

Abstract
Human metapneumovirus (hMPV) is a leading cause of respiratory infections in pediatric populations globally, with no prophylactic or therapeutic measures. Recently, a recombinant hMPV lacking the M2-2 protein (rhMPV-ΔM2-2) demonstrated reduced replication in the respiratory tract of animal models, making it a promising live vaccine candidate. However, the exact nature of the interaction between the M2-2 protein and host cells that regulates viral infection/propagation is largely unknown. By taking advantage of the available reverse genetics system and ectopic expression system for viral protein, we found that M2-2 not only promotes viral gene transcription and replication but subverts host innate immunity, therefore identifying M2-2 as a novel virulence factor, in addition to the previously described hMPV G protein. Since we have shown that the RIG-I/MAVS pathway plays an important role in hMPV-induced signaling in airway epithelial cells, we investigated whether M2-2 antagonizes the host cellular responses by targeting this pathway. Reporter gene assays and coimmunoprecipitation studies indicated that M2-2 targets MAVS, an inhibitory mechanism different from what we previously reported for hMPV G, which affects RIG-I- but not MAVS-dependent gene transcription. In addition, we found that the domains of M2-2 responsible for the regulation of viral gene transcription and antiviral signaling are different. Our findings collectively demonstrate that M2-2 contributes to hMPV immune evasion through the inhibition of MAVS-dependent cellular responses.
INTRODUCTION
Human metapneumovirus (hMPV) is the first and only identified human pathogen belonging to the genus Metapneumovirus in the Pneumovirinae subfamily of the Paramyxoviridae family (50). It was discovered in 2001 and quickly recognized as a leading cause of lower respiratory tract disease in children, the elderly, and immunocompromised patients worldwide (17, 18, 55). hMPV encodes nine proteins. Among them, phosphoprotein P, glycoprotein G, and small hydrophobic protein SH have been shown to modulate hMPV-induced innate immune response, the first line of host defense against invading pathogens (5, 6, 23, 33). Whether or not other hMPV proteins are involved in the regulation of host cellular responses is currently unknown.
hMPV M2 encodes two overlapping proteins: M2-1 and M2-2. The M2-1 open reading frame (ORF) of strain CAN 97-83 is assumed to start with the first AUG at nucleotide (nt) position 14 and encodes a protein of 187 amino acids. The M2-2 ORF possibly initiates with the AUGs at positions 525 and 537, overlapping the M2-1 ORF by 53 or 41 nucleotides, respectively (13, 49). The M2-1 protein of hMPV is not essential for virus recovery using the reverse genetic system in vitro, in contrast to respiratory syncytial virus (RSV) M2-1 protein, which is essential for full viral transcription (13, 27, 45). The role of hMPV M2-2 protein in regulating viral replication, both in vitro and in vivo, using a rodent and a primate model of infection, was recently investigated (10, 13, 41). Compared to its counterpart rhMPV-WT, rhMPV-ΔM2-2 exhibited increased viral gene transcription and no change in viral genome accumulation (13, 41). This contradicts the results obtained using a minigenome reporter system, which demonstrated that hMPV M2-2 protein inhibited viral genome replication in addition to viral gene transcription (32). The initial aim of our study was to define the exact role of M2-2 in viral RNA synthesis. To accomplish this, we first generated a mutant recombinant hMPV, which has an intact M2-1 and genome length but lacks M2-2 expression, by site-directed mutagenesis. We found that accumulation of viral RNAs, both viral messenger and genomic RNAs, was significantly inhibited by M2-2 deletion, demonstrating that M2-2 promotes viral RNA synthesis.
Virus-induced innate immune signaling is regulated by viral RNA recognition through the Toll-like receptors (TLRs) and/or two DExD/H box RNA helicases, retinoic acid-inducible gene I (RIG-I) and MDA5 (reviewed in references 1 and 30). MAVS, a mitochondrial protein, links RIG-I/MDA5 to downstream kinases tumor necrosis factor receptor-associated factors (TRAFs)/IκB kinases (IKKs), responsible for NF-κB and interferon (IFN) regulatory factor (IRF) activation, leading to proinflammatory and antiviral gene expression (43, 44). Recently, we have shown that hMPV infection of alveolar epithelial cells, the primary target of respiratory viruses, induces cytokine, chemokine, and type I IFN via RIG-I/MAVS-dependent signaling, but not via TLR-3- and MDA5-dependent pathways (34). In this study, we discovered that the M2-2 protein antagonizes MAVS-mediated innate antiviral response.
This new function of M2-2 in blocking host innate immunity was initially considered because of results showing that rhMPV-ΔM2-2-infected cells produced higher levels of beta interferon (IFN-β) and other immune mediators than did rhMPV-WT-infected cells. Although the expression of hMPV G protein (a previously described virulence protein) was suppressed by the lower growth of rhMPV-ΔM2-2, which might contribute indirectly to the enhancement of innate immunity by M2-2 deletion, ectopic expression of G in rhMPV-ΔM2-2-infected cells at a higher level than that of G in rhMPV-WT-infected cells only partially reversed the enhancement, suggesting that M2-2 contributed to hMPV immune evasion as well.
In reporter gene assays, the M2-2 protein, but not other soluble hMPV proteins, inhibited the MAVS-activated IFN-β promoter but not the one specifically mediated by downstream signaling molecules, suggesting that MAVS is a target of M2-2. Coimmunoprecipitation (co-IP) studies, either in an overexpression system or in the context of viral infection, showed an association of M2-2 with MAVS, further supporting the idea that M2-2 targets MAVS.
In this study, we also identified the domains of M2-2 responsible for the regulation of viral gene transcription, viral replication, and RIG-I-mediated signaling. We found that the first 25 amino acids of M2-2 are critical to promote viral gene transcription but not involved in the regulation of viral replication and hMPV-induced signaling. In contrast, the domains spanning from amino acids 26 to 69 are dispensable for the regulation of viral gene transcription but responsible for RIG-I signaling inhibition and viral replication facilitation. Of note, two M2-2 deletion mutants (Δ26–54 and Δ55–69), which exhibited regulation on viral gene transcription similar to that of wild-type (WT) M2-2, did not suppress IFN-β secretion as well as did M2-2 in response to rhMPV-ΔM2-2 infection, suggesting a G-independent inhibition of host innate immunity by M2-2. In reporter gene assays, these two mutants also failed to block the MAVS-activated IFN-β promoter, confirming the importance of these domains in suppressing hMPV-induced immune responses.
MATERIALS AND METHODS
Cell lines and antibodies.
LLC-MK2 and Vero cells (ATCC, Manassas, VA) were maintained in minimal essential medium (MEM) (Invitrogen Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS), 100 IU/ml penicillin, and 100 μg/ml streptomycin. A549, human alveolar type II-like epithelial cells, and 293, a human embryonic kidney epithelial cell line (ATCC), were maintained in F-12K medium and MEM, respectively, containing 10% (vol/vol) FBS, 10 mM glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin. BSR T7/5 cells, baby hamster kidney cells that constitutively express the T7 RNA polymerase, were a gift from Karl-Klaus Conzelmann, Munich, Germany. They were maintained in Glasgow minimal essential medium (GMEM) supplemented with 1% amino acids, 10% FBS, 12 mg/liter tryptose phosphate broth, 1 mg/ml of Geneticin, 100 U/ml of penicillin, and 100 U/ml of streptomycin. Cells derived from mouse embryonic fibroblasts (MEFs) were cultured in Dulbecco modified Eagle medium (DMEM) containing 10% (vol/vol) FBS, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Monoclonal antibodies against lamin b and Flag were obtained from Sigma-Aldrich (St. Louis, MO). The antibody against V5 was obtained from Invitrogen (Carlsbad, CA). The polyclonal rabbit anti-hMPV and anti-hMPV G protein antibodies (gifts from Antonella Casola at the University of Texas Medical Branch, TX) were raised against purified hMPV and full-length G protein, respectively. The polyclonal rabbit anti-MAVS antibody was a gift from Ilkka Julkunen (National Public Health Institute, Finland). Primary antibodies against phosphorylated IRF-3, p50, and p65 were purchased from Millipore (Billerica, MA). Fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit antibody was from Zymed (South San Francisco, CA). Primary antibody against IRF-3 and horseradish-coupled secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Construction of ΔM2-2 antigenome and viral recovery.
A plasmid encoding wild-type hMPV antigenome was constructed as described previously (5, 6, 11). Construction of M2-2 mutant cDNA was done as illustrated in Fig. 1A. In brief, two start sites of M2-2 were deleted by a first-step multiple-site mutagenesis, which did not affect M2-1 codons in the corresponding sites (silent mutations). We then introduced two stop codons at the 13th and 19th amino acid sites of M2-2 by a second multiple-site mutagenesis to disable M2-2 expression. By doing so, the M2-1 gene expression was kept intact, while M2-2 expression was completely blocked. The sequence of the primer for generating ΔM2-2 antigenome is available upon request.
Fig 1.

Overview of the hMPV M2-2 gene deletion. (A) Map of hMPV antigenome and introduced mutations. Overlapped ORFs of M2-1 and M2-2 are shown as a gray rectangle with amino acid sites given above. Symbolized amino acids of overlapped M2-1 and M2-2 are listed under antigenome codons. The mutated sites were generated by site-directed mutagenesis. Mutations involved in silencing the M2-2 ORF include two abolished start codons (stars) and two introduced stop codons (triangles). These mutations did not affect the amino acid coding of M2-1 (comparison of M2-1 amino acids in the rectangles of WT/M2-1 with those for ΔM2-2/M2-1). (B) The genome length of rhMPV. Viral RNAs from purified rhMPV-WT, -ΔM2-2, or -ΔSH were prepared and subsequently subjected to RT-PCR. First-strand synthesis was done with a first-strand primer spanning hMPV nt 6896 to 6917. PCR was performed using paired primers matching the 5′ end of N (forward) and the 3′ end of G (reverse) to clone the hMPV fragment spanning N, P, M, F, M2-2, and G. The products were separated on a 1% agarose gel. (C) Confirmation of M2-2 deletion. Purified virus particles, rhMPV-WT or rhMPV-ΔM2-2, were loaded into a 4 to 20% SDS-PAGE gel and subjected to Western blotting using an anti-hMPV antibody. Data are representative of two independent experiments.
To recover recombinant hMPV, confluent BSR T7/5 cells in six-well dishes were transfected with 5 μg of antigenomic plasmid corresponding to rhMPV-WT or -ΔM2-2, together with plasmids encoding support proteins for recombinant virus recovery as previously described (5, 6). Trypsin (Worthington, Lakewood, NJ) was added on day 3 posttransfection to a final concentration of 1 μg/ml, and then cell-medium mixtures were passed onto fresh LLC-MK2 cells and incubated at 35°C. Typical viral cytopathic effect (CPE) was usually observed around day 5 to 6 postinfection (p.i.). Recombinant virus lacking M2-2 was confirmed by restriction enzyme digestion, sequencing of viral RNA, and Western blotting using an anti-hMPV antibody. The recovered viruses were then amplified for two passages in LLC-MK2 cells and saved as stock viral preparations. Viruses with no more than 4 to 5 passages were used in all experiments.
Viral preparation and infection.
The naive hMPV and its derived recombinant viruses were propagated in LLC-MK2 cells at 35°C in the absence of serum and in the presence of 1 μg/ml of trypsin and were sucrose purified, as previously described (5, 6). Viral titer was determined by immunostaining in LLC-MK2 cells, as previously described (5, 6). To characterize the rhMPV-ΔM2-2 growth pattern, LLC-MK2 or Vero cell monolayers in a 6-well plate were infected with rhMPV, WT or mutant, at a multiplicity of infection (MOI) of 0.1. An equivalent amount of sucrose solution was added to uninfected LLC-MK2 or Vero cells, as a control (mock infection). After initial absorption, viral inoculum was removed and cells were supplied with fresh serum-free medium with trypsin. Viruses were harvested at different times p.i., and viral titer was determined by immunostaining in LLC-MK2 cells, as previously described (5, 6).
To investigate the role of M2-2 in regulating innate antiviral signaling at the acute phase of infection, A549 cell monolayers were infected with rhMPV-WT or -ΔM2-2, at an MOI of 2. Mock infection was used as a negative control. Supernatants were harvested at different times p.i., and the concentrations of cytokines and chemokines were determined by enzyme-linked immunosorbent assay (ELISA) or a multiplex immunoassay. Total cells were lysed to prepare nuclear and cytosolic fractions, as previously described (5, 6).
Plasmid construction.
For protein expression studies, the M2-2 gene was cloned from the hMPV antigenome template. PCR was carried out using Pfu DNA polymerase (Stratagene, La Jolla, CA) according to the manufacturer's instructions. A V5 tag was added using the following primers: forward, 5′-ACGCgaattcATGACTCTTCATATGCCCTGCAAGACAGT-3′, and reverse, 5′-TctcgagTCACGTAGAATCGAGACCGAGGAGAGGGTTAGGGATAGGCTTACCACTTAAGTAAGCCTTGACATATATAATTTCTATGTTTTC-3′. Italicized lowercase letters indicate the restriction enzyme site; underlined letters indicate V5 sequence; bold letters indicate start or stop codons. The V5-tagged M2-2 was first cloned into the TOPO cloning vector and then cut by EcoRI and XhoI and subcloned into the pCAGGS vector.
To characterize the functional domains of M2-2 in MAVS-mediated signaling, truncated forms of M2-2 were constructed. To construct M2-2 lacking the first 25 amino acids (ΔN25), a primer, 5′-AAGTGCAGTGAGCATGGTCCTGAATTCATGACTATAGAGGTTGATGAAATG-3′, was used in a site-directed mutagenesis reaction (kit provided by Agilent, Santa Clara, CA) to create an EcoRI site at the Val22 site by mutating T65T66 to AA and to generate a start codon, ATG. The mutagenesis product was then cut by EcoRI, followed by a self-ligation. To generate an M2-2 mutant with deletion of Ile26-Asp54 (Δ26–54), a primer, 5′-GAGCATGGTCCTGTTTTCATTACCAATTGGGTTGATGAAATGATATGGACTC-3′, was used to create a MunI site (CAATTG) around Thr25. The letters in bold are mutated nucleotides. The created MunI site was then used for self-ligation with an endogenous MunI site around Asn55 of M2-2. To generate a an M2-2 lacking Asn55 to Tyr69 (Δ55–69), a primer, 5′-GTGAAGTCTCACACCAACATTTACTTAAGTTATTTAGAAAACATAGAAATT-3′, was used to generate an AflII site (CTTAAG) around Asn55 and Cys56 of M2-2. Letters in bold are mutated nucleotides. Lightface italic letters in sequences are part of created restriction enzyme digestion sites. The new AflII site was then used for self-ligation with the AflII site around Tyr69 of M2-2.
To synthesize hMPV N transcripts, which were used to generate the standard curve for the absolute N gene transcription assays, a T7-driven plasmid encoding hMPV N was constructed. Briefly, the open reading frame for hMPV N was amplified using PCR primers that introduced unique BamHI (5′) and SalI (3′) restriction sites. This DNA was inserted into a transcription vector, pT7PA, which was digested with BglII and SalI. The pT7PA vector directed transcription of synthetic transcripts using a T7 bacteriophage promoter. A 39-base adenine sequence was added to the 3′ end of the multiple-cloning region in this vector. The final constructs were verified by sequencing performed by the protein chemistry core laboratory at the University of Texas Medical Branch (UTMB).
Reverse transcriptase PCR (RT-PCR).
Viral RNA from purified viruses was extracted using the QIAamp viral RNA kit (Qiagen, Alameda, CA). The first-strand cDNA was then generated using Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA). PCR was carried out using Pfu DNA polymerase according to the manufacturer's instructions. Primers used to clone the fragment of N-P-M-F-M2-SH-G were 5′-ATGTCTCTTCAAGGGATTCA-3′ (forward) and 5′-GCATTGTGCTTACAGATGCCTG-3′ (reverse). The cloned fragments were either sent for sequencing or subjected to electrophoresis.
Quantitative RT-PCR (qRT-PCR).
We used two methods for viral gene transcription assays, absolute versus relative, for different experimental purposes. The absolute assay used synthetic viral gene transcripts to generate the standard curve to calculate the absolute copy numbers of unknown samples. The relative method quantifies differences in the expression level of a specific target (gene) between different samples. The data output is expressed as a fold change or a fold difference of expression levels. For the absolute N transcript assays, synthetic transcripts of the N gene were generated using the T7 MegaScript kit from Ambion (Austin, TX), treated with Turbo DNase, and purified according to the MegaScript kit protocol. The RT primer to measure the transcription of the hMPV N gene is 5′-CGTCTCAGCCAATCCCTGGTTTTTTTTTTTTAATTACTC-3′.
Primers were designed to incorporate a “tag” (underlined letters) as part of the assay due to self-priming exhibited by viral RNA (4). The tag sequence was derived from the bacterial chloramphenicol resistance (Cmr) gene. The sequence with bold letters is complementary to poly(A) tails of the transcribed hMPV N gene. The sequence in italic is N gene specific. At a 25°C annealing temperature, the 8 nucleotides (nt) matching N-specific sequences would not be sufficient for a stable efficient priming of cDNA from an antigenome of hMPV (positive strand). On the other hand, 20 nucleotides matching transcribed N (12 T's and N-gene-specific nucleotides) are able to attain stable annealing to the transcribed N gene. For the unknown samples, 1 μg of RNA was used. For the synthetic RNA, 104 to 107 transcripts were used to generate the standard curve. All reactions were performed under the following conditions: 25°C, 10 min; 48°C, 30 min; 95°C, 5 min. Quantitative PCRs (QPCRs) were performed with 2 μl cDNA and 300 nM N- and tag-specific primers using the FastStart Universal SYBR green master (ROX) (Roche). The hMPV N forward primer was 5′-CACAGACTATTTTCGCAGCAG-3′, and the reverse primer against hMPV tag was 5′-CGTCTCAGCCAATCCCTGG-3′. QPCRs were run in the ABI 7500 sequence detection system under the standard default conditions: initial steps of 50°C for 2 min and 95°C for 10 min and PCR steps of 95°C for 15 s and 60°C for 1 min, for 40 cycles. Similar strategies were used to design the primers for the relative gene transcription assays for hMPV F and G. Information on primers is available upon request.
To quantify viral antigenomic copies in the context of hMPV infection, synthetic transcripts of the genome were generated from Topo plasmid containing N-P-M genes, using the T7 MegaScript kit, following the digestion with PmeI. The reaction mixture was then treated with Turbo DNase and purified using the MegaScript kit. Primers were designed to span the N and P regions of the viral genome and incorporated a Cmr tag. First-strand cDNA was transcribed with a P-specific primer, 5′-CGTCTCAGCCAATCCCTGGTGATTATGAGTAATTAAAAAATGGGACAAG-3′.
The underlined letters indicate the Cmr tag sequence. QPCRs were performed using the following primers: forward, 5′-CGTCTCAGCCAATCCCTGG-3′, and reverse, 5′-GCTTCATTACCCATGAAAAGAATATC-3′. RT-PCRs and QPCRs were performed as described above.
Reporter gene assays.
To investigate the role of M2-2 in mediating host antiviral responses to hMPV infection, logarithmically growing 293 cells were transfected in triplicate with luciferase reporter gene plasmids containing IFN-β promoter (designated IFN-β-Luc) or multiple copies of NF-κB binding sites (Kb-5-Luc) or IRF-3 binding sites (IRF-3-Luc), together with plasmids encoding M2-2 and/or other viral proteins or their empty vector using FuGene 6 (Roche, Indianapolis, IN), as previously described (5, 6). Cells were infected with recombinant hMPV at 24 h posttransfection at an MOI of 2. Uninfected plates served as a control. At various times p.i., cells were lysed to measure independently luciferase and β-galactosidase reporter activity. Luciferase was normalized to the internal control β-galactosidase activity. In experiments where the role of M2-2 in modulating the RIG-I/MAVS/TRAF/IKK signaling pathway was investigated, A549 cells or 293 cells were cotransfected with a plasmid encoding RIG-I or expression plasmids for its downstream signaling molecules MAVS/TRAFs/IKKs, a plasmid encoding M2-2 or its mutants or control vectors, and a luciferase reporter plasmid, IFN-β-Luc/Kb-5-Luc/IRF-3-Luc. Cells were then lysed to measure luciferase as described above.
Coimmunoprecipitation.
Logarithmically growing 293 cells in 6-well plates were cotransfected with 2 μg of pEF-TAK containing Flag-tagged MAVS and 2 μg of either pCAGGS encoding V5-tagged M2-2 protein or the empty vector, a V5-tagged negative control. Cells were harvested 30 h after transfection, and immunoprecipitation was carried out using an immunoprecipitation kit from Roche (catalog no. 11719386001). In brief, 6 × 106 cells were lysed using 1.5 ml of lysis buffer. A preclearing step was performed by incubating the sample with 50 μl of the protein A/G-agarose for 3 h at 4°C on a rocking platform. Precleared samples were exposed to 5 μg of antibody against either V5 or Flag or to an isotype antibody control, for 1 h at 4°C. Fifty microliters of the protein A/G-agarose was added to the samples and incubated overnight at 4°C. The immunoprecipitated (IP) complexes were recovered by centrifugation and washed three times using buffers provided with the kit. The IP complexes were eluted from the beads and subjected to SDS-PAGE followed by Western blot analysis.
To investigate the specificity of M2-2 in the association with endogenous MAVS, logarithmically growing A549 cells in 6-well plates were transfected with a plasmid encoding V5-tagged hMPV M2-2 or RSV NS1 (a negative control) or their control plasmid for 40 h. Cells were lysed and immunoprecipitated using an anti-MAVS antibody or isotype control antibody, as described above. The presence of M2-2 protein in the complex was then detected using an anti-V5 antibody.
RIG-I–MAVS complex formation is critical for signaling transduction (31). To investigate whether MAVS–M2-2 interaction is through the binding of M2-2 to RIG-I, MAVS−/− MEFs in 6-well plates were transfected with a plasmid encoding Flag-tagged RIG-I or its control vector, a plasmid encoding V5-tagged M2-2 or G (a positive control), or their control plasmid for 40 h. Cells were harvested for immunoprecipitation using an anti-V5 antibody to pull down M2-2 or G. The presence of RIG-I in the IP complex was then determined with an anti-Flag antibody. Plasmids encoding MAVS were also cotransfected into cells expressing M2-2 and/or RIG-I, followed by the investigation on whether RIG-I is present in the MAVS–M2-2 complex.
We also investigated the interaction between endogenous MAVS and M2-2 in the context of hMPV infection. To do that, confluent A549 cells in 6-well plates were mock infected or infected with rhMPV-ΔM2-2 at an MOI of 2 for 15 h. Cells were lysed and immunoprecipitated using an anti-MAVS antibody or isotype control antibody, as described above. The presence of M2-2 protein in the complex was then detected using an anti-hMPV antibody.
Western blot analysis.
The cytosol and nuclear extracts of uninfected and infected cells were prepared using hypotonic/nonionic detergent lysis as described previously (6, 40), according to the protocol of Schaffner and colleagues (42). The lysates were collected and quantified with a protein quantification kit from Bio-Rad. Nuclear extracts were fractionated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. Membranes were blocked with 5% milk in Tris-buffered saline (TBS)–Tween 20 and incubated with the proper primary antibodies according to the manufacturer's instructions.
Intracellular staining for hMPV.
Confluent A549 cells in a 6-well plate were infected with WT or ΔM2-2 at an MOI of 2. Mock infection was used as a control. At 1 h p.i., cell supernatant was removed and replaced with fresh serum-free medium with trypsin. Cells were harvested at 3 h p.i., following a treatment with Accutase solution (Sigma, St. Louis, MO) for 5 min at 37°C. Cells were collected by centrifugation at 1,500 rpm for 5 min and resuspended in 200 μl of Cytofix/Cytoperm solution (Becton, Dickinson, San Jose, CA). After incubation for 20 min at room temperature, cells were washed with Perm/Wash buffer twice, followed by incubation with rabbit anti-hMPV antibodies at a dilution of 1:1,000 in 100 μl of Perm/Wash buffer at 4°C for 30 min. FITC-conjugated goat anti-rabbit antibody (1:100) was used as a secondary antibody for an additional 30 min at 4°C, after primary antibody removal. Cell acquisition was performed on a FACScan flow cytometer equipped with CellQuest software (both from Becton, Dickinson). Result analysis was performed using FlowJo software (Tree Star, La Jolla, CA).
Cytokine and chemokine quantification in A549 cell supernatants.
The levels of IFN-β, interleukin-8 (IL-8), and RANTES proteins in A549 cell supernatants after infection were quantified by ELISA (IFN-β, PBL Biomedical Laboratories, Piscataway, NJ; IL-8 and RANTES, R&D, Minneapolis, MN), while the remaining chemokines and cytokines shown in Fig. 3A were quantified using the Multi-Analyte Profiling human cytokine/chemokine kit (Bio-Rad, Hercules, CA), according to the manufacturer's instructions. Data were analyzed using the Milliplex Analyst software from Bio-Rad.
Fig 3.

Effect of M2-2 protein deletion on type I IFN, cytokine, and chemokine secretion. (A) Immune mediator induction by rhMPV. A549 cells in triplicate were mock infected or infected with rhMPV-WT or rhMPV-ΔM2-2, at an MOI of 2, for various times as indicated. The secretion of cytokines and chemokines in cell supernatants was measured by Bio-Plex and/or ELISA. Data shown are from two independent experiments and are expressed as means ± SEs. *, P < 0.05, and **, P < 0.01, relative to rhMPV-WT-infected A549 cells. (B) G protein overexpression partially converted the enhancement of cellular responses by M2-2 deletion. A549 cells in 6-well plates were transfected with a plasmid encoding G or its control vector (2 μg/well) At 24 h posttransfection, cells were mock infected or infected with WT or ΔM2-2 as indicated. At 15 h p.i., supernatant was harvested for cytokine/chemokine measurement by Bio-Plex. Three representative mediators were shown. The G protein in cell pallets was analyzed by Western blotting using an anti-hMPV G antibody. Data are representative of two independent experiments. (C) Both G and M2-2 contributed to hMPV immune evasion. 293 cells in triplicate were transfected with a luciferase reporter plasmid (IFN-β-Luc; 0.1 μg/well) and a plasmid encoding hMPV M2-2 and/or G or a control vector which either is empty or encodes N protein. Vectors (0.2 μg/well) encoding indicated viral proteins were transfected individually or in a combination. For cells which were transfected with one hMPV protein, 0.2 μg/well of control empty vectors was cotransfected to ensure equal loading. After 24 h, cells were mock infected or infected with WT or ΔM2-2. At 15 h p.i., cells were harvested for luciferase activity measurement. *, P < 0.05, and **, P < 0.01, relative to ΔM2-2 + CV. CV, control vector for M2-2, G, or N expression.
Statistical analysis.
Statistical significance was analyzed using analysis of variance (ANOVA). A P value of less than 0.05 was considered significant. Means ± standard errors (SEs) are shown.
RESULTS
Recovery of recombinant hMPV lacking the M2-2 protein.
To investigate the role of the hMPV M2-2 protein in viral RNA synthesis, we generated rhMPV-WT, as well as hMPV lacking M2-2 protein (rhMPV-ΔM2-2), using a reverse genetic system approach (5, 6, 11, 38). The overall strategy to abolish M2-2 expression is depicted in Fig. 1A. We used mutagenesis to eliminate the translation initiation codons of M2-2. This mutagenesis (labeled with an asterisk) did not likely affect the expression of M2-1, as it created only silent mutations in the corresponding sites of M2-1. Two in-frame stop codons (labeled with triangles) were then introduced into the 13th and 19th codons of M2-2 by a second round of mutagenesis. These changes did not alter the length of the M2 gene but completely abolished M2-2 expression (Fig. 1B and C). To verify that the length of the viral M2 gene was unaffected by the mutagenesis, viral RNAs were prepared and subsequently subjected to reverse transcriptase PCR (RT-PCR) using paired primers matching the 5′ end of N (forward) and the 3′ end of G (reverse), as described in Materials and Methods. As expected, there was no difference in the band size corresponding to the N-P-M-F-M2-SH-G fragment cloned from viral RNAs of rhMPV-WT and rhMPV-ΔM2-2, while the RT-PCR product of rhMPV-ΔSH, used as a control, showed a reduced size (Fig. 1B). To verify whether the M2-2 expression was completely abolished, purified recombinant viruses, WT or ΔM2-2, were suspended in 3× SDS sample buffer, followed by protein separation in a 4 to 20% Tris-glycine (TG) gel and Western blotting using an anti-hMPV antibody. As shown in Fig. 1C, there was a band slightly below 12 kDa, which is specific to rhMPV-WT. According to the gene size (M2-2 is the only gene smaller than M2-1), as well as the fact that M2-1 is reported to produce a protein of 21 kDa (13), we believe that the WT-specific band around 12 kDa represents the M2-2 protein.
The role of M2-2 in viral gene transcription and genome replication.
Previously, we have demonstrated that the replication of rhMPV-WT was similar to that of the parental hMPVCAN-83 isolate in LLC-MK2 cells (6). To investigate whether M2-2 plays a role in regulating hMPV replication, LLC-MK2 cells were infected with recombinant hMPV, WT or ΔM2-2, at an MOI of 0.1. A multicycle growth of WT and ΔM2-2 in these cells was then investigated by immune staining using anti-hMPV antibody (a gift from MedImmune, Gaithersburg, MD). We found that viral titers of rhMPV-ΔM2-2 were about a log- to a half-log-fold less than those of rhMPV-WT. Attenuated replication of rhMPV-ΔM2-2 was also observed in Vero cells, a cell line deficient in type I IFN genes, following multicycle growth (see Fig. S1 in the supplemental material).
To investigate whether M2-2 elimination affects the ability of hMPV to infect cells, A549 cells were infected with rhMPV-WT or -ΔM2-2 at an MOI of 2 and harvested at 3 h p.i. Flow cytometry using anti-hMPV polyclonal antibody was performed. As shown in Fig. 2A, the percentages of cells infected with WT and ΔM2-2 were similar, suggesting equal infectivities of WT and ΔM2-2 of airway epithelial cells. However, as infection progressed, there was significantly lower viral protein expression in rhMPV-ΔM2-2-infected cells than in rhMPV-WT-infected cells (Fig. 2B), indicating a role of M2-2 in promoting viral gene transcription in hMPV infection. Real-time PCR using primers against the N gene confirmed that viral gene transcription was reduced by M2-2 elimination in A549 cells (Fig. 2C, left panel).
Fig 2.
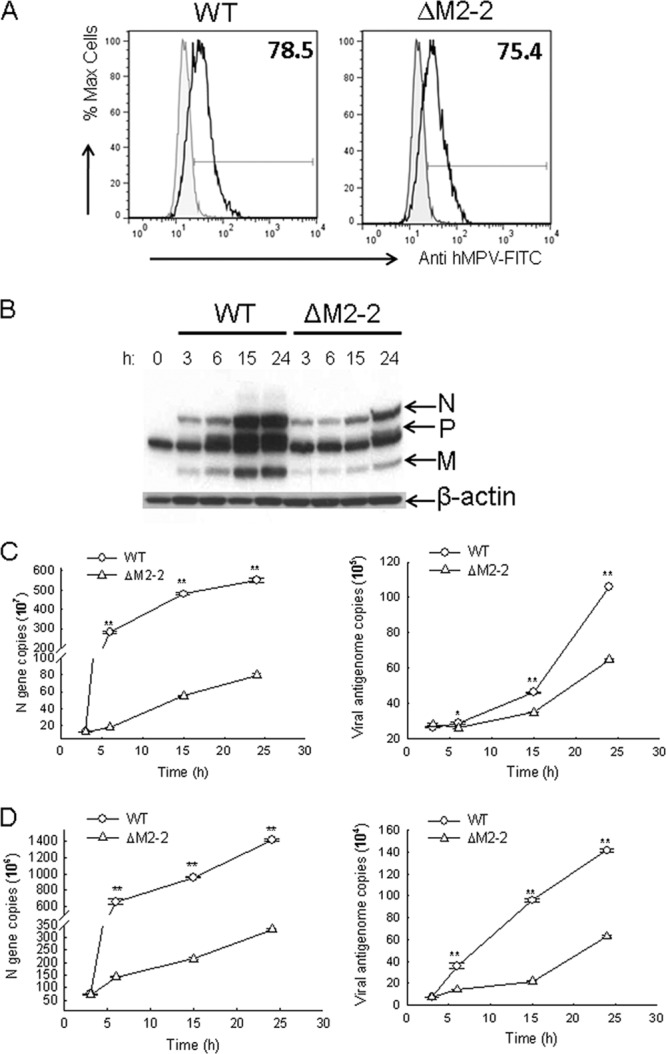
Replication and gene transcription characterization of recombinant viruses. (A) Intracellular staining of hMPV in WT- or ΔM2-2-infected cells. A549 cells were mock infected or infected with rhMPV, WT, or ΔM2-2, at an MOI of 2. After 1 h, supernatant was replaced with new serum-free medium containing 1 μg/ml trypsin. Cells were harvested at 3 h p.i. and stained with an anti-hMPV antibody as described in Materials and Methods. Samples were run on a FACScan flow cytometer equipped with BD FACSDiva software. Analysis was performed using FlowJo software (version 7.2.2). Corrected mean fluorescence intensities of WT- and ΔM2-2-infected samples were compared. (B) Viral protein expression analysis in infected cells. A549 cells were infected with rhMPV-WT or rhMPV-ΔM2-2 at an MOI of 2. After 1 h, supernatant was replaced with new serum-free medium containing trypsin. The cells were then harvested to prepare total cell lysates at the indicated times. Equal amounts of protein were subjected to SDS-PAGE, followed by Western blotting using a polyclonal antibody against hMPV. Membranes were stripped and reprobed for β-actin, as a control for equal loading of the samples. The results are representative of two independent experiments. (C and D) Copy number analysis of N gene and viral genome. A549 cells (C) or Vero cells (D) were mock infected or infected with rhMPV at an MOI of 2 for various periods of time as indicated, followed by total RNA extraction using TRIzol. The extracted RNAs in triplicate were then subjected to real-time PCR to assay viral N gene transcription (left panels) or genomic RNAs (right panels). The results are representative of two independent experiments and are expressed as means ± SEs of absolute copy numbers of transcribed N gene or viral genome. **, P < 0.01, relative to rhMPV-WT-infected A549 or Vero cells.
To investigate the role of M2-2 in regulating genomic RNA synthesis, viral genome copies in infected A549 cells were also measured by real-time PCR. As shown in the right panel of Fig. 2C, the number of antigenomic RNAs in WT-infected cells was significantly higher than that in ΔM2-2-infected cells at all time points tested, with the exception of 3 h p.i., indicating a role of M2-2 in facilitating hMPV genome replication. The attenuation of viral transcription and replication was also observed in Vero cells, suggesting that the attenuation is type I IFN independent (Fig. 2D; see also Fig. S2 in the supplemental material).
Regulation of hMPV-induced antiviral and proinflammatory molecules by M2-2.
Aside from the above-described restriction of viral RNA accumulation by M2-2 elimination, another mechanism(s) may also contribute to the attenuation of rhMPV-ΔM2-2. It is known that many viruses use their proteins to counteract antiviral IFN production and/or signaling pathways to favor viral replication (2, 3, 6). Since hMPV replication is sensitive to type I IFN (25), we investigated whether the M2-2 protein antagonizes type I IFN production to promote hMPV replication. To accomplish this, A549 cells were infected with either WT or ΔM2-2 at an MOI of 2 and cell supernatants were harvested at various times p.i. to measure IFN-β production by ELISA. As shown in Fig. 3A, infection of A549 cells with rhMPV-ΔM2-2 significantly enhanced IFN-β secretion, compared to infection with rhMPV-WT. The difference of induction started at 6 h p.i., peaked at 15 h p.i. (23-fold increase), and then declined at 24 h p.i. (9-fold increase).
To determine whether M2-2 elimination had a broader effect on hMPV-induced secretion of proinflammatory and immunoregulatory molecules, we compared the secretion patterns of chemokines and cytokines in A549 cells, infected with either rhMPV-WT or rhMPV-ΔM2-2, using a combination of ELISA and Bio-Plex assays (Fig. 3A). We found that rhMPV-ΔM2-2 induced significantly higher levels of the cytokine IL-6, the CXC chemokines IL-8 and IP-10, and the CC chemokines MCP-1 and RANTES at 6, 15, and 24 h p.i. than did hMPV-WT. A significant difference in IL-6, RANTES, MCP-1, and IP-10 induction between rhMPV-WT- and rhMPV-ΔM2-2-infected cells was noted as early as 3 h p.i., when the difference in viral gene transcription and replication was not detectable between WT- and ΔM2-2-infected A549 cells, suggesting a role of M2-2 in regulating early innate immune signaling.
Transcription factors of the IRF family play an essential role in virus-induced expression of type I IFN genes, as well as several other immune regulatory genes, including RANTES and IP-10 (reviewed in reference 8). Among the IRF family, IRF-3 is necessary for IFN-β and RANTES gene expression in response to paramyxovirus infections (15, 35, 46). As expected, there was significantly higher induction of IRF-3-dependent gene transcription in cells infected with ΔM2-2 than in those infected with WT (see Fig. S3A in the supplemental material). Expression of M2-2 reversed gene transcription inhibition in response to ΔM2-2 infection, suggesting the ability of M2-2 to inhibit virus-induced cellular gene expression. Consistently, there was a significant increase in IRF-3 nuclear translocation in rhMPV-ΔM2-2-infected cells compared to rhMPV-WT-infected cells (see Fig. S3B). Other important transcription factors are members of the NF-κB superfamily, such as p50 and p65 (7, 34). It has been previously shown that both p65 and p50 are induced by paramyxovirus infection of airway epithelial cells. The induction of p65 and p50 is necessary for expression of a variety of virus-induced chemokine and cytokine genes (21, 48). Compared to rhMPV-WT infection, infection with rhMPV-ΔM2-2 resulted in significantly higher NF-κB-driven gene transcription at 15 h p.i., which was reversely blocked by hMPV M2-2 expression (see Fig. S4A), suggesting the inhibitory effect of M2-2 in NF-κB activation. The significant role of the M2-2 protein in modulating hMPV-induced NF-κB activation was also confirmed by enhanced p65 and p50 nuclear translocation in ΔM2-2-infected A549 cells, compared to that in rhMPV-WT-infected cells (see Fig. S4B).
Previously, we have shown that hMPV glycoprotein G is an important virulence factor to counteract the innate immunity in A549 cells (6). Therefore, the enhanced cytokine/chemokine induction by ΔM2-2 infection at later times p.i. might indirectly result from decreased transcription of the G protein (see Fig. S2 in the supplemental material). To address that, A549 cells were transfected with a plasmid encoding the G protein, followed by ΔM2-2 infection. WT and ΔM2-2 infections in the absence of G expression were used as controls. We found that even when G was expressed at a higher level than in WT-infected cells, ΔM2-2 infection still induced more cytokines and chemokines than did that with WT, demonstrating the contribution of M2-2 to defeating hMPV-induced host innate immunity (Fig. 3B).
Consistent with the enhanced IFN-β secretion by M2-2 elimination (Fig. 3A), hMPV-activated IFN-β promoter was also increased by M2-2 elimination. As shown in Fig. 3C, ΔM2-2 infection stimulated cells to produce higher luciferase activity than did WT infection (lane 3 versus lane 2). The enhanced luciferase activity was reversed by ectopic G expression (lane 4 versus lane 3) and by M2-2 expression to a greater extent (lane 5 versus lane 4). In addition, coexpression of G and M2-2 exhibited significantly more inhibition than did individual proteins (lane 6 versus lanes 5 and 4). Collectively, the experiments shown in Fig. 3B and C supported the idea that both G and M2-2 are inhibitory to hMPV-induced cellular responses.
M2-2 inhibits MAVS-mediated antiviral signaling.
In addition to the recognition of viral RNA through the Toll-like receptors (TLRs), two DExD/H box RNA helicases, RIG-I and MDA5, have been shown to be essential for IFN induction in several viruses (reviewed in references 1 and 30). MAVS, a mitochondrial protein, is a master switch for RIG-I/MDA5 signaling. It interacts with RIG-I/MDA5 and subsequently splits signals into two paths, e.g., the TRAF3/IKKε/TBK-1 path for IRF activation and TRAF6/IKKα/IKKβ for NF-κB activation (43, 44).
We have recently shown that hMPV activates a RIG-I-dependent, but not MDA5- and TLR-3-dependent, signaling pathway in airway epithelial cells to induce chemokines/cytokines (34). We have also shown that expression of a MAVS protein lacking the N-terminal CARD domain, which acts as a dominant negative mutant, significantly decreased hMPV-induced IRF- and NF-κB-dependent gene transcription, suggesting a critical role of MAVS in hMPV-induced signaling pathways (34). In this study, we compared the expression levels of cytokines/chemokines in hMPV-infected wild-type (WT) and MAVS-deficient (MAVS−/−) MEFs. We found that the induction of KC, RANTES, and IFN-β was significantly dependent on MAVS expression, confirming the role of MAVS in hMPV-induced cellular signaling (see Table S1 in the supplemental material).
To investigate whether M2-2 targets this pathway to broadly inhibit the synthesis of immune mediators following hMPV infection, A549 cells were transfected with RIG-I or MAVS expression plasmids and a luciferase reporter plasmid, IFN-β-Luc. Individual expression of RIG-I and MAVS significantly induced IFN-β transcription, which was inhibited by M2-2 protein expression in the absence of viral infection (Fig. 4A), confirming an inhibitory role of M2-2 in MAVS-mediated signaling. The inhibition of M2-2 on the RIG-I-mediated pathway was dose dependent (see Fig. S5 in the supplemental material) and also viral protein specific, as the nucleoprotein (N) of hMPV did not have a similar inhibitory effect (Fig. 4A). We also did not find the evidence of other hMPV proteins, such as SH, F, and M2-1, being inhibitory to MAVS-mediated signaling (see Fig. S6). Previously, we have shown that hMPV G protein inhibits type I IFN synthesis by targeting RIG-I (6). The host targets of hMPV M2-2 and G protein seemed different, as the M2-2 protein showed significant inhibition on MAVS-induced IFN-β transcription, while the G protein did not (Fig. 4A). Overall, it is becoming recognized as a common strategy that viruses use two distinct viral proteins to target molecules belonging to the same cellular signaling pathway (37, 51, 53).
Fig 4.

Inhibition of MAVS-mediated signaling by M2-2 protein. (A) Inhibition of MAVS-induced IFN-β transcription by M2-2. A549 cells in triplicates (24-well plate) were transfected with a luciferase reporter plasmid (IFN-β-Luc), plasmids encoding either RIG-I (0.5 μg/ml) or MAVS (0.2 μg/ml) or their control vectors (CV), or a plasmid expressing hMPV M2-2 or control proteins or the empty vector (0.2 μg/well). Cells were harvested 30 h posttransfection to measure luciferase activity. (B) Inhibition of MAVS-induced NF-κB- or IRF-3-dependent gene transcription by M2-2. HEK 293 cells in triplicates were transfected with a luciferase reporter plasmid (Kb-5-Luc or IRF-3-Luc), plasmids encoding MAVS or its control vector, and a plasmid expressing hMPV M2-2 or the empty vector (0.2 μg/well). Cells were harvested 30 h posttransfection to measure luciferase activity. (C) TRAF6/IKKβ-induced NF-κB-dependent gene transcription was not affected by M2-2. HEK 293 cells in triplicates were transfected with a luciferase reporter plasmid (Kb-5-Luc); plasmids encoding TRAF6, IKKβ, or their control vector; and a plasmid expressing hMPV M2-2 or the empty vector (0.2 μg/well). Cells were harvested 30 h posttransfection to measure luciferase activity. (D) IKKε/IRF-3 (5D)-induced IRF-3-dependent gene transcription was not affected by M2-2. HEK 293 cells in triplicates were transfected with a luciferase reporter plasmid (IRF-3-Luc); plasmids encoding IKKε, the constitutively active form of IRF-3, or their control vector; and a plasmid expressing hMPV M2-2 or the empty vector (0.2 μg/well). Cells were harvested 30 h posttransfection to measure luciferase activity. For all these experiments in panels A to D, luciferase was normalized to the β-galactosidase reporter activity. Data are representative of two to three independent experiments and are expressed as means ± SEs of normalized luciferase activity. *, P < 0.05 relative to signal inducer + pCAGGS group.
To investigate the mechanism by which M2-2 inhibits MAVS-mediated IFN-β transcription, we first examined the effects of M2-2 on MAVS-activated NF-κB and IRF-3. 293 cells were transfected with MAVS expression plasmid and a luciferase reporter plasmid, Kb-5-Luc/IRF-3-Luc. We found that both NF-κB-dependent gene transcription and IRF-3-dependent gene transcription, which were induced by MAVS overexpression, were significantly inhibited by M2-2 protein expression (Fig. 4B).
It is commonly believed that MAVS activates NF-κB and IRF-3 via its downstream signaling molecules TRAF6/IKKα/β and TRAF3/IKKε/TBK-1, respectively (1, 30). To precisely dissect the target(s) of M2-2, we overexpressed TRAF6 and IKKβ individually and investigated the effect of M2-2 on TRAF6/IKKβ-induced NF-κB-dependent gene expression. As shown in Fig. 4C, the activation of NF-κB by TRAF6 or IKKβ was not affected by M2-2 protein expression. Similarly, we found that IRF-3-dependent gene transcription, which was induced by IKKε or constitutively active IRF-3 (IRF-3 5D, a gift from Rongtuan Lin, McGill University, Canada), was not affected by M2-2 overexpression (Fig. 4D). In addition, the expression levels of TRAF3 were comparable between WT- and ΔM2-2-infected cells (data not shown). All the results in Fig. 4 suggest MAVS as the putative target of M2-2.
We also differentiated the targeting mechanisms of M2-2 and G by comparing their inhibitory efficiencies in poly(I·C)-induced IFN-β transcription in 293 cells. 293 cells normally do not express any TLRs (52) and, therefore, no signals through TLR-3 activation. It is known that MDA5 is a sensor for double-stranded RNA (29). Poly(I·C) also has some capability for RIG-I activation (56). Therefore, poly(I·C) likely induces IFN-β transcription through the activation of MDA5 and RIG-I. As shown in Fig. S7 in the supplemental material, M2-2 was significantly more potent than G against poly(I·C)-induced signaling (lane 3 versus lane 4), suggesting that M2-2 might attack a common signaling molecule(s) of MDA5 and RIG-I pathways, therefore inhibiting the cellular signaling mediated by these two pathways, while G counteracts RIG-I only (6), leaving MDA5-mediated signaling intact and therefore less inhibitory to poly(I·C)-induced signaling. Taking these findings together with the results of Fig. 4, we deduced that MAVS is the common signaling molecule targeted by M2-2. The difference in targeting mechanisms of M2-2 and G was further confirmed and analyzed by investigating the presence of MAVS and RIG-I in the immunoprecipitated M2-2 or G complex as described below.
M2-2 forms a complex with MAVS.
To confirm that MAVS is a target of M2-2, we investigated whether the M2-2 protein physically interacts with MAVS, since this kind of physical interaction was recently shown for hepatitis C virus NS3/4A protein and the influenza virus PB1-F2 protein (44, 51). 293 cells were transfected with V5-tagged M2-2 and Flag-tagged MAVS expression plasmids. Vectors expressing V5 or Flag only were used as negative controls. After 30 h of transfection, cells were lysed, followed by immunoprecipitation using an anti-V5 antibody (Fig. 5A, middle panel). The immunoprecipitated complex was separated by SDS-PAGE and transferred onto a polyvinylidene difluoride (PVDF) membrane. Western blotting using an anti-Flag antibody revealed that MAVS coprecipitated with M2-2 protein. Reverse immunoprecipitation, using anti-Flag to precipitate expressed MAVS and then using an anti-V5 antibody for Western blotting, also confirmed that M2-2 was present in the immunoprecipitated complex (Fig. 5A, bottom panel).
Fig 5.
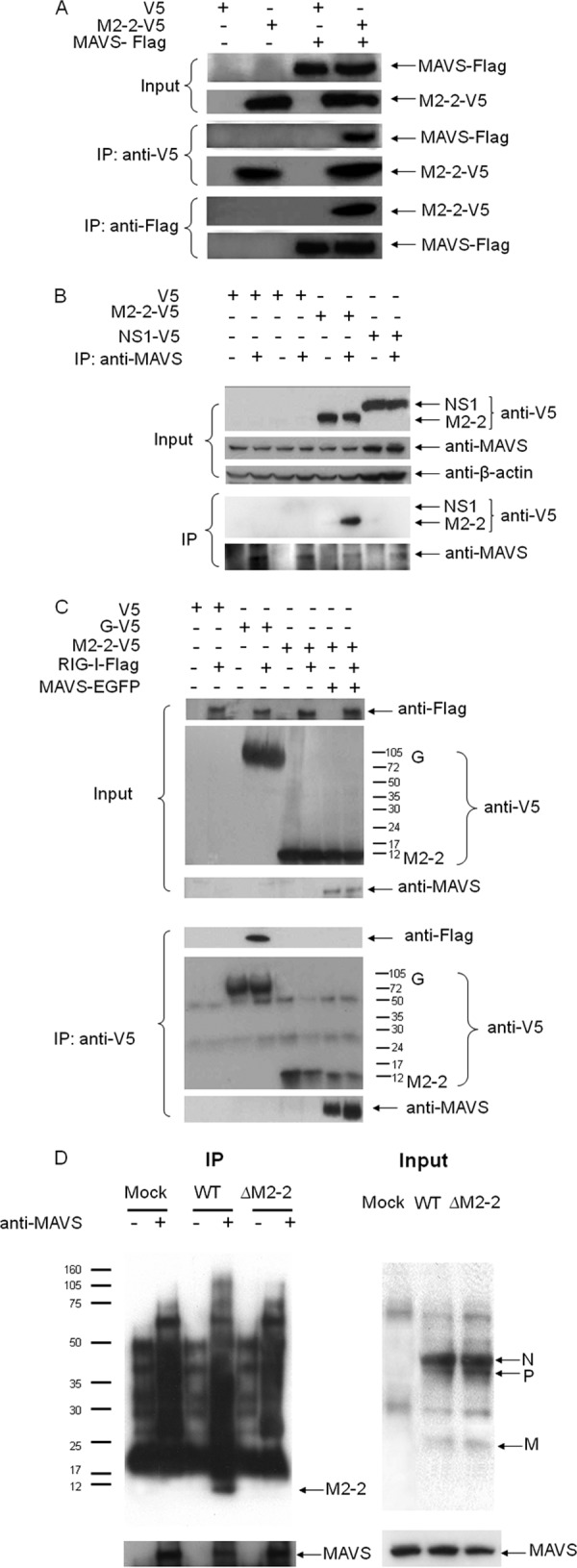
M2-2 interacts with MAVS. (A) M2-2 forms a complex with MAVS in the overexpression system. 293 cells were transfected with plasmids encoding Flag-tagged MAVS and V5-tagged M2-2 or their control vectors. Total cell lysates were immunoprecipitated with an anti-V5 antibody followed by Western blotting using an anti-Flag antibody to detect MAVS. Reverse immunoprecipitation was also done, where MAVS was immunoprecipitated using an anti-Flag antibody and M2-2 protein was then detected using an anti-V5 antibody. Membranes were stripped and reprobed to check for proper immunoprecipitation of M2-2 and MAVS. A small aliquot was also prepared before the IP for a Western blot for equal input of MAVS and proper expression of M2-2. (B) Overexpressed M2-2 interacts with endogenous MAVS. A549 cells at 50% confluence were transfected with a plasmid encoding V5-tagged hMPV M2-2 or RSV NS1 (negative control) or their common control vector. After 40 h, cells were harvested to prepare total cell lysates. Samples were subjected to immunoprecipitation using an anti-MAVS antibody or a control isotype. The immunoprecipitated complexes were then subjected to 10% SDS-PAGE followed by Western blotting using an anti-V5 antibody. The membrane was then stripped and reprobed with an anti-MAVS antibody to determine levels of immunoprecipitated MAVS. A small aliquot was also prepared before the IP for a Western blot for equal input of MAVS and proper expression of hMPV M2-2 or RSV NS1 using 4 to 20% SDS-PAGE. (C) M2-2 does not bind to RIG-I. MAVS−/− MEFs were transfected with a plasmid encoding Flag-tagged RIG-I or its control vector, a plasmid expressing enhanced green fluorescent protein (EGFP)-tagged MAVS, and a plasmid expressing V5-taggged M2-2 or G or their common control vector. At 40 h posttransfection, cells were lysed followed by IP using an anti-V5 antibody. The association of RIG-I with M2-2 or G in the IP complex was investigated by Western blotting using an anti-Flag antibody. The membrane was then stripped and reprobed with an anti-MAVS or anti-V5 antibody to determine levels of coimmunoprecipitated MAVS, M2-2, or G. (D) Viral M2-2 binds to endogenous MAVS in the context of hMPV infection. A549 cells were mock infected or infected with rhMPV-WT or -ΔM2-2, at an MOI of 2, and harvested at 6 h p.i. to prepare total cell lysates. Samples were subjected to immunoprecipitation using an anti-MAVS antibody or control isotype. The immunoprecipitated complexes were then subjected to SDS-PAGE followed by Western blotting using an anti-hMPV antibody. The membrane was then stripped and reprobed with an anti-MAVS antibody to determine levels of immunoprecipitated MAVS.
To confirm the specificity of M2-2 in associating with MAVS, A549 cells were transfected with a plasmid encoding V5-tagged M2-2. Vectors expressing V5 only or V5-tagged RSV NS1 protein were used as negative controls. RSV NS1 protein is a soluble protein with a molecular weight similar to that of M2-2. Recently, we have demonstrated that RSV NS1 protein inhibits IFN-β synthesis via direct targeting of IRF-3 but not MAVS (40). After transfection, half of the sample was coimmunoprecipitated using an anti-MAVS antibody to pull down the endogenous MAVS, while the other half was exposed to an isotype antibody to rule out nonspecific protein binding during the immunoprecipitation. In immunoprecipitated MAVS complex from cells expressing V5-tagged M2-2, but not in the complex from the cells expressing V5 or V5-tagged RSV NS1, a specific band around 12 kDa was recognized by the anti-V5 antibody (Fig. 5B, bottom panel), suggesting the association of MAVS with M2-2, but not with RSV NS1. Therefore, we concluded that the interaction of M2-2 with MAVS is viral protein specific.
It is possible that M2-2 interacts with MAVS through its association with other signaling proteins, such as RIG-I, in MAVS signalosome. To investigate that, MAVS−/− MEFs were transfected with a plasmid encoding RIG-I or its control vector and a plasmid encoding M2-2 or G or their common control vector. After 40 h of transfection, cells were lysed followed by immunoprecipitation using an anti-V5 antibody to pull down M2-2 or G complex. The immunoprecipitated complex was separated on a 4 to 20% SDS-PAGE gel and transferred onto a PVDF membrane. Western blotting using an anti-Flag antibody revealed that RIG-I was coprecipitated with G but not with M2-2, demonstrating that the MAVS–M2-2 interaction does not require RIG-I (Fig. 5C, bottom panel, first blot, lane 4 versus lane 6). Both Fig. 5A and B showed that MAVS formed a complex with M2-2. It is also known that RIG-I–MAVS interaction is necessary for the signaling transduction. Therefore, to further explore the interactive relationship among RIG-I, MAVS, and M2-2, we questioned whether RIG-I is in the complex of MAVS–M2-2. To address that, cells were cotransfected with MAVS plasmids and plasmids encoding M2-2 and/or RIG-I. Consistent with the results of Fig. 5A and B, MAVS was coprecipitated with M2-2. However, the RIG-I was not present in the MAVS–M2-2 complex (Fig. 5C, bottom panel, first blot, lane 8), suggesting that an interruption of M2-2 on RIG-I–MAVS association may be one of the mechanisms contributing to the inhibitory effect of M2-2 on RIG-I-mediated signaling.
The interaction between the M2-2 protein and MAVS was also investigated in the context of WT and ΔM2-2 infection. A549 cells, mock infected or infected with WT and ΔM2-2 at an MOI of 2, were harvested after 6 h p.i. Total cell lysates were subjected to immunoprecipitation using anti-MAVS or isotype antibodies. The immunoprecipitated complex was separated by SDS-PAGE and transferred onto a PVDF membrane. Western blotting using an anti-hMPV antibody revealed that MAVS coprecipitated with a protein, which is present only in WT-infected samples and corresponds to the size of identified M2-2 in WT virus particles (Fig. 5D), demonstrating that M2-2 associated with endogenous MAVS in the context of hMPV infection. In our experiments, the anti-hMPV antibody was able to detect M2-2 protein of purified hMPV particles (Fig. 1C). However, it did not detect M2-2 from WT-infected cell lysate, possibly due to its low expression level. IP-enriched M2-2 detected by anti-MAVS antibody likely met the detection threshold of this anti-hMPV antibody.
Functional domains of M2-2.
To identify the functional domain(s) of M2-2 for the inhibition of MAVS-mediated signaling, we first analyzed M2-2 using motif prediction software online (22) (http://elm.eu.org). Interestingly, we found that the M2-2 protein has five PDZ domains, which are a common structural domain in signaling proteins for signal transduction (39). As illustrated in Fig. 6A, three M2-2 deletion mutants were constructed. The first mutant, lacking the first 25 amino acids in the N terminus, is named ΔN25. The deleted region of ΔN25 does not have PDZ domains. The second mutant is called Δ26–54, as it has a deletion of amino acids from Ile26 to Tyr54. The first three PDZ domains were deleted in this mutant. The third mutant is named Δ55–69, a construct lacking amino acids from Asn55 to Tyr69. This mutant does not have the last two PDZ domains. All of these constructs were well expressed. As shown in Fig. 6B, ΔN25, but not Δ26–54 and Δ55–69, exhibited an inhibitory effect on MAVS-induced IFN-β transcription similar to that of WT M2-2, demonstrating that the N terminus is not essential for the inhibitory effect of M2-2 on MAVS. In the future, the importance of the PDZ domain(s) of M2-2 in cellular signaling will be investigated by site-directed mutagenesis studies.
Fig 6.
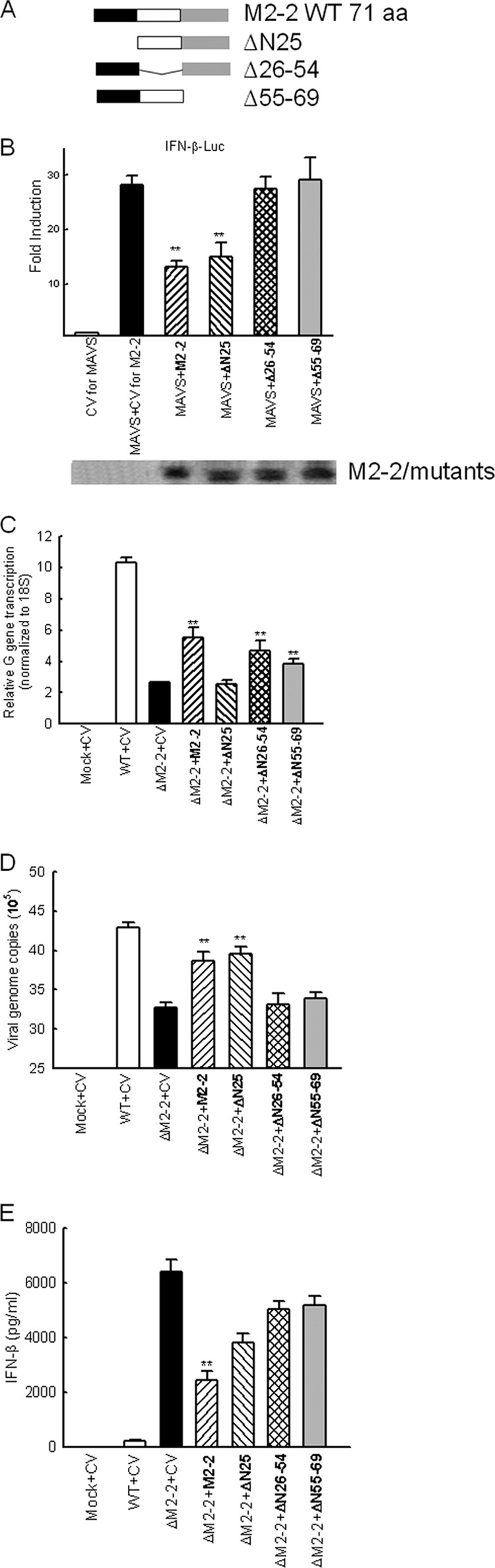
M2-2 domains responsible for MAVS-mediated signaling. (A) Schematic drawing of WT M2-2 and its deletion mutants. aa, amino acids. (B) The N-terminal portion of M2-2 is not responsible for the inhibitory effect of M2-2 on MAVS-mediated signaling. 293 cells in triplicates were transfected with a luciferase reporter plasmid (IFN-β-Luc; 0.1 μg/well), a plasmid encoding Flag-tagged MAVS (0.1 μg/ml) or its control vectors, and a plasmid expressing WT M2-2 or indicated M2-2 mutants or the empty vector. Cells were harvested 30 h posttransfection to measure luciferase activity. For each plate, luciferase was normalized to the β-galactosidase reporter activity. (C to E) The regulation of viral gene transcription, viral replication, and IFN-β induction by M2-2 domains. A549 cells in triplicates were transfected with a plasmid encoding M2-2 or its mutants or the empty vector as indicated. At 24 h posttransfection, cells were mock infected or infected with WT and ΔM2-2 for 15 h. Cell pallets were harvested to prepare RNA for the measurement of G gene transcription (C) and viral replication (D) by real-time PCR, while supernatant was used for IFN-β quantification (E). Data are representative of three independent experiments and are expressed as means ± SEs. *, P < 0.05, and **, P < 0.01, relative to MAVS + CV. CV, control vector for M2-2 and its mutants.
We then investigated whether these domains exhibit regulatory functions in viral gene transcription. As shown in Fig. 6C, M2-2 deletion led to the gene transcription inhibition as expected (lane 3 versus lane 2), which was reversed by M2-2 overexpression (lane 4 versus lane 3). Interestingly, ΔN25, but not Δ26–54 and Δ55–69, failed to reverse the inhibition of gene transcription (lanes 5, 6, and 7 versus lane 4), suggesting that the first 25 amino acids are critical for viral gene transcription regulation while other domains are not.
The regulatory roles of M2-2 and its mutants in viral replication were also compared. Our data suggested that the domain in charge of gene transcription is different from the ones responsible for the regulation of viral replication. As shown in Fig. 6D, the domains after the first 25 amino acids are important for M2-2-promoted viral replication. Currently, it is not well understood why some viruses use the same domain(s), while others have distinct ones, to regulate viral gene transcription and viral replication (16, 19).
We then studied the effect of the above-described domains on IFN-β induction. As shown in Fig. 6E, the enhanced IFN-β synthesis by M2-2 deletion (lane 3 versus lane 2) was reversed most effectively by M2-2 (lanes 4, 5, 6, and 7 versus lane 3). Compared to WT M2-2, Δ26–54 and Δ55–69 had equivalent abilities to promote G gene transcription (Fig. 6C) and an inability to recover the viral replication (Fig. 6D). However, the cells transfected with these mutants produced significantly more IFN-β than did cells expressing WT M2-2 in response to ΔM2-2 infection, demonstrating that they are responsible for the anticellular signaling of M2-2, which is consistent with the results of Fig. 6B.
DISCUSSION
The innate immune response functions as a first line of host defense against invading pathogens, as well as a critical component in regulating adaptive immune responses. The effectiveness of innate immune response against viral infection depends on the interactive nature of virus components with the host innate antiviral immune systems, including the type I interferon synthesis system (1). hMPV is a major cause of epidemic respiratory infections in infants, as well as in the elderly and immunocompromised patients. As it is a recently identified virus, little is known about the role of individual hMPV proteins in modulating host cell responses. Reverse genetic systems were recently developed for hMPV, providing an important tool for characterizing hMPV protein function and for designing live-attenuated hMPV vaccines (5, 6, 10, 12–14, 41). Currently, recombinant ΔM2-2 virus is listed as a live vaccine candidate, as it is attenuated, immunogenic, and protective against hMPV challenge in both African green monkeys and hamsters (10, 13, 41). In this study, we focused on the possible mechanism(s) underlying ΔM2-2 attenuation. We found that M2-2 elimination reduced the accumulation of viral genomic and messenger RNAs and enhanced antiviral signaling. All of these likely contribute to the attenuation of ΔM2-2.
The ability to inhibit the production of type I IFN appears to be a common feature of paramyxoviruses (20). Upon hMPV infection, the airway epithelial cells activate the RIG-I/MAVS, but not TLR-3 and MDA5, signaling pathway for the expression of important antiviral molecules (34) (see also Table S1 in the supplemental material). In the meantime, hMPV is also able to develop strategies to evade the host defense. Previously, we showed that hMPV G protein interacts with RIG-I and inhibits RIG-I-dependent gene transcription (6). In the present study, we demonstrate that hMPV M2-2 protein also plays a significant role in inhibiting host innate immunity. However, the mechanisms underlying the inhibitory role of M2-2 and G seemed different. M2-2 blocked IFN-β transcription likely through targeting MAVS because (i) the inhibitory effect of M2-2 on RIG-I signaling stopped at the level of MAVS (Fig. 4), (ii) M2-2 did not bind to RIG-I, and (iii) M2-2 interacted with MAVS (Fig. 5), which prevented the recruitment of MAVS to RIG-I (Fig. 5C), while G inhibits IFN-β transcription by attacking RIG-I as (i) G inhibits IFN-β transcription at the level of RIG-I as the signaling induced by RIG-I downstream molecules was not affected by G expression (Fig. 4A) (6) and (ii) G associated with RIG-I, which is MAVS independent (Fig. 5C) (6). There are still many questions that need to be addressed for a better understanding of the molecular mechanisms underlying the regulation of MAVS by M2-2. Besides blocking MAVS–RIG-I association, does M2-2 also prevent MAVS from binding to its adaptors? It has been recently reported that the phosphorylation of MAVS is important for MAVS activity (54); does M2-2 affect the phosphorylation of MAVS? In addition, studying the cellular localization of M2-2, MAVS, and MAVS interactive signaling molecules, in response to hMPV infection, is also critical to dissect the regulation mechanisms in further detail.
The inhibitory effect of M2-2 on antiviral signaling is M2-2 specific, as other hMPV proteins, such as N, P, SH, and M2-1 proteins, did not have similar inhibitory effects (Fig. 4A; see also Fig. S6 in the supplemental material), and the inhibitory effect of M2-2 was dose dependent (see Fig. S5) and protein structure dependent (Fig. 6B). Collectively, these series of experiments from overexpression systems highlighted the specificity of M2-2 for antiviral signaling.
Compared to WT-infected cells, ΔM2-2-infected cells induced more IFN-β transcription, which was reversed by M2-2, but not by N, overexpression (Fig. 3C). As shown in Fig. 3C and also in Fig. S2 in the supplemental material, ΔM2-2 attenuation also led to reduced G expression, suggesting that the cellular responses promoted by M2-2 elimination might indirectly result from the reduced G expression (hMPV G protein is a known virulence factor antagonizing host innate immunity by attacking RIG-I [6]). However, we found that ectopic expression of G only partially blocked ΔM2-2-enhanced cytokine/chemokine secretion, suggesting that M2-2 contributed to the signaling inhibition as well (Fig. 3B). In this study, we also identified the domains responsible for the regulation of viral gene transcription, viral replication, and RIG-I signaling. Of note, two M2-2 mutants, Δ26–54 and Δ55–69, had an inhibition of hMPV G transcription similar to that of WT M2-2 (Fig. 6C). In addition, these two mutants promoted less viral replication than did WT M2-2 (Fig. 6D), meaning less RIG-I activation, and yet reduced the suppression of ΔM2-2-induced IFN-β synthesis by M2-2 (Fig. 6E), highlighting a G-independent role of M2-2 in antiviral signaling. In a luciferase reporter study, these two domains were also responsible for the suppression of MAVS-induced signaling by M2-2 (Fig. 6B). Collectively, results from both overexpression and recombinant virus systems complementarily revealed a novel function of M2-2 as an innate immunity regulator and provided additional insight into the attenuation mechanism of ΔM2-2.
The mechanism by which viruses use two distinct viral proteins to target molecules belonging to the same cellular signaling pathway is becoming recognized as a common strategy to evade host immune defenses. For example, the influenza virus uses its NS1 protein to target RIG-I (26, 37) and its PB1-F2 and PB2 proteins to interact with MAVS (24, 51). In the case of respiratory syncytial virus (RSV), the NS2 protein of RSV antagonizes the activation of IFN-β transcription by interaction with RIG-I (36), and we recently found that NS1 protein inhibits IFN-β synthesis by associating with RIG-I downstream transcription factor IRF-3 and its transcriptional coactivator CBP (40). Why multiple viral proteins are needed to antagonize the same antiviral signaling is currently unknown but is an interesting topic.
The functions of the M2-2 protein have been extensively identified for RSV (9, 28, 47). Deletion of RSV M2-2 resulted in an increase of viral mRNA and a decreased production of viral genome. Thus, RSV M2-2 is involved in regulating the balance between transcription and genome replication. Reports on the role of hMPV M2-2 in regulating viral RNAs are controversial. Recombinant viruses with deleted M2-2 sequence showed enhanced viral gene transcription but no changes in the accumulation of viral genomic RNAs (13, 41). A different study using an hMPV minigenome reporter system demonstrated that the M2-2 protein inhibited both viral transcription and replication (32). In the present study, we observed that hMPV M2-2 elimination reduced accumulation of both viral mRNAs and genomic RNAs during hMPV infection. Differences in the strategies to generate recombinant ΔM2-2 (deletion versus site-directed mutagenesis), research systems, and/or viral strain may account for the observed discrepancy. In this study, we used site-directed mutagenesis to manipulate start and stop codons of M2-2 for gene silencing. Therefore, the whole genomic length of hMPV was unlikely changed (Fig. 1B). We also found that reduced viral RNA accumulation by M2-2 elimination is IFN-β independent, as such reduction was present in Vero cells, a cell line deficient in type I IFN genes (Fig. 1D). On the other hand, the inhibition of antiviral signaling by M2-2 does not require the presence of viral replication (Fig. 4). In the future, we will continue to dissect domains in further detail. Since the domains after the first 25 amino acids are critical for the regulation of viral replication and antiviral signaling, we will determine whether there are subdomains which can separately regulate viral replication and cellular signaling. It will be ideal to generate a recombinant hMPV mutant(s) whose M2-2 proteins are deficient in viral RNA synthesis regulation but maintain the full function of M2-2 to antagonize antiviral signaling, to elucidate the exact molecular mechanisms by which the M2-2 protein inhibits MAVS function. Nevertheless, this study reveals a novel function of hMPV M2-2 protein in regulating host antiviral response, providing an additional molecular mechanism underlying the attenuation of ΔM2-2.
Supplementary Material
ACKNOWLEDGMENTS
All authors concur that there are no conflicts of interest associated with this published work.
This work was supported by grants from the National Institutes of Health, National Institute of Allergy and Infectious Diseases (KAI074829A); the American Lung Association (RG232529N); and the American Heart Association (12BGIA12060008) to X.B.
We thank Cynthia Tribble for assistance with manuscript editing and submission. We also thank Antonella Casola and Roberto Garofalo for their comments on the manuscript.
Footnotes
Published ahead of print 26 September 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801 [DOI] [PubMed] [Google Scholar]
- 2. Alff PJ, et al. 2006. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. J. Virol. 80:9676–9686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andrejeva J, et al. 2004. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. U. S. A. 101:17264–17269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bannister R, et al. 2010. Use of a highly sensitive strand-specific quantitative PCR to identify abortive replication in the mouse model of respiratory syncytial virus disease. Virol. J. 7:250 doi:10.1186/1743-422X-7-250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bao X, et al. 2008. Human metapneumovirus small hydrophobic protein inhibits NF-kappaB transcriptional activity. J. Virol. 82:8224–8229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bao X, et al. 2008. Human metapneumovirus glycoprotein G inhibits innate immune responses. PLoS Pathog. 4:e1000077 doi:10.1371/journal.ppat.1000077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bao X, et al. 2007. Airway epithelial cell response to human metapneumovirus infection. Virology 368:91–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barnes B, Lubyova B, Pitha PM. 2002. On the role of IRF in host defense. J. Interferon Cytokine Res. 22:59–71 [DOI] [PubMed] [Google Scholar]
- 9. Bermingham A, Collins PL. 1999. The M2-2 protein of human respiratory syncytial virus is a regulatory factor involved in the balance between RNA replication and transcription. Proc. Natl. Acad. Sci. U. S. A. 96:11259–11264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Biacchesi S, et al. 2005. Infection of nonhuman primates with recombinant human metapneumovirus lacking the SH, G, or M2-2 protein categorizes each as a nonessential accessory protein and identifies vaccine candidates. J. Virol. 79:12608–12613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Biacchesi S, et al. 2004. Recovery of human metapneumovirus from cDNA: optimization of growth in vitro and expression of additional genes. Virology 321:247–259 [DOI] [PubMed] [Google Scholar]
- 12. Biacchesi S, et al. 2004. Recombinant human metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: deletion of G yields a promising vaccine candidate. J. Virol. 78:12877–12887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Buchholz UJ, et al. 2005. Deletion of M2 gene open reading frames 1 and 2 of human metapneumovirus: effects on RNA synthesis, attenuation, and immunogenicity. J. Virol. 79:6588–6597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buchholz UJ, Nagashima K, Murphy BR, Collins PL. 2006. Live vaccines for human metapneumovirus designed by reverse genetics. Expert Rev. Vaccines 5:695–706 [DOI] [PubMed] [Google Scholar]
- 15. Casola A, Henderson A, Liu T, Garofalo RP, Brasier AR. 2002. Regulation of RANTES promoter activation in alveolar epithelial cells after cytokine stimulation. Am. J. Physiol. Lung Cell. Mol. Physiol. 283:L1280–L1290 [DOI] [PubMed] [Google Scholar]
- 16. De BP, Hoffman MA, Choudhary S, Huntley CC, Banerjee AK. 2000. Role of NH(2)- and COOH-terminal domains of the P protein of human parainfluenza virus type 3 in transcription and replication. J. Virol. 74:5886–5895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Englund JA, et al. 2006. Brief communication: fatal human metapneumovirus infection in stem-cell transplant recipients. Ann. Intern. Med. 144:344–349 [DOI] [PubMed] [Google Scholar]
- 18. Falsey AR, Erdman D, Anderson LJ, Walsh EE. 2003. Human metapneumovirus infections in young and elderly adults. J. Infect. Dis. 187:785–790 [DOI] [PubMed] [Google Scholar]
- 19. Ferguson MK, Botchan MR. 1996. Genetic analysis of the activation domain of bovine papillomavirus protein E2: its role in transcription and replication. J. Virol. 70:4193–4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fontana JM, Bankamp B, Rota PA. 2008. Inhibition of interferon induction and signaling by paramyxoviruses. Immunol. Rev. 225:46–67 [DOI] [PubMed] [Google Scholar]
- 21. Garofalo RP, et al. 1996. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J. Virol. 70:8773–8781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gould CM, et al. 2010. ELM: the status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res. 38:D167–D180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goutagny N, et al. 2010. Cell type-specific recognition of human metapneumoviruses (HMPVs) by retinoic acid-inducible gene I (RIG-I) and TLR7 and viral interference of RIG-I ligand recognition by HMPV-B1 phosphoprotein. J. Immunol. 184:1168–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Graef KM, et al. 2010. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J. Virol. 84:8433–8445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guerrero-Plata A, et al. 2005. Activity and regulation of alpha interferon in respiratory syncytial virus and human metapneumovirus experimental infections. J. Virol. 79:10190–10199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo Z, et al. 2007. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 36:263–269 [DOI] [PubMed] [Google Scholar]
- 27. Herfst S, et al. 2004. Recovery of human metapneumovirus genetic lineages a and B from cloned cDNA. J. Virol. 78:8264–8270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jin H, et al. 2000. Recombinant respiratory syncytial viruses with deletions in the NS1, NS2, SH, and M2-2 genes are attenuated in vitro and in vivo. Virology 273:210–218 [DOI] [PubMed] [Google Scholar]
- 29. Kato H, et al. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105 [DOI] [PubMed] [Google Scholar]
- 30. Kawai T, Akira S. 2007. Antiviral signaling through pattern recognition receptors. J. Biochem. 141:137–145 [DOI] [PubMed] [Google Scholar]
- 31. Kawai T, Akira S. 2008. Toll-like receptor and RIG-I-like receptor signaling. Ann. N. Y. Acad. Sci. 1143:1–20 [DOI] [PubMed] [Google Scholar]
- 32. Kitagawa Y, et al. 2010. Human metapneumovirus M2-2 protein inhibits viral transcription and replication. Microbes Infect. 12:135–145 [DOI] [PubMed] [Google Scholar]
- 33. Kolli D, et al. 2011. Human metapneumovirus glycoprotein G inhibits TLR4-dependent signaling in monocyte-derived dendritic cells. J. Immunol. 187:47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liao S, et al. 2008. Role of retinoic acid inducible gene-I in human metapneumovirus-induced cellular signalling. J. Gen. Virol. 89:1978–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin R, Heylbroeck C, Genin P, Pitha PM, Hiscott J. 1999. Essential role of interferon regulatory factor 3 in direct activation of RANTES chemokine transcription. Mol. Cell. Biol. 19:959–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ling Z, Tran KC, Teng MN. 2009. Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J. Virol. 83:3734–3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Opitz B, et al. 2007. IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell. Microbiol. 9:930–938 [DOI] [PubMed] [Google Scholar]
- 38. Pham QN, et al. 2005. Chimeric recombinant human metapneumoviruses with the nucleoprotein or phosphoprotein open reading frame replaced by that of avian metapneumovirus exhibit improved growth in vitro and attenuation in vivo. J. Virol. 79:15114–15122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ranganathan R, Ross EM. 1997. PDZ domain proteins: scaffolds for signaling complexes. Curr. Biol. 7:R770–R773 [DOI] [PubMed] [Google Scholar]
- 40. Ren J, et al. 2011. A novel mechanism for inhibition of IRF-3-dependent gene expression by human respiratory syncytial virus NS1 protein. J. Gen. Virol. 92:2153–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schickli JH, et al. 2008. Deletion of human metapneumovirus M2-2 increases mutation frequency and attenuates growth in hamsters. Virol. J. 5:69 doi:10.1186/1743-422X-5-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schreiber E, Matthias P, Muller MM, Schaffner W. 1989. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 17:6419 doi:10.1093/nar/17.15.6419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682 [DOI] [PubMed] [Google Scholar]
- 44. Sun Q, et al. 2006. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 24:633–642 [DOI] [PubMed] [Google Scholar]
- 45. Sutherland KA, Collins PL, Peeples ME. 2001. Synergistic effects of gene-end signal mutations and the M2-1 protein on transcription termination by respiratory syncytial virus. Virology 288:295–307 [DOI] [PubMed] [Google Scholar]
- 46. Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. 2001. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 19:623–655 [DOI] [PubMed] [Google Scholar]
- 47. Teng MN, et al. 2000. Recombinant respiratory syncytial virus that does not express the NS1 or M2-2 protein is highly attenuated and immunogenic in chimpanzees. J. Virol. 74:9317–9321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tian B, et al. 2002. Identification of NF-kB dependent gene networks in respiratory syncytial virus-infected cells. J. Virol. 76:6800–6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van den Hoogen BG, Bestebroer TM, Osterhaus AD, Fouchier RA. 2002. Analysis of the genomic sequence of a human metapneumovirus. Virology 295:119–132 [DOI] [PubMed] [Google Scholar]
- 50. van den Hoogen BG, et al. 2001. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 7:719–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Varga ZT, et al. 2011. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 7:e1002067 doi:10.1371/journal.ppat.1002067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang J, et al. 2006. The functional effects of physical interactions among Toll-like receptors 7, 8, and 9. J. Biol. Chem. 281:37427–37434 [DOI] [PubMed] [Google Scholar]
- 53. Wang X, et al. 2000. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J. Virol. 74:11566–11573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wen C, et al. 2012. Identification of tyrosine-9 of MAVS as critical target for inducible phosphorylation that determines activation. PLoS One 7:e41687 doi:10.1371/journal.pone.0041687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Williams JV, et al. 2006. The role of human metapneumovirus in upper respiratory tract infections in children: a 20-year experience. J. Infect. Dis. 193:387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yoneyama M, et al. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730–737 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.