Brucellosis—new aspects of an old disease. J. Appl. Microbiol. 98:1270–1281 [DOI] [PubMed] [Google Scholar]
- 16. Das G, Sheridan S, Janeway CA., Jr 2001. The source of early IFN-gamma that plays a role in Th1 priming. J. Immunol. 167:2004–2010 [DOI] [PubMed] [Google Scholar]
- 17. Elzer PH, et al. 1994. Antibody-mediated protection against Brucella abortus in BALB/c mice at successive periods after infection: variation between virulent strain 2308 and attenuated vaccine strain 19. Immunology 82:651–658 [PMC free article] [PubMed] [Google Scholar]
- 18. Fernandes DM, Baldwin CL. 1995. Interleukin-10 downregulates protective immunity to Brucella abortus. Infect. Immun. 63:1130–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fernandes DM, Jiang X, Jung JH, Baldwin CL. 1996. Comparison of T cell cytokines in resistant and susceptible mice infected with virulent Brucella abortus strain 2308. FEMS Immunol. Med. Microbiol. 16:193–203 [DOI] [PubMed] [Google Scholar]
- 20. Ficht TA, Kahl-McDonagh MM, Arenas-Gamboa AM, Rice-Ficht AC. 2009. Brucellosis: the case for live, attenuated vaccines. Vaccine 27(Suppl 4):D40–D43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giambartolomei GH, Scian R, Acosta-Rodriguez E, Fossati CA, Delpino MV. 2012. Brucella abortus-infected macrophages modulate T lymphocytes to promote osteoclastogenesis via IL-17. Am. J. Pathol. 181:887–896 [DOI] [PubMed] [Google Scholar]
- 22. Gill N, Finlay BB. 2011. The gut microbiota: challenging immunology. Nat. Rev. Immunol. 11:636–637 [DOI] [PubMed] [Google Scholar]
- 23. Godfroid J, et al. 2005. From the discovery of the Malta fever's agent to the discovery of a marine mammal reservoir, brucellosis has continuously been a re-emerging zoonosis. Vet. Res. 36:313–326 [DOI] [PubMed] [Google Scholar]
- 24. Goenka R, Parent MA, Elzer PH, Baldwin CL. 2011. B cell-deficient mice display markedly enhanced resistance to the intracellular bacterium Brucella abortus. J. Infect. Dis. 203:1136–1146 [DOI] [PubMed] [Google Scholar]
- 25. Grillo MJ, Blasco JM, Gorvel JP, Moriyon I, Moreno E. 2012. What have we learned from brucellosis in the mouse model? Vet. Res. 43:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Happel KI, et al. 2005. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J. Exp. Med. 202:761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hemmers S, Mowen KA. 2009. T(H)2 bias: Mina tips the balance. Nat. Immunol. 10:806–808 [DOI] [PubMed] [Google Scholar]
- 28. Huang W, Na L, Fidel PL, Schwarzenberger P. 2004. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J. Infect. Dis. 190:624–631 [DOI] [PubMed] [Google Scholar]
- 29. Izadjoo MJ, et al. 2000. Impaired control of Brucella melitensis infection in Rag1-deficient mice. Infect. Immun. 68:5314–5320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kawabe T, et al. 1994. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1:167–178 [DOI] [PubMed] [Google Scholar]
- 31. Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11:115–122 [DOI] [PubMed] [Google Scholar]
- 32. Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD. 2003. Interleukin-4 and interleukin-13 signaling connections maps. Science 300:1527–1528 [DOI] [PubMed] [Google Scholar]
- 33. Khader SA, et al. 2007. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 8:369–377 [DOI] [PubMed] [Google Scholar]
- 34. Khader SA, et al. 2005. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J. Immunol. 175:788–795 [DOI] [PubMed] [Google Scholar]
- 35. Kim J, et al. 2008. Beta 2-microglobulin deficient mice catabolize IgG more rapidly than FcRn- alpha-chain deficient mice. Exp. Biol. Med. (Maywood) 233:603–609 [DOI] [PubMed] [Google Scholar]
- 36. Kitamura D, Roes J, Kuhn R, Rajewsky K. 1991. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 350:423–426 [DOI] [PubMed] [Google Scholar]
- 37. Ko J, Gendron-Fitzpatrick A, Splitter GA. 2002. Susceptibility of IFN regulatory factor-1 and IFN consensus sequence binding protein-deficient mice to brucellosis. J. Immunol. 168:2433–2440 [DOI] [PubMed] [Google Scholar]
- 38. Kreymborg K, et al. 2007. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J. Immunol. 179:8098–8104 [DOI] [PubMed] [Google Scholar]
- 39. Leonard WJ, Spolski R. 2005. Interleukin-21: a modulator of lymphoid proliferation, apoptosis and differentiation. Nat. Rev. Immunol. 5:688–698 [DOI] [PubMed] [Google Scholar]
- 40. Lieberman LA, et al. 2004. IL-23 provides a limited mechanism of resistance to acute toxoplasmosis in the absence of IL-12. J. Immunol. 173:1887–1893 [DOI] [PubMed] [Google Scholar]
- 41. Locksley RM, et al. 1991. Induction of Th1 and Th2 CD4+ subsets during murine Leishmania major infection. Res. Immunol. 142:28–32 [DOI] [PubMed] [Google Scholar]
- 42. Locksley RM, Heinzel FP, Sadick MD, Holaday BJ, Gardner KD., Jr 1987. Murine cutaneous leishmaniasis: susceptibility correlates with differential expansion of helper T-cell subsets. Ann. Inst. Pasteur Immunol. 138:744–749 [DOI] [PubMed] [Google Scholar]
- 43. Locksley RM, Scott P. 1991. Helper T-cell subsets in mouse leishmaniasis: induction, expansion and effector function. Immunol. Today 12:A58–A61 [DOI] [PubMed] [Google Scholar]
- 44. Magram J, et al. 1996. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity 4:471–481 [DOI] [PubMed] [Google Scholar]
- 45. Mangan PR, et al. 2006. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441:231–234 [DOI] [PubMed] [Google Scholar]
- 46. Martirosyan A, Moreno E, Gorvel JP. 2011. An evolutionary strategy for a stealthy intracellular Brucella pathogen. Immunol. Rev. 240:211–234 [DOI] [PubMed] [Google Scholar]
- 47. Mattner F, et al. 1996. Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur. J. Immunol. 26:1553–1559 [DOI] [PubMed] [Google Scholar]
- 48. Mielke ME. 1991. T cell subsets in granulomatous inflammation and immunity to L. Monocytogenes and B. abortus. Behring Inst. Mitt., p 99–111 [PubMed] [Google Scholar]
- 49. Mombaerts P, et al. 1992. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68:869–877 [DOI] [PubMed] [Google Scholar]
- 50. Montaraz JA, Winter AJ. 1986. Protection against Brucella abortus in mice with O-polysaccharide-specific monoclonal antibodies. Infect. Immun. 51:961–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Murphy EA, Parent M, Sathiyaseelan J, Jiang X, Baldwin CL. 2001. Immune control of Brucella abortus 2308 infections in BALB/c mice. FEMS Immunol. Med. Microbiol. 32:85–88 [DOI] [PubMed] [Google Scholar]
- 52. Murphy EA, Sathiyaseelan J, Parent MA, Zou B, Baldwin CL. 2001. Interferon-gamma is crucial for surviving a Brucella abortus infection in both resistant C57BL/6 and susceptible BALB/c mice. Immunology 103:511–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nakae S, et al. 2002. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 17:375–387 [DOI] [PubMed] [Google Scholar]
- 54. Oliveira SC, Giambartolomei GH, Cassataro J. 2011. Confronting the barriers to develop novel vaccines against brucellosis. Expert Rev. Vaccines 10:1291–1305 [DOI] [PubMed] [Google Scholar]
- 55. Oliveira SC, Splitter GA. 1995. CD8+ type 1 CD44hi CD45 RBlo T lymphocytes control intracellular Brucella abortus infection as demonstrated in major histocompatibility complex class I- and class II-deficient mice. Eur. J. Immunol. 25:2551–2557 [DOI] [PubMed] [Google Scholar]
- 56. Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV. 2006. The new global map of human brucellosis. Lancet Infect. Dis. 6:91–99 [DOI] [PubMed] [Google Scholar]
- 57. Pasquevich KA, et al. 2011. An oral vaccine based on U-Omp19 induces protection against B. abortus mucosal challenge by inducing an adaptive IL-17 immune response in mice. PLoS One 6:e16203 doi:10.1371/journal.pone.0016203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pavlov H, Hogarth M, McKenzie IF, Cheers C. 1982. In vivo and in vitro effects of monoclonal antibody to Ly antigens on immunity to infection. Cell. Immunol. 71:127–138 [DOI] [PubMed] [Google Scholar]
- 59. Perkins SD, Smither SJ, Atkins HS. 2010. Towards a Brucella vaccine for humans. FEMS Microbiol. Rev. 34:379–394 [DOI] [PubMed] [Google Scholar]
- 60. Rolan HG, Tsolis RM. 2007. Mice lacking components of adaptive immunity show increased Brucella abortus virB mutant colonization. Infect. Immun. 75:2965–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rolan HG, Xavier MN, Santos RL, Tsolis RM. 2009. Natural antibody contributes to host defense against an attenuated Brucella abortus virB mutant. Infect. Immun. 77:3004–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sathiyaseelan J, et al. 2006. Treatment of Brucella-susceptible mice with IL-12 increases primary and secondary immunity. Cell. Immunol. 243:1–9 [DOI] [PubMed] [Google Scholar]
- 63. Schulz SM, et al. 2008. Protective immunity to systemic infection with attenuated Salmonella enterica serovar Enteritidis in the absence of IL-12 is associated with IL-23-dependent IL-22, but not IL-17. J. Immunol. 181:7891–7901 [DOI] [PubMed] [Google Scholar]
- 64. Shahinian A, et al. 1993. Differential T cell costimulatory requirements in CD28-deficient mice. Science 261:609–612 [DOI] [PubMed] [Google Scholar]
- 65. Skyberg JA, et al. 2011. Murine and bovine gammadelta T cells enhance innate immunity against Brucella abortus infections. PLoS One 6:e21978 doi:10.1371/journal.pone.0021978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stevens MG, Pugh GW, Jr, Tabatabai LB. 1992. Effects of gamma interferon and indomethacin in preventing Brucella abortus infections in mice. Infect. Immun. 60:4407–4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ubeda C, et al. 2012. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J. Exp. Med. 209:1445–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Umemura M, et al. 2007. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J. Immunol. 178:3786–3796 [DOI] [PubMed] [Google Scholar]
- 69. Vance RE, Isberg RR, Portnoy DA. 2009. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6:10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Van Kaer L, Ashton-Rickardt PG, Ploegh HL, Tonegawa S. 1992. TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4-8+ T cells. Cell 71:1205–1214 [DOI] [PubMed] [Google Scholar]
- 71. Weiss DS, Takeda K, Akira S, Zychlinsky A, Moreno E. 2005. MyD88, but not Toll-like receptors 4 and 2, is required for efficient clearance of Brucella abortus. Infect. Immun. 73:5137–5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Winter AJ, Duncan JR, Santisteban CG, Douglas JT, Adams LG. 1989. Capacity of passively administered antibody to prevent establishment of Brucella abortus infection in mice. Infect. Immun. 57:3438–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ye P, et al. 2001. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am. J. Respir. Cell Mol. Biol. 25:335–340 [DOI] [PubMed] [Google Scholar]
- 74. Zhan Y, Cheers C. 1993. Endogenous gamma interferon mediates resistance to Brucella abortus infection. Infect. Immun. 61:4899–4901 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.