

Abstract
The bacteriophage vB_YecM-ϕR1-37 (ϕR1-37) is a lytic yersiniophage that can propagate naturally in different Yersinia species carrying the correct lipopolysaccharide receptor. This large-tailed phage has deoxyuridine (dU) instead of thymidine in its DNA. In this study, we determined the genomic sequence of phage ϕR1-37, mapped parts of the phage transcriptome, characterized the phage particle proteome, and characterized the virion structure by cryo-electron microscopy and image reconstruction. The 262,391-bp genome of ϕR1-37 is one of the largest sequenced phage genomes, and it contains 367 putative open reading frames (ORFs) and 5 tRNA genes. Mass-spectrometric analysis identified 69 phage particle structural proteins with the genes scattered throughout the genome. A total of 269 of the ORFs (73%) lack homologues in sequence databases. Based on terminator and promoter sequences identified from the intergenic regions, the phage genome was predicted to consist of 40 to 60 transcriptional units. Image reconstruction revealed that the ϕR1-37 capsid consists of hexameric capsomers arranged on a T=27 lattice similar to the bacteriophage ϕKZ. The tail of ϕR1-37 has a contractile sheath. We conclude that phage ϕR1-37 is a representative of a novel phage type that carries the dU-containing genome in a ϕKZ-like head.
INTRODUCTION
Bacteriophages, the viruses that infect bacteria, are the most abundant organisms on Earth, and it is estimated that for each microbial isolate at least 10 different phages exist (19, 35). Present knowledge indicates that phages are extremely diverse in nature (7a). Studies on bacteriophages have escalated, since they are excellent targets for genomic and evolutionary research and as models for systems biology studies; in addition, they are important vehicles in horizontal gene transfer. Phages are used as tools in bacterial genetics, and phage gene products are used as tools in molecular biology. Furthermore, their potential as therapeutic agents during the increasing emergence of antibiotic resistance is being reexamined (45, 46). In summary, a thorough knowledge of the bacteriophage and its biology is considered essential to all phage research.
We have isolated several Yersinia enterocolitica-specific bacteriophages that use different parts of lipopolysaccharide (LPS) as receptors and used them to study the molecular biology and genetics of LPS biosynthesis (2, 23, 24, 33, 34, 46–48). The bacteriophage vB_YecM-ϕR1-37 (ϕR1-37) was isolated from sewage based on its ability to infect Y. enterocolitica strain YeO3-R1, an O-polysaccharide (O-PS)-lacking Y. enterocolitica serotype O:3 strain (44, 48). The host range of ϕR1-37, as well as genetic and structural data, showed that the LPS outer core (OC) hexasaccharide of Y. enterocolitica O:3 is the phage receptor (23, 37, 38, 48).
Electron microscopy and analysis of its genome indicated that ϕR1-37 is an exceptionally large-tailed phage with an estimated genome size of 270 kb (23). Structural studies on large bacteriophages with contractile tails are currently limited to the Pseudomonas aeruginosa phage ϕKZ. The icosahedrally ordered ϕKZ head has a diameter of 145 nm, and it consists mainly of hexamers formed by the 65-kDa major capsid protein Gp120 arranged on a T=27 lattice. The pentameric vertices are occupied by complexes composed of several other capsid proteins. The tail of phage ϕKZ is contractile and approximately 200 nm long (14, 16).
The nucleotide composition of the ϕR1-37 DNA is unusual, with all thymidines replaced by deoxyuridines (dU) (23). Very few bacteriophages with such characteristics have been encountered and studied; therefore, in this work, we elucidated further the biological, structural, and genomic features of ϕR1-37.
MATERIALS AND METHODS
Bacterial strains, phage isolation, and growth conditions.
The Escherichia coli strains CJ236 [FΔ(HindIII)::cat (Tra+ Pil+ Camr)/ung-1 relA1 dut-1 thi-1 spoT1 mcrA] and KT8052 [Δ(pro-lac) thi ara trpE9777 ung-1/F′ (proAB lacIqZΔM15)] were used for propagating phage shotgun libraries. Bacteriophage ϕR1-37 was propagated in Y. enterocolitica O:3 strain YeO3-R1 (44, 48) as described previously (23), and large-scale isolation and purification were performed as described previously (33, 42). E. coli strains were grown in lysogeny broth (LB) (4) at 37°C. LB agar plates (LA plates), LB supplemented with 1.5% Bacto agar, were used for all solid cultures. Y. enterocolitica strain was grown in tryptic soy broth (TSB) or LB medium at room temperature (22°C). Chloramphenicol (20 μg ml−1) or ampicillin (150 μg ml−1) was added to the media when required.
For the preparation of ϕR1-37 for cryo-electron microscopy (cryo-EM) studies, ϕR1-37 phage particles were prepared as described previously (21). Alternatively, cell debris from 500 ml of Y. enterocolitica O:3 strain YeO3-R1 infected with ϕR1-37 was pelleted by low-speed centrifugation (Sorvall SLA-1500 rotor) (8,500 rpm, 20 min, 4°C). Phage particles were precipitated with 1 M NaCl and 10% polyethylene glycol (PEG) 8000 at 4°C with stirring for 60 min. The precipitated phage particles were harvested by low-speed centrifugation (Sorvall SLA-1500 rotor) (8,500 rpm, 20 min, 4°C). Phage particles were resuspended in TM buffer (50 mM Tris [pH 7.8], 10 mM MgSO4) and extracted along with an equal volume of chloroform. After low-speed centrifugation (3,000 rpm, 15 min, 4°C), the aqueous phase was collected and loaded onto a linear 15 to 35% glycerol gradient in TM buffer (Beckman SW 41 Ti rotor) (24,000 rpm, 35 min, 4°C). The light-scattering bands were collected and used immediately for preparation of the cryo-EM samples.
Genome sequencing.
Two phage DNA genomic libraries were constructed to obtain the complete genomic sequence of phage ϕRI-37. Purified phage DNA was partially digested with the restriction enzyme TasI (Fermentas), and 1.5- to 2.5-kb DNA fragments were ligated into a pUC19 vector (58) digested with restriction enzyme EcoRI. In addition, a second library was constructed where partially TasI-digested 2.5- to 3-kb DNA fragments were inserted into a pBAD30 vector (17), also digested with EcoRI. Plasmid libraries were propagated in E. coli strain CJ236 or KT8052, and the plasmid clones were purified with an Edge Biosystems SeqPrep 96 kit (Edge BioSystems) or the E.Z.N.A. plasmid minikit I (Omega Bio-Tek), respectively. For sequencing, universal forward and reverse primers were used for the pUC19 library clones, whereas the primers for the pBAD30 library clones were BadFor (5′-CTACCTGACGCTTTTTATCGCAAC-3′) and BadRev (5′-GCAAATTCTGTTTTATCAGACCGC-3′). Primer walking with purified phage DNA and primers designed based on the known phage sequence was used to close gaps between the contigs. Both DNA strands were sequenced at least once. Sequencing reactions were done with an Applied Biosystems dye terminator (v.3.1) kit and run on a 3100 capillary sequence analyzer. The average length of the sequence reads was 741 bp, and the whole phage genome project database contained 1,544 individual reads altogether; thus, the average sequence coverage was 1,544 × 741 bp/262,391 bp = 4.36×.
Characterization of the genome ends.
Bal31 digestion to identify the phage genome physical ends was performed at 30°C with 0.5 units of Bal31 (New England BioLabs) and 30 μg of ϕRI-37 DNA following the supplier's instructions. The samples were removed at different time points during incubation, and the reactions were stopped by chilling them on ice. The DNA was extracted with phenol, precipitated with ethanol, and used for KpnI restriction digestions. The samples were analyzed on 0.7 to 1.0% agarose gels.
Southern and Northern blot analysis.
To identify whether the phage replicates as a linear or circular molecule during infection, Southern hybridization was performed using digoxigenin (DIG)-labeled phage-specific PCR fragments located near the identified physical ends of the phage genome. The PCR fragments were amplified from the phage genome with the primers fR-645 (5′-AGCTACTAAACGGATGGAAGAA-3′), fR-373 (5′-GGTATTCAGCAAATTCGTATAAGG-3′), fR-301 (5′-GAACTTTCCGGTTAGTGGTCA-3′), and fR-525 (5′-TGCAGATGCAACATGATTGTAATA-3′) and labeled by digoxigenin using the DIG High Prime DNA labeling and detection starter kit II (Roche Diagnostics, Mannheim, Germany). YeO3-R1 bacteria grown to the logarithmic phase were infected with ϕR1-37 (at an MOI of 10), the surface-adsorbed phage particles were eliminated after 5 min of incubation by washes with 0.5 M and 1 M NaCl, and the infected bacteria were resuspended in LB. One-milliliter samples were withdrawn at different time points for DNA isolation. The total genomic DNA was isolated from all the samples using the Jet Flex genomic DNA isolation kit (Genomed GmbH) and subjected to restriction digestion with KpnI. The digested products were transferred to positively charged nylon membranes after agarose gel electrophoresis. After UV cross-linking, the blotted membrane was incubated with the DIG-labeled probes for 16 h at 42°C in a formamide-containing hybridization solution. After high-stringency washes (0.1× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate]), the bound DIG-labeled probes were detected according to the instructions of the manufacturer (Roche, Germany).
The bacteriophage transcriptome was analyzed with Northern blotting. The YeO3-R1 culture was grown to the logarithmic phase, and after removing a sample for RNA isolation (time zero), the bacteria were infected with ϕR1-37 (MOI value of 1), and the ϕR1-37-infected samples were taken at 7-min intervals up to 42 min total duration. Total RNA was isolated as described previously (3, 21). Five to 10 μg of total RNA was denatured for 30 min at 50°C with glyoxal load dye (NorthernMax-Gly; Ambion), and electrophoresis was performed according to the manufacturer's instructions. The DNA probes for the Northern blots were amplified with PCR using phage gene-specific primers. To create single-stranded DNA probes, digoxigenin-11-dUTP labeling was performed with a second PCR by using just the reverse primers. The blotting, hybridization, and DIG detection were performed with the DIG system (Roche Diagnostics GmbH) according to the manufacturer's instructions.
Computational analysis.
For general DNA analysis, the EMBOSS package was used (40) as well as other Web-based services (see below). Sequence assembly and analysis were done with the Staden software package (http://staden.sourceforge.net/). The Glimmer, RBSfinder, and GeneMarkS software programs were used for the prediction of open reading frames and the putative start sites of genes (5, 11, 51). The TransTerm program predicted the rho-independent transcription terminators (13). The PHIRE program was used to identify repeat sequences in the phage genome (27). The identification of bacterial promoter sequences was carried out online (http://linux1.softberry.com/berry.phtml) using the BPROM program. The BLASTP (http://www.ncbi.nlm.nih.gov/BLAST/), FASTA3 (http://www.ebi.ac.uk/fasta33/), and HHPred (http://toolkit.tuebingen.mpg.de/hhpred/) programs were used to search putative homologies and proteins sharing similarities with predicted phage proteins. The Artemis genome-browsing and annotation tool (http://www.sanger.ac.uk/resources/software/artemis/) was used for genome annotation (41).
Mass-spectrometric identification of phage proteins.
SDS-PAGE-separated proteins were identified by mass spectrometry (MS). For this, the gel was stained with Coomassie brilliant blue or MS-compatible silver stain (20). The individual separated protein bands or the gel regions of proteins that were run only a few centimeters into the gel were cut from the gels and “in gel” digested as described by Shevchenko et al. (43). The proteins were reduced with dithiothreitol and alkylated with iodoacetamide before digestion with trypsin (sequencing grade modified trypsin, catalog no. V5111; Promega). The peptides generated by enzymatic cleavage were analyzed by matrix-assisted laser desorption ionization–time-of-flight (MALDI-TOF/TOF) MS using an Ultraflex TOF/TOF instrument (Bruker Daltonik GmbH, Bremen, Germany) or by liquid chromatography-electrospray ionization-tandem MS (LC-ESI-MS/MS) using an UltiMate 3000 Nano-LC system (Dionex) coupled to a QSTAR Elite hybrid quadrupole TOF mass spectrometer (Applied Biosystems/MDS Sciex) with a nano-ESI source (16). The peptide mass and fragment ion mass data were identified using the Mascot version 2.2 (Matrix Science) against an in-house database of the ORF set of ϕR1-37 using the BioTools 3.0 (Bruker-Daltonik) and ProteinPilot 2.0.1 (Applied Biosystems) interfaces. The search criteria for both Mascot searches were as follows: trypsin digestion with one missed cleavage allowed, carbamidomethyl modification of cysteine as a fixed modification, and oxidation of methionine as a variable modification. For the peptide mass fingerprint spectra, the maximum peptide mass tolerance was ±80 ppm. For the LC-ESI-MS/MS spectra, the maximum precursor ion mass tolerance and the MS/MS fragment ion mass tolerance were both 0.2 Da, and a peptide charge state of +1, +2, +3 was used. An identification was reported when a significant match (P < 0.05) was obtained. In addition, to consider the LC-ESI-MS/MS identification reliable, a minimum of two peptides with an ion score of at least 40 was required.
Cryo-EM.
Aliquots of phage ϕR1-37 (3 μl) were vitrified on holey carbon film-coated grids (Quantifoil R 2/2) in liquid ethane as described previously (1). The specimens were imaged at −180°C in a FEI Tecnai F20 field emission gun transmission electron microscope operating at 200 kV and using a Gatan 626 cryoholder. Micrographs were recorded on Kodak SO163 film under low-dose conditions at a nominal magnification of ×62,000. The film was developed in full-strength Kodak D19 film developer for 12 min. When we assessed the internal structure of ϕR1-37, the data were collected on a Gatan UltraScan 4000 charge-coupled-device (CCD) camera under low-dose conditions at a nominal magnification of ×68,000. All electron microscopy data were collected in the Biocenter Finland National Cryo-EM unit, Institute of Biotechnology, University of Helsinki.
Image processing.
Films were digitized at 7-μm intervals on a Zeiss Photoscan TD scanner, resulting in a nominal sampling of 0.113 nm pixel−1, and the images were further binned by four to 0.452 nm pixel−1 to speed up orientation search and refinement. CTFFIND3 (30) was used to estimate the contrast transfer function. Drifted and astigmatic micrographs were discarded. ETHAN (25) was used to locate the virus particles, and particles were extracted in the EMAN1 program Boxer (29). Bsoft (20) was used for further image processing unless stated otherwise. Auto3DEM was used to determine the particle orientations and origins and to calculate the reconstructions (57). The resolution was estimated by comparing two half-data sets using a Fourier shell correlation cutoff of 0.5 from Harauz and van Heel (18). The reconstructions were visualized in Chimera (36).
Database accession numbers.
The annotated nucleotide sequence data for ϕR1-37 have been submitted to the EMBL/GenBank databases under the accession number AJ972879. The full (DNA-containing) and empty (DNA-lacking) ϕR1-37 reconstructions have been deposited in the EMDB (Electron Microscopy Data Bank) with the accession codes EMDB-2159 and EMDB-2160, respectively.
RESULTS
The ϕR1-37 genome sequence.
Two separate shotgun libraries of the double-stranded (ds) ϕR1-37 DNA were sequenced and combined with the previously obtained 76 kb of sequence (23), resulting in ca. 191 kb of sequence across 68 contigs. The gaps between the contigs were closed by primer walking using purified ϕR1-37 DNA as the template to assemble a circular molecule of 262,391 bp. Both strands were sequenced over the entire genome.
The genome of ϕR1-37 is particularly A-dU rich, with the G+C content being 32.91%. A total of 367 open reading frames (ORFs) were predicted from the genome, and the genes were named in a clockwise fashion by the number and prefix “g,” and the corresponding gene products were given designations beginning with “Gp.” The transcriptional direction of 284 of the genes (77.2%) was clockwise, and that of 84 genes (22.8%) was counterclockwise. The average gene density per kilobase of the phage genome was 1.406. The physical map of the genome is shown in Fig. 1.
Fig 1.

Map of the phage ϕR1-37 genome based on the nucleotide sequence deposited in nucleotide sequence databases under the EMBL/GenBank accession number AJ972879. The defined left end of the genome, starting at nucleotide position 258,600, is shown at the top, and the undefined, redundant right end at the bottom, with a dashed line representing the redundancy. The genes are shown by different-colored arrows, and every second gene number is indicated above the arrow. The arrow directions indicate the coding directions of the genes. The genes encoding proteins identified by the MS analysis from the phage particles are colored brown, the genes encoding proteins involved in nucleotide metabolism are yellow, and genes encoding proteins with some other function are red. The predicted functions of the gene products are indicated above the arrows (see also Table 1). The locations of tRNA-encoding genes at 67.7 and 71.5 kb are shown as blue cloverleaf outlines. The putative ϕR1-37-specific (see Table S3 in the supplemental material) and the σ70 promoters (see Table S4 in the supplemental material) are indicated as red and green bent arrows, respectively. The putative antisense promoter at 163.2 kb is indicated by a blue bent arrow. The predicted factor-independent terminators (see Table S5 in the supplemental material) are indicated by hairpins.
Mass-spectrometric fingerprinting of structural proteins.
Altogether, 69 structural proteins were identified by two approaches (Table 1). First, proteins in individual bands excised from silver-stained SDS-PAGE gel (Fig. 2) were digested by trypsin, and the identification was performed by peptide mass fingerprint or MS/MS analysis of the tryptic peptides; this was followed by matching the identified peaks to the predicted protein sequences of the ϕR1-37 genome using the Mascot software (Table 1). Seventeen proteins were identified by this approach, and the identified proteins migrated in the gel nicely according to their calculated sizes; for example, the proteins Gp196, Gp207, and Gp234 (calculated masses, 95 to 102 kDa) were identified from the 94-kDa band, Gp300 (calculated mass, 78 kDa) from the 80-kDa band, and Gp149 (65 kDa) from the 67- to 70-kDa band. An additional 52 virion proteins were identified by the second approach, via the analysis of tryptic peptides from gel slices of short SDS-PAGE runs by LC-ESI-MS/MS. We used a Mascot score of 40 as the lower limit for the positive identification of proteins. The peptide coverages of the identified virion proteins are shown in Table S1 in the supplemental material.
Table 1.
ϕR1-37 orthologsa and virion structural proteins identified by MS analysis of tryptic peptides
| Gp (aa) | ϕR1-37 virion protein Mascot scoreb | Ortholog, function, protein family, comments | Organism | Protein accession no.c (size, aa) | Identity/similarity % (aa overlap) | E value (HHpredd probability %) |
|---|---|---|---|---|---|---|
| Virion structural proteins | ||||||
| Gp045 (368) | 388 | |||||
| Gp046 (202) | 50 | Peptidoglycan recognition protein-LC isoform LCa | Drosophila melanogaster | 2f2l (PDB), (167) | (117) | 0.27 (91.2) |
| Gp047 (154) | 322 | |||||
| Gp048 (212) | 112 | |||||
| Gp074 (164) | 51 | |||||
| Gp079 (266) | 720 | Prohead core protein protease Pfam: peptidase_U9 (PF03420) | Enterobacteria phage RB43 | YP_239201.1 (215) | 28/43 (191) | 4e−10 |
| Gp081 (463) | 181 | |||||
| Gp082 (192) | 197 | |||||
| Gp083 (2,553) | 555 (102 kDa, 40) | Tail tape-measure protein, transglycolase, Pfam: SLT (PF01464) | Pseudomonas phage ϕ-12 | NP_690829.1 (247) | 36/48 (168) | 9e−14 |
| Gp085 (457) | 114 | Muramoyl-pentapeptide carboxypeptidase | Streptomyces albus | 1lbu (PDB) (213) | (118) | 7.3e−24 (99.9) |
| Gp088 (86) | 72 | Crossover junction endodeoxyribonuclease RusA | Escherichia coli | 2h8e (PDB) (120) | 1.1e−31 (99.97) | |
| Gp089 (364) | 119 | peptidase | Bacteroides | E1YNY6 (138) | 30/48 (124) | 0.067 |
| Gp094 (121) | 400 | |||||
| Gp099 (1,393) | 976 (101 kDa, 50) | Structural protein, putative RNA polymerase β subunit | Pseudomonas phage ϕKZ | NP_803744.1 (1,451) | 24/42 (1009) | 2e−50 |
| Gp101 (347) | 128 | Polyadenylate-binding protein 1; RNA-binding | Homo sapiens | 4f25 (PDB), (115) | (114) | 1.7e−17 (99.7) |
| Gp128 (440) | 249 | |||||
| Gp129 (210) | 158 | Ran GTPase-activating protein 1 | Schizosaccharomyces pombe | 2ca6 (PDB), (386) | (46) | 0.076 (91.7) |
| Gp130 (201) | 51 | |||||
| Gp131 (120) | 128 | Bacterial DNA-binding protein, Pfam: Bac_DNA_binding (PF00216) | Staphylococcus pseudintermedius | F0P5K0 (90) | 44/64 (75) | 1e−06 |
| Gp133 (154) | 92 | Gluconate kinase | Escherichia coli | 1knq (PDB), (175) | (148) | 5.2e−13 (99.5) |
| Gp134 (228) | 372 | |||||
| Gp135 (518) | 2,760 (45 kDa, 16) | Major capsid protein sp46 precursor; also detected from 28-, 35-, 43-, 47-, 94-, and 102-kDa bands | ||||
| Gp146 (243) | 79 | |||||
| Gp148 (232) | 140 | |||||
| Gp149 (590) | 831 (67 kDa, 31) | Tail sheath protein Pfam: phage_sheath_1 (PF04984) | Ralstonia phage RSL1 | B2ZXZ8 (648) | 28/48 (285) | 3e−12 |
| Gp150 (250) | 2,625 (30 kDa, 9) | Structural protein sp31; N-terminal sequence | ||||
| Gp160 (878) | 363 | Head portal vertex protein | Stenotrophomonas phage | A1XGY4 (531) | 25/47 (160) | 0.005 |
| Gp161 (344) | 72 | |||||
| Gp162 (340) | 107 | |||||
| Gp163 (174) | 225 | |||||
| Gp168 (159) | 894 | |||||
| Gp174 (259) | 57 | |||||
| Gp176 (301) | 84 | |||||
| Gp178 (472) | 240 | Structural protein, RNA polymerase β′ subunit | Pseudomonas phage 201ϕ2–1 | B3FJD6 (550) | 34/51 (171) | 2e−16 |
| Gp196 (850) | 76 (94 kDa, 18) | |||||
| Gp197 (282) | 754 | |||||
| Gp198 (263) | 460 | |||||
| Gp199 (259) | 7,977 (24 kDa, 8) | Structural protein sp24 precursor; also present in 22- and 24-kDa bands | CAI99861.1 (259) | 100/100 (259) | 4e−83 | |
| Gp203 (248) | 158 | Pseudomonas phage ϕKZ | NP_803715.1(224) | 27/46 (236) | 4e−09 | |
| Gp204 (505) | 431 | Probable protease HtpX homolog | Vibrio parahaemolyticus | 3cqb (PDB), (107) | (37) | 0.00085 (96.4) |
| Gp205 (386) | 186 | |||||
| Gp206 (324) | 427 | |||||
| Gp207 (863) | 180 (94 kDa, 31) | |||||
| Gp208 (336) | 80 | |||||
| Gp209 (374) | 855 (28 kDa, 25) | |||||
| Gp210 (363) | 1132 | |||||
| Gp229 (135) | 182 | Pre-mRNA-processing protein PRP40 | Saccharomyces cerevisiae | 2b7e (PDB), (59) | (45) | 0.029 (92.4) |
| Gp231 (424) | 340 | Structural protein, DNA-directed RNA polymerase, β subunit | Nitrosococcus oceani | B6BZI8 (1,403) | 25/44 (346) | 1e−08 |
| Gp233 (228) | 1,147 (25 kDa, 9) | |||||
| Gp234 (616) | 120 (94 kDa, 13) | |||||
| Gp236 (140) | 50 | NF-YCe; histone-like Pair | Homo sapiens | 1n1j (PDB), (97) | (48) | 0.23 (82.7) |
| Gp237 (236) | 184 | Structural protein, putative Vsr/MutH/archaeal HJRf family endonuclease | Lausannevirus | F2WLN5 (528) | 30/42 (168) | 0.002 |
| Gp239 (131) | 123 | |||||
| Gp240 (155) | 49 | |||||
| Gp244 (290) | 69 | |||||
| Gp270 (318) | 1,004 (35 kDa, 16) | |||||
| Gp271 (591) | 590 (70 kDa, 29) | |||||
| Gp272 (345) | 107 | |||||
| Gp275 (234) | 539 | |||||
| Gp280 (118) | 280 | Oxidative stress DPS DNA binding | Lactococcus lactis | 1zuj (PDB), (179) | (115) | 8.2e−15(99.6) |
| Gp281 (129) | 448 | |||||
| Gp285 (288) | 58 | Type VI secretion system component | E. coli | 2p5z (PDB), (491) | (274) | 4.1e−23 (99.9) |
| Gp294 (1,302) | 233 (100 kDa, 38) | Kelch-like protein 12 | Homo sapiens | 2vpj (PDB), (568) | (499) | 2e−8 (98.9) |
| Gp295 (1,372) | 156 (100 kDa, 32) | Kelch-like ECH-associated protein 1 | Homo sapiens | 1zgk (PDB), (624) | (333) | 3.9e−8 (99.0) |
| Gp297 (228) | 117 | Caudovirales tail fiber assembly protein, Pfam: Caudo_TAP (PF02413) | Dickeya dadantii | E0SB04 (206) | 36/54 (205) | 4e−31 |
| Gp298 (704) | 589 (70 kDa, 19) | Phage tail collar protein, Pfam: collar (PF07484) | Yersinia aldovae | C4UEH4 (387) | 40/51 (310) | 1e−37 |
| Gp299 (173) | Conserved hypothetical phage tail fiber protein | Cronobacter phage ENT90 | F1BUP0 (157) | 37/60 (162) | 3e−21 | |
| Gp300 (731) | 142 (80 kDa, 18) | Phage tail collar domain protein | Cronobacter phage ENT90 | F1BUP1 (408) | 33/56 (107) | 2e−10 |
| Gp326 (445) | 56 | |||||
| Gp333 (239) | 75 | |||||
| ATPases | ||||||
| Gp049 (261) | ATPase, Pfam: PhoH-like protein (PF02562) | Prochlorococcus marinus | YP_001009873.1 (318) | 41/63 (212) | 2e−36 | |
| Gp061 (929) | ATPase, Pfam: AAA_23 (PF13476) | Leptospira biflexa phage LE1 | CAE14781.1 (729) | 21/44 (322) | 3e−07 | |
| Gp097 (219) | ATP-dependent Clp protease, Pfam: CLP_protease (PF00574) | Hyphomonas neptunium | YP_760784. 1(207) | 29/43 (148) | 2e−08 | |
| Gp304 (430) | ATPase, Pfam: AAA_5 (PF07728) | Sulfuricurvum kujiense | E4TZU6 (331) | 33/54 (207) | 7e−22 | |
| Nucleotide metabolism | ||||||
| Gp056 (180) | Guanylate kinase, Pfam: guanylate kinase (PF00625) | Parvularcula bermudensis | ZP_01018099.1 (211) | 40/60 (60) | 8e−05 | |
| Gp060 (145) | Phosphohydrolase, Pfam: NUDIX (PF00293) | Mycobacterium marinum | YP_001851419 (155) | |||
| Gp068 (274) | Phosphoribosyltransferase, Pfam: Pribosyltran_N (PF13793) + Pribosyltran (PF00156) | Salmonella phage PVP-SE1 | G3BLW2 (266) | 42/59 (264) | 1e−51 | |
| Gp070 (584) | Nicotinate phosphoribosyltransferase, Pfam: NAPRTase (PF04095) | Salmonella phage PVP-SE1 | G3BLW1 (564) | 48/67 (570) | 1e−137 | |
| Gp100 (358) | Noncanonical purine NTP pyrophosphatase, Pfam: rdgB/Ham1p-like (PF01725) | Sulfurospirillum deleyianum | D1B4Y5 (196) | 32/62 (94) | 2e−06 | |
| Gp103 (171) | CMP/dCMP deaminase, zinc-binding, Pfam: dCMP_cyt_deam_1 (PF00383) | Flavobacterium johnsoniae | YP_001192884.1 (143) | 35/54 (119) | 4e−13 | |
| RNA polymerase and RNA interaction (includes also Gp099, Gp178, and Gp231found among virion structural proteins, see above) | ||||||
| Gp102 (603) | Putative DNA-directed RNA polymerase, β′ subunit | Campylobacter fetus subsp. fetus | YP_892467.1 (1508) | 32/47 (158) | 5e−10 | |
| Gp213 (229) | RNase H, Pfam RNase_H (PF00075) | Vibrio coralliilyticus | C9NNH4 (154) | 44/53 (165) | 2e−23 | |
| Gp261 (658) | DNA-directed RNA polymerase β subunit | E. coli | 3lu0 (PDB), (1,342) | (629) | 5.8e−149 (100) | |
| Gp274 (452) | DNA-directed RNA polymerase β′ subunit | E. coli | 3lu0 (PDB), (1,407) | (451) | 3e−111 (100) | |
| Gp284 (98) | Putative RNA-binding protein | Lactobacillus plantarum | 3kwr (PDB), (97) | (74) | 4.2e−18 (99.8) | |
| Toxin-antidote proteins | ||||||
| Gp308 (130) | Antidote protein, Pfam: HTH_3 (PF01381) | Deferribacter desulfuricans | D3PC74 (99) | 36/55 (103) | 1e−06 | |
| Gp309 (104) | Plasmid maintenance system antidote protein, Pfam: HTH_3 (PF01381) | Phaeobacter gallaeciensis | A9FYI4 (91) | 41/61 (71) | 1e−09 | |
| Peptidoglycan lysis proteins (includes also Gp083 found among virion structural proteins, see above) | ||||||
| Gp084 (119) | Muramoyl peptidase, Pfam: Peptidase_M15_3 (PF05291) | Enterobacteria phage K1E | YP_425023.1 (114) | 59/74 (113) | 9e−38 | |
| Gp289 (251) | Endo-type membrane-bound lytic murein transglycosylase | E. coli | 3t21 (PDB), (206) | (205) | 1.1e−33 (99.9) | |
| Gp331 (238) | Endo-type membrane-bound lytic murein transglycosylase | E. coli | 3t21 (PDB), (206) | (144) | 1.7e−26 (99.9) | |
| DNA replication and repair (includes also Gp237 found among virion structural proteins, see above) | ||||||
| Gp069 (217) | HNH homing endonuclease | Bacillus phage SPO1 | 1u3e (PDB), (174) | (110) | 4e−07 (97.91) | |
| Gp086 (178) | Holliday junction resolvase, endonuclease, Pfam: RusA (PF05866) | Bacillus RusA | G4NT32 (151) | 39/56 (157) | 3e−18 | |
| Gp096 (690) | Reverse transcriptase, Pfam: RVT_1 (PF00078) | Synechocystis sp. PCC 6803 | NP_440337.1 (508) | 26/48 (202) | 4e−05 | |
| Gp107 (175) | HNH endonuclease, Pfam: HNH-3 (PF13392) | Rhizobium leguminosarum | B5ZXT2 (162) | 41/60 (83) | 2e−12 | |
| Gp121 (511) | Type III restriction enzyme, Pfam: ResIII (PF04851) | Lachnospiraceae bacterium | F3AGK2 (1,000) | 23/43 (450) | 3e−07 | |
| Gp143 (210) | Terminase large subunit, helicases | Pseudomonas phage ϕKZ | Q8SDD7 (717) | 33/51 (209) | 8e−17 | |
| Gp145 (469) | DNA helicase, terminase large subunit, Pfam: Terminase_6 (PF03237) | Salmonella phage SPN3US | G5DF03 (703) | 22/41 (403) | 1e−09 | |
| Gp151 (585) | DNA mismatch endonuclease, Pfam: Vsr (PF03852) | Xanthomonas phage OP1 | Q2NPH5 (510) | 33/53 (519) | 8e−68 | |
| Gp164 (242) | DNA polymerase II | E. coli | 3k59 (PDB), (786) | 0.14 (92.0) | ||
| Gp167 (420) | DNA polymerase, Pfam: DNA_pol_B (PF00136) | Organic Lake phycodnavirus 1 | F2Y1S6 (1,208) | 39/53 (117) | 3e−07 | |
| Gp170 (576) | DNA polymerase, Pfam: DNA_pol_B (PF00136) | Cyanobacteria phage Syn9 | YP_717843.1 (832) | 24/45 (285) | 8e−10 | |
| Gp171 (561) | Similarity to ϕR1-37 g230, a putative homing endonuclease | Campylobacter phage CP81 | G0LWK8 (541) | 33/47 (530) | 8e−53 | |
| Gp187 (203) | Phosphoesterase, Pfam: metallophos_2 (PF12850) | Shigella phage ϕSboM-AG3 | C8XUC7 (186) | 37/55 (182) | 1e−25 | |
| Gp230 (469) | Putative homing endonuclease, similarity to ϕR1-37 Gp171 | Campylobacter phage NCTC 12673 | F4YAA6 (484) | 33/51 (377) | 6e−39 | |
| Gp247 (353) | DNA primase | Aquifex aeolicus | 2au3 (PDB), (407) | (299) | 3.3e−14 (99.5) | |
| Gp250 (407) | Protein RECA recombinase A | E. coli | 3cmu (PDB), (2,050) | (372) | 1.3e−50 (100.0) | |
| Gp267 (697) | DNA ligase | E. coli | 2owo (PDB), (671) | (687) | 1.8e−165 (100) | |
| Gp269 (110) | Single-stranded DNA-binding protein | Mycobacterium leprae | 3afp (PDB), (168) | (107) | 2.5e−31 (99.98) | |
| Gp286 (477) | Exonuclease V alpha chain, Pfam: AAA_30 (PF13604), UvrD_C_2 (PF13538) | Brucella ceti | C9TG04 (373) | 34/52 (412) | 4e−55 | |
| Gp287 (225) | Homing endonuclease, Pfam: HNH_3 (PF13392) | Acinetobacter baumannii | F5IJP1 (198) | 34/53 (158) | 1e−11 | |
| Gp311 (441) | DNA gyrase B, Pfam: HATpase_c (PF02518), DNA_gyrase_B (PF00204) | Thermotoga sp. | B1LCJ8 (636) | 38/57 (447) | 3e−65 | |
| Gp313 (248) | VSR endonuclease | E. coli | 1vsr (PDB), (136) | (111) | 2.8e−17 (99.7) | |
| Gp314 (357) | DNA gyrase subunit B | E. coli | 3nuh (PDB), (420) | (356) | 9.2e−100 (100) | |
| Gp327 (70) | DNA topoisomerase IV subunit A, Pfam: DNA_topoisoIV (PF00521) | Geobacillus | C5DAU4 (814) | 46/63 (72) | 4e−10 | |
| Gp328 (311) | Homing endonuclease I-DMOI | Desulfurococcus mobilis | 2vs7 (PDB), (199) | (178) | 1.4e−23 (99.9) | |
| Gp329 (749) | DNA gyrase A, Pfam: DNA_topoisoIV (PF00521), four repeats of DNA_gyrase_A_c (PF03989) | Thermovibrio ammonificans | E8T2L0 (809) | 29/51 (759) | 1e−68 | |
| Gp334 (413) | Exonuclease | Thermotoga maritima | 3tho (PDB), (379) | (380) | 1.8e−36 (100) | |
| Gp335 (251) | DNAB replication FORK helicase | Thermus aquaticus | 2q6t (PDB), (444) | (209) | 0.012 (96.5) | |
| Gp336 (282) | VSR endonuclease | E. coli | 1vsr (PDB), (136) | (121) | 4.5e−17 (99.7) | |
| Gp337 (357) | DNAB-like replicative helicase | Bacillus phage SPP1 | 3bgw (PDB), (444) | (268) | 2.3e−30 (100) | |
| Uncharacterized | ||||||
| Gp095 (169) | Uncharacterized protein | Cupriavidus spp. | B3R3H5 (124) | 37/58 (73) | 2e−04 | |
| Gp154 (240) | Uncharacterized protein | Klebsiella sp. | F3Q626 (250) | 34/56 (203) | 2e−25 | |
| Gp179 (310) | Uncharacterized protein | Paenibacillus polymyxa | E3EJZ7 (452) | 28/45 (310) | 2e−15 | |
| Gp190 (394) | Uncharacterized protein | Labrenzia aggregata IAM 12614 | A0NP95 (401) | 42/66 (391) | 4e−85 | |
| Gp191 (440) | Uncharacterized protein | Serratia proteamaculans 568 | A8GD92 (463) | 36/48 (125) | 1e−10 | |
| Gp224 (166) | Uncharacterized protein | Klebsiella phage KP15 | D5JF55 (146) | 29/44 (129) | 2e−04 | |
| Gp263 (191) | Uncharacterized protein | Enterobacteria phage RB16 | D9ICD9 (189) | 41/58 (186) | 4e−27 | |
| Gp265 (166) | Uncharacterized protein | Enterobacteria phage CC31 | E5DIB3 (150) | 31/50 (165) | 5e−05 | |
| Gp276 (253) | Uncharacterized protein | Fusobacterium sp. | C3WL22 (202) | 23/49 (194) | 4e−05 | |
| Gp278(295) | Uncharacterized protein, similarity to Gp282 of ϕR1-37 | ϕR1-37 | G4KKM0(225) | 35/61(150) | 5e21 | |
| Gp282 (225) | Uncharacterized protein, similarity to Gp278 of ϕR1-37 | ϕR1-37 | G4KKL6 (295) | 35/61 (150) | 2e−21 | |
| Gp293 (179) | Uncharacterized protein | Syntrophus aciditrophicus SB | YP_460612 | 2e−05 | ||
| Diverse | ||||||
| Gp077 (131) | Lincosamide antibiotic adenylyltransferase LinA | Staphylococcus haemolyticus | 4e8j (PDB), (161) | (108) | 6.4e−17 (99.7) | |
| Gp106 (30) | BH1478 protein; unknown function | Bacillus halodurans | 2qup (PDB), (145) | (21) | 0.13 (92.1) | |
| Gp142 (95) | PAAR repeat containing protein, Pfam: PAAR_motif (PF05488) | Acidithiobacillus caldus | C6NTR4 (98) | 58/68 (95) | 2e−23 | |
| Gp219 (119) | Hypothetical protein rv5-gp079 | Escherichia phage rv5 | B3RGL8 (116) | 36/51 (122) | 3e−08 | |
| Gp243 (574) | CPN60 (GroEL); chaperonin, chaperone, GroEL, HSP60 | Thermus thermophilus | 1we3 (PDB), (543) | 1.1e−92 (100) | ||
| Gp296 (928) | Kelch-like protein 12 | Homo sapiens | 2vpj (PDB), (568) | (366) | 1e−6 (98.6) | |
| Gp305 (611) | Uncharacterized protein with a domain of unknown function, Pfam: DUF2201 (PF09967) | Sulfurovum sp. | A6QAE6 (396) | 33/54 (181) | 9e−17 |
Orthologs were identified by BLASTP, Fasta3, and HHPred searches.
Phage particle proteins were identified from individual bands (size indicated in parentheses, together with the number of matching peptides) excised from the silver-stained SDS-PAGE gel (Fig. 2) or from gel slices cut from a gel lane where total phage particle proteins were run 3 to 4 cm into the SDS-PAGE gels. The protein samples were treated with trypsin, and the peptides were analyzed by MS. The peptide coverages of the identified proteins are shown in Table S1 in the supplemental material.
Accession numbers are fromEMBL/GenBank unless noted otherwise.
HHpred, homology detection and structure prediction by HMM-HMM comparison.
NF-YC, nuclear transcription factor Y subunit gamma.
HJR, Holliday junction resolvase.
Fig 2.
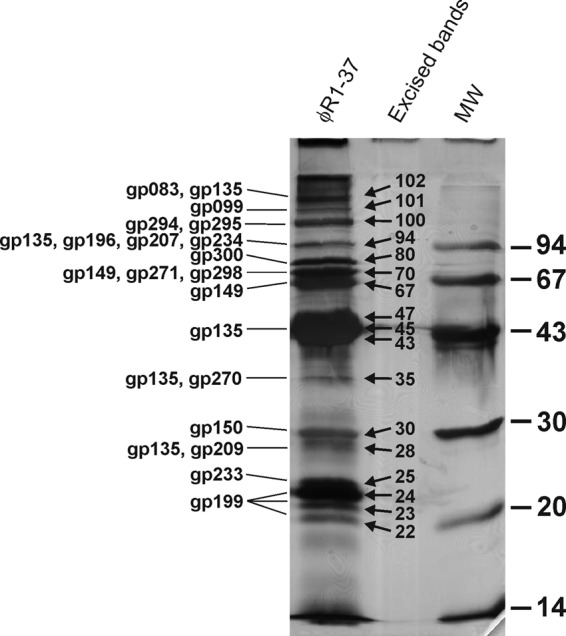
Silver-stained SDS-PAGE analysis of ϕR1-37 virion proteins. The clearly visible individual protein bands that were excised for trypsin digestion and MS identification are indicated (according to their relative masses in kDa) between the sample and molecular weight standard (MW) lanes. The gene products identified in these excised bands are indicated on the left. For the identification of the other possible virion proteins, a shorter gel was run and systematically sliced, and each slice was subjected to MS identification of its proteins.
The identified virion proteins are encoded by genes scattered around the genome, which in many instances are apparently organized into operons, i.e., g045 to g048, g081 to g079 (transcribed from reverse strand), g082 to g090, g128 to g131, g132 to g135, g146 to g150, g163 to g160, g173 to g178, g208 to g197, g232 to g229, g236 to g233, g237 to g244, g272 to g270, g281 to g280, and g300 to g294 (Fig. 1). Many of these operons encode gene products which were not identified in MS analysis but which may in the end prove to be present as minor constituents in the phage particles.
Finally, only 18 of the identified 69 virion proteins showed similarities to known proteins (Table 1). Gp079, Gp083, Gp099, Gp135, Gp150, Gp178, Gp199, and Gp231 are discussed in more detail below. Of the others, Gp131 showed similarity to DNA-binding proteins and could therefore participate in DNA packaging, Gp149 is suggested to be a tail sheath-forming protein, and Gp160 is a portal protein. Gp298 and Gp300 are suggested to participate in the formation of the tail collar, and while Gp299 is predicted to form the tail fiber, it was not identified by the MS analysis.
RNA polymerase domains.
Three gene products (Gp099, Gp178, and Gp231) among the structural proteins showed weak but significant similarities to the DNA-directed RNA polymerase β and β′ subunits (Table 1; see also Table S2 in the supplemental material). While only the size of Gp099 is that of a full-length β subunit polypeptide (ca. 1,400 residues), the others align to different parts of the β′ subunit (Fig. 3). Gp099 was also identified as a >100-kDa band in the SDS-PAGE analysis (see Gp099 in Fig. 2). Furthermore, another three gene products (Gp102, Gp261, and Gp274), which were not identified from the phage particles by the MS analysis, showed similarities to different domains of the RNA polymerase β′ subunit (Table 1; Fig. 3). Gp274 and Gp178, which aligned with the same part of the β′ subunit, showed similarities with each other (23% identity). Similarly, Gp231 and Gp102 showed local similarities over the Rpb1_5 domain of the β′ subunit, with 27% identity over 147 amino acids (aa).
Fig 3.
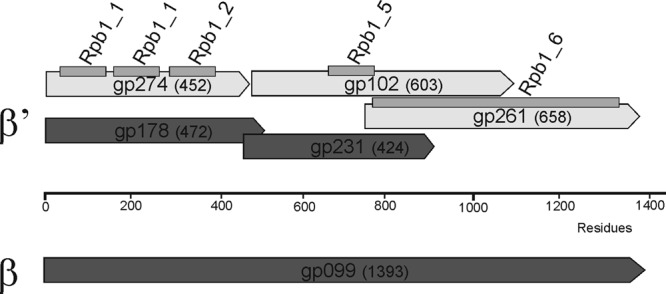
Alignment of the RNA polymerase β and β′ subunit homologous polypeptides of ϕR1-37.
Genome annotation.
Altogether, 140 of the 367 predicted ϕR1-37 proteins (38%) could be annotated based on significant amino acid sequence similarities to other proteins in databases and/or they were identified as structural proteins by mass spectrometry (Table 1; see also Table S2 in the supplemental material). Based on the available information, the gene products were annotated and grouped to the following classes.
(i) DNA replication and repair.
A number of genes were found to code for proteins similar to the known enzymes of the DNA replication machinery (Table 1; see also Table S2 in the supplemental material): Gp086 showed similarity to a RusA family Holliday junction resolvase, Gp131 to a bacterial DNA-binding protein, Gp247 to DNA primase, Gp267 to DNA ligase, Gp269 to a single-stranded DNA (ssDNA)-binding protein, and Gp335 to a DnaB replication fork helicase. Gp213 showed similarity to the RNase H enzyme, and it might function in the degradation of the RNA primer during replication initiation. The remaining identified replication machinery-encoding genes appeared to either be incomplete or encode only some of the domains found in their homologs. For example, the 210- and 469-aa proteins Gp143 and Gp145 showed joint similarities to ∼700-aa DNA helicases, suggesting that they might constitute different domains of the helicase. Gp313 appeared to be a group I intron that separates two domains of the gyrase/topoisomerase-encoding genes g311 and g314, since the in silico-joined Gp311-Gp314 product aligned over its entire length to a number of DNA gyrase B subunits. Finally, the in silico-joined Gp327-Gp329 polypeptide also aligned over the full length with the DNA gyrase A subunit. On the other hand, the 420- and 576-aa Gp167 and Gp170 proteins organized in divergent operons showed similarities to much longer (ca. 1,000-aa) DNA polymerases. The 690-aa Gp096 protein showed similarity to a reverse transcriptase, but at present we anticipate that it functions as a DNA polymerase replicating the dU-containing DNA. Gp121 is predicted to be a type III restriction endonuclease, and Gp286 to be an exonuclease V, and both might be partly responsible for host DNA degradation. Gp151 is a putative DNA mismatch endonuclease, and Gp187 may be a phosphoesterase, both involved in DNA repair. Finally, Gp237 is a structural protein predicted to have endonuclease activity (Table 1) and could function during the headful packaging of the phage genome. Gp171, Gp230, and Gp287 are putative homing endonucleases.
(ii) Nucleotide metabolism.
Only a few genes coding for enzymes predicted to participate in nucleotide metabolism were identified in the ϕR1-37 genome (Table 1; see also Table S2 in the supplemental material): Gp56 is predicted to be a guanylate kinase, Gp068 a phosphoribosyltransferase, Gp070 a nicotinate phosphoribosyltransferase, and Gp103 a CMP/dCMP deaminase, and Gp100 showed similarity to a purine nucleoside triphosphate (NTP) pyrophosphorylase.
(iii) ATPases.
Gp049, Gp061, and Gp304 are predicted to be ATPases, albeit in different protein families (Table 1). Gp097 is predicted to be a Clp-like ATP-dependent protease. Gp250 showed similarity to the RecA ATPases that mediate homologous recombination (see Table S2 in the supplemental material).
(iv) Toxin antidote system and lysis proteins.
Gp308 and Gp309 showed similarities to the helix-turn-helix 3 (HTH3-3) family of antidote proteins (Table 1). They could function early in the infection process to overcome a bacterial abortive infection system (26), remotely resembling the Dmd antidote protein of T4 (32). Gp084, Gp289, and Gp331 were predicted to act on peptidoglycan (Table 1); thus, they might be involved in the lysis of bacteria.
High-GC content genes.
While the average GC content of the phage genome is ∼33%, a number of genes had significantly higher GC percentages, sharply rising in the GC plot from the average level. Interestingly, 9 of the 13 >40%-GC genes (g094*, g101*, g129*, g131*, g135*, g142, g144, g149*, g168*, g199*, g241, g304, g326*) encoded phage particle structural proteins (indicated with an asterisk) (Table 1), indicating that either the genes might be recent acquisitions or the higher GC content would allow a more host-suited codon usage and therefore a higher expression level.
Specific features of the genome.
The TransTerm program predicted five tRNA genes in the genome (Fig. 1) encoding tRNA-Arg (anticodon TCT), tRNA-Leu (TAG), tRNA-Leu (TAA), tRNA-Met (CAT), and tRNA-Asn (GTT). While the tRNA-Asn gene is located between genes g115 and g116, the other four genes are located between genes g105 and g107. Gp352 and Gp363 showed some similarities to tRNA synthetases (see Table S2 in the supplemental material).
The PHIRE program was used to identify repeat sequences in the genome sequence of 20 bp with a maximum of 4 mismatches. Two types of repeats were identified. First, a few locations with (GXX)n repeats, where X is A or dU. These repeats occurred within genes g101, g130, and g278, resulting when translated in peculiar poly(E) or poly(DE) sequences; however, no specific functions for these gene products were identified.
The second type of repeats included 59 A-dU-rich repeats scattered around the genome (see Table S3 in the supplemental material). As most of them were located in intergenic regions (Fig. 1), they might represent ϕR1-37-specific promoters. Sequence analysis of the 59 repeats (Fig. 4) revealed a consensus sequence of 5′-uuuuannnAUAUAUuAUuannuga-3′ (u=dU) for the putative ϕR1-37-specific promoters (the uppercase letters indicate >90% probability for this base in this position, the lowercase letters indicate a preferred base, and n indicates any base).
Fig 4.

Sequence logo of the putative phage ϕR1-37 promoters generated using the sequence logo generator (10) (http://weblogo.threeplusone.com/) and the data in Table S3 in the supplemental material. The upper logo shows the residue probabilities, and the lower logo shows the information content at each position.
The PHIRE search identified putative divergent ϕR1-37 promoters from the g169-g170, g273-g274, g282-g283, and g333-g334 intergenic regions (Fig. 1) but not from the g081-g082, g094-g095, g099-g100, g116-g117, g167-g168, g208-g209, and g236-g237 intergenic regions. On the other hand, the BPROM prediction tool identified putative σ70 promoters from the latter locations, suggesting that these genes may be transcribed by the host RNA polymerase (RNAP) (see Table S4 in the supplemental material). In general, the intergenic regions of ϕR1-37 predicted to contain a promoter are, as a rule, very A-dU rich (in most such intergenic regions, the GC percentages are 10 to 25), and the BPROM prediction tool could easily identify putative σ70 promoters from these sequences. Further experimental evidence is needed to validate the promoters.
In an attempt to confirm the promoter status of the PHIRE-identified repeats, we cloned a number of the intergenic regions into a luciferase reporter plasmid. The obtained reporter plasmids were introduced into Y. enterocolitica strain YeO3-R1 to test whether the putative promoters would be induced upon infection by ϕR1-37. To our disappointment, this approach was not successful, as in all constructs the reporter activity rapidly disappeared after phage infection (data not shown), suggesting that a strong DNase was likely produced upon phage infection, resulting in reporter plasmid degradation. The promoter reporter plasmids produced interesting results in noninfected control cultures. The g236-g237 intergenic region (that should contain divergent promoters; see Fig. 1), when cloned into the luciferase reporter plasmid, functioned as a strong host RNAP promoter and produced a high luciferase activity also found in E. coli. This supported the BPROM prediction tool results, which identified σ70 promoter motifs in both directions from this sequence (see Table S4 in the supplemental material). The g233-g232 intergenic region, carrying a putative promoter sequence for genes g232 and g231, on the other hand, strongly suppressed the background luciferase activity in both E. coli and Y. enterocolitica, indicating the presence of considerable transcriptional silencing potential. Detailed analysis of the silencing revealed that it was due to antisense promoter activity directed against g233 (L. Kalin-Mänttäri and M. Skurnik, unpublished observations), and indeed, BPROM identified an antisense σ70 promoter motif from this sequence (Fig. 1; see also Table S4). We speculate that the host RNAP-transcribed antisense RNA would block the g233 expression in the early phase of infection, and this would be released later. As Gp233 is a phage structural protein (Table 1), it indeed should be expressed later in infection.
Terminator-like hairpins were identified with a PHIRE search using settings to detect repeats of 30 bp, allowing for 6 mismatches. These, combined with analysis of the ϕR1-37 sequence with the TransTerm program, resulted in the identification of 100 terminator-like hairpins all together. The locations of the terminators are indicated in Fig. 1 and are listed in Table S5 in the supplemental material.
The circular map of the genome gave no indication of where the ends of the linear genome were. Restriction enzyme digestions of the phage DNA revealed fragments that were not predicted from the circular sequence; e.g., KpnI gave extra 2.7- and 5.7-kb fragments that likely represent linear genome ends (Fig. 5A). The simplest explanation for the extra fragments is the presence of a >8.4-kb terminal redundancy. The controlled Bal31 digestion prior to KpnI digestion caused the disappearance of the 5.7- and 2.7-kb bands (Fig. 5A). This indicated that the genome is a nonpermuted linear molecule. On the other hand, digestion with SphI produced an extra 6-kb fragment. Analysis of the possibilities of the sequence-predicted 33 KpnI and 6 SphI sites fitting the extra fragments revealed that only the SphI site at position 2,540 could accommodate the extra KpnI fragments such that the left end of the phage would be located at ca. 258,600 and the right end at ca 4,600 of the circular map. Indeed, sequencing the phage genomic DNA or the 6-kb SphI fragment using reverse primers (3′ ends at 258,727 and 258,973) identified the left end unequivocally at nt 258,670 (Fig. 5B); however, the right end could not be mapped at position 4,600 or elsewhere despite the use of several different restriction enzymes and sequencing primers. Further experimental work is needed to elucidate the nature of the right end of the genome.
Fig 5.

Physical ends of the phage genome. (A) The phage genomic DNA was treated with Bal31 for 0, 30, and 60 min and thereafter digested with KpnI. The 5.7- and 2.7-kb KpnI fragments that disappear in the 30- and 60-min samples are indicated. M lanes, molecular mass markers. (B) Snapshots of sequencing reads using whole-phage genome or the isolated 6-kb SphI fragment as the template, demonstrating the end of the template. In the whole genome read, the sequence intensity drops to half due to the end of the template in the left end of the genome, while the primers that started the reads from the redundant right end of the genome continue further. (C) Strategy to detect the circular form of the phage genome by Southern blotting. A probe covering nt 258,737 to 259,767 would hybridize only to the 15.7-kb KpnI fragment if the replicating form of the genome was circular but to 5.7- and 15.7-kb KpnI fragments if it was linear.
It is possible that the phage DNA circularizes during infection based on the terminal redundancy of the genome. To address this experimentally, we designed a DIG-labeled probe that hybridizes to the 15.7-kb KpnI fragment that overlaps the 5.7-kb left end of the genome. The total DNA isolated from the phage-infected bacterial samples digested with KpnI was analyzed by Southern hybridization. If the phage replicates in circular form, the left-end 5.7-kb KpnI fragment should not be seen with Southern hybridization; only the 15.7-kb KpnI fragment should be detected (Fig. 5C). Both the 5.7- and the 15.7-kb fragments were present at all the time points, indicating that the phage genome is linear throughout the infection area (data not shown). These data, however, cannot exclude the possibility that a fraction of the replicating genomes take a circular form during infection.
Finally, there is a case of possible frameshift for g040 and g041. The ORFs overlap by over 40 bp, giving the possibility for a ribosomal slippage over a stretch of poly(A). A similar phenomenon has been described for several phages; an example is the Bacillus subtilis phage SPO1 and its relatives, where a +1 frameshift over an AAAG sequence results in a protein facilitating the assembly of the tail tube around the tail length tape measure protein (49). While no function could be predicted for the g040 and g041 product(s), their locations are similar to that of SPO1, being directly upstream of the genes that encode structural proteins.
Snapshots of the ϕR1-37 transcriptome.
To evaluate the temporal expression of the ϕR1-37 genome, we performed a Northern blot analysis using probes targeting selected genes encoding products with a significant similarity to known proteins (Table 2). Since the burst time leading to the lysis of phage ϕR1-37-infected bacteria is ca. 70 min (23), the total RNA from infected bacteria was extracted every 7 min after infection up to a total duration of 42 min. Northern blotting results after low-stringency washes showed that in most cases, a single major band hybridized with the probe (Table 2). The exceptions were the g070 to g072 and g103 probes, which showed a wide range of signals with high-molecular-weight bands in the early time point and an accumulation of shorter species later, and the g329 probe, which hybridized to three bands, of which the middle band (4 kb) was of the size predicted to carry the g329 transcript (data not shown). Apparently, in these multicistronic operons, the mRNA was degraded to smaller fragments with different half-lives. The Northern blotting results demonstrated that the transcription of the nucleotide metabolism-associated genes g70 and g103 had started in the 21-min samples, as was also the case with the g329 gene, encoding the gyrase A subunit, while the DNA replication-related g145 (helicase) transcription had started a little later, at 35 min, and that of the phage structural proteins only at 42 min. The earliest (at 7 min) transcription was detected for g231, encoding an RNAP β subunit-like protein.
Table 2.
Summary of the Northern blot results for expression of selected ϕR1-37 transcripts
| Gene | Size of transcript(s) (kb)a | Time after infection (min) | Gene product similarities (see Table 1) |
|---|---|---|---|
| g048–g049 | ∼1.2 | 42 | Gp048 is a phage structural protein and Gp049 is similar to PhoH-like phosphate starvation-inducible ATPase |
| g070–g072 | ∼0.7 to ∼10 | 21–42 | Nicotinate phosphoribosyltransferase |
| g103 | ∼3–8 | 21–42 | Zinc-binding CMP/dCMP deaminase |
| g145 | ∼3 | 35–42 | ATP-dependent DNA helicase |
| g170 | ∼3 | 21–42 | DNA polymerase type B family |
| g231 | ∼10 | 7–28 | DNA-directed RNA polymerase, β subunit |
| g281 | ∼3 | 42 | Phage (tail) structural protein |
| g298 | ∼5 | 42 | Phage tail collar protein |
| g329 | ∼3, 4, and 5 | 21–42 | DNA gyrase subunit A |
The sizes of the transcripts were estimated with the help of the RNA markers and rRNA bands.
Cryo-EM and image reconstruction of ϕR1-37.
We analyzed purified ϕR1-37 viral particles using cryo-EM and three-dimensional image reconstruction in order to further characterize the virus. Half of the particles observed were intact DNA-filled virions (Fig. 6A, black arrow). The other half of the particles comprised empty DNA-lacking viruses (Fig. 6A, white arrow). Both particle types were copurified in a glycerol gradient. Of the DNA-containing virions, 97% were tailed (n = 177), compared to the empty-type particles, of which merely 25% were tailed (n = 219). The DNA-filled virion shown in Fig. 6A has an uncontracted tail (black arrowhead), and the empty particle has a contracted tail (white arrowhead). A closer inspection of the previous EM analyses (23) revealed that the measurements were based on an inaccurate calibration of the electron microscope and that the reported dimensions were 1.3- to 1.5-fold too small. Indeed, the Gp83 is the tail tape measure protein (TMP) of 2,553 aa. According to other TMPs, each amino acid contributes ca 1.5 Å to the tail length (22, 49); thus, the calculated tail length would be 383 nm, which is in stark contrast to the 246 nm that we reported earlier (23). A reevaluation of the values based on cryo-EM measurements resulted in the following corrected dimensions for ϕR1-37 (Fig. 6B). The length of the uncontracted tail is 310 ± 10 nm (n = 12), whereas the length of the sheath on the contracted tail is 130 ± 10 nm (n = 12), with the inner tube extending 170 nm outside the contracted sheath (Fig. 6A). The neck between the phage head and tail is approximately 15 ± 5 nm long. The elongated tail tip has clear 75-nm-long tail fibers (Fig. 6A, asterisks; shown schematically in Fig. 6B), as was observed by Kiljunen et al. (23).
Fig 6.
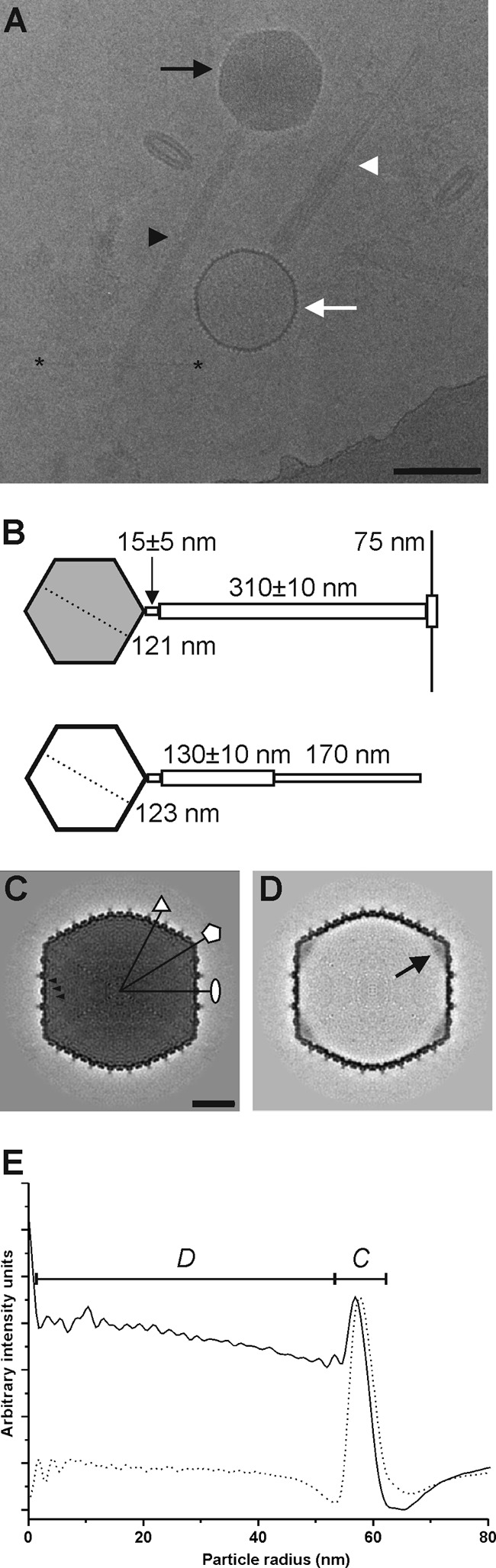
Organization of ϕR1-37. (A) Electron cryomicrograph (2.5 μm underfocus) of ϕR1-37 showing intact DNA-containing virions (black arrow) with extended tails (black arrowhead) and empty DNA-lacking particles (white arrow) with contracted tail sheaths (white arrowhead). The clear tail fibers are visible starting at the tip of the extended tail and reaching to the sides (the ends of two tail fibers are indicated with asterisks). Bar, 100 nm. (B) Schematic representation of an intact (gray head) and an empty (white head) ϕR1-37 particle with uncontracted and contracted tails, respectively. The numbers indicate the sizes obtained in this study. (C) A 0.45-nm-thick central section through the 23.8-Å-resolution DNA-containing virion reconstruction. The symmetry axes are indicated with a white ellipse (2-fold), triangle (3-fold), and pentagon (5-fold). The black arrowheads point to three successive layers of packaged DNA. Bar, 20 nm. (D) A 0.45-nm-thick central section through the 23.4-Å-resolution DNA-lacking particle reconstruction. A black arrow points to the connector density at one of the 5-fold vertices. (E) Radial density profiles of the icosahedral reconstructions of the DNA-containing (solid line) and DNA-lacking (dashed line) viral particles. The capsid density is indicated with a C, and the DNA with a D.
We calculated icosahedrally symmetric three-dimensional reconstructions of both particle types (Fig. 6C to E and 7). The reconstruction of the intact virion was calculated to a 23.8-Å resolution from 1,028 particle images using 297 micrographs (underfocus range, 0.05 to 5.76 μm) (Fig. 6C and 7A). The reconstruction of the empty viral particle was calculated to a 23.4-Å resolution from 1,022 particle images using 322 micrographs (underfocus range, 0.07 to 5.15 μm) (Fig. 6D and 7B). Both particle types were angular in appearance, with the empty particles having an approximately 9-Å-larger radius than the DNA-filled virions, as measured from the radial intensity profiles of the reconstructions (Fig. 6E). The diameters of the intact ϕR1-37 virion are 138 nm from vertex to vertex and 121 nm from facet to facet. Similarly, the sizes of the empty ϕR1-37 particle are 138 nm from vertex to vertex and 123 nm from facet to facet (Fig. 6B and E). The average thickness of the capsid in both particle types is 4.5 nm (Fig. 6E). Based on the internal volume of the ϕR1-37 virion and assuming that the genome fully occupies the cavity (as seems to be apparent from central sections and radial profiles; Fig. 6C and E), the packaging density is 0.385 bp nm−3. The concentric rings of DNA are packaged at an average spacing of 2.7 nm (Fig. 6C and E), which is comparable to the 2.4-nm spacing of bacteriophage ϕKZ (16).
Fig 7.
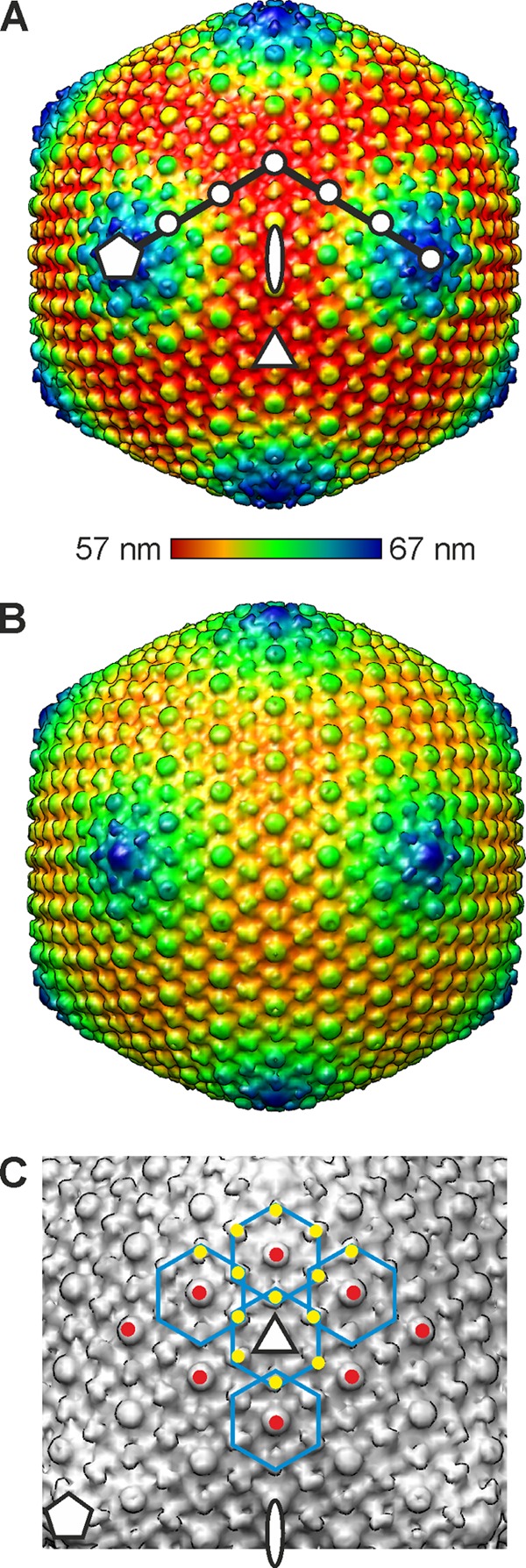
Structure and capsomer organization of ϕR1-37. (A) A radially colored isosurface representation of the DNA-filled virion drawn at 2σ above the mean viewed down a 2-fold symmetry axis. The symmetry axes are indicated with a white ellipse (2-fold), triangle (3-fold), and pentagon (5-fold). The triangulation number (T) of a virus describes the geometrical arrangement of the capsomers and is given by the relationship T = h2 + hk + k2 (8). The integers h and k that define the lattice points for a T=27 lattice (h = 3 and k = 3) are indicated with white dots. The color key indicates radii (57 to 67 nm). (B) Surface representation of the DNA-lacking particle drawn at 4σ above the mean viewed down a 2-fold symmetry axis. (C) Blow-up of a facet of the ϕR1-37 virion viewed down a 3-fold axis of symmetry. The symmetry axes are indicated as described for panel A. Five hexameric capsomers are outlined in blue for clarity. The type I protrusions on the capsomers are indicated with red dots, and the type II protrusions with yellow dots. The type II protrusions are shared by adjacent capsomers.
The capsomers of ϕR1-37 are arranged on a T=27 lattice (Fig. 7A), formed by the major capsid protein Gp135 (Table 1). A similar T number has been observed in the myovirus bacteriophage ϕKZ of Pseudomonas aeruginosa (14, 16), where the major capsid protein is predicted to have the HK97 fold, and the icosahedral satellite virus Sputnik of the giant mimivirus (50) is predicted to have a double β-barrel fold. The surface of ϕR1-37 is covered by protrusions similar to those of the bacteriophage ϕKZ (16), with the type I protrusions located at the center of the hexameric capsomers on the quasi-6-fold axes (Fig. 7A, red dots) and the larger, elongated, and rotated type II protrusions located at the periphery of the hexamers on local 2-fold axes of symmetry (Fig. 7A, yellow dots).
Fuzzy density on the inside of the capsid for the connector was evident in the icosahedrally averaged reconstruction of the empty particles (Fig. 6D, black arrow). Each vertex appears to contain this density, although it is most probably located only at the vertex where the tail attaches (see where the contracted tail enters the empty head in Fig. 6A). In bacteriophage ϕKZ, the core of the icosahedral head is radiation sensitive (56) and thus can be distinguished in heavily irradiated samples. However, the core of ϕR1-37 is no more radiation sensitive than is the rest of the particle, so no distinct structure could be detected by repeatedly exposing the virus to an increasing electron dose (data not shown).
DISCUSSION
We have characterized the structure, proteome, and genome of yersiniophage ϕR1-37. The genome includes 367 ORFs and 5 tRNAs. We could identify the physical left end of the genome to the nucleotide (Fig. 5) but not the right end, even though the Bal31 digest (Fig. 5A) clearly demonstrated that two KpnI bands started to disappear at the same time, indicating that the Bal31 enzyme is able to access the genome from both ends simultaneously and, consequently, that the right end features a fixed structure. Thus, we have to conclude that the right end of the genome and the length of the terminal redundancy remain unresolved, but it appears to be >8 kb. Furthermore, the findings suggest that the phage DNA may not be packed by the conventional pac-type headful mechanism (31). On the other hand, we estimated earlier that the phage genome size was 270 kb (14), which is 103% of the full-length genomic sequence reported here. This is a typical percentage for headful-packaged genomes. The presence of a fixed left end would suggest a novel DNA-packaging strategy. If correct, it would indicate that the genome packaging into the heads would always start from the unique left end and that the right end would be (semi)randomly cleaved after headful packaging. At the same time, it would indicate that the phage might not use the concatemer strategy in DNA packaging (6). Our Southern blot experiments on DNA isolated from infected bacteria at different time points after infection revealed the presence of the left-end terminal 5.7-kb KpnI fragment in all samples, further suggesting that most of the replicating DNA is linear in form. Based on these genome properties and the unique DNA composition, ϕR1-37 appears to represent a novel phage type.
The ϕR1-37 genome encodes 140 proteins that are either similar to known proteins or were detected as structural virion proteins by MS analysis (Table 1), thus leaving 227 proteins without a function. The phage gene products predicted to participate in nucleotide metabolism are part of the normal nucleotide biosynthesis pathway and did not explain the strategy by which ϕR1-37 controls the host nucleotide metabolism and redirects it from dTTP to dUTP. The only phage for which this has been studied to any extent is the Bacillus subtilis phage PBS2. This phage expresses a distinct set of proteins and enzymes that participate in the synthesis of the dU DNA, but only one of these, a protein called uracil-DNA glycosylase inhibitor (Ugi), has been characterized in detail (55). Ugi inhibits the breakdown of dU DNA in the cell by inhibiting the uracil-DNA glycosylase, the first enzyme of the base excision pathway (BER) (28). The ϕR1-37 genome did not code for proteins showing sequence similarity to PBS2 Ugi, illustrating that these two phages apparently do not share the same mechanism for maintaining the dU DNA or that the proteins are too evolutionarily distant to be detected by amino acid sequence alignment.
Sequence analysis identified several genes encoding proteins that show significant similarity to DNA-directed RNA polymerase β and β′ subunits (Table 1; Fig. 4), indicating that the phage expresses its own RNAP. Indeed, from the intergenic regions predicted to carry a promoter, we identified likely phage RNAP-specific promoter motifs (see Table S4 in the supplemental material). Although the reporter plasmid-based promoter identification approach failed, our preliminary RNA-sequencing results of the transcriptome of phage-infected bacteria show great promise for promoter identification. In the MS analysis, we identified three RNAP β and β′ subunit homologs from the phage particles. Thus, it is possible that the phage particle introduces the β and β′ subunits of the phage RNAP and/or other RNAP components to the host cell already during injection of the nucleic acid into the host in order to take over transcription at the early stage of infection. This scenario is supported by similar findings of RNA polymerase domains both for the giant phage myovirus 201ϕ2-1 (52) and for the above-mentioned B. subtilis phage PBS2 (9). Thomas et al. (53) suggested that having the β subunits in several distinct fragments would allow their injection with DNA into the host, while a full-length β or β′ subunit might be too large to pass through the tail tube.
A general feature in bacteriophage capsid maturation is a cascade, where the structural proteins are produced as preproteins that are converted to functional proteins by dedicated proteases; for example, a number of capsid proteins of bacteriophage ϕKZ are processed by the Gp175 protease of ϕKZ (54). In the MS analysis, the processed proteins were identified based on the fact that the eliminated parts of the processed proteins do not produce tryptic peptides. An analysis of the peptide coverage data of ϕR1-37 proteins with a Mascot score of >200 revealed that N-terminal tryptic peptides were missing from Gp150 (however, see below), Gp198 and Gp204, C-terminal peptides from Gp079, and both N- and C-terminal peptides from Gp206 and Gp275 (see Table S1 in the supplemental material). Furthermore, several <200-score proteins also lacked N- and C-terminal tryptic peptides. These data strongly suggest that proteolysis also plays a role in ϕR1-37 head morphogenesis. The candidate protease is Gp079, which was annotated as a phage structural protein with similarity to Pfam of prohead core protein proteases (Table 1). The head proteases are, in general, processed by autocleavage to release a C-terminal fragment (54). In line with this, the 89 C-terminal residues of Gp079 lacked peptide coverage (see Table S1 in the supplemental material); the last identified tryptic peptide ended at residue 179 of Gp079, suggesting that the plausible autocleavage site is the LAE sequence (residues 198 to 200) that perfectly conforms to the consensus T4 protease recognition sequence I/L-X-E (7). The active site residues of the proteases (54) are also conserved in Gp079; hence, it is also likely that it similarly has T4 protease-like activity.
We earlier determined the N-terminal sequences of four structural proteins, sp69, sp46, sp31, and sp24, of which sp69 and sp46 appeared to be two different polypeptides encoded by the same gene (23). Here, we identified the sp46/sp69-encoding gene as g135. g135 can code for a 518-aa polypeptide (predicted mass, 57.8 kDa). The N-terminal sequence of sp46 (23) was identified, starting at Ser87 of Gp135, thus leaving a 432-aa polypeptide with a predicted mass of 47.76 kDa that agrees nicely with the size observed with SDS-PAGE (Fig. 2). Most tryptic peptides identified by MS analysis were located in the 432-aa polypeptide; among residues 1 to 86, only the N-terminal peptide was identified (see Table S1 in the supplemental material), albeit with a relatively poor score. Furthermore, bioinformatic analyses identified a very good ribosomal binding site upstream of g135, suggesting that Gp135 is produced as a preprotein that is proteolytically cleaved during capsid maturation at the LQE sequence upstream of Ser87 (7). Our data do not explain the sp69 band identification made earlier (23). In the present MS analysis, Gp135 was not identified from the 67- and 70-kDa bands but was present in the 94-kDa band (Fig. 3). Thus, it is likely that Gp135 contaminated the sp69 band used for N-terminal sequencing previously (23).
The gene encoding sp31 was identified as g150, and the N-terminal sequence determined earlier indicated that Gp150, in contrast to the prediction made above, was not processed. This was also in line with the absence of a protease recognition sequence in Gp150. The gene encoding sp24 was identified as g199, and the fact that the N-terminal sequence determined earlier (23) started at Glu56 preceded by LTE suggested that it was also proteolytically processed (7). This is also supported by the SDS-PAGE analysis, where Gp199 was identified from 22- to 24-kDa bands (Fig. 2), while the calculated mass of the preprotein was 27.2 kDa (see Table S1 in the supplemental material).
The empty DNA-lacking particles of ϕR1-37 are slightly expanded compared to the DNA-containing virions (Fig. 6E, 7A, and 6B), as seen in bacteriophage T5 (12). As the majority of the empty particles (75%) lack tails, it is difficult to predict whether these particles are true procapsids or merely virions that have lost their tails, and with them, their DNA. There are two gross structural similarities between ϕR1-37 and ϕKZ that currently indicate a possible evolutionary relationship between these two bacteriophages. The capsomer arrangements of ϕR1-37 (Fig. 7C) and ϕKZ are similar. It has been suggested that the major capsid protein of ϕKZ has a fold similar to that of the myovirus HK97 (16). As the capsids of ϕKZ and ϕR1-37 appear to be similar at low resolution, it is possible that the ϕR1-37 major capsid protein Gp135 has the HK97 fold, as do all other tailed viruses that have been studied so far (39), despite very limited sequence identity. Furthermore, the type I and II protrusions on the ϕR1-37 surface are similar to those of bacteriophage ϕKZ (Fig. 7C) (16). Similar decorating proteins sitting at the center of the hexameric capsomers have been identified in the well-studied tailed bacteriophages T4 (hoc) and T5 (Gp10) as well (12, 15). These are the only similarities that indicate a possible evolutionary relationship between these two bacteriophages. In contrast to ϕKZ, we could find no evidence that the genome packaging of ϕR1-37 is organized around an off-axis spindle (56). Hence, as ϕR1-37 and Pseudomonas phage ϕKZ have different DNA nucleotide compositions, different tail types, and only a handful of homologous proteins, we conclude that these phages belong to distinct classes.
Supplementary Material
ACKNOWLEDGMENTS
We thank T. Kunkel for kindly providing E. coli strain KT8052 and Anita Liljegren, Pasi Laurinmäki, and Eevakaisa Vesanen for their technical assistance. We also thank the Biocenter Finland National Cryo-EM Unit and the CSC for providing facilities.
This work was supported by the Academy of Finland (grants 1104361 and 1114075 to M.S.), Academy of Finland Centre of Excellence Programme in Virus Research (2006-2011) grant 1129684 (to S.J.B.), and a grant from the Viikki Doctoral Programme in Molecular Biosciences (to L.J.H.).
Footnotes
Published ahead of print 12 September 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Adrian M, Dubochet J, Lepault J, McDowall AW. 1984. Cryo-electron microscopy of viruses. Nature 308:32–36 [DOI] [PubMed] [Google Scholar]
- 2. Al-Hendy A, Toivanen P, Skurnik M. 1991. Expression cloning of Yersinia enterocolitica O:3 rfb gene cluster in Escherichia coli K12. Microb. Pathog. 10:47–59 [DOI] [PubMed] [Google Scholar]
- 3. Ausubel FM, et al. 1987. Current protocols in molecular biology. John Wiley & Sons, Inc., New York, NY [Google Scholar]
- 4. Bertani G. 2004. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol. 186:595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Besemer J, Lomsadze A, Borodovsky M. 2001. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29:2607–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Black LW. 1995. DNA packaging and cutting by phage terminases: control in phage T4 by a synaptic mechanism. Bioessays 17:1025–1030 [DOI] [PubMed] [Google Scholar]
- 7. Black LW, Showe MK, Steven AC. 1993. Morphogenesis of the T4 head, p 218–258 In Karam JD. (ed), Molecular biology of bacteriophage T4. ASM Press, Washington, DC [Google Scholar]
- 7a. Casjens SR. 2008. Diversity among the tailed-bacteriophages that infect the Enterobacteriaceae. Res. Microbiol. 159:340–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Caspar DL, Klug A. 1962. Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol. 27:1–24 [DOI] [PubMed] [Google Scholar]
- 9. Clark S, Losick R, Pero J. 1974. New RNA polymerase from Bacillus subtilis infected with phage PBS2. Nature 252:21–24 [DOI] [PubMed] [Google Scholar]
- 10. Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27:4636–4641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Effantin G, Boulanger P, Neumann E, Letellier L, Conway JF. 2006. Bacteriophage T5 structure reveals similarities with HK97 and T4 suggesting evolutionary relationships. J. Mol. Biol. 361:993–1002 [DOI] [PubMed] [Google Scholar]
- 13. Ermolaeva MD, Khalak HG, White O, Smith HO, Salzberg SL. 2000. Prediction of transcription terminators in bacterial genomes. J. Mol. Biol. 301:27–33 [DOI] [PubMed] [Google Scholar]
- 14. Fokine A, et al. 2007. Cryo-EM study of the Pseudomonas bacteriophage phiKZ. Structure 15:1099–1104 [DOI] [PubMed] [Google Scholar]
- 15. Fokine A, et al. 2004. Molecular architecture of the prolate head of bacteriophage T4. Proc. Natl. Acad. Sci. U. S. A. 101:6003–6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fokine A, et al. 2005. A three-dimensional cryo-electron microscopy structure of the bacteriophage phiKZ head. J. Mol. Biol. 352:117–124 [DOI] [PubMed] [Google Scholar]
- 17. Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harauz G, van Heel M. 1986. Exact filters for general geometry three dimensional reconstruction. Optik 73:146–156 [Google Scholar]
- 19. Hendrix RW. 2002. Bacteriophages: evolution of the majority. Theor. Popul. Biol. 61:471–480 [DOI] [PubMed] [Google Scholar]
- 20. Heymann JB. 2001. Bsoft: image and molecular processing in electron microscopy. J. Struct. Biol. 133:156–169 [DOI] [PubMed] [Google Scholar]
- 21. Hyytiäinen H, Sjöblom S, Palomäki T, Tuikkala A, Tapio Palva E. 2003. The PmrA-PmrB two-component system responding to acidic pH and iron controls virulence in the plant pathogen Erwinia carotovora ssp. carotovora. Mol. Microbiol. 50:795–807 [DOI] [PubMed] [Google Scholar]
- 22. Katsura I, Hendrix RW. 1984. Length determination in bacteriophage lambda tails. Cell 39:691–698 [DOI] [PubMed] [Google Scholar]
- 23. Kiljunen S, et al. 2005. Yersiniophage ϕR1-37 is a tailed bacteriophage having a 270 kb DNA genome with thymidine replaced by deoxyuridine. Microbiology 151:4093–4102 [DOI] [PubMed] [Google Scholar]
- 24. Kiljunen S, Vilen H, Pajunen M, Savilahti H, Skurnik M. 2005. Nonessential genes of phage ϕYeO3-12 include genes involved in adaptation to growth on Yersinia enterocolitica serotype O:3. J. Bacteriol. 187:1405–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kivioja T, Ravantti J, Verkhovsky A, Ukkonen E, Bamford D. 2000. Local average intensity-based method for identifying spherical particles in electron micrographs. J. Struct. Biol. 131:126–134 [DOI] [PubMed] [Google Scholar]
- 26. Labrie SJ, Samson JE, Moineau S. 2010. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8:317–327 [DOI] [PubMed] [Google Scholar]
- 27. Lavigne R, Sun WD, Volckaert G. 2004. PHIRE, a deterministic approach to reveal regulatory elements in bacteriophage genomes. Bioinformatics 20:629–635 [DOI] [PubMed] [Google Scholar]
- 28. Lindahl T, Ljungquist S, Siegert W, Nyberg B, Sperens B. 1977. DNA N-glycosidases: properties of uracil-DNA glycosidase from Escherichia coli. J. Biol. Chem. 252:3286–3294 [PubMed] [Google Scholar]
- 29. Ludtke SJ, Baldwin PR, Chiu W. 1999. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128:82–97 [DOI] [PubMed] [Google Scholar]
- 30. Mindell JA, Grigorieff N. 2003. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142:334–347 [DOI] [PubMed] [Google Scholar]
- 31. Oliveira L, Alonso JC, Tavares P. 2005. A defined in vitro system for DNA packaging by the bacteriophage SPP1: insights into the headful packaging mechanism. J. Mol. Biol. 353:529–539 [DOI] [PubMed] [Google Scholar]
- 32. Otsuka Y, Yonesaki T. 2012. Dmd of bacteriophage T4 functions as an antitoxin against Escherichia coli LsoA and RnlA toxins. Mol. Microbiol. 83:669–681 [DOI] [PubMed] [Google Scholar]
- 33. Pajunen M, Kiljunen S, Skurnik M. 2000. Bacteriophage ϕYeO3-12, specific for Yersinia enterocolitica serotype O:3, is related to coliphages T3 and T7. J. Bacteriol. 182:5114–5120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pajunen MI, Kiljunen SJ, Söderholm ME, Skurnik M. 2001. Complete genomic sequence of the lytic bacteriophage ϕYeO3-12 of Yersinia enterocolitica serotype O:3. J. Bacteriol. 183:1928–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pedulla ML, et al. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113:171–182 [DOI] [PubMed] [Google Scholar]
- 36. Pettersen EF, et al. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 37. Pinta E, et al. 2009. Identification and role of a 6-deoxy-4-keto-hexosamine in the lipopolysaccharide outer core of Yersinia enterocolitica serotype O:3. Chemistry 15:9747–9754 [DOI] [PubMed] [Google Scholar]
- 38. Pinta E, et al. 2010. Characterization of the six glycosyltransferases involved in the biosynthesis of Yersinia enterocolitica serotype O:3 lipopolysaccharide outer core. J. Biol. Chem. 285:28333–28342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rao VB, Black LW. 2010. Structure and assembly of bacteriophage T4 head. Virol. J. 7:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16:276–277 [DOI] [PubMed] [Google Scholar]
- 41. Rutherford K, et al. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945 [DOI] [PubMed] [Google Scholar]
- 42. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 43. Shevchenko A, et al. 1996. Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc. Natl. Acad. Sci. U. S. A. 93:14440–14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Skurnik M. 1984. Lack of correlation between the presence of plasmids and fimbriae in Yersinia enterocolitica and Yersinia pseudotuberculosis. J. Appl. Bacteriol. 56:355–363 [DOI] [PubMed] [Google Scholar]
- 45. Skurnik M, Pajunen M, Kiljunen S. 2007. Biotechnological challenges of phage therapy. Biotechnol. Lett. 29:995–1003 [DOI] [PubMed] [Google Scholar]
- 46. Skurnik M, Strauch E. 2006. Phage therapy: facts and fiction. Int. J. Med. Microbiol. 296:5–14 [DOI] [PubMed] [Google Scholar]
- 47. Skurnik M, Venho R, Bengoechea JA, Moriyón I. 1999. The lipopolysaccharide outer core of Yersinia enterocolitica serotype O:3 is required for virulence and plays a role in outer membrane integrity. Mol. Microbiol. 31:1443–1462 [DOI] [PubMed] [Google Scholar]
- 48. Skurnik M, Venho R, Toivanen P, Al-Hendy A. 1995. A novel locus of Yersinia enterocolitica serotype O:3 involved in lipopolysaccharide outer core biosynthesis. Mol. Microbiol. 17:575–594 [DOI] [PubMed] [Google Scholar]
- 49. Stewart CR, et al. 2009. The genome of Bacillus subtilis bacteriophage SPO1. J. Mol. Biol. 388:48–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sun S, et al. 2010. Structural studies of the Sputnik virophage. J. Virol. 84:894–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Suzek BE, Ermolaeva MD, Schreiber M, Salzberg SL. 2001. A probabilistic method for identifying start codons in bacterial genomes. Bioinformatics 17:1123–1130 [DOI] [PubMed] [Google Scholar]
- 52. Thomas JA, et al. 2007. Complete genomic sequence and mass spectrometric analysis of highly diverse, atypical Bacillus thuringiensis phage 0305phi8-36. Virology 368:405–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thomas JA, et al. 2008. Characterization of Pseudomonas chlororaphis myovirus 201ϕ2-1 via genomic sequencing, mass spectrometry, and electron microscopy. Virology 376:330–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thomas JA, et al. 2012. Extensive proteolysis of head and inner body proteins by a morphogenetic protease in the giant Pseudomonas aeruginosa phage φKZ. Mol. Microbiol. 84:324–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Z, Mosbaugh DW. 1988. Uracil-DNA glycosylase inhibitor of bacteriophage PBS2: cloning and effects of expression of the inhibitor gene in Escherichia coli. J. Bacteriol. 170:1082–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wu W, Thomas JA, Cheng N, Black LW, Steven AC. 2012. Bubblegrams reveal the inner body of bacteriophage phiKZ. Science 335:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yan X, Sinkovits RS, Baker TS. 2007. AUTO3DEM—an automated and high throughput program for image reconstruction of icosahedral particles. J. Struct. Biol. 157:73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.