

Abstract
Five genes (cps2E, cps2T, cps2F, cps2G, and cps2I) are predicted to encode the glycosyltransferases responsible for synthesis of the Streptococcus pneumoniae serotype 2 capsule repeat unit, which is polymerized to yield a branched surface structure containing glucose-glucuronic acid linked to a glucose-rhamnose-rhamnose-rhamnose backbone. Cps2E is the initiating glycosyltransferase, but experimental evidence supporting the functions of the remaining glycosyltransferases is lacking. To biochemically characterize the glycosyltransferases, the donor substrate dTDP-rhamnose was first synthesized using recombinant S. pneumoniae enzymes Cps2L, Cps2M, Cps2N, and Cps2O. In in vitro assays with each of the glycosyltransferases, only reaction mixtures containing recombinant Cps2T, dTDP-rhamnose, and the Cps2E product (undecaprenyl pyrophosphate glucose) generated a new product, which was consistent with lipid-linked glucose-rhamnose. cps2T, cps2F, and cps2I deletion mutants produced no detectable capsule, but trace amounts of capsule were detectable in Δcps2G mutants, suggesting that Cps2G adds a nonbackbone sugar. All Δcps2F, Δcps2G, and Δcps2I mutants contained different secondary suppressor mutations in cps2E, indicating that the initial mutations were lethal in the absence of reduced repeat unit synthesis. Δcps2T mutants did not contain secondary mutations affecting capsule synthesis. The requirement for secondary mutations in mutants lacking Cps2F, Cps2G, and Cps2I indicates that these activities occur downstream of the committed step in capsule synthesis and reveal that Cps2T catalyzes this step. Therefore, Cps2T is the β1-4 rhamnosyltransferase that adds the second sugar to the repeat unit and, as the committed step in type 2 repeat unit synthesis, is predicted to be an important point of capsule regulation.
INTRODUCTION
Streptococcus pneumoniae (the pneumococcus) is a significant human pathogen (13) that generally persists as a commensal colonizer of the nasopharynx but has the capacity to transition to a virulent state, potentially causing pneumonia, meningitis, or bacteremia. The capsular polysaccharide (CPS) plays an integral role in this process. High levels of CPS serve as a barrier against host complement-mediated opsonophagocytosis and limit access to underlying bacterial surface components during infection, but only low levels are required to reduce clearance during nasopharyngeal colonization (2, 7, 24, 28, 38, 44, 57, 60). S. pneumoniae strains that are deficient in CPS production are avirulent (2, 24, 28, 38), indicating that the capsule, its synthesis, and regulation are all critical processes for pneumococcal pathogenesis.
Three pathways for synthesizing CPS in bacteria have been identified and are based on their similarities to the lipopolysaccharide O-antigen biosynthetic mechanisms: the Wzy-, ABC transporter-, and synthase-dependent pathways (48, 52, 59, 62). This study focuses on the Wzy-dependent pathway, which is the predominant mechanism for CPS synthesis in Gram-positive bacteria. Here, the capsule repeat unit is systematically synthesized in the cytoplasm on a membrane lipid acceptor (undecaprenyl phosphate [Und-P]) by multiple glycosyltransferases. Once the repeat unit is formed, it is enzymatically transferred across the cytoplasmic membrane by the Wzx flippase and polymerized by the Wzy polymerase (Fig. 1). In addition to CPS and O antigens, the Wzy-dependent pathway is also used to synthesize many exopolysaccharides (25). Characterization of the enzymes in this pathway may thus reveal biosynthetic and regulatory properties that are conserved among polysaccharides produced by bacteria of medical and industrial importance.
Fig 1.

S. pneumoniae type 2 genetic locus, capsular polysaccharide repeat unit structure, and Wzy-dependent pathway. (Top) cps2 genetic locus. The arrow indicates the direction of transcription and the predicted operon. (Middle) Repeat unit structure. Letters below each glycosidic linkage represent the glycosyltransferase known or predicted to catalyze the linkage. Predicted enzymes are in parentheses and are based on previous work (1), where the specificities of Cps2G and Cps2I were unclear. Glc, glucose; Rha, rhamnose; GlcUA, glucuronic acid. E, Cps2E; T, Cps2T; F, Cps2F; G, Cps2G; I, Cps2I. (Bottom) Wzy-dependent model for synthesis of the type 2 repeat unit. Synthesis of the capsule repeat unit occurs through the activity of multiple glycosyltransferases. The completed repeat unit is transferred to the outer face of the cytoplasmic membrane, is polymerized, and then remains associated with the membrane or is released or attached to peptidoglycan. With the exception of GalU (the UDP-glucose pyrophosphorylase encoded by galU), all gene products are encoded in the cps2 locus. (Adapted from reference 63.)
Over 90 distinct capsular serotypes have been reported for S. pneumoniae, each differentiated by their polysaccharide composition and/or glycosidic bonds (5, 9, 29, 53). Two of these serotypes synthesize capsule via the synthase-dependent pathway, whereas the remaining serotypes utilize the Wzy-dependent pathway (5, 15, 37, 62). As a model for studying the Wzy-dependent pathway in S. pneumoniae, our laboratory uses serotype 2 (Fig. 1). Previous work in our laboratory characterized the initiating glycosyltransferase, Cps2E (12). This integral membrane enzyme transfers glucose-1-phosphate (Glc-1-P) to a membrane-bound polyprenyl phosphate acceptor with properties consistent with those of Und-P (12). In addition to cps2E, 4 other open reading frames within the type 2 capsule locus (cps2T, cps2F, cps2G, and cps2I) are predicted to encode glycosyltransferases (1, 32, 39), but biochemical evidence supporting their functions is lacking.
Donor substrates for glycosyltransferase reactions are usually activated in the form of nucleoside diphosphate sugars (6, 10, 11, 36, 56). dTDP rhamnose (dTDP-Rha) is one of the nucleotide-sugar donors utilized by S. pneumoniae capsular glycosyltransferases. dTDP-Rha is not commercially available but can be synthesized from Glc-1-P via four enzymatic reactions: α-d-Glc-1-P → dTDP–α-d-Glc → dTDP–6-deoxy-α-d-xylo-4-hexulose → dTDP–6-deoxy-β-l-lyxo-4-hexulose → dTDP–β-l-Rha (20–22, 35, 46). These reactions are predicted to be catalyzed by 4 enzymes encoded within the type 2 locus: Cps2L, Cps2N, Cps2M, and Cps2O, respectively (32). cpsLMNO from serotype 19 encode enzymes that can functionally complement an Escherichia coli strain where the homologous dTDP-Rha biosynthesis genes, rfbBDAC, are deleted (42). In vitro activities and synthesis of dTDP-Rha, however, have not been demonstrated for the serotype 19 enzymes. Biochemical analyses of dTDP-Rha biosynthesis enzymes from Pseudomonas aeruginosa and Escherichia coli have been reported (21, 35, 46).
In a previous study, we found that mutants unable to synthesize UDP glucuronic acid (UDP-GlcUA), the nucleotide-sugar donor for the terminal sugar in the type 2 repeat unit, not only lacked GlcUA in the repeat unit but also produced low levels of polysaccharide that failed to be transferred to the cell wall. Each of these isolates also contained a secondary suppressor mutation (61). The suppressor mutations consistently mapped to genes within the capsule locus, primarily cps2E, and were indicative of lethality in the absence of reduced capsule synthesis. Suppressor mutations also occurred with primary mutations that eliminated transport of repeat units by the Wzx flippase or their polymerization by the Wzy polymerase (61). In each case, the lethality was attributed to a buildup of lipid-linked repeat units which either destabilized the membrane or sequestered Und-P in the capsule pathway and away from essential pathways such as the peptidoglycan synthesis pathway.
Here, we use a genetic approach to identify the first committed, physiologically irreversible step in serotype 2 capsule synthesis. We also provide biochemical evidence for the functions of Cps2L, Cps2N, Cps2M, Cps2O, and Cps2T (also annotated WchF [1, 39]). The findings indicate that Cps2T is the β1-4 rhamnosyltransferase that adds the second sugar of the repeat unit, and this is the committed step in type 2 capsule synthesis.
MATERIALS AND METHODS
Materials.
UDP-[14C]Glc (293 mCi/mmol) was obtained from Amersham Biosciences. Inorganic pyrophosphatase, NAD+, NADH, dTTP, Glc-1-P, and dTDP-Glc were obtained from Sigma-Aldrich. Nonidet P-40 (NP-40) was obtained from Pierce Chemical Company. Solid-phase extraction (SPE) carbograph (150 mg/4 ml) columns were obtained from Grace Davison Discovery Sciences.
Bacterial strains and growth conditions.
S. pneumoniae strains (Table 1) were grown at 37°C in THY (Todd-Hewitt broth supplemented with 0.5% yeast extract) or on blood agar plates (BBL [Difco] or blood agar base [Becton Dickinson] containing 3% defibrinated sheep blood [Colorado Serum Company]). Broth cultures of S. pneumoniae were grown statically at 37°C. Plate cultures were incubated at 37°C in a candle jar. E. coli strains (see Table S1 in the supplemental material) were grown at 37°C in L broth (1% tryptone, 0.5% yeast extract, 0.5% NaCl, 0.1% glucose) with shaking or on L-agar plates (15 g agar/liter L broth). The plasmids and primers used in this study are listed in Tables S1 and S2 in the supplemental material, respectively. Media were supplemented with the following antibiotics when appropriate: ampicillin (Ap; 100 μg/ml), erythromycin (Em; 15 μg/ml for E. coli DB11 and 0.3 μg/ml for S. pneumoniae), and kanamycin (Km; 250 μg/ml).
Table 1.
S. pneumoniae strains used in this study
| Strain(s) | Propertiesa | Reference or source |
|---|---|---|
| AM1000 | D39 Δ(cps2A to cps2I) Cps− | 38 |
| BX515 | D39 Δcps2K repair; Cpsr; retains cps2E5924G→T (G303V) | 61 |
| D39 | Type 2 parent strain; Cps+ | 3 |
| DJ901, DJ902, DJ910, DJ911, DJ973, DJ975, DJ977 | pDJ078 × D39; Cps−; Δcps2T mutants isolated from five independent transformations; Kmr | This study |
| DJ904, DJ914, DJ916 | pDJ086 × D39; Cps−; Δcps2F mutants with respective cps2E mutations cps2E5766insA (E240*frameshift), cps2E5924insG (G293*frameshift), and cps2E5718A→G (H258R); isolated from two independent transformations; Kmr | This study |
| DJ918, DJ919, DJ921 | pDJ131 × D39; Cpsr; Δcps2G mutants with respective cps2E mutations cps2E6059G→C (M338I), cps2E5964C→A (R307S), and cps2E5529G→T (A162S); isolated from two independent transformations; Kmr | This study |
| DJ930, DJ943, DJ944 | pDJ184 × D39; Cps−; Δcps2I mutants with respective cps2E mutations cps2E6267C→T (Q408*stop), cps2E6222C→A (Q393K), and cps2E5221insCT (M59*frameshift); isolated from two independent transformations; Kmr | This study |
| DJ960 | pDJ192 × DJ910; Cps+ ; Δcps2T repair | This study |
| DJ965 | pDJ192 × DJ901; Cps+ ; Δcps2T repair | This study |
cps2E superscripts indicate locations of mutations compared to the sequence with GenBank accession no. AF026471. Amino acid changes are indicated in parentheses. Cpsr, reduced capsule levels; del, deletion; ins, insertion; →, nucleotide change; Kmr, kanamycin resistant; Apr, ampicillin resistant; Emr, erythromycin resistant. Plasmids are listed in Table S1 in the supplemental material.
Generation of glycosyltransferase mutants.
In-frame deletions of the glycosyltransferase-encoding genes were generated by allelic replacement with a nonpolar Km resistance (aphA-3) cassette. The entire upstream and downstream genes flanking the target gene were individually amplified from S. pneumoniae D39 chromosomal DNA by PCRs. The PCR products were individually ligated into pCR2.1-TOPO vectors as directed in the TOPO TA cloning kit manual (Invitrogen). The fidelity of each PCR product was confirmed by DNA sequencing (University of Alabama [UAB] Center for AIDS Research [CFAR] DNA sequencing core or the Genomics Core Facility of the Heflin Center for Genomic Science). The native ribosomal binding sites were included in the downstream flanking genes. The resulting flanking genes were ligated to either side of the Km resistance cassette and subcloned into the S. pneumoniae suicide vector pJY4164 in a single reaction. The ligation reaction mixture was transformed into chemically competent E. coli DB11 cells. Deletion constructs were generated using the following primer pairs: for the Δcps2T construct, cps2E flank (E54/E55) and cps2F flank (T12/F12); for the Δcps2F construct, cps2T flank (T10/T11) and cps2G flank (F7/G8); for the Δcps2G construct, cps2F flank (F8/F9) and cps2H flank (G9/H7); and for the Δcps2I construct, cps2H flank (H9/H8) and cps2J flank (I9/J9). aphA-3 was amplified from the pneumococcal shuttle vector pSF151 using primers DJ-01 and DJ-02. Upstream flanks were digested with KpnI and NotI, downstream flanks were digested with XhoI and BamHI, aphA-3 was digested with NotI and XhoI, and pJY4164 was digested with KpnI and BamHI.
For pneumococcal transformations, deletion constructs were transformed into competent D39 cells (27) and Km-resistant isolates were selected. Isolates were screened for Em sensitivity to identify transformants that resulted from allelic replacement (Kmr, Ems) rather than insertion of the suicide plasmid (Kmr, Emr).
The glycosyltransferase mutations were repaired by allelic replacement using constructs that contained the relevant gene and full-length upstream and downstream genes. PCR products were ligated into pCR2.1-TOPO vectors, sequenced, subcloned into pJY4164, and transformed into E. coli DB11, as described above. Primer pairs and restriction enzymes for the Δcps2T repair construct were E8/T14 and NdeI/XhoI.
For pneumococcal transformations, repair constructs were transformed into the relevant mutant background. Transformation mixtures were plated in the absence of selection. Isolates were screened for Km sensitivity, indicative of allelic replacement of the insertion.
Immunoblot and capsule analyses.
For immunoblot analyses, proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–12% PAGE) and transferred onto 0.22-μm-pore-size nitrocellulose membranes (GE Healthcare) using a semidry transfer apparatus (Fisher). Detection of Cps2E was performed as previously described (12). Other protein immunoblots were reacted with a tetra-His monoclonal antibody (MAb) (1:4,000 dilution; Qiagen), a MAb to the β subunit of E. coli RNA polymerase (1:2,000 dilution; Neoclone), or a MAb to the β subunit of ATP synthase (1:2,000 dilution; Abcam), as indicated. The secondary antibody was a goat anti-mouse immunoglobulin directly conjugated to alkaline phosphatase (1:5,000 dilution; Southern Biotech). Blots were washed and developed as previously described (4).
S. pneumoniae fractionation and subsequent capsule immunoblot assays were performed as previously described (4), except that a 1:1,000 dilution of goat anti-rabbit immunoglobulin directly conjugated to alkaline phosphatase was used instead of a streptavidin-biotin conjugate. Polyclonal antiserum to type 2 capsule (Statens Serum Institut) was used in the Quellung reaction (45) to visualize capsule microscopically.
Methylpentose assays were performed as previously described (61).
Purification and characterization of S. pneumoniae dTDP-Rha biosynthesis enzymes.
To characterize dTDP-Rha biosynthesis enzymes, the sequences for cps2L, cps2N, cps2M, and cps2O were individually amplified from S. pneumoniae D39 chromosomal DNA by PCR using the following primer pairs: for cps2L, L6/L7; for cps2N, N4/N5; for cps2M, M4/M5; and for cps2O, O3/O4. The PCR products were ligated into pCR2.1-TOPO vectors and sequenced as described above. The respective sequences were subcloned into pET-16b plasmids (facilitating generation of N-terminal His tags) and transformed into chemically competent E. coli BL21-AI cells. The resulting strains, DJ017 (cps2L), DJ014 (cps2M), DJ015 (cps2N), and DJ016 (cps2O), were used for expression of the respective proteins. The pET plasmids used in this study contain a T7 RNA polymerase promoter. E. coli BL21-AI contains a chromosomally encoded T7 RNA polymerase under the control of an arabinose-inducible promoter.
For protein expression, 250-ml cultures were grown under the appropriate antibiotic selection to mid-exponential phase (cell density, approximately 4.5 × 108 CFU/ml) and then induced for 4 h with 0.2% arabinose. Cultures were centrifuged at 3,000 × g for 20 min, and the pellet was suspended in 5 ml of wash buffer (300 mM NaCl, 50 mM NaH2PO4, pH 7.0). Cells were sonicated on ice, using a Sonic Dismembrator 300 apparatus (Fischer) at 50% power, 5 times for 10 s each time with 30-s pause intervals. The lysate was then centrifuged at 8,000 × g for 10 min to pellet cellular debris. The cell-free lysate was passed over a Talon cobalt-metal affinity resin (Clontech) to purify the respective proteins. Native purification procedures were followed as directed in the manufacturer's specifications. Eluates containing purified His-tagged proteins were concentrated using Amicon Ultra 10-kDa centrifugal filters (Millipore). The retained proteins were washed twice with 500 μl of 50 mM Tris-acetate, pH 7.6, and then suspended in 100 μl of 30% glycerol in 50 mM Tris-acetate, pH 7.6. Protein concentrations were calculated from absorbances at 280 nm using Beer's law. Protein extinction coefficients were calculated using the Scripps Research Institute Protein Calculator Program (http://www.scripps.edu/∼cdputnam/protcalc.html). Enzymes were stored at −80°C and were active for several months.
To assay Glc-1-P thymidylyltransferase (Cps2L) activity, 300-μl reaction mixtures containing 30 mM HEPES, pH 7.6, 3 mM dTTP, 3 mM Glc-1-P, 9 mM MgCl2, and 0.3 μM Cps2L were incubated for 1 h at 37°C. Proteins were removed using Amicon Ultra 10-kDa centrifugal filters. The filtrate was analyzed by high-performance liquid chromatography (HPLC) with a CarboPac PA-100 or PA-1 column utilizing UV detection at 260 nm and a gradient of ammonium acetate (0 to 1 M) as an eluent.
To assay dTDP–d-Glc-4,6-dehydratase (Cps2N) activity, 300-μl reaction mixtures containing 30 mM HEPES, pH 7.6, 3 mM dTDP-Glc, 1 mM NAD+, and 0.3 μM Cps2N were incubated for 1 h at 37°C. Proteins were removed, and HPLC analysis was performed as described above.
To further characterize Cps2L and Cps2N activities, spectrophotometric analyses were used to monitor NADH accumulation as a by-product of dTDP–6-deoxy-α-d-xylo-4-hexulose synthesis. Synthesis of dTDP–6-deoxy-α-d-xylo-4-hexulose using dTDP–d-Glc-4,6-dehydratase yields a characteristic maximum absorbance at 320 nm in an alkaline solution (46). Products were generated in a 500-μl reaction mixture containing 30 mM HEPES, pH 7.6, 3 mM dTTP, 3 mM Glc-1-P, and 9 mM MgCl2 with 0.3 μM Cps2L and/or 0.3 μM Cps2N. The reaction mixtures were incubated at 37°C. At 30-min intervals, 25-μl aliquots were removed and placed in 775 μl of 100 mM NaOH. Incubations were continued for an additional 15 min before determining the absorbance at 320 nm.
dTDP–6-deoxy-d-xylo-4-hexulose-3,5-epimerase (Cps2M) and dTDP–6-deoxy-l-lyxo-4-hexulose-reductase (Cps2O) activities were analyzed in a combined spectrophotometric assay measuring the oxidation of NADH at 340 nm. Kinetic experiments were carried out with dTDP–6-deoxy-α-d-xylo-4-hexulose (filtrate from the above-mentioned Cps2N reaction), 1 mM NADH, 0.3 μM Cps2M, and/or 0.3 μM Cps2O. The reactions were carried out in 300-μl volumes, and the reaction mixtures were incubated for 1 h at 37°C.
Synthesis, purification, and quantification of dTDP-Rha.
dTDP-Rha was synthesized in two steps. First, dTDP-Glc was synthesized in a 350-μl reaction mixture containing 50 mM HEPES, pH 7.6, 30 mM MgCl2, 3.4 mM dTTP, 3.4 mM Glc-1-P, 2 units of inorganic pyrophosphatase, and 1 μM Cps2L. After an incubation of 1 h at 37°C, the proteins were removed by filtration as described above. The filtrate was used in the second step and supplemented with 0.1 mM NAD+, 6 mM NADH, 1 μM Cps2N, 1 μM Cps2M, and 1 μM Cps2O in a final volume of 350 μl. The reaction mixture was incubated for 1 h at 37°C, the proteins were filtered, and dTDP-Rha was separated by HPLC as described above. Fractions containing dTDP-Rha were pooled and then desalted on an Extract-Clean SPE carbograph (150 mg/4 ml) column. The column was first equilibrated by adding 4 ml of 80% acetonitrile–0.1% trifluoroacetic acid, followed by 10 ml of water. The sample was then added to the column, followed by another 10 ml of water. dTDP-Rha was eluted with 1.5 ml of 25% acetonitrile–0.1% trifluoroacetic acid. The effluent was freeze-dried using a SpeedVac apparatus (Savant). The dried sample was suspended in 0.5 ml anhydrous methanol and again freeze-dried. This process was repeated 5 times, and the dried sample was stored at −20°C. For use in glycosyltransferase assays, samples were resuspended in 100 mM Tris-acetate, pH 7.6, and dTDP-Rha concentrations were calculated from the absorbances at 260 nm using Beer's law and the thymidine extinction coefficient (7.4 × 103 M−1 cm−1). For electrospray ionization tandem mass spectrometry (ESI-MS/MS) analysis (see below), samples were suspended in the mobile phase prior to analysis.
GC/MS and ESI-MS/MS analyses.
The identity of dTDP-Rha was confirmed using gas chromatography-mass spectrometry (GC-MS) and ESI-MS/MS. A 1-μl aliquot of a 10 mM solution of Glc and Rha standards, along with 1 μl of purified dTDP-Rha (approximately 1 mM), was dried in a SpeedVac apparatus for sugar component analyses. Dried samples were subjected to methanolysis (500 μl of 3 N methanolic HCl at 80°C) for 16 h, dried, and washed 5 times with 500 μl of anhydrous methanol. The dried samples were trimethylsilylated by reacting them with 100 μl Tri-Sil reagent (Pierce) for 30 min at 80°C. The reaction products were analyzed on a GC-MS (Varian 4000; Agilent Technologies, Santa Clara, CA) fitted with a 30-m (inner diameter, 0.25 mm) VF-5ms capillary column. The column temperature was maintained at 160°C for 3 min and then increased to 260°C at 3°C/min and finally held at 260°C for 2 min. The effluent was analyzed by MS using the electron impact ionization mode.
For ESI-MS/MS, 1 μl of purified dTDP-Rha (approximately 1 mM) was injected by direct infusion into an API4000 (Applied Biosystems) triple-quadrupole mass spectrometer operated under the negative electrospray ionization mode. The ion spray voltage was set to −4,000 V. The flow injection mobile phase was 50% acetonitrile–0.1% formic acid, and the flow rate was 20 μl/min. The collision energy, collision gas, declustering potential, and nebulizing gas parameters were set to −40, 4, −40, and 15, respectively.
Glycosyltransferase expression and isolation.
Sequences for cps2E, cps2T, cps2F, cps2G, and cps2I were individually amplified from S. pneumoniae D39 chromosomal DNA by PCR using the following primer pairs: for cps2E, E8/E11; for cps2T, T13/T14; for cps2F, F10/F12; for cps2G, G10/G11; and for cps2I, I7/I8 (or J8/I6). The GTG start of cps2F was changed to ATG to facilitate proper in vitro expression. Cloning with primer pair I7/I8 requires a partial digest with NdeI to acquire full-length Cps2I. PCR products ligated into pCR2.1-TOPO were sequenced for verification. Appropriate sequences were subcloned into pET-16b or pET-20b and transformed into chemically competent E. coli BL21-AI cells as described above. The resulting strains were used for expression of the respective proteins. Strains KJ4152 (cps2E), DJ009 (cps2T), DJ204 (cps2F), DJ011 (cps2G), and DJ005 (cps2I) containing the non-His-tagged proteins were used in glycosyltransferase assays, while DJ052 (cps2T), DJ205 (cps2F), DJ056 (cps2G), and DJ089 (cps2I) containing the His-tagged proteins were used in protein localization experiments.
Cps2T, Cps2F, Cps2G, and Cps2I were expressed as described for the dTDP-Rha biosynthetic enzymes. Following centrifugation of the cultures, cell pellets were frozen and subsequently used to isolate membrane and cytoplasmic fractions. Cultures expressing Cps2E were processed as previously described (12). For all, frozen cell pellets were thawed at room temperature and suspended in 6 ml of spheroplast buffer (10 mM Tris-HCl [pH 7.5], 20% sucrose, 10 mM EDTA) containing 0.4 mg/ml lysozyme, 0.5 μg/ml pepstatin, 0.7 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1 mM dithiothreitol (DTT). The cellular suspension was incubated at 4°C with constant mixing for 4 h. Spheroplasts were sedimented by centrifugation at 8,000 × g for 10 min and lysed by suspension in 6 ml of sterile water containing 10 mM EDTA, 1 mM PMSF, and 1 mM DTT. Suspensions were sonicated on ice as described above. Cellular debris was sedimented by centrifugation at 8,000 × g for 20 min. The supernatant, containing both the cytoplasmic and membrane fractions, was centrifuged at 50,000 × g for 20 min. The supernatant was saved as the cytoplasmic fraction and centrifuged once more at 50,000 × g for 20 min to reduce residual membranes. The pellet (membranes) was washed two times with 3 ml of 100 mM Tris-acetate (pH 7.5) containing 10% glycerol and 1 mM DTT. The final membrane pellet was suspended in 500 μl of 100 mM Tris-acetate (pH 7.5) containing 10% glycerol and 1 mM DTT. Total cytoplasmic and membrane proteins were quantified using the Bradford Bio-Rad protein dye assay.
The interactions of the glycosyltransferases with the membrane were tested by suspending pelleted membranes in 30 μl of 100 mM Na2CO3 (pH 11.5). The mixture was centrifuged at 50,000 × g for 20 min, and the entire supernatant was saved. This process was repeated twice; after the final wash was collected, the remaining membranes were suspended in 30 μl of 100 mM Na2CO3.
Glycosyltransferase assays.
Glycosyltransferase assays were modified from those described elsewhere (12, 34). Briefly, membranes containing 3 μg of total protein were incubated for 1 h at 10°C in reaction mixtures containing 10 mM MnCl2, 1 mM DTT, 0.025 μCi UDP-[14C]Glc (293 mCi/mmol), and, where indicated, 0.1 mM dTDP-Rha. The final volume was brought to 75 μl with 100 mM Tris-acetate, pH 7.6. For reaction mixtures containing independent membrane preparations, membranes were preincubated with 0.008% NP-40 (Pierce) for 10 min, prior to the addition of metal ions and nucleotide-sugar donors. Reactions were stopped, and the organic phase was extracted as described previously (12). Glycolipids were either preserved for later use, as described for Und-P-P-[14C]Glc (see below), or subjected to mild-acid hydrolysis to liberate the saccharide moiety for separation by thin-layer chromatography (TLC), as has been used previously to analyze glycosyltransferase activity (12, 34, 47). Hydrolysis was performed by suspending the dried organic phase in 80 μl of 20 mM HCl and incubating at 70°C for 20 min. Hydrolyzed samples were dried, suspended in 10 μl of chloroform-methanol (1:1), and applied as spots on silica-coated TLC (Whatman) plates. TLC plates were chromatographed for 8 h in butanol-ethanol-water (5:3:2). Dried plates were exposed to a phosphor screen (General Electric Healthcare) for 15 h, and bands were visualized in a Storm 820 phosphorimager (General Electric Healthcare).
Synthesis of Und-P-P-[14C]Glc and assay of Cps2T activity in the absence of Cps2E.
Und-P-P-[14C]Glc was synthesized as previously described (12) with a few modifications. Briefly, membranes (10 μg total protein) containing Cps2E were incubated as described above for the glycosyltransferase assays. Extracted glycolipids were dried and suspended in 100 μl of water (or 100 mM Tris-acetate, pH 7.6) containing 0.1% NP-40. This solution was used as the source of Und-P-P-[14C]Glc in subsequent glycosyltransferase reactions. Membranes (3 μg total protein) from DJ009 containing non-His-tagged Cps2T or from RC124, a vector-only E. coli strain, were incubated in reaction mixtures containing 0.008% NP-40, 10 mM MnCl2, 1 mM DTT, 25 μl of the Und-P-P-[14C]Glc solution, and 0.1 mM dTDP-Rha. Reaction products were visualized as described above.
RESULTS
Genetic evidence that Cps2T performs the committed step in serotype 2 capsule synthesis.
From our previous study (61), we knew that mutations resulting in loss of either the terminal sugar (a GlcUA branched from the backbone), Wzx-mediated transport, or Wzy-mediated assembly of the repeat unit could be obtained only in the presence of suppressor mutations that reduced repeat unit synthesis. The primary mutations were postulated to be lethal because they resulted in the accumulation of lipid-linked repeat units that could not be readily disassembled; i.e., capsule synthesis in these mutants had proceeded beyond a committed step. Since suppressor mutations were required to obtain the Wzx mutants, the committed step must occur prior to transport of the repeat unit across the membrane and thus be performed by a glycosyltransferase. To identify the glycosyltransferase that performs the committed step, we took advantage of the requirement for secondary suppressor mutations to prevent the lethal accumulation of lipid-linked repeat units. We hypothesized that mutations affecting enzymes downstream of the committed step would be obtainable only in the presence of suppressor mutations, whereas mutation of the enzyme performing the committed step should not require suppressor mutations.
We first constructed deletions of cps2T, cps2F, cps2G, and cps2I in S. pneumoniae by replacing the respective open reading frames with a nonpolar Km resistance cassette. For each gene, multiple mutants were derived from at least two independent transformation reactions with D39. All mutants displayed the small, rough colony morphology indicative of loss of capsule, and no capsule was detected on any of the mutants using type 2-specific antiserum with intact cells in the Quellung reaction (data not shown). During growth in liquid medium, the mutant cultures exhibited an abrupt decrease in optical density, as did other capsule-negative mutants, compared to that for the parent strain (shown in Fig. 2 for Δcps2T and Δcps2G). These mutants aggregated and precipitated out of the culture medium, consistent with contact of exposed hydrophobic cell surfaces resulting from the absence of a parental capsule.
Fig 2.
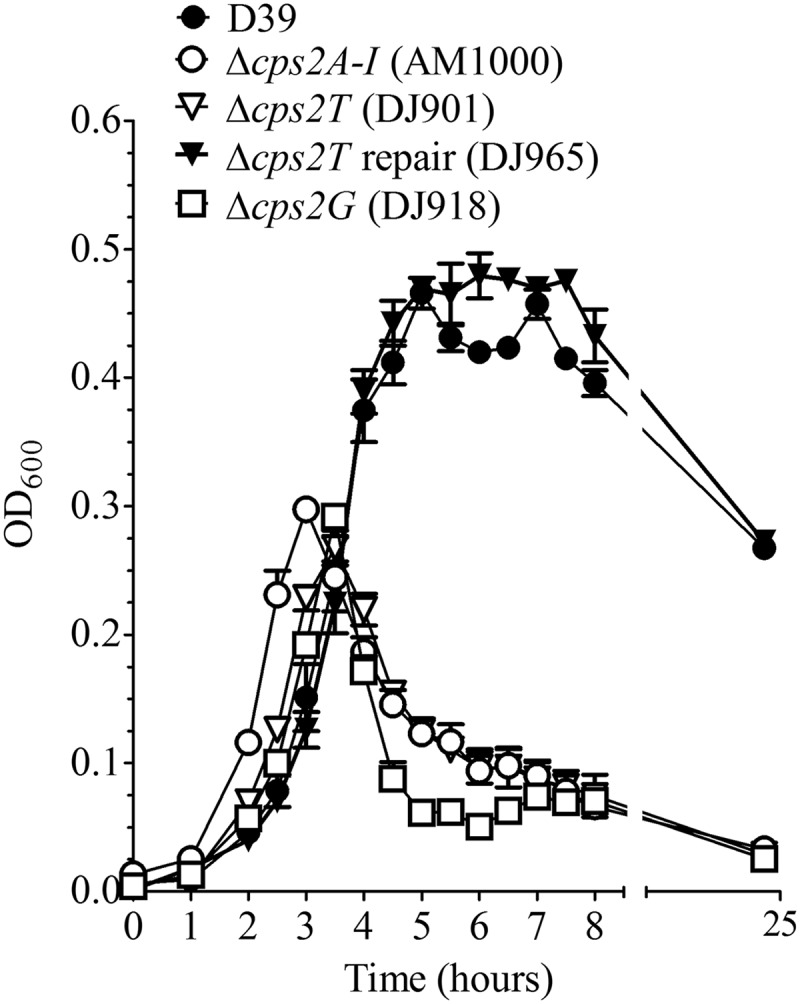
Growth of D39 and derivatives. Cultures grown in THY to an optical density at 600 nm (OD600) of 0.15 were diluted 1/10 and grown in THY. A600 readings were taken at the indicated time points. Data are the means and standard errors from two cultures. AM1000 is a capsule-negative mutant of D39.
To further investigate whether any capsule was detectable in specific cellular factions, the protoplast (membrane-containing) and cell wall fractions were individually extracted, separated by SDS-PAGE, transferred to a nitrocellulose membrane, and reacted with type 2-specific antiserum. Here, no capsule was detected in independent Δcps2T, Δcps2F, or Δcps2I mutants (shown in Fig. 3 for a Δcps2T mutant). For the Δcps2G mutants, a very small amount of capsule was detected in the protoplast fraction, but none was detected in the cell wall fraction, even when the starting material was concentrated an additional 5-fold (Fig. 3 and data not shown). The reduction of CPS in the Δcps2G mutants was further demonstrated by analysis of the methylpentose (rhamnose) levels of polymers extracted from whole cells. The methylpentose levels in strains D39 (parent, Cps+), DJ918 (Δcps2G), and AM1000 (Cps−) were 1.4 ± 0.024, 0.13 ± 0.017, and 0.11 ± 0.013 μg/108 CFU, respectively. The values represent the means ± standard errors from two assays performed in duplicate. The level for D39 was significantly different from the levels for cps2G mutant DJ918 and nonencapsulated strain AM1000 (P < 0.001), which were not different from each other, as determined using one-way analysis of variance with Bonferroni posttests. These results indicate that the immunoblot detection of capsule in cps2G mutants represents reduced CPS levels and not altered antibody reactivity from loss of an epitope. The ability of Δcps2G mutants to synthesize polymers indicated that Cps2G does not add a backbone sugar.
Fig 3.

Capsule production by D39 and derivatives. Protoplast (membrane-containing) and cell wall fractions were extracted as described in Materials and Methods. Samples were separated by SDS–12% PAGE and transferred to a nitrocellulose membrane. Capsule was detected using polyclonal antiserum against the type 2 capsule. Protoplasts (PP) were normalized to 30 μg of total protein, and cell wall (CW) fractions were loaded relative to their protoplast counterparts. AM1000 is a capsule-negative mutant of D39.
In our previous study, most of the suppressor mutations mapped to cps2E (61). We therefore sequenced this gene from multiple isolates of the glycosyltransferase mutants. All of the Δcps2F, Δcps2G, and Δcps2I mutants (three from two independent reactions for each mutant) contained mutations in cps2E (Table 1). In each case, the cps2E mutations were different. Sequencing of cps2E from the Δcps2T mutants (seven obtained from five independent reactions) revealed no mutations. Repair of independent Δcps2T mutants resulted in restoration of the capsule, level of growth, and colony morphology to those for the parent (Fig. 2 and 3 and data not shown), demonstrating that these mutants did not contain secondary mutations that affected capsule synthesis. In contrast, repair of other deletion mutations did not restore parental phenotypes (data not shown). These results indicated that the phenotypes observed in the latter mutants were not solely due to the primary deletion mutations but were affected by the cps2E mutations and that suppressor mutations were required to obtain mutants lacking Cps2F, Cps2G, and Cps2I but not Cps2T, thus identifying the Cps2T reaction as the committed step.
During construction of the glycosyltransferase mutants, the Δcps2G and Δcps2I mutants were obtained at very low frequencies compared to those for the Δcps2T and Δcps2F mutants (Table 2). The frequencies for the former were increased when a recipient that already contained a cps2E point mutation that reduced Cps2E activity was used (Table 2), underscoring the need for a suppressor mutation to obtain cps2G and cps2I mutants. The fact that Δcps2F mutants were obtained at relatively high frequencies, which were not increased further by a preexisting cps2E mutation, suggests differences in the detrimental effects of the cps2F mutation relative to those of cps2G and cps2I. These observations are considered in more detail in the Discussion.
Table 2.
Frequencies of acquiring deletion mutationsa
| Mutation | Transformant frequencyb |
|
|---|---|---|
| D39 | BX515 | |
| Δcps2Tc | 1.2 × 10−2 (9.2 × 10−3) | 1.2 × 10−2 (4.2 × 10−3) |
| Δcps2Fc | 4.2 × 10−3 (1.1 × 10−3) | 2.8 × 10−3 (8.4 × 10−5) |
| Δcps2Gc | 3.2 × 10−5 (1.1 × 10−6) | 2.6 × 10−3 (5.4 × 10−4)d |
| Δcps2Ic | 2.9 × 10−5 (1.7 × 10−5) | 3.1 × 10−4 (3.2 × 10−5)d |
Recipients were the D39 parent and the D39 derivative BX515, which contains a cps2E point mutation.
Frequencies are the number of transformants per recipient, i.e., number of antibiotic-resistant CFU/total number of CFU, expressed as the means (± standard errors) from two independent transformations. The complete data sets are provided in Table S3 in the supplemental material.
Donor DNAs used for deletions were obtained from the E. coli constructs indicated in Table S1 in the supplemental material.
Significantly different (P < 0.05) from D39, as determined using a two-tailed unpaired Student t test.
Synthesis of dTDP-rhamnose and biochemical evidence for Cps2L, Cps2N, Cps2M, and Cps2O functions.
To biochemically demonstrate activities for the dTDP-Rha biosynthetic enzymes (Fig. 4A) and to generate a source of dTDP-Rha for use in in vitro glycosyltransferase assays, cps2L, cps2M, cps2N, and cps2O were individually cloned from S. pneumoniae D39 and expressed with N-terminal His tags in E. coli (Fig. 4B). Enzymatic activities of the purified proteins were initially demonstrated using indirect assays that followed reduction of NAD+ to NADH in reaction mixtures containing Cps2L and Cps2N (see Fig. S1A in the supplemental material) or oxidation of NADH in reaction mixtures containing Cps2M and Cps2O (see Fig. S1B in the supplemental material). The products generated from individual reactions were analyzed using HPLC (Fig. 4C). The Cps2L product was confirmed to be dTDP-Glc by its retention time, which was similar to that of the standard dTDP-Glc. The Cps2N and Cps2M reaction products had a delayed retention time compared to that of their precursors, similar to other dTDP-keto intermediates analyzed with these methods (16). The shorter retention time for the Cps2O product relative to dTDP-Glc is consistent with that reported for dTDP-Rha in other publications (23, 50). Sugar component analysis of the purified HPLC peak corresponding to the Cps2O reaction product confirmed the presence of Rha and the absence of Glc (Fig. 4D), while the mass of the purified Cps2O reaction product was confirmed through ESI-MS/MS to be that of dTDP-Rha (see Fig. S2 in the supplemental material).
Fig 4.

dTDP-Rha biosynthesis. (A) dTDP-Rha biosynthesis pathway in S. pneumoniae type 2. (B) dTDP-Rha biosynthesis enzymes were expressed in E. coli and purified using an incorporated N-terminal His tag. Purified proteins (0.1 μg) were separated by SDS–12% PAGE and stained with Coomassie brilliant blue. Lanes: 1, Cps2L (calculated molecular mass, 34.7 kDa); 2, Cps2N (41.5 kDa); 3, Cps2M (24.8 kDa); 4, Cps2O (34.8 kDa). Apparent molecular masses from standards are presented on the left. (C) HPLC analyses of the Cps2L, Cps2N, Cps2M, and Cps2O reaction products. Samples analyzed, listed from the top to bottom, are dTDP-Glc standard, Cps2L, Cps2N, Cps2M, and Cps2O reaction products. (D) GC-MS sugar component analyses of Glc standard, Rha standard, and purified Cps2O product.
cps2T encodes the β1-4 rhamnosyltransferase that adds the second sugar to the repeat unit.
We previously demonstrated that Cps2E initiates serotype 2 capsule repeat unit synthesis by transferring Glc-1-P to Und-P, generating Und-P-P-Glc (12). With the premise of a stepwise addition for each sugar residue, the next step is the addition of a single β1-4 Rha residue (Fig. 1). To determine which of the predicted glycosyltransferases accomplishes this step, we first cloned and expressed Cps2T, Cps2F, Cps2G, and Cps2I in E. coli. We then performed cellular fractionation experiments and found that Cps2T, Cps2F, Cps2G, and Cps2I were contained in the membrane fraction (Fig. 5A and B). In contrast to Cps2E, a known integral membrane protein, large amounts of Cps2T, Cps2F, Cps2G, and, to a lesser extent, Cps2I dissociated from the membranes by washing with Na2CO3 (pH 11.5), a procedure that releases peripherally associated membrane proteins (19) (see Fig. S3 in the supplemental material).
Fig 5.

Cps2T, Cps2F, Cps2G, and Cps2I localization in E. coli. (A) N-terminal His-tagged glycosyltransferases were expressed in E. coli. The cytoplasmic and membrane fractions were extracted and normalized as described in Materials and Methods. Three micrograms of total protein for each fraction was separated by SDS–12% PAGE, transferred to a nitrocellulose membrane, and probed with anti-tetra-His antibody. The calculated molecular masses for His-tagged Cps2T, Cps2F, Cps2G, and Cps2I are 47.6, 38.7, 42.9, and 46.8 kDa, respectively. (B) Fractionation controls, using representative preparations from DJ052 (Cps2T), were probed with MAb to the β subunit of E. coli RNA polymerase (150 kDa) for cytoplasmic protein detection (left) and with MAb to the β subunit of ATP synthase (50 kDa) for membrane protein detection (right). C, cytoplasmic fraction; M, membrane fraction.
To assess glycosyltransferase activities, we used Cps2E-containing membranes in reaction mixtures with membranes containing the respective glycosyltransferases and nucleotide sugars. Under these conditions, we found it essential to add a small amount of nonionic detergent (0.008% NP-40) for enzyme activity. Glycolipids formed in the reactions were extracted and subjected to mild-acid hydrolysis to liberate the saccharide moiety, and the products were separated by TLC. In the presence of UDP-[14C]Glc only, a single product consistent with the incorporation of Glc was observed for all reactions (Fig. 6A). This product was not observed when membranes containing Cps2E were absent from the reactions (data not shown), suggesting that only Cps2E was capable of adding Glc to a lipid acceptor. When UDP-[14C]Glc and dTDP-Rha were added to membranes that contained Cps2E and each of the other glycosyltransferases, only the reaction that contained Cps2T resulted in the formation of a new [14C]Glc-labeled product (Fig. 6B). The slower migration of this product relative to [14C]Glc is consistent with the addition of a sugar residue(s) (34). Formation of this product only upon addition of dTDP-Rha and Cps2T indicates that Cps2T is the β1-4 rhamnosyltransferase that adds the second sugar to the repeat unit.
Fig 6.

Cps2T rhamnosyltransferase activity. Cps2E-containing membranes (3 μg total membrane protein) were incubated with membranes (3 μg total protein) containing the indicated non-His-tagged glycosyltransferases or membrane controls (MC; 3 μg total protein from vector-only E. coli strain RC124) and UDP-[14C]Glc only (A) or UDP-[14C]Glc and dTDP-Rha (B). Reaction mixtures (total volume, 75 μl) were incubated for 1 h at 10°C and contained 0.008% NP-40, 10 mM MnCl2, 1 mM DTT, 0.025 μCi UDP[14C]Glc (293 mCi/mmol), and, for panels B and C, 0.1 mM dTDP-Rha. Glycolipids were extracted, mild-acid hydrolyzed to liberate the saccharide moiety, and separated by TLC as described in Materials and Methods. The migration of the product designated Glc was equivalent to that of a [14C]Glc standard. (C and D) Cps2T activity in the absence of Cps2E. (C) The extracted Cps2E glycolipid product (Und-P-P-[14C]Glc) was incubated with membranes containing non-His-tagged Cps2T or membranes from the vector-only E. coli strain (MC). Reaction mixtures contained 3 μg of total membrane protein, 0.008% NP-40, 10 mM MnCl2, 1 mM DTT, 25 μl of the Und-P-P-[14C]Glc solution (see Materials and Methods), and 0.1 mM dTDP-Rha. Reactions were processed as described above. (D) Cps2E immunoblot demonstrating controls for extracted glycolipids. Lanes: 1, 3 μg of total membrane protein before extraction; 2, extracted organic phase containing glycolipids; 3, interface between organic and aqueous phase containing extracted proteins.
Because both Cps2E and Cps2T were present in the glycosyltransferase reactions, we next tested whether Cps2T could add Rha to Und-P-P-Glc in the absence of Cps2E. Here, membranes containing Cps2E were used to synthesize Und-P-P-[14C]Glc, which was then extracted and incubated with dTDP-Rha and membranes containing Cps2T. As shown in Fig. 6C, Cps2T was capable of adding Rha to Und-P-P-[14C]Glc independently of Cps2E, which was undetectable in the glycolipid fraction by immunoblotting (Fig. 6D). The formation of this product in the absence of free UDP-[14C]Glc further confirms the Cps2T addition of Rha to a Cps2E-dependent glycolipid.
DISCUSSION
Bacterial polysaccharide capsules are commonly assembled by the Wzy-dependent mechanism, where multiple glycosyltransferases synthesize the capsule repeat unit on a lipid acceptor. cps2T was first predicted to encode a rhamnosyltransferase by Iannelli et al. (32). Later bioinformatic analyses showed that each of the 14 solved capsule structures for serotypes containing a cpsT homologue has a β1-4 Rha at the second sugar position, leading to the prediction that Cps2T (WchF) is a β1-4 rhamnosyltransferase (1, 39). Each of these serotypes also has a cps2E homologue, and as a result, the first two sugars and linkages in their capsule repeat unit backbone (Rhaβ1-4Glcβ1-) are conserved. The serotypes differ, however, in the remaining composition of sugars and linkages. All but two (serotypes 2 and 7B) lack a Rha residue at the third sugar position, strongly suggesting that Cps2T adds only a single Rha residue. In the type 2 capsule structure, two identical inverting linkages (α1-3Rha-α1-3 Rha) are present, but the cps2 locus contains only one gene for an inverting transferase (cps2F, also annotated as wchG). Biochemical data presented here confirm the assignment of Cps2T as the β1-4 rhamnosyltransferase that adds the first Rha, and genetic evidence discussed below indicates that Cps2F adds one or both of the subsequent Rha residues.
Knowledge of the mechanisms that regulate the repeat unit assembly process is limited, but it is a common property of many pathways that the committed step serves as an important point of regulation. The committed step is generally considered to reflect the first irreversible step in a pathway. In capsule synthesis, this step leads to the formation of a lipid-linked saccharide product, the accumulation of which can be lethal if the pathway is stalled prior to transfer of the sugars from Und-P to another acceptor. The lethality is thought to be due to the sequestering of the limited quantities of Und-P in the cell (41) that are required for other essential pathways or to the destabilization of the membrane (8, 14, 33, 49, 61, 64). Based on the results of a previous study, we anticipated that the committed step in type 2 capsule synthesis might not be the addition of the first Glc. There, mutations resulting in partial repeat unit formation due to a lack of the terminal GlcUA or mutations that inhibit repeat unit transport (Wzx flippase mutants) or polymerization (Wzy polymerase mutants) were obtained only in the presence of secondary suppressor mutations (61). Although most of these suppressor mutations mapped to cps2E, one was an insertion mutation in cps2L that was likely polar on cps2M, cps2N, and cps2O, thereby eliminating synthesis of dTDP-Rha and capsule. Cps2E activity from the latter strain was comparable to that of the parent in in vitro assays, and no mutations were present in cps2E or other capsule genes. The results of the present study are consistent with the notion that the Cps2E-catalyzed addition of the first sugar (Glc) is not the committed step, as elimination of Cps2T activity did not require suppressor mutations. Cps2E activity in vivo therefore either is reversible (as has been demonstrated for the in vitro reaction [12]) or requires the presence of the subsequent substrate and/or enzyme to proceed efficiently. An alternate hypothesis is that Und-P-P-Glc is not toxic because it can be used in another pathway, such as glycoprotein synthesis (31). However, if Und-P-P-Glc does occur in other S. pneumoniae pathways, it is unavailable for use in capsule synthesis, as Δcps2E mutants do not produce CPS. The presence of secondary mutations in only the Δcps2F, Δcps2G, and Δcps2I mutants provides genetic evidence that Cps2F, Cps2G, and Cps2I act downstream of the committed step and Cps2T performs this step.
A requirement for suppressor mutations to prevent lethality resulting from mutations affecting later-acting steps has similarly been reported for lipopolysaccharide O-antigen and exopolysaccharide mutants in Gram-negative bacteria (8, 33, 49, 64) and teichoic acid mutants in the Gram-positive bacterium Staphylococcus aureus (14). The S. pneumoniae capsule system seems to differ from some of these systems, however, in that addition of the second sugar can be eliminated without apparent detriment. It remains unclear why most secondary mutations occur in cps2E (30 among 33 mutants in our studies to date), unless it provides a target larger and more effective than the genes for other enzymes. In both this and our previous study (61), mutations in either the Cps2E cytoplasmic loop, which encodes the glycosyltransferase activity, or extracytoplasmic loop were effective in suppressing the effects of the primary mutation. Both domains thus provide important functions, though the role of the extracytoplasmic loop remains unknown.
Deletion of cps2G or cps2I required suppressor mutations for viable mutants, and the observation that these mutants were acquired at a low frequency supports the notion that these gene products are essential in the parental background. Despite the requirement for a suppressor mutation, Δcps2F mutants were obtained at a higher frequency than Δcps2G or Δcps2I mutants and at a frequency more similar to that for Δcps2T mutants. This result suggests that these suppressor mutations arose at a different point in time than those in the rarely obtained Δcps2G and Δcps2I mutants and may reflect a different stringency for tolerance of the initial mutation. If loss of cps2G or cps2I is rapidly lethal, viability of these mutants may require the initial transformation to occur in those rare recipients that already contain a cps2E mutation. In contrast, if Δcps2F mutants are viable for many generations, the deletion mutation could be obtained at a high frequency and the necessary cps2E mutation could occur in any of the transformants over many generations. Scenarios consistent with this possibility are a less efficient Cps2T when Cps2F is missing and/or feedback inhibition of the dTDP-Rha biosynthetic enzymes (40) as dTDP-Rha accumulates in the Δcps2F mutants. Either effect would slow the accumulation of Und-P-P-Glc-Rha compared to that in the Δcps2G and Δcps2I mutants, with a consequent reduction in time to death. These results are also consistent with Cps2F activity immediately succeeding that of Cps2T, as suggested by the assignment of Cps2F as the sole inverting transferase in the type 2 locus (1, 39). Because the type 2 capsule has two identical inverting linkages (α1-3 Rha–α1-3 Rha), Cps2F may catalyze two sequential reactions, a dual functionality that was first described in eukaryotes (18) but only recently characterized for prokaryotic glycosyltransferases (51). We are currently testing this possibility, in addition to identifying the functions of the other glycosyltransferases.
Polymerization of the Und-P-P-Glc-Rha-Rha-Rha-Glc-GlcUA repeat unit results in the formation of a Glc-GlcUA side chain in the type 2 polymer (Fig. 1). Detection of a small amount of CPS in the Δcps2G mutants must mean that the cps2E suppressor mutations in these strains do not completely abolish Cps2E activity in vivo, resulting in sublethal amounts of partial repeat units that can be translocated and polymerized. A similar capsule phenotype was observed with the Δcps2K UDP-Glc dehydrogenase mutants, which also contained suppressor mutations that primarily mapped to cps2E (61). These results suggest that cps2G encodes the glycosyltransferase that adds the Glc or GlcUA, and its elimination does not preclude synthesis of the backbone. Cps2I is expected to add the other sugar in the resulting side chain, but we have not been able to detect any polymer with the Δcps2I mutants. It is not yet known whether this result is due to its true absence, to polymer production below our limit of detection, or to an inactive Cps2E in each of the mutants obtained to date. Capsule transfer to the cell wall in the Δcps2G and Δcps2K mutants has also not been detected (61; this study). Although this result may be due to detection limits, failure to complete capsule transfer to the cell wall could contribute to the lethality that requires a suppressor mutation for survival. Reduced efficiencies of the Wzx flippase and Wzy polymerase in recognizing the incomplete repeat units may also be important, as the Wzx flippase for Salmonella enterica group B and D2 O antigen demonstrates a higher efficiency of translocation for the complete repeat unit than for one lacking a side-branch sugar (30).
When expressed in E. coli, Cps2T, as well as Cps2F, Cps2G, and Cps2I, behaved as a peripheral membrane protein. Glycosyltransferases responsible for synthesizing O antigens and other polysaccharide capsules have similarly been reported to localize to the membrane (48). This location is likely important in their ability to interact with the growing lipid-linked repeat unit and possibly with the other glycosyltransferases. In vitro, Cps2T catalyzed the formation of Und-P-P-Glc-Rha from Und-P-P-Glc and dTDP-Rha in the absence of Cps2E. The rhamnosyltransferase activity of Cps2T therefore does not require interaction with Cps2E, but we cannot rule out the possibility that interaction enhances in vivo activity and repeat unit formation, as suggested above.
Homologues of Cps2T are present in at least 27 S. pneumoniae serotypes (1, 5, 39). This abundance elevates cpsT to one of the most prevalent S. pneumoniae glycosyltransferase-encoding genes in the cps locus, second only to cpsE homologues. As a result of this work, we predict that Cps2T homologues in each of these S. pneumoniae serotypes are responsible for the committed step in capsule synthesis. The addition of Rha to Und-P-P-Glc as the committed step raises the possibility that regulation of Cps2T rhamnosyltransferase activity is a critical point of control. A principal factor regulating the action of glycosyltransferases is the availability of their nucleotide-sugar donor (54). The synthesis of dTDP-Rha is resource intensive, requiring the activity of four enzymes and two cofactors (Fig. 4A). Biochemical studies by Melo and Glaser (40) demonstrated that the dTDP-Rha biosynthetic pathway in P. aeruginosa possesses a feedback regulation where the end product, dTDP-Rha, competitively and noncompetitively inhibits the activity of the first enzyme in the pathway, which is a Cps2L homologue. These enzymes are also pyrophosphorylases, capable of converting dTDP-Glc to dTTP and Glc-1-P (Fig. 4A) (40). Cps2L could therefore be an important cellular target for regulating dTDP-Rha concentrations, Cps2T activity, and ultimately, capsule levels. Interestingly, recent findings in Caulobacter crescentus have identified homologues of Cps2L and Cps2M (the third enzyme in the dTDP-Rha biosynthesis pathway) to be phosphorylated at serine and/or threonine residues in response to carbon starvation and rich environments, respectively (26). If Cps2L and/or Cps2M is phosphorylated in S. pneumoniae, these modifications could affect dTDP-Rha synthesis and, correspondingly, capsule levels. Homologues of cpsLNMO are present in at least 38 S. pneumoniae serotypes (5). S. pneumoniae possesses a tyrosine phosphorylation system, encoded by cpsBCD, which is known to influence capsule levels but through an unidentified mechanism (4, 43, 58). Regulation of nucleotide-sugars as a modulator of capsule levels has been observed in S. pneumoniae serotype 3 strains that utilize a synthase-dependent mechanism for capsule synthesis (17, 55) but thus far has not been described in Wzy-dependent synthesis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants to J.Y. from the National Institutes of Health (AI28457), the Mizutani Foundation for Glycoscience, and the UAB Health Services Foundation. Funds for the operation of the Targeted Metabolomics and Proteomics (TMP) core facility come in part from the UAB Center for Nutrient-Gene Interaction (U54 CA 100949), the Purdue-UAB Botanicals Center for Age-Related Disease (P50 AT00477), the UAB O'Brien Acute Kidney Injury Center (P30 DK079337), the UAB Skin Disease Research Center (P30 AR50948), and the UAB Lung Health Center. DNA sequencing was performed by the UAB CFAR DNA sequencing core (P30A127767) or the Genomics Core Facility of the Heflin Center for Genomic Science.
We thank Doyle Ray Moore II from the University of Alabama at Birmingham TMP core facility for assistance with ESI-MS/MS analyses and Kellie Putnam for generating E. coli strain KJ4152.
Footnotes
Published ahead of print 21 September 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Aanensen DM, Mavroidi A, Bentley SD, Reeves PR, Spratt BG. 2007. Predicted functions and linkage specificities of the products of the Streptococcus pneumoniae capsular biosynthetic loci. J. Bacteriol. 189:7856–7876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Avery OT, Dubos R. 1931. The protective action of a specific enzyme against type III pneumococcus infection in mice. J. Exp. Med. 54:73–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Avery OT, MacLeod CM, McCarty M. 1944. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: induction of transformation by a deoxyribonucleic acid fraction isolated from pneumococcus type III. J. Exp. Med. 79:137–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bender MH, Cartee RT, Yother J. 2003. Positive correlation between tyrosine phosphorylation of CpsD and capsular polysaccharide production in Streptococcus pneumoniae. J. Bacteriol. 185:6057–6066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bentley SD, et al. 2006. Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS Genet. 2:e31 doi:10.1371/journal.pgen.0020031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Breton C, Snajdrova L, Jeanneau C, Koca J, Imberty A. 2006. Structures and mechanisms of glycosyltransferases. Glycobiology 16:29R–37R [DOI] [PubMed] [Google Scholar]
- 7. Brown EJ, Joiner KA, Cole RM, Berger M. 1983. Localization of complement component 3 on Streptococcus pneumoniae: anti-capsular antibody causes complement deposition on the pneumococcal capsule. Infect. Immun. 39:403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burrows LL, Lam JS. 1999. Effect of wzx (rfbX) mutations on A-band and B-band lipopolysaccharide biosynthesis in Pseudomonas aeruginosa O5. J. Bacteriol. 181:973–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Calix JJ, Nahm MH. 2010. A new pneumococcal serotype, 11E, has a variably inactivated wcjE gene. J. Infect. Dis. 202:29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Caputto R, Leloir LF, Cardini CE, Paladini AC. 1950. Isolation of the coenzyme of the galactose phosphate-glucose phosphate transformation. J. Biol. Chem. 184:333–350 [PubMed] [Google Scholar]
- 11. Cardini CE, Paladini AC, Caputto R, Leloir LF. 1950. Uridine diphosphate glucose: the coenzyme of the galactose-glucose phosphate isomerization. Nature 165:191 [Google Scholar]
- 12. Cartee RT, Forsee WT, Bender MH, Ambrose KD, Yother J. 2005. CpsE from type 2 Streptococcus pneumoniae catalyzes the reversible addition of glucose-1-phosphate to a polyprenyl phosphate acceptor, initiating type 2 capsule repeat unit formation. J. Bacteriol. 187:7425–7433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Centers for Disease Control and Prevention 7 May 2012, revision date Epidemiology and prevention of vaccine-preventable diseases, 12th ed, second printing, p 233–248 Public Health Foundation, Washington, DC: http://www.cdc.gov/vaccines/pubs/pinkbook/table-of-contents.html [Google Scholar]
- 14. D'Elia MA, et al. 2006. Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J. Bacteriol. 188:4183–4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dillard JP, Vandersea MW, Yother J. 1995. Characterization of the cassette containing genes for type 3 capsular polysaccharide biosynthesis in Streptococcus pneumoniae. J. Exp. Med. 181:973–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dong S, et al. 2010. Characterization of the enzymes encoded by the anthrose biosynthetic operon of Bacillus anthracis. J. Bacteriol. 192:5053–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Forsee WT, Cartee RT, Yother J. 2009. A kinetic model for chain length modulation of Streptococcus pneumoniae cellubiuronan capsular polysaccharide by nucleotide sugar donor concentrations. J. Biol. Chem. 284:11836–11844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frank CG, Aebi M. 2005. ALG9 mannosyltransferase is involved in two different steps of lipid-linked oligosaccharide biosynthesis. Glycobiology 15:1156–1163 [DOI] [PubMed] [Google Scholar]
- 19. Fujiki Y, Hubbard AL, Fowler S, Lazarow PB. 1982. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93:97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giraud MF, Naismith JH. 2000. The rhamnose pathway. Curr. Opin. Struct. Biol. 10:687–696 [DOI] [PubMed] [Google Scholar]
- 21. Glaser L, Kornfeld S. 1961. The enzymatic synthesis of thymidine-linked sugars. II. Thymidine diphosphate l-rhamnose. J. Biol. Chem. 236:1795–1799 [PubMed] [Google Scholar]
- 22. Graninger M, Kneidinger B, Bruno K, Scheberl A, Messner P. 2002. Homologs of the Rml enzymes from Salmonella enterica are responsible for dTDP-beta-l-rhamnose biosynthesis in the gram-positive thermophile Aneurinibacillus thermoaerophilus DSM 10155. Appl. Environ. Microbiol. 68:3708–3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Graninger M, Nidetzky B, Heinrichs DE, Whitfield C, Messner P. 1999. Characterization of dTDP-4-dehydrorhamnose 3,5-epimerase and dTDP-4-dehydrorhamnose reductase, required for dTDP-l-rhamnose biosynthesis in Salmonella enterica serovar Typhimurium LT2. J. Biol. Chem. 274:25069–25077 [DOI] [PubMed] [Google Scholar]
- 24. Griffith F. 1928. The significance of pneumococcal types. J. Hyg. (Lond.) 27:113–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guo H, Yi W, Song JK, Wang PG. 2008. Current understanding on biosynthesis of microbial polysaccharides. Curr. Top. Med. Chem. 8:141–151 [DOI] [PubMed] [Google Scholar]
- 26. Ham BM. 2011. Prokaryotic phosphorylation of serine, threonine, and tyrosine, p 181–244 Proteomics of biological systems. John Wiley & Sons, Inc, New York, NY [Google Scholar]
- 27. Hardy GG, Caimano MJ, Yother J. 2000. Capsule biosynthesis and basic metabolism in Streptococcus pneumoniae are linked through the cellular phosphoglucomutase. J. Bacteriol. 182:1854–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hardy GG, Magee AD, Ventura CL, Caimano MJ, Yother J. 2001. Essential role for cellular phosphoglucomutase in virulence of type 3 Streptococcus pneumoniae. Infect. Immun. 69:2309–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Henrichsen J. 1995. Six newly recognized types of Streptococcus pneumoniae. J. Clin. Microbiol. 33:2759–2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hong Y, Cunneen MM, Reeves PR. 2012. The Wzx translocases for Salmonella enterica O-antigen processing have unexpected serotype specificity. Mol. Microbiol. 84:620–630 [DOI] [PubMed] [Google Scholar]
- 31. Hug I, Feldman MF. 2011. Analogies and homologies in lipopolysaccharide and glycoprotein biosynthesis in bacteria. Glycobiology 21:138–151 [DOI] [PubMed] [Google Scholar]
- 32. Iannelli F, Pearce BJ, Pozzi G. 1999. The type 2 capsule locus of Streptococcus pneumoniae. J. Bacteriol. 181:2652–2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Katzen F, et al. 1998. Xanthomonas campestris pv. campestris gum mutants: effects on xanthan biosynthesis and plant virulence. J. Bacteriol. 180:1607–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kolkman MA, van der Zeijst BA, Nuijten PJ. 1997. Functional analysis of glycosyltransferases encoded by the capsular polysaccharide biosynthesis locus of Streptococcus pneumoniae serotype 14. J. Biol. Chem. 272:19502–19508 [DOI] [PubMed] [Google Scholar]
- 35. Kornfeld S, Glaser L. 1961. The enzymic synthesis of thymidine-linked sugars. I. Thymidine diphosphate glucose. J. Biol. Chem. 236:1791–1794 [PubMed] [Google Scholar]
- 36. Lairson LL, Henrissat B, Davies GJ, Withers SG. 2008. Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. 77:521–555 [DOI] [PubMed] [Google Scholar]
- 37. Llull D, Garcia E, Lopez R. 2001. Tts, a processive beta-glucosyltransferase of Streptococcus pneumoniae, directs the synthesis of the branched type 37 capsular polysaccharide in pneumococcus and other gram-positive species. J. Biol. Chem. 276:21053–21061 [DOI] [PubMed] [Google Scholar]
- 38. Magee AD, Yother J. 2001. Requirement for capsule in colonization by Streptococcus pneumoniae. Infect. Immun. 69:3755–3761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mavroidi A, et al. 2007. Genetic relatedness of the Streptococcus pneumoniae capsular biosynthetic loci. J. Bacteriol. 189:7841–7855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Melo A, Glaser L. 1965. The nucleotide specificity and feedback control of thymidine diphosphate d-glucose pyrophosphorylase. J. Biol. Chem. 240:398–405 [PubMed] [Google Scholar]
- 41. Mengin-Lecreulx D, Texier L, Rousseau M, van Heijenoort J. 1991. The murG gene of Escherichia coli codes for the UDP-N-acetylglucosamine: N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase involved in the membrane steps of peptidoglycan synthesis. J. Bacteriol. 173:4625–4636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morona JK, Morona R, Paton JC. 1997. Characterization of the locus encoding the Streptococcus pneumoniae type 19F capsular polysaccharide biosynthetic pathway. Mol. Microbiol. 23:751–763 [DOI] [PubMed] [Google Scholar]
- 43. Morona JK, Paton JC, Miller DC, Morona R. 2000. Tyrosine phosphorylation of CpsD negatively regulates capsular polysaccharide biosynthesis in Streptococcus pneumoniae. Mol. Microbiol. 35:1431–1442 [DOI] [PubMed] [Google Scholar]
- 44. Nelson AL, et al. 2007. Capsule enhances pneumococcal colonization by limiting mucus-mediated clearance. Infect. Immun. 75:83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neufeld F. 1902. Ueber die Agglutination der Pneumokokken und über die Theorieen der Agglutination. Med. Microbiol. Immunol. 40:54 [Google Scholar]
- 46. Okazaki R, Okazakit J, Strominger L, Michelson AM. 1962. Thymidine diphosphate 4-keto-6-deoxy-d-glucose, an intermediate in thymidine diphosphate l-rhamnose synthesis in Escherichia coli strains. J. Biol. Chem. 237:3014–3026 [PubMed] [Google Scholar]
- 47. Pelosi L, Boumedienne M, Saksouk N, Geiselmann J, Geremia RA. 2005. The glucosyl-1-phosphate transferase WchA (Cap8E) primes the capsular polysaccharide repeat unit biosynthesis of Streptococcus pneumoniae serotype 8. Biochem. Biophys. Res. Commun. 327:857–865 [DOI] [PubMed] [Google Scholar]
- 48. Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71:635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rick PD, et al. 2003. Evidence that the wzxE gene of Escherichia coli K-12 encodes a protein involved in the transbilayer movement of a trisaccharide-lipid intermediate in the assembly of enterobacterial common antigen. J. Biol. Chem. 278:16534–16542 [DOI] [PubMed] [Google Scholar]
- 50. Stern RJ, et al. 1999. Conversion of dTDP-4-keto-6-deoxyglucose to free dTDP-4-keto-rhamnose by the rmlC gene products of Escherichia coli and Mycobacterium tuberculosis. Microbiology 145(Pt 3):663–671 [DOI] [PubMed] [Google Scholar]
- 51. Troutman JM, Imperiali B. 2009. Campylobacter jejuni PglH is a single active site processive polymerase that utilizes product inhibition to limit sequential glycosyl transfer reactions. Biochemistry 48:2807–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Valvano MA. 2003. Export of O-specific lipopolysaccharide. Front. Biosci. 8:s452–s471 [DOI] [PubMed] [Google Scholar]
- 53. van Dam JE, Fleer A, Snippe H. 1990. Immunogenicity and immunochemistry of Streptococcus pneumoniae capsular polysaccharides. Antonie Van Leeuwenhoek 58:1–47 [DOI] [PubMed] [Google Scholar]
- 54. Varki A, Esko CRJ, Freeze H, Hart G, Marth J. 1999. Glycosyltransferases, 1st ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 55. Ventura CL, Cartee RT, Forsee WT, Yother J. 2006. Control of capsular polysaccharide chain length by UDP-sugar substrate concentrations in Streptococcus pneumoniae. Mol. Microbiol. 61:723–733 [DOI] [PubMed] [Google Scholar]
- 56. Weadge JT, Palcic MM, Begley TP. 2007. Glycosyltransferases, chemistry of. John Wiley & Sons, Inc, New York, NY [Google Scholar]
- 57. Weiser JN, Austrian R, Sreenivasan PK, Masure HR. 1994. Phase variation in pneumococcal opacity: relationship between colonial morphology and nasopharyngeal colonization. Infect. Immun. 62:2582–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Weiser JN, et al. 2001. Changes in availability of oxygen accentuate differences in capsular polysaccharide expression by phenotypic variants and clinical isolates of Streptococcus pneumoniae. Infect. Immun. 69:5430–5439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Whitfield C. 2006. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu. Rev. Biochem. 75:39–68 [DOI] [PubMed] [Google Scholar]
- 60. Wood WB, Jr, Smith MR. 1949. The inhibition of surface phagocytosis by the capsular slime layer of pneumococcus type III. J. Exp. Med. 90:85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xayarath B, Yother J. 2007. Mutations blocking side chain assembly, polymerization, or transport of a Wzy-dependent Streptococcus pneumoniae capsule are lethal in the absence of suppressor mutations and can affect polymer transfer to the cell wall. J. Bacteriol. 189:3369–3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yother J. 2011. Capsules of Streptococcus pneumoniae and other bacteria: paradigms for polysaccharide biosynthesis and regulation. Annu. Rev. Microbiol. 65:563–581 [DOI] [PubMed] [Google Scholar]
- 63. Yother J. 2006. Integration of capsular polysaccharide biosynthesis with metabolic and virulence pathways in Streptococcus pneumoniae, p 51–65 In Brogden KA, et al. (ed), Virulence mechanisms of bacterial pathogens, 4th ed ASM Press, Washington, DC [Google Scholar]
- 64. Yuasa R, Levinthal M, Nikaido H. 1969. Biosynthesis of cell wall lipopolysaccharide in mutants of Salmonella. V. A mutant of Salmonella typhimurium defective in the synthesis of cytidine diphosphoabequose. J. Bacteriol. 100:433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.