

Abstract
Listeria monocytogenes is a Gram-positive intracellular pathogen that causes meningitis and septicemia in immunocompromised individuals and spontaneous abortion in pregnant women. The innate immune response against L. monocytogenes is primarily mediated by neutrophils and monocytes. Interleukin-23 (IL-23) is an important proinflammatory cytokine well known for its role in neutrophil recruitment in various infectious and autoimmune diseases. We have previously shown that IL-23 is required for host resistance against L. monocytogenes and for neutrophil recruitment to the liver, but not the spleen, during infection. Despite efficient neutrophil recruitment to the spleen, IL-23p19 knockout (KO) mice have an increased bacterial burden in this organ, suggesting that IL-23 may regulate the recruitment/function of another cell type to the spleen. In this study, we show that specific depletion of neutrophils abrogated the differences in bacterial burdens in the livers but not the spleens of C57BL/6 (B6) and IL-23p19 KO mice. Interestingly, L. monocytogenes-infected IL-23p19 KO mice had fewer monocytes in the spleen than B6 mice, as well as a reduction in the monocyte-recruiting chemokines CCL2 and CCL7. Additionally, the overall concentrations of tumor necrosis factor alpha (TNF-α) and nitric oxide (NO•), as well as the percentages and total numbers of monocytes producing TNF-α and NO•, were reduced in IL-23p19 KO mice compared to levels in B6 mice, leading to increased bacterial burdens in the spleens of L. monocytogenes-infected IL-23p19 KO mice. Collectively, our data establish that IL-23 is required for the optimal recruitment of TNF-α- and NO•-producing inflammatory monocytes, thus revealing a novel mechanism by which this proinflammatory cytokine provides protection against bacterial infection.
INTRODUCTION
IL-23 is a heterodimeric proinflammatory cytokine secreted by activated macrophages and dendritic cells (14, 26). The innate production of interleukin-23 (IL-23) enhances inflammatory responses by regulating the generation and maintenance of IL-17-producing T cells (14, 27). The IL-17 family members (IL-17A and IL-17F) promote the mobilization of neutrophils through the induction of cytokines and chemokines, including IL-6, granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), CXCL1, CXCL2, and CXCL8 (17). Studies in mice lacking IL-23 have demonstrated that this cytokine is not only involved in inducing protective immune responses against pathogens but can also be detrimental during inflammatory and autoimmune disorders (19, 23, 25, 42, 43).
Protection mediated by the IL-23/IL-17 axis has been studied with several pathogens, including Klebsiella pneumoniae (13), Citrobacter rodentium (22), Salmonella enterica (11), Mycobacterium tuberculosis (16), and Toxoplasma gondii (20). Our lab has recently established that the IL-23/IL-17 axis is important during systemic infection with Listeria monocytogenes (24). L. monocytogenes is a Gram-positive intracellular pathogen that causes spontaneous abortions in pregnant women and septicemia and meningitis in immunocompromised individuals. L. monocytogenes is a widely accepted model pathogen to study host-pathogen immune interactions. In the mouse model of systemic infection, L. monocytogenes primarily localizes to the spleen and liver, triggering a local immune response (29). Complete eradication of L. monocytogenes is dependent on CD8+ T cell-mediated immunity (18, 32); however, the initial innate immune response is required for containing the bacterial burden, thus influencing adaptive immunity and the outcome of infection.
Systemic infection with L. monocytogenes induces a robust innate immune response primarily mediated by neutrophils and monocytes (29). Inflammatory mediators, including tumor necrosis factor alpha (TNF-α) and nitric oxide (NO•), produced by these cell types are required for antilisterial defense, and mice lacking TNF-α or inducible nitric oxide synthase (iNOS) display increased susceptibility to L. monocytogenes infection (30, 41). Our recent data indicate that neutrophils are required for protection against L. monocytogenes in the liver at all doses tested but are required for clearance of L. monocytogenes from the spleen only at high infectious doses. Depletion of both neutrophils and monocytes leads to increased susceptibility compared to depletion of neutrophils alone, suggesting that monocytes are also required for protection against L. monocytogenes (5).
Studies on immune-mediated protection in the spleen have shown that L. monocytogenes induces the recruitment of inflammatory monocytes, also known as TNF-α- and iNOS-producing dendritic cells (TipDCs) (35). These Ly6Chi CD11bint CCR2+ (where Ly6Chi indicates high levels of Ly6C expression and CD11bint indicates intermediate levels of CD11b) inflammatory monocytes emigrate from the bone marrow to the blood through the binding of CCR2 ligands, CCL2 (MCP-1) and CCL7 (MCP-3) (15, 34). It has been shown that inflammatory monocytes produce TNF-α and NO• at the site of infection, thereby contributing to the eradication of L. monocytogenes (35). Additionally, the absence of CCR2, CCL2, or CCL7 leads to diminished recruitment of inflammatory monocytes to the spleen and impaired clearance of L. monocytogenes (15, 35). Thus, the protective response against L. monocytogenes in the spleen may be predominantly mediated by inflammatory monocytes.
We have previously demonstrated that mice lacking IL-23 (IL-23p19 KO, where KO indicates knockout) are more susceptible to L. monocytogenes infection than C57BL/6 (B6) mice. Furthermore, IL-23p19 KO mice have higher bacterial burdens in both the spleen and liver than B6 mice (24). In the liver, neutrophil-mediated protection is required for resistance against L. monocytogenes infection (5), and the recruitment of neutrophils is regulated by the IL-23/IL-17 axis (24). Surprisingly, despite efficient neutrophil recruitment to the spleens of IL-23p19 KO mice, bacterial burdens increased, suggesting that a different mechanism for IL-23-mediated immune protection might exist in this organ. However, this notion has not been previously investigated. In the current study, we demonstrate that during a systemic L. monocytogenes infection, IL-23 is required for the optimal recruitment of inflammatory monocytes to the spleen, as well as the optimal production of TNF-α and NO•.
MATERIALS AND METHODS
Mice.
B6 mice were purchased from the National Cancer Institute. IL-23p19 KO mice were originally obtained from Nico Ghilardi and backcrossed on a B6 background for at least 8 generations (10). Age (6 to 12 weeks)- and gender-matched mice were used for all experiments. Mice were provided food and water ad libitum and were housed in sterile microisolator cages with sterile bedding at the University of North Texas Health Science Center animal facility accredited by the American Association for the Accreditation of Laboratory Animal Care. Animal studies were performed under the approval of the Institutional Animal Care and Use Committee at the University of North Texas Health Science Center.
L. monocytogenes infection and quantification of bacterial burden.
L. monocytogenes 10403s was grown on brain heart infusion (BHI) agar plates, and virulent stocks were maintained by passage through B6 mice. For infections, log-phase cultures of L. monocytogenes were grown in BHI broth, washed twice, and diluted to the required concentration in sterile phosphate-buffered saline (PBS). Unless otherwise stated, mice were intravenously (i.v.) injected with ∼ 104 L. monocytogenes CFU. To determine bacterial burdens, spleen and liver homogenates were resuspended in sterile double-distilled water and serially diluted, and 50 μl of each dilution was plated on BHI agar plates. After 24 h of incubation at 37°C, the number of CFU of L. monocytogenes was counted.
Neutrophil depletions.
Neutrophils were depleted using anti-Ly6G antibody (clone 1A8) (Bio-X-Cell, West Lebanon, NH), which has been previously shown to exclusively and completely deplete neutrophils (5). For all depletion experiments, mice were injected intraperitoneally with 500 μg of 1A8 antibody or isotype control antibody at 1 day prior to infection and at 3 days postinfection (p.i.).
Organ processing and tissue culture.
For collecting serum, blood was obtained from the mice, kept on ice for 8 h, and centrifuged at 18,000 × g for 30 min. Peripheral blood leukocytes were isolated from peripheral blood collected from the lateral tail vein as previously described (24). Splenocytes and liver leukocytes were isolated as previously described (24). Bone marrow cells were obtained as previously described (39). Briefly, mouse femurs and tibiae were flushed with Hanks balanced salt solution (HBSS) containing 2% fetal bovine serum (FBS) (Atlanta Biologicals, Norcoss, GA), and the isolated bone marrow cells were incubated in HBSS containing 2% FBS, 3 mg/ml collagenase D (Roche Diagnostics, Indianapolis, IN), and 20 U/ml DNase I (Roche Diagnostics, Indianapolis, IN) for 60 min at 37°C; red blood cells were lysed in Tris-ammonium chloride (pH 7.2). For culture, splenocytes were resuspended in phenol red-free Dulbecco's modified Eagle medium supplemented with 10% FBS, l-glutamine, vitamins, and penicillin-streptomycin (Invitrogen-Gibco, Carlsbad, CA) (2). The splenocytes were cultured overnight with heat-killed L. monocytogenes (HKLM) at a multiplicity of infection (MOI) of 50:1 or left unstimulated. Where indicated in the figure legends, splenocytes were cultured overnight with 10 ng/ml recombinant IL-23 (rIL-23) (Biolegend, San Diego, CA). Cell cultures were performed at 37°C in humidified air containing 5% CO2. For organ homogenates, spleens were homogenized in ice-cold PBS containing 0.01% Triton X-100, centrifuged at 10,000 × g for 30 min, and supernatants were collected for analysis (15).
Flow cytometry.
For cell staining, the following antibodies were used: anti-CD16/CD32 (2.4G2), anti-Ly6G-phycoerythrin ([PE] 1A8), anti-CD11b-biotin or PE-Cy7 (M1/70), anti-TNF-α PE-Cy7 (MP6-XT22) (all from BD Biosciences, San Diego, CA), anti-Ly6C Alexa Fluor 647 (HK1.4; Biolegend, San Diego, CA), streptavidin PE-Texas Red (PE-TR) (Caltag Laboratories/Invitrogen, Carlsbad, CA), goat-anti-iNOS (M-19; Santa Cruz Biotechnology), goat IgG isotype control and anti-goat IgG-PE (Jackson ImmunoResearch Laboratories, West Grove, PA), and anti-CD45 energy-coupled dye (ECD) (I3/2.3; Beckman Coulter, Brea, CA). For direct ex vivo staining, cells were incubated with saturating amounts of antibodies in staining buffer (PBS plus 2% FBS and 0.1% sodium azide) for 15 min at 4°C. After cell surface staining, the cells were fixed in BD stabilizing fixative. To measure intracellular TNF-α, splenocytes were cultured with HKLM in the presence of GolgiPlug containing brefeldin A (BD Pharmingen) for 3 h. The cells were stained for cell surface markers and then fixed and permeabilized for 20 min at 4°C with a kit from BD Biosciences. After cells were washed with permeabilization buffer, they were stained with saturating amounts of anti-TNF-α for 15 min at 4°C. For iNOS staining, cells were stained for surface markers, fixed and permeabilized, and incubated with saturating amounts of goat anti-iNOS or goat isotype control antibody, followed by anti-goat IgG-PE for 15 min at 4°C. Data were acquired and analyzed on a Beckman Coulter FC500 flow cytometer.
Quantification of TNF-α, CCL2, CCL7, and NO·.
TNF-α enzyme-linked immunosorbent assays (ELISAs) were performed with antibody pairs and recombinant TNF-α from eBioscience (San Diego, CA). To measure chemokine levels, the following ELISA kits were used: a CCL2 kit from BD Biosciences and CCL7 kit from Bender Med-Systems (Burlingame, CA). NO• concentrations were determined with a Nitric Oxide Quantitation Kit (Active Motif, Carlsbad, CA). Cytokine/chemokine levels were determined by comparison with standard curves, using a Biotek EL808 spectrophotometer.
Statistical analyses.
Analyses of variance (ANOVAs) or Student's t tests were conducted on the data where appropriate. For posthoc analyses, Bonferroni t tests were used. L. monocytogenes CFU data were log transformed prior to analysis and are represented as such in the figures. The survival curves between groups were compared using Kaplan-Meier plots and log rank tests. A P value of 0.05 or less was considered significant in all cases.
RESULTS
Depletion of neutrophils further increases the susceptibility of IL-23p19 KO mice to L. monocytogenes infection.
We have recently established that IL-23 is required for protection against systemic L. monocytogenes infection and that IL-23p19 KO mice have reduced recruitment of neutrophils to the liver compared to B6 mice (24). In order to determine if the reduced neutrophil recruitment to the liver results in increased susceptibility of IL-23p19 KO mice, neutrophils were depleted from B6 and IL-23p19 KO mice, and a survival study was performed. Neutrophil depletions were performed using an anti-Ly6G antibody that is highly specific for neutrophils (5, 7), and depletions were confirmed in the spleen, liver, and blood by performing flow cytometric analysis (see Fig. S1 in the supplemental material). In agreement with our previous publications, there was a reduced recruitment of neutrophils to the livers, but not the spleens, of IL-23p19 KO mice compared to B6 mice (24), and treatment with the anti-Ly6G antibody efficiently depleted neutrophils from the spleen, liver, and blood in both B6 and IL-23p19 KO mice (5) (see Fig. S1). As shown in Fig. 1, there were no differences in survival rates between neutrophil-depleted B6 mice, isotype-treated B6 mice, and isotype-treated IL-23p19 KO mice. However, the percentage of neutrophil-depleted IL-23p19 KO mice that succumbed to infection (75%) was much higher than that of the other groups. These data indicate that depletion of neutrophils in IL-23p19 KO mice increases their susceptibility to infection. Importantly, the fact that neutrophil-depleted B6 and IL-23p19 KO mice do not show equivalent susceptibilities to L. monocytogenes suggests that differences other than deficient neutrophil recruitment must exist between B6 and IL-23p19 KO mice.
Fig 1.
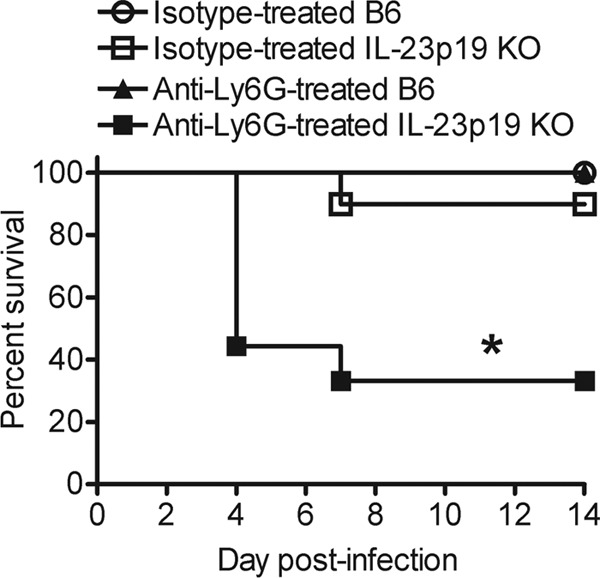
Depletion of neutrophils increases the susceptibility of IL-23p19 KO mice to L. monocytogenes infection. B6 and IL-23p19 KO mice were infected with ∼104 L. monocytogenes CFU. These mice were treated with anti-Ly6G or isotype antibody at 1 day prior to infection and at 3 days p.i. The susceptibility of the mice was monitored for 14 days p.i. These data are combined from two independent experiments (n = 9 to 10/group). A log rank analysis was performed to determine statistical differences between groups. An asterisk (*) indicates a significant difference from all the other groups.
Depletion of neutrophils abrogates the differences in bacterial burdens in the livers, but not the spleens, of B6 and IL-23p19 KO mice during L. monocytogenes infection.
We have previously demonstrated that IL-23p19 KO mice have higher bacterial burdens in the spleen and liver than B6 mice at day 5 post-L. monocytogenes infection (24). To investigate if the increased bacterial burden in the IL-23p19 KO mice is due to deficient neutrophil recruitment, spleen and liver CFU counts from L. monocytogenes-infected neutrophil-depleted B6 and IL-23p19 KO mice were determined. Because most of the neutrophil-depleted IL-23p19 KO mice succumb to infection by day 4 p.i. at ∼104 L. monocytogenes CFU, a sublethal dose of ∼3,000 L. monocytogenes CFU was used for this study. As shown in Fig. 2, in accordance with our previous study (24), the isotype-treated IL-23p19 KO mice had higher CFU counts in the spleen and liver than the isotype-treated B6 mice. Additionally, depletion of neutrophils in both the B6 and IL-23p19 KO mice compromised their ability to efficiently clear bacteria, compared to the respective isotype-treated control mice (5). However, it is important to note that the depletion of neutrophils resulted in significantly higher bacterial burdens in the spleens of IL-23p19 KO mice than in B6 mice, in contrast to the livers, where depletion equalized the bacterial burdens between the B6 and IL-23p19 KO mice (Fig. 2). This enhanced bacterial burden in the spleens of neutrophil-depleted IL-23p19 KO mice could lead to increased susceptibility of these mice to L. monocytogenes infection. Collectively, these data strengthen our previous findings that the reduced neutrophil recruitment to the livers of IL-23p19 KO mice is causal for the increased L. monocytogenes burden in this organ (24). However, in the spleen, IL-23 may regulate the recruitment or function of an additional cell type, which may account for the differences in spleen CFU counts between L. monocytogenes-infected IL-23p19 KO and B6 mice.
Fig 2.
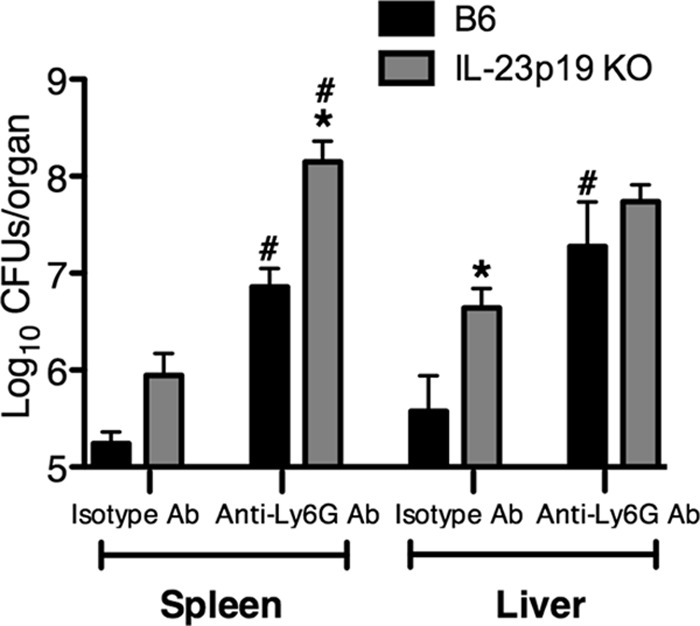
Depletion of neutrophils eliminates the differences in bacterial burden in the livers, but not the spleens, of B6 and IL-23p19 KO mice during L. monocytogenes infection. B6 and IL-23p19 KO mice were infected with ∼3,000 L. monocytogenes CFU. The mice were treated with anti-Ly6G or isotype antibody (Ab) at 1 day prior to infection and at 3 days p.i. At day 5 p.i., spleens and livers were harvested, and the bacterial CFU were enumerated in the spleen and the liver. A two-way ANOVA was performed to determine statistical differences between groups (*, significantly different from B6 mice; #, significantly different from isotype-treated controls). All data are expressed as means ± standard errors of the means (n = 5/group). These data are representative of two independent experiments.
IL-23 is required for the optimal recruitment of monocytes to the spleen during L. monocytogenes infection.
Inflammatory monocytes are recruited to the spleen during L. monocytogenes infection and are required for resistance (35). To investigate whether IL-23 is required for the recruitment of monocytes, mice were left uninfected or were infected for 1 or 3 days with L. monocytogenes, and percentages of monocytes in the blood, spleen, and liver were determined by flow cytometric analysis. Prior to infection, monocyte percentages were decreased in the blood of IL-23p19 KO mice compared to levels in B6 mice (Fig. 3A). However, there were no differences in monocyte percentages in the spleens or livers (Fig. 3A) or in total numbers of monocytes in the spleens (Fig. 3B) between uninfected B6 and IL-23p19 KO mice. Upon L. monocytogenes infection, there was a reduction in the monocyte percentages in the blood, spleens, and livers of IL-23p19 KO mice compared to levels in B6 mice at day 1 p.i. (Fig. 3C). Likewise, the total numbers of monocytes in the spleens of IL-23p19 KO mice was reduced compared to levels in B6 mice (Fig. 3D). However, at day 3 p.i., the monocyte percentages did not differ in the blood and livers of B6 and IL-23p19 KO mice, but both the percentages and total numbers of monocytes in the spleens were reduced in the IL-23p19 KO mice compared to levels in B6 mice (Fig. 3E and F). The representative dot plots (Fig. 3G) from day 1 L. monocytogenes-infected splenocytes depict monocytes that were identified based on the expression of the surface markers Ly6C and CD11b as Ly6Chi CD11bint cells, which were clearly reduced in the spleens of L. monocytogenes-infected IL-23p19 KO mice. In order to confirm that IL-23 was required for monocyte recruitment at lower infectious doses, B6 and IL-23p19 KO mice were infected with the same dose (∼3,000 CFU) of L. monocytogenes used in the experiment shown in Fig. 2. Indeed, mice lacking IL-23 showed reduced monocyte recruitment to the spleen at days 1 and 3 p.i. compared to B6 mice (see Fig. S2 in the supplemental material). These results indicate that IL-23 is required for the optimal recruitment of monocytes to the spleen during infection with L. monocytogenes.
Fig 3.
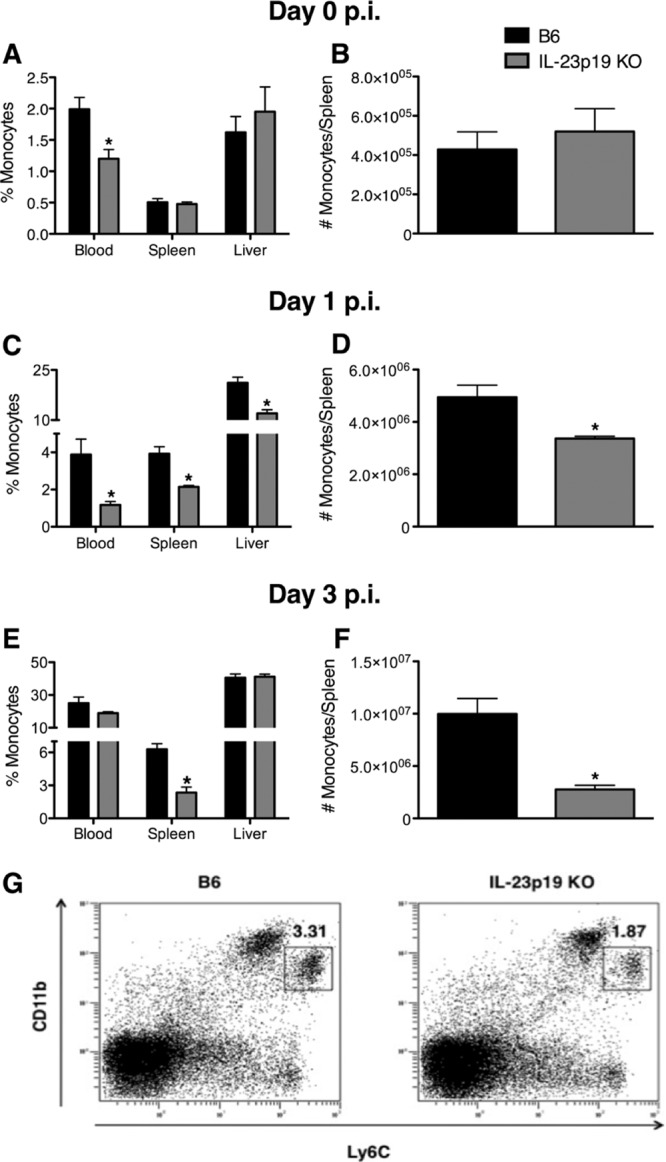
IL-23 is required for the optimal recruitment of monocytes during L. monocytogenes infection. B6 and IL-23p19 KO mice were infected with ∼104 L. monocytogenes CFU. The monocyte percentages in the blood, spleen, and liver were determined at day 0 (A), day 1 (C), and day 3 p.i. (E). The monocyte numbers in the spleen were examined at day 0 (B), day 1 (D), and day 3 p.i. (F). Monocytes were identified as cells expressing high levels of Ly6C and intermediate levels of CD11b (Ly6Chi CD11bint) in splenocytes from day 1 L. monocytogenes-infected mice (G). A t test was performed to determine statistical differences between groups (*, significantly different from B6 mice). All data are expressed as means ± standard errors of the means (n = 5/group). These data are representative of two independent experiments.
IL-23 is required for the optimal production of monocyte-recruiting chemokines during L. monocytogenes infection.
Reports have identified CCL2 and CCL7 as the key chemokines responsible for the emigration of monocytes from the bone marrow to the blood during L. monocytogenes infection (15, 34). Therefore, B6 and IL-23p19 KO mice were infected for 1 day with L. monocytogenes, and CCL2 and CCL7 concentrations were measured in serum, spleen homogenates, and supernatants from splenocytes and bone marrow cells stimulated overnight with HKLM or left unstimulated. There were no significant differences in CCL2 production between uninfected B6 and IL-23p19 KO mice in the serum, spleen homogenates, or overnight-culture supernatants from spleen and bone marrow (see Fig. S3 in the supplemental material). However, upon L. monocytogenes infection, there was a significant reduction in the amount of CCL2 and CCL7 in the serum of infected IL-23p19 KO mice compared to that in B6 mice (Fig. 4A). Additionally, CCL2 concentrations were reduced in the spleen homogenates from IL-23p19 KO mice compared to those from B6 mice (Fig. 4B). There were no significant differences between B6 and IL-23p19 KO mice in the levels of CCL2 in the serum and spleen homogenates or in CCL7 levels in the serum at day 3 p.i. (data not shown). Although the production of CCL2 did not differ between B6 and IL-23p19 KO mice under unstimulated culture conditions, it was significantly reduced in the supernatants from HKLM-stimulated splenocytes and bone marrow cells of IL-23p19 KO mice compared to those of B6 mice (Fig. 4C and D). Collectively, these data demonstrate that early production of the monocyte-recruiting chemokines CCL2 and CCL7 is regulated by IL-23 during L. monocytogenes infection.
Fig 4.
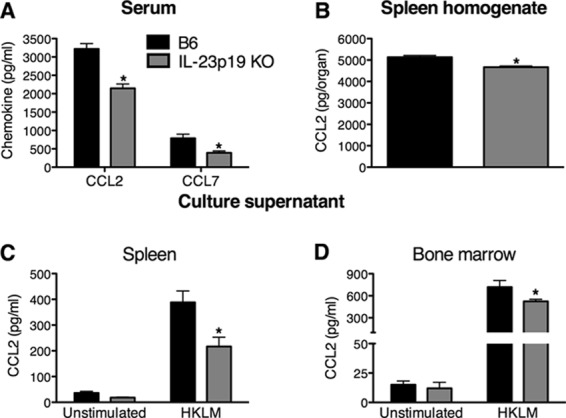
IL-23 is required for the optimal production of CCL2 and CCL7 during L. monocytogenes infection. B6 and IL-23p19 KO mice were infected with ∼104 L. monocytogenes CFU for 1 day. ELISAs were performed to determine the concentrations of CCL2 and CCL7 in the serum (A), CCL2 in spleen homogenates (B), and CCL2 in supernatants from overnight cultures of splenocytes (C) and bone marrow cells (D) that were stimulated with HKLM or left unstimulated. A two-way ANOVA (A) and t tests (B, C, and D) were used to determine statistical differences between groups (*, significantly different from B6 mice). All data are expressed as means ± standard errors of the means (n = 5/group). These data are representative of two independent experiments.
IL-23 is required for the optimal production of TNF-α and NO• in the spleen during L. monocytogenes infection.
The effective eradication of L. monocytogenes is dependent upon several effector molecules, including TNF-α and NO·. Since mice lacking IL-23 display increased severity of L. monocytogenes infection, it was of interest to investigate the effect of IL-23 on the production of TNF-α and NO·. To determine if IL-23 was required for the production of TNF-α and NO• from splenocytes, B6 and IL-23p19 KO mice were infected with L. monocytogenes for 1 or 3 days, and ELISAs were performed on cultured splenocyte supernatants. There was a reduction in the overall concentration of TNF-α in the culture supernatants from IL-23p19 KO mice compared to those from B6 mice at both days 1 and 3 p.i. (Fig. 5A). Although the concentrations of NO• did not differ between the groups at day 1 p.i., the NO• concentration was significantly reduced in the supernatants from IL-23p19 KO mice compared to those of B6 mice at day 3 p.i. (Fig. 5B). To further examine if this reduction in TNF-α and NO• was due to a direct effect of IL-23, splenocytes from L. monocytogenes-infected B6 mice were cultured in the presence of rIL-23. Addition of rIL-23 did not directly induce or enhance the production of TNF-α or NO• from L. monocytogenes-infected B6 splenocytes (Fig. 5C and D). These results indicate that IL-23 mediates the optimal production of TNF-α or NO• in the spleen via an indirect mechanism during L. monocytogenes infection.
Fig 5.
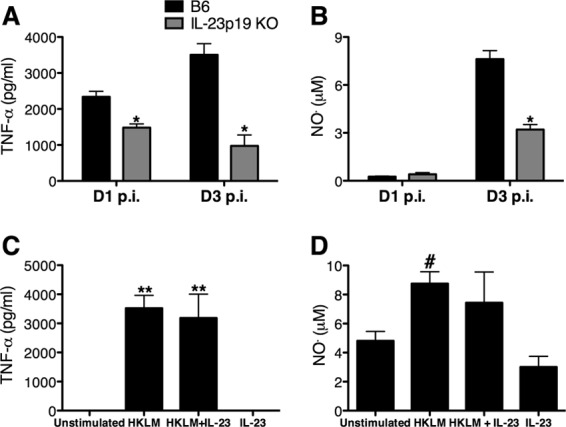
IL-23 is required for the optimal production of TNF-α and NO• in the spleen during L. monocytogenes infection. B6 and IL-23p19 KO mice were infected with ∼104 L. monocytogenes CFU for 1 or 3 days (D1 or D3, respectively). The overall production of TNF-α and NO• was determined by performing ELISAs on supernatants from overnight cultures of splenocytes stimulated with HKLM (A and B) and B6 splenocytes from day 3 p.i. stimulated with rIL-23 in the presence or absence of HKLM (C and D). A two-way ANOVA (A and B) and one-way ANOVA (C and D) were performed to determine statistical differences between groups (*, significantly different from B6 mice; **, significantly different from unstimulated and rIL-23-stimulated mice; #, significantly different from rIL-23 stimulation. All data are expressed as means ± standard errors of the means (n = 5/group). These data are representative of two independent experiments.
Optimal recruitment of TNF-α- and iNOS-producing inflammatory monocytes to the spleen is dependent on IL-23 during L. monocytogenes infection.
It has been shown that L. monocytogenes infection induces the differentiation of TNF-α- and iNOS-producing inflammatory monocytes in the spleen (35). Therefore, to determine if IL-23 impacts inflammatory monocyte-derived TNF-α and iNOS (35), intracellular cytokine staining for TNF-α and iNOS was performed on splenocytes isolated from B6 and IL-23p19 KO mice infected with L. monocytogenes for 1 or 3 days. The deficient recruitment of monocytes to the spleens of IL-23p19 KO mice compared to levels in B6 mice resulted in fewer monocytes producing TNF-α, with a decrease in the percentage (Fig. 6A and B) and total numbers of TNF-α-producing monocytes at both days 1 and 3 p.i. (Fig. 6C). There were no significant differences in the expression levels of iNOS between B6 and IL-23p19 KO mice at day 1 p.i. (data not shown). However, at day 3 p.i. there was a reduction in the percentages (Fig. 6D and E) and numbers of iNOS-expressing monocytes in the IL-23p19 KO mice compared to those in B6 mice (Fig. 6F). These results indicate that the decreased recruitment of TNF-α- and iNOS-expressing inflammatory monocytes to the spleen in IL-23p19 KO mice during L. monocytogenes infection causes a reduction in the overall amounts of these inflammatory mediators.
Fig 6.
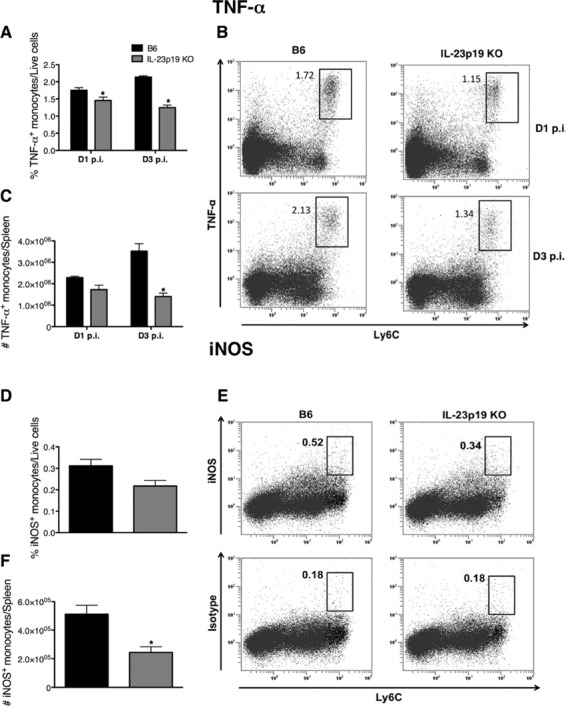
Optimal recruitment of TNF-α- and iNOS-producing inflammatory monocytes to the spleen is dependent on IL-23 during L. monocytogenes infection. B6 and IL-23p19 KO mice were infected with ∼104 L. monocytogenes CFU for 1 or 3 days. Flow cytometric analysis was performed on isolated splenocytes to determine the expression of TNF-α or iNOS by monocytes (Ly6G− Ly6Chi). The percentages (A) and numbers (C) of TNF-α-producing monocytes in the spleen at days 1 and 3 p.i. were determined from dot plots (B). The percentages (D) and numbers (F) of iNOS-expressing monocytes in the spleen at day 3 p.i. were obtained after the corresponding isotype control values determined from dot plots (E) were subtracted. A two-way ANOVA (A and C) and t tests (D and F) were performed to determine statistical differences between groups (*, significantly different from B6 mice). All data are expressed as means ± standard errors of the means (n = 5/group). These data are representative of two independent experiments.
DISCUSSION
The IL-23/IL-17 axis has been shown to be critical for bacterial clearance and neutrophil recruitment during infection with L. monocytogenes and other pathogens (9, 11, 24, 40, 45, 46). In the current study, we present novel data that identify an additional role for IL-23 in enhancing the recruitment of monocytes to the spleen during L. monocytogenes infection. Furthermore, a reduction in the monocyte percentages in IL-23p19 KO mice coincides with a reduction in the levels of known monocyte-recruiting chemokines (CCL2 and CCL7). The overall production of TNF-α and NO• is reduced in IL-23p19 KO mice, which corresponds with reduced numbers and percentages of TNF-α- and iNOS-expressing monocytes in the spleens of infected IL-23p19 KO mice. Taken together, our data suggest that the lack of efficient recruitment of TNF-α- and NO•-producing monocytes to the spleens during the early stages of L. monocytogenes infection could lead to increased bacterial burden and, therefore, enhanced susceptibility of IL-23p19 KO mice to L. monocytogenes infection.
Our findings demonstrate that IL-23 can play divergent roles in mediating host protection against L. monocytogenes in the spleen versus the liver. IL-23 is required for the optimal production of IL-17A and IL-17F in the spleen and liver during L. monocytogenes infection (24), and IL-17 has been shown to facilitate the mobilization of neutrophils to sites of infection by inducing the production of neutrophil chemoattractants, such as CXCL1, CXCL2, and CXCL8 (17). Moreover, IL-17RA KO mice display a reduction in the percentages and total numbers of neutrophils in the liver during L. monocytogenes infection (24). Our neutrophil depletion studies show that a lack of neutrophil recruitment in IL-23p19 KO mice is likely causal for increased liver CFU counts. These observations are in line with our previous findings that the lack of IL-23 results in diminished recruitment of neutrophils to the liver (24) and that neutrophils are required for bacterial clearance in the liver via TNF-α production (5). A recent publication suggested that neutrophils are not required for mediating protection against L. monocytogenes infection (38). The discrepancy in the conclusions between these two studies could be due to the differences in experimental variables, including the source of mice, dose of L. monocytogenes, and timing and amount of the depleting antibody used. Based on our observations, we conclude that during systemic L. monocytogenes infection, neutrophils confer protection in the liver, and the IL-23/IL-17 axis mediates the recruitment of neutrophils to this organ.
Our data add another dimension to the role of IL-23 in recruiting cell types in addition to neutrophils. This is supported by the finding that depletion of neutrophils did not abrogate the differences in CFU counts in the spleens of IL-23p19 KO mice and B6 mice. Furthermore, the absence of IL-23 results in deficient recruitment of inflammatory monocytes due to a reduction in the circulating amounts of CCL2 and CCL7, indicating that there is an IL-23-dependent mechanism for monocyte mobilization during L. monocytogenes infection. It is interesting that IL-23 impacts the production of CCL2 during L. monocytogenes infection but not under homeostatic conditions. This could be attributed to the infection-induced production of proinflammatory cytokines that may act to enhance CCL2 production during L. monocytogenes infection. Genome analysis has identified a TNF-α-responsive enhancer region in the CCL2 locus, suggesting that this proinflammatory cytokine can cause the upregulation of CCL2 expression (3). In a rheumatoid arthritis model, IL-17 mediates monocyte recruitment both directly, by acting as a chemoattractant (36), and indirectly, by inducing the production of CCL2 (37), further supporting the notion of an IL-23/IL-17/CCL2/monocyte axis. While it is known that inflammatory monocyte-derived TNF-α and NO• are required for antilisterial defense (35), we now show that the lack of IL-23 results in a reduction in the overall amounts of these effector molecules in the spleen.
Our data indicate that rIL-23 does not directly induce or enhance TNF-α or NO• production from splenocytes cultured in vitro with HKLM. Our current data favor a model whereby IL-23 modulates the levels of TNF-α and NO• via an indirect mechanism during L. monocytogenes infection (i.e., by increasing the mobilization of inflammatory monocytes to sites of infection). Previous studies have shown that peritoneal macrophages upregulate TNF-α mRNA following in vivo administration of rIL-23 (6), and transgenic expression of IL-23p19 results in increased serum levels of TNF-α (44). However, these studies do not establish whether IL-23 directly or indirectly induces TNF-α. Further studies are required to fully understand how IL-23 may lead to the production of proinflammatory molecules in cells of the myeloid lineage during different disease states.
The regulation of immune responses by IL-23 depends upon its downstream cytokines, IL-17 and IL-22, and their respective effector functions. IL-23 can induce the production of IL-22 from different cells, including T cells, natural killer cells, and dendritic cells (28, 47). IL-22 is known to stimulate the production of antimicrobial peptides as well as protect tissues from damage (28). The IL-23/IL-22/antimicrobial peptide axis has been shown to be important during extracellular bacterial infections with Citrobacter rodentium (47) and Klebsiella pneumoniae (1). Interestingly, although infection with L. monocytogenes induces IL-23-dependent production of IL-22, this cytokine is not necessary for bacterial clearance in the spleen or liver (12). In addition to the IL-23/IL-17/neutrophil and IL-23/IL-22/antimicrobial peptide pathways, our data uncover a novel IL-23/monocyte pathway that protects against bacterial infection. Considering that monocytes are protective against bacterial, protozoan, and viral pathogens (21, 33), but detrimental in diseases such as breast cancer (31), atherosclerosis (4), and experimental autoimmune encephalomyelitis (8), it is crucial to gain a complete understanding of the mechanisms governing their recruitment and function.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nico Ghilardi at Genentech, Inc., for the generous donation of the IL-23p19 KO mice, Xavier Edoh for technical assistance, and Alexandra Witter for critical reading of the manuscript. Flow cytometry was performed in the Flow Cytometry and Laser Capture Microdissection Core Facility at the University of North Texas Health Science Center.
This research was funded by a Texas Norman Hackerman Advanced Research Program Grant (000130-0025-2007) and NIH grant AI09951 (R.E.B.). A.N.S. was supported by NIH AI072946.
Footnotes
Published ahead of print 10 September 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Aujla SJ, et al. 2008. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 14:275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berg RE, Crossley E, Murray S, Forman J. 2003. Memory CD8+ T cells provide innate immune protection against Listeria monocytogenes in the absence of cognate antigen. J. Exp. Med. 198:1583–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bonello GB, et al. 2011. An evolutionarily conserved TNF-alpha-responsive enhancer in the far upstream region of human CCL2 locus influences its gene expression. J. Immunol. 186:7025–7038 [DOI] [PubMed] [Google Scholar]
- 4. Boring L, Gosling J, Cleary M, Charo IF. 1998. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 394:894–897 [DOI] [PubMed] [Google Scholar]
- 5. Carr KD, et al. 2011. Specific depletion reveals a novel role for neutrophil-mediated protection in the liver during Listeria monocytogenes infection. Eur. J. Immunol. 41:2666–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cua DJ, et al. 2003. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421:744–748 [DOI] [PubMed] [Google Scholar]
- 7. Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. 2008. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 83:64–70 [DOI] [PubMed] [Google Scholar]
- 8. Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. 2000. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J. Exp. Med. 192:899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Freitas A, et al. 2009. IL-17 receptor signaling is required to control polymicrobial sepsis. J. Immunol. 182:7846–7854 [DOI] [PubMed] [Google Scholar]
- 10. Ghilardi N, et al. 2004. Compromised humoral and delayed-type hypersensitivity responses in IL-23-deficient mice. J. Immunol. 172:2827–2833 [DOI] [PubMed] [Google Scholar]
- 11. Godinez I, et al. 2009. Interleukin-23 orchestrates mucosal responses to Salmonella enterica serotype Typhimurium in the intestine. Infect. Immun. 77:387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Graham AC, et al. 2011. IL-22 production is regulated by IL-23 during Listeria monocytogenes infection but is not required for bacterial clearance or tissue protection. PLoS One 6:e17171 doi:10.1371/journal.pone.0017171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Happel KI, et al. 2005. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J. Exp. Med. 202:761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hunter CA. 2005. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat. Rev. Immunol. 5:521–531 [DOI] [PubMed] [Google Scholar]
- 15. Jia T, et al. 2008. Additive roles for MCP-1 and MCP-3 in CCR2-mediated recruitment of inflammatory monocytes during Listeria monocytogenes infection. J. Immunol. 180:6846–6853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khader SA, et al. 2005. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J. Immunol. 175:788–795 [DOI] [PubMed] [Google Scholar]
- 17. Kolls JK, Linden A. 2004. Interleukin-17 family members and inflammation. Immunity 21:467–476 [DOI] [PubMed] [Google Scholar]
- 18. Ladel CH, Flesch IE, Arnoldi J, Kaufmann SH. 1994. Studies with MHC-deficient knock-out mice reveal impact of both MHC I- and MHC II-dependent T cell responses on Listeria monocytogenes infection. J. Immunol. 153:3116–3122 [PubMed] [Google Scholar]
- 19. Langrish CL, et al. 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201:233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lieberman LA, et al. 2004. IL-23 provides a limited mechanism of resistance to acute toxoplasmosis in the absence of IL-12. J. Immunol. 173:1887–1893 [DOI] [PubMed] [Google Scholar]
- 21. Lim JK, et al. 2011. Chemokine receptor Ccr2 is critical for monocyte accumulation and survival in West Nile virus encephalitis. J. Immunol. 186:471–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mangan PR, et al. 2006. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441:231–234 [DOI] [PubMed] [Google Scholar]
- 23. McKenzie BS, Kastelein RA, Cua DJ. 2006. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 27:17–23 [DOI] [PubMed] [Google Scholar]
- 24. Meeks KD, Sieve AN, Kolls JK, Ghilardi N, Berg RE. 2009. IL-23 is required for protection against systemic infection with Listeria monocytogenes. J. Immunol. 183:8026–8034 [DOI] [PubMed] [Google Scholar]
- 25. Murphy CA, et al. 2003. Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 198:1951–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oppmann B, et al. 2000. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13:715–725 [DOI] [PubMed] [Google Scholar]
- 27. Ouyang W, Kolls JK, Zheng Y. 2008. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 28:454–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ouyang W, Valdez P. 2008. IL-22 in mucosal immunity. Mucosal. Immunol. 1:335–338 [DOI] [PubMed] [Google Scholar]
- 29. Pamer EG. 2004. Immune responses to Listeria monocytogenes. Nat. Rev. Immunol. 4:812–823 [DOI] [PubMed] [Google Scholar]
- 30. Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. 1996. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 184:1397–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Qian BZ, et al. 2011. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475:222–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Roberts AD, Ordway DJ, Orme IM. 1993. Listeria monocytogenes infection in beta 2 microglobulin-deficient mice. Infect. Immun. 61:1113–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Serbina NV, Jia T, Hohl TM, Pamer EG. 2008. Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 26:421–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Serbina NV, Pamer EG. 2006. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7:311–317 [DOI] [PubMed] [Google Scholar]
- 35. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. 2003. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity 19:59–70 [DOI] [PubMed] [Google Scholar]
- 36. Shahrara S, Pickens SR, Dorfleutner A, Pope RM. 2009. IL-17 induces monocyte migration in rheumatoid arthritis. J. Immunol. 182:3884–3891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shahrara S, et al. 2010. IL-17-mediated monocyte migration occurs partially through CC chemokine ligand 2/monocyte chemoattractant protein-1 induction. J. Immunol. 184:4479–4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shi C, et al. 2011. Ly6G+ neutrophils are dispensable for defense against systemic Listeria monocytogenes infection. J. Immunol. 187:5293–5298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shi C, et al. 2011. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating Toll-like receptor ligands. Immunity 34:590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. 2007. Resident Vδ1+ γδ T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J. Immunol. 178:4466–4472 [DOI] [PubMed] [Google Scholar]
- 41. Shiloh MU, et al. 1999. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity 10:29–38 [DOI] [PubMed] [Google Scholar]
- 42. Tato CM, Cua DJ. 2008. Reconciling id, ego, and superego within interleukin-23. Immunol. Rev. 226:103–111 [DOI] [PubMed] [Google Scholar]
- 43. Thakker P, et al. 2007. IL-23 is critical in the induction but not in the effector phase of experimental autoimmune encephalomyelitis. J. Immunol. 178:2589–2598 [DOI] [PubMed] [Google Scholar]
- 44. Wiekowski MT, et al. 2001. Ubiquitous transgenic expression of the IL-23 subunit p19 induces multiorgan inflammation, runting, infertility, and premature death. J. Immunol. 166:7563–7570 [DOI] [PubMed] [Google Scholar]
- 45. Ye P, et al. 2001. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 194:519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang X, et al. 2009. A MyD88-dependent early IL-17 production protects mice against airway infection with the obligate intracellular pathogen Chlamydia muridarum. J. Immunol. 183:1291–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zheng Y, et al. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14:282–289 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.