

Abstract
Intrinsically disordered proteins (IDP) are a broad class of proteins with relatively flat energy landscapes showing a high level of functional promiscuity, which are frequently regulated through posttranslational covalent modifications. Histone tails, which are the terminal segments of the histone proteins, are prominent IDPs that are implicated in a variety of signaling processes, which control chromatin organization and dynamics. Although a large body of work has been done on elucidating the roles of posttranslational modifications in functional regulation of IDPs, molecular mechanisms behind the observed behaviors are not fully understood. Using extensive atomistic molecular dynamics simulations, we found in this work that H4 tail mono-acetylation at LYS-16, which is a key covalent modification, induces a significant reorganization of the tail’s conformational landscape, inducing partial ordering and enhancing the propensity for alpha-helical segments. Furthermore, our calculations of the potentials of mean force between the H4 tail and a DNA fragment indicate that contrary to the expectations based on simple electrostatic reasoning, the Lys-16 mono-acetylated H4 tail binds to DNA stronger than the unacetylated protein. Based on these results, we propose a molecular mechanism for the way Lys-16 acetylation might lead to experimentally observed disruption of compact chromatin fibers.
Keywords: histone tails, atomistic simulations, polyelectrolytes
Many proteins of higher organisms are found in the “no-man’s-land” between the well-folded globular proteins, which function through their delicately assembled architectures, and the more disordered peptides, which function by either relying on localized sequence signals or on their generic polymer properties. The physical principles behind folding of globular proteins have become well understood based on the concepts of the energy landscape theory, in particular the idea of the folding funnel (1). Namely, the topography of the folding landscape is correlated in such a way that conformational transitions leading to more native-like contacts tend to lower the conformational free energy, while deep traps have been eliminated by evolution so to prevent glassy kinetics of folding. On the other extreme there are functional proteins that are disordered or in a random-coil-like state with the energy landscapes that are relatively flat and sprinkled with some “random” traps (2).
Both theoretical considerations and experiments suggest that most proteins are expected to be found between these two extremes—the perfectly funneled and flat landscapes. For example, almost all globular proteins still have significant residual entropy and may be described by a hierarchical organization of conformational substates within their native basins (3–7). This entropy, however, is rather small compared to large conformational entropy of proteins called IDPs (8, 9). The latter polypeptide chains are found much closer to the extreme scenario of flat landscapes with random traps. However, one of us recently suggested that IDPs that contain truly flat landscapes should be distinguished from IDPs that have significant organization of their energy landscape, for example with one or multiple shallow funnels (2). In particular, our recent work has showed that most histone tails, which mediate compaction of genomic DNA in nuclei of eukaryotic cells, are such IDPs with significant organization of their energy landscapes (10). When discussed in this light, it is not surprising that posttranslational modifications of histone tails may significantly remodel their energy landscapes, allowing for rich functional control over their behaviors (11–14). In this manuscript, we first provide a broad-strokes review of some of the theoretical issues pertaining to understanding IDPs, followed by reporting on our atomistic computer simulations of the H4 tail binding and folding upon interaction with DNA, which we found to be modulated in some unexpected ways upon acetylation of the Lys-16 residue.
It is now widely accepted that IDPs play a major role in biology, in particular for higher organisms (8, 9). While many chemical transformations in the cell do require overall rigidity for the tertiary structure, for example to achieve a precise geometric orientation of the catalytic site in enzymatic catalyses (15), the signaling and transcriptional regulatory processes in general benefit from more flexible conformations of proteins because of the ease of regulation, fast turnover rate, high binding promiscuity, and larger capture radius (9). The notion of functionally viable IDPs has emerged relatively recently and embodies a radically new way of thinking in protein science by challenging the classical structural thinking and having a statistical or ensemble behavior as a determinant for the ultimate functional outcome. Nevertheless, mechanistic explorations of IDPs have largely focused on the possibility of coupled binding and folding, according to which an unstructured protein chain acquires a unique three-dimensional structure upon coming in contact with a specific binding partner (16, 17). Both the kinetic and thermodynamic aspects of coupling of binding and folding have been extensively studied in the last decade or so (16–20). The idea of coupling between binding and folding swiftly gained popularity, partly because of its intuitive appeal to the old “structure-function” dogma, which acknowledges the existence of in vivo disordered conformations but nonetheless requires a prompt disorder-to-order transition for proper functioning.
On the other hand, the processes where functional IDPs fail to fold into unique structures upon binding remained largely ignored until very recently (9, 21, 22). It has turned out that perfectly functional cases of uncoupled binding and folding do exist and are likely to be widespread, especially for higher organisms, where there are unique advantages for having a “permanent disorder” (22). Recently, the complexes where proteins retain various degrees of disorder in the bound state have been identified and brought to the attention of the biophysics community (21–23). These novel phenomena of structural polymorphism (or fuzziness) (22, 23) of the bound IDPs reinforces the idea that structural spectrum of functional proteins is truly continuous (2) and indicates that in certain cases the binding strength and structural rigidity can be compromised for better evolutional adaptability, when the flexibility of chains and ease of regulation is of primary importance for function.
Despite significant recent progress, full understanding of physics and biology of IDPs has not yet been achieved. Many conceptual as well as technical challenges need to be overcome to gain deeper insights into the synergy between intense fluctuations of IDPs and their corresponding functional behaviors. In this work, we investigate the extent of coupling between binding and folding of histone tails, which are IDPs that mediate compaction of DNA in the eukaryotic cells. Our previous work has showed that histone tails are mostly molten globules (see Fig. 1), not random coils, and are characterized by significant organization of their energy landscapes, containing multiple well-defined conformational basins of attraction (10). In this light, one would expect that binding to DNA could substantially alter this landscape (2, 16), restructuring the ensemble of thermodynamically dominant conformations. Furthermore, IDPs, including the histone tails, are heavily regulated by posttranslational modifications. This regulation is very complex, both from biological and physical viewpoints. From the latter perspective, the multifunnel landscape of histone tails may be significantly remodeled upon posttranslational modifications, such as lysine acetylation. The latter modification has been traditionally viewed from the polyelectrolyte perspective as a simple reduction of histone tail’s net positive charge, that diminishes binding to negatively charged DNA. In this work, we show that this view may be too simplistic and that the internal conformational transitions induced by posttranslational modifications may sometimes completely “overrule” direct electrostatic effects.
Fig. 1.

A phase diagram showing denatured, molten globular and native globular regions for proteins, with the histone tails indicated with the filled circles, was estimated based on the results from ref. 10.
From the biological perspective, histones occupy a special place in the protein universe due to their unique architectures, incorporating folded globular core with the flanking intrinsically disordered N- or C-terminal tails (24). The posttranslational covalent modifications of the histone proteins are routes that are widely exploited by eukaryotic cells for regulating variety of important cellular processes (25). While the cores of histones are essential for packaging the genomic DNA inside the cell nucleus, which is further mediated by the terminal histone tails, the highly specific posttranslational modifications of both histone core and tail residues allows the cell to achieve a broad control of the accessibility of the genetic information.
The following modifications of histone tails have been observed among others: methylation, acetylation, ubiquitinations, and sumoylation (26). The different combinations of these are recognized as signals for activating or suppressing particular biochemical events (11, 26). There are two ways that covalent modifications can modulate the timing of the gene expression (26): One is by recruiting specific binding agents to the modified sites, and the other is by direct physical changes in histone tail-nucleosome interactions. Our present work deals with the acetylation of the H4 tail, the mode of action of which in part belongs to the second category (27). Some of the end effects of histone tail acetylations are now well known and documented (26, 28–30). However, there seems to be little if any molecular-level rationale about how the acetylation of the H4 tail induces the observed dramatic changes in the chromatin organization (12, 31). Here we propose a molecular level mechanism by which the acetylations may regulate the functions of the H4 tail, which may have implications for the functional regulation of other IDPs based on similar posttranslational modification strategies.
Using explicit solvent all atom simulations and analyses tools borrowed from the spin glass theory, we show the ways that the H4 tail acetylation can affect the conformational and binding propensities of the histone tails. The H4 tail acetylations have been implicated in the transcriptional activation (29, 32). From in vivo experiments, it was found that acetylation weakens the chromatin packing, allowing the transcriptional factors to access the specific gene sequences (29). In another landmark in vitro experiment with the reconstituted nucleosomal assays, the homogeneous mono-acetylation of H4 tails at the LYS-16 residue leads to the massive disruption of the dense 30 nm chromatin fibers (12). Therefore, acetylation mediates the destabilizing effect of the acetylated (AC) H4 tail through a physical mechanism that weakens chromatin fibers. This is commonly thought to be a simple consequence of electrostatics charge reduction upon acetylation (33). To investigate whether the latter hypothesis is well-justified, we generated the conformational ensemble for LYS-16 AC H4, by performing 3-microseconds-long replica exchange molecular dynamics (REMD) simulations following the same protocol as outlined in the previous work (10). Our prior study of wild-type (WT) histone tails (10) revealed that LYS-16 (see Fig. S1), which resides in the sterically crowded beta turn, might destabilize the transient beta hairpins. In the present study, we find that in the LYS-16 AC H4 tail the beta hairpin content is indeed diminished relative to the WT H4 tail (see Fig. 2 and also Figs. S2 and S3). We employed the distribution of the pairwise structural overlap values, q, to dissect the intrinsic conformational preferences of AC H4 tail. The parameter 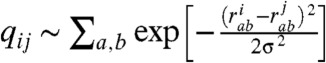 , originally introduced for the study of spin glasses (34), has subsequently found wide applications in the folding studies of the native globular proteins. It quantifies the structural similarity of conformations i and j on a (qmin = 0,qmax = 1) scale using pairwise comparison of all the inter-atomic (Cαs in our case) distances, with a resolution set by the parameter σ (which is 2 Å in the present study). The higher values of q mean higher structural resemblance between two structures, which are being compared. Histogramming q among all pairs of conformational snapshots produces P(q), where the shape of the distribution characterizes the structural heterogeneity of the conformational ensemble. The comparison of the P(q) distributions for the WT and the AC H4 tails immediately reveals that the latter contains a subset of significantly structured (Fig. 2) and also slightly more compact conformations (Fig. 3). The chain compaction is not surprising, because acetylation reduces the positive charge of the chain and, hence, the polyelectrolyte’s self-repulsion. It also adds to the hydrophobic interactions within the chain. However, significant structuring of chain upon acetylation is the truly remarkable feature that is worthy of a greater attention here. What we find, though histograms like Fig. 2, is that the acetylation induces a transition in the conformational ensemble of the H4 tail, changing its IDP class (see ref. 2 for the formal definition) and turning the more or less uniformly random conformational ensemble of unmodified H4 tail into a bimodal distribution with a subset of significantly more structured conformations (see the peak at q ∼ 0.7 - 0.8 in Fig. 2). This partial-ordering phase transition, where the chain entropy is expected to be reduced (but still large) for the AC H4 tail, is a key reason for the wildly divergent binding pathways of the WT and AC forms of H4 tail, which is elaborated below. Interestingly, if only the data for the radii of gyration were compared between the H4 WT and AC tails (see below), one would not expect much change between the chain entropies, indicating that radius of gyration is a too-coarse order parameter to address this issue for polypeptide chains. On the other hand, P(q) plots clearly reveal additional configurational correlations in the underlying energy landscapes, hence demonstrating their utility as a powerful general tool for analyzing IDPs and their posttranslational modifications. In summary, the introduction of the acetyl group leads to slightly more compact globules, which are significantly more structured, with diminished beta hairpin content and enhanced alpha helical flickering elements (see Fig. 2 and Figs. S2 and S3). Some of the qualitative trends of the conformational changes upon acetylation of LYS-16, observed from simulations, is consistent with the prior CD experiments, which reported steady increase in the alpha helical content of the H4 tails as a function of the number of added acetyl groups (35, 36).
, originally introduced for the study of spin glasses (34), has subsequently found wide applications in the folding studies of the native globular proteins. It quantifies the structural similarity of conformations i and j on a (qmin = 0,qmax = 1) scale using pairwise comparison of all the inter-atomic (Cαs in our case) distances, with a resolution set by the parameter σ (which is 2 Å in the present study). The higher values of q mean higher structural resemblance between two structures, which are being compared. Histogramming q among all pairs of conformational snapshots produces P(q), where the shape of the distribution characterizes the structural heterogeneity of the conformational ensemble. The comparison of the P(q) distributions for the WT and the AC H4 tails immediately reveals that the latter contains a subset of significantly structured (Fig. 2) and also slightly more compact conformations (Fig. 3). The chain compaction is not surprising, because acetylation reduces the positive charge of the chain and, hence, the polyelectrolyte’s self-repulsion. It also adds to the hydrophobic interactions within the chain. However, significant structuring of chain upon acetylation is the truly remarkable feature that is worthy of a greater attention here. What we find, though histograms like Fig. 2, is that the acetylation induces a transition in the conformational ensemble of the H4 tail, changing its IDP class (see ref. 2 for the formal definition) and turning the more or less uniformly random conformational ensemble of unmodified H4 tail into a bimodal distribution with a subset of significantly more structured conformations (see the peak at q ∼ 0.7 - 0.8 in Fig. 2). This partial-ordering phase transition, where the chain entropy is expected to be reduced (but still large) for the AC H4 tail, is a key reason for the wildly divergent binding pathways of the WT and AC forms of H4 tail, which is elaborated below. Interestingly, if only the data for the radii of gyration were compared between the H4 WT and AC tails (see below), one would not expect much change between the chain entropies, indicating that radius of gyration is a too-coarse order parameter to address this issue for polypeptide chains. On the other hand, P(q) plots clearly reveal additional configurational correlations in the underlying energy landscapes, hence demonstrating their utility as a powerful general tool for analyzing IDPs and their posttranslational modifications. In summary, the introduction of the acetyl group leads to slightly more compact globules, which are significantly more structured, with diminished beta hairpin content and enhanced alpha helical flickering elements (see Fig. 2 and Figs. S2 and S3). Some of the qualitative trends of the conformational changes upon acetylation of LYS-16, observed from simulations, is consistent with the prior CD experiments, which reported steady increase in the alpha helical content of the H4 tails as a function of the number of added acetyl groups (35, 36).
Fig. 2.

Intrinsic conformational preferences of WT (blue) and LYS-16 AC (blue) H4 tails are shown using their P(q) distributions. The small peak on the right side shows the emergence of more structured and also slightly more compact states in the conformational ensemble of the AC H4 tail. The inset shows how the percentages of structures with the secondary structural elements are affected by the acetylation of LYS-16. Secondary structures were estimated based on the DSSP algorithm, where only structures with more than four residues in either alpha or beta conformations were considered.
Fig. 3.

The dependence of the block-averaged Rg on the pairwise q values. The used block size is δq = 0.025, which corresponds to > 103 configurations in each block. The robustness of results was tested by varying the block sizes in the range of δq = 0.01 - 0.1, which yields quantitatively similar plots to the one shown on the present figure. The confidence intervals are estimated to be in the range of +/-3% with respect to the mean.
Next, we probed the impact of the H4 tail acetylation on the DNA binding propensity. Through model building and an extensive atomistic simulation of the whole nucleosome with tails (see Fig. 4), we identified the most probable binding modes for the H4 tails, which we later used in all of our subsequent simulations. We chose a 20 bp DNA sequence as a mimic of a segment of the nucleosomal DNA and simulated the binding processes for the WT and AC H4 tails by performing two sets of mutually independent umbrella sampling simulations (see Fig. 5) for the unmodified and Lys-16 AC tails (see Materials and Methods for more details). To ascertain that our subsequent umbrella simulations with restrained DNA-protein distances are not biased toward unrealistic conformations, we carried out control simulations of the H4 tails with the DNA for approximately 200 ns. After binding to the DNA segment, the WT H4 tail undergoes less structural fluctuations, which is expected for a highly charged polymer, although the bound H4 tail is still rather flexible and retains significant fraction of disorder, which is also seen in our umbrella sampling simulations. The residues that are in contact with the DNA are all part of the transiently flickering beta hairpin, which also includes the LYS-16. In the nucleosome, the C terminals of H4 tails are structurally in close proximity to the DNA ramp of the nucleosome (see Fig. 4). Hence, it is a natural choice to use the center of mass distance between the target DNA base pair and the C-terminal segment of the H4 tail centered around Lys-16. From the umbrella sampling simulations we extracted the potentials of mean force (PMFs) as a function of the above-mentioned DNA-protein distance. To account for the finite size effects, we calibrated the right-most points of all PMF curves using continuum electrostatics estimates, as outlined in Materials and Methods.
Fig. 4.

A snapshot from the explicit solvent nucleosome simulation, showing a representative conformation of the H4 tail as an integral part of the nucleosome. The LYS-16 is colored red in both structures.The inset shows the high resolution view of the typical binding mode of the H4 tail, with the LYS-16 bound inside the nucleosomal DNA’s groove.
Fig. 5.

Comparison of the PMF profiles of the DNA binding for the WT (blue) and the LYS-16 AC (red) H4 histone tails are shown, computed from two independent simulations. Simulations 1 are indicated with circles, and Simulations 2 are indicated with diamonds. The vertical offsets of the rightmost points of all “curves” are calculated using Debye–Huckel estimations of 50 snapshots from the corresponding farthermost umbrella windows using the full set of partial atomic charges of the DNA chain and the protein.
From the PMF plots of WT H4-DNA binding (see the blue symbols in Fig. 5), we find approximately 2-kBT stabilization of the DNA bound configurations at the PMF minimum compared with the completely dissociated conformations. This indicates that thermal fluctuations can frequently break the DNA-tail contacts, making the complex highly dynamic. This would allow the WT H4 tail to be flexible at reasonably fast time scales and easily explore extended chain conformations needed for reaching linker DNA and other nucleosomes. On the other hand, the free energy stabilization of the DNA bound states of the AC H4, at the PMF minimum, is more significant, being in the range between 5 and 6 kBT (see the red symbols in Fig. 5). The latter is large enough to keep the AC H4 tail frequently “glued” on the surface of the DNA. Interestingly, in the WT form, the LYS-16 is intercalated in the DNA groove, but the AC LYS-16, due to the increased side-chain bulkiness and overall neutral charge prefers to sample states that are outside of DNA grooves (see the inset in Fig. 6). However, this is compensated by collapsing the chain and bringing other lysins closer to the DNA’s surface compared to the WT form, hence, the overall electrostatic attraction is basically unchanged, as discussed below. In terms of compaction, the DNA segment amplifies the initially small difference in the sizes of the tails’ conformational ensembles, driving the conformational distributions of the WT and AC H4 tails in divergent directions (see Fig. 6).
Fig. 6.

The extent of poly-electrolytic collapse of the H4 tails near the DNA chain are indicated by the distributions of the radius of gyration for the bound states of the WT and AC H4 tails. The inset shows representative snapshots from the DNA-bound configurations of the WT (left) and AC (right) H4 tails, corresponding to the minima in the respective PMF plots on the Fig. 5. All lysins are shown as vdW models, with the LYS-16 highlighted by element-based coloring.
These observations, in turn, explain the considerable difference in the PMF depths between the WT and AC forms: Despite the fact that acetyl group is reducing the overall positive charge on the H4 tail, the electrostatic stabilizations are nevertheless similar, while some additional nonpolar interactions favor the AC form. Thus, the acetylation of the LYS-16 affects binding to DNA indirectly by changing the conformational ensemble of WT through mediating interactions with the remaining residues of the chain, which ultimately leads to a different binding pathway. In fact, one can see from the structures on Fig. 6, that acetylation has a dramatic impact on the DNA binding mode of the H4 tail. Our present findings are qualitatively consistent with the experiments on acetylation of the disordered C-terminal segment of the p53 protein, where a significant increase in its DNA binding affinity was observed (13) (see also refs. 37 and 38).
To gain further insights into thermodynamic consequences of the H4 tail mono-acetylation, we consider next forces that drive protein–DNA binding. The favorable binding free energy can generally be broken down into three distinct contributions (39): electrostatic interactions, hydrogen bonding, and hydrophobic forces. The electrostatic component alone tends to create nonspecific DNA–protein complexes (40), which are diffuse associations (41), utilized for instance by facilitating the rapid scanning of the genome for the target sites (42). On the other hand, the specific association with the DNA is obtained by forming extensive network of hydrogen bonds, which is further assisted by the hydrophobically driven clustering of nonpolar residues near the binding surface. Furthermore, the electrostatic calculations (43) and thermodynamic analysis of numerous DNA–protein complexes (40) shows that the major driving force for binding often originates not from nonspecific polyelectrolyte interactions but from hydrophobic forces and hydrogen bonding (17). To support our reasoning with quantitative data, we performed detailed structural analysis of the H4–DNA contacts in the bound complex of the WT and AC H4 tails (see Table 1). Counting the number of contacts between various atoms that are classified as “neutral,’, “acidic,” and “basic,” we see that the overall number of atomic contacts are comparable, with the AC H4 tail forming an approximately similar number of overall interfacial contacts, which is also consistent when comparing the surface accessible areas of the two forms (see Table 1). However, the number of contacts of mainly hydrophobic origin (indicated as neutral-neutral in SI Text and Table 1) is significantly enhanced upon acetylation. Another interesting observation is made when comparing the electrostatic components of the binding free energy of the AC and WT H4 tails, by employing simple Debye–Huckel calculations of bound state snapshots, which take into account partial charges on all atoms. While the electrostatic free energy unsurprisingly favors the binding of the more positively charged WT H4 tail (see Table 1), the net gain itself is marginal, on the order of 0.1 kBT. Thus, the difference in binding affinities of WT and AC H4 tails toward DNA originates largely from nonelectrostatic contributions. More specifically, the enhanced binding affinity of the AC H4 tail can be attributed to its collapsed and more hydrophobic nature that makes more contacts with the surface of the DNA. Another likely contribution that would favor the higher affinity for the AC H4 tail is the diminution of the entropy loss upon binding to DNA, because some chain entropy is already lost upon acetylation of the free WT tail, as discussed above.
Table 1.
Summarization of structural analyses of DNA bound state for WT and AC forms of the H4 tails. Further details are elaborated in SI Text
| Contact type |
WT |
AC |
| No. of total contacts | 150 | 150* |
| No. of atoms in contact | 110 | 110 |
| No. of neutral–neutral contacts | 38 | 45 |
| No. of acid–acid contacts | 11 | 11 |
| No. of acid–base contacts | 9.0 | 8.0 |
| No. of acid–neutral contacts | 23 | 23 |
| No. of base–neutral contacts | 41 | 39 |
| No. of base–neutral contacts | 29 | 28 |
| ΔSAS (Å2) | 1,000 | 1,000 |
| Debye–Huckel free energy (kBT) | -9.3 | -9.2 |
*The global cutoff was set to 3.5 Å.
The stronger interaction of the AC H4 with the DNA is expected to have a noticeable structural impact on the local internucleosomal configurations, which would then propagate down further by ultimately changing the chromatin architecture on a much larger scale (12). The latter assertion is supported by the FRET measurements of DNA labeled nucleosomes (44, 45), which reveal distinct structural changes on the mononucleosomal level that are driven solely by H4 acetylation. Furthermore, these FRET experiments indicate that H4 acetylation results in tightening of the free DNA ends coming out of the nucleosome (44, 45). The latter result may be potentially consistent with the idea of stronger binding of AC H4 tail to its own nucleosomal DNA, which is the main suggestion of the current work. The unique role of the H4 histone tail in maintaining the chromatin architectural organization has been established by sedimentation experiments (30), which showed that, from all the tails, only the absence of H4, or equivalently its acetylation, can cause an irreversible disruption of the chromatin fiber. One commonly accepted explanation states (14) that all of the WT core histone tail domains have well-pronounced preference to bind the linker DNA—the short segment that joins two adjacent nucleosomes (44, 46). Coarse-grained molecular simulations also seem to stress the importance of the so-called “tail bridging” effect in increasing the attraction between the nucleosome cores (47). The acetylation of LYS-16 on H4 tail might therefore oppose the tail bridging effect, leading to weakened nucleosome–nucleosome interactions.
In the context of the chromatin folding our results suggest a simple molecular level mechanism that might explain how the mono-acetylation of Lys-16 of the H4 tail can cause the local unraveling of the chromatin fibers (see Fig. 7). We speculate that Lys-16 acetylation results in partial ordering and the subsequently tighter binding of the H4 tail to its own nucleosomal DNA, which, in turn, would significantly suppress the access of H4 tail’s N-terminal segment to the regions outside of the nucleosome and, hence, disallow the H4 tails to mediate stabilizing interactions with the neighboring linker DNA and nucleosomes. As a consequence, local bulges may form within the dense poly-nucleosomal arrays. Hence, we hypothesize that the WT H4 tail can freely explore its conformational space, while acetylation is an autoinhibitory molecular switch that leads to H4 tail’s sequestration by its own nucleosome. Our mechanism may provide a molecular explanation for the experiments conducted by Widom and coworkers (48, 49), who have observed similar rates for transcriptional elongation by either acetylation or complete removal of the H4 histone tails. From the broader perspective of the general mechanisms for regulating IDPs, our work indicates that posttranslational modifications significantly remodel IDPs’ energy landscapes and may induce somewhat complicated disorder-to-less-disorder conformational phase transitions. This, in turn, primes the IDP for stronger interactions with specific binding targets, allowing for great functional plasticity and complexity of regulation. Given the combinatorially large number of posttranslational modifications that are used in biology, including for modulating the function of histone tails, a great deal of future research is needed to elucidate the physical principles used to achieve specificity in binding and folding pathways of various IDPs. In particular, it will be interesting to investigate how binding specificity depends on the degree and combinatorics of multiply AC histone tails, including the H4 tail. For instance, we expect a nonmonotonous dependence on the degree of the H4 tail acetylation, where binding to DNA is first enhanced for less-acetylated forms but is subsequently diminished upon further hyper-acetylation because of electrostatic consequences of acetylation eventually becoming more dominant. Future experiments and computer simulations should address these interesting possibilities.
Fig. 7.

We propose a speculative molecular level explanation of the H4 tail acetylation induced chromatin fiber decompaction. A schematic drawing of the polynucleosomal array is shown, with nucleosomes represented as red cylinder and H4 histone tails as gray lines. Only the N terminals of the H4 tails are shown for clarity purposes. The yellow sphere at the end of the H4 tail indicates the N-terminal region. We hypothesize that upon acetylation the H4 tails undergo collapse and, subsequently, their binding to adjacent nucleosomes is compromised, which results in weakening of the effective inter nucleosomal contacts.
Materials and Methods
All the simulations reported in present contribution were carried out with the aid of SANDER and PMEMD molecular dynamics engines, which are the part of AMBER11 code. The amino acid sequence for the H4 tail was chosen based on the physical definition of histone tails (see Fig. S1) first given in the previous work (10). The nucleotide sequence for the DNA was chosen in semirandom way as a mimic of a typical fragment of the nucleosomal DNA sequence (ss: TGATTCTCCAGGGCGGCCAG). The MM force field used in our study consists of the following force fields: ff99SB* for proteins (50), parmbsc0 for nucleic acids (51), TIP3P for water molecules (52), and Joung–Cheatham model for ions (53). The WT and Lys-16 AC H4 tails were first equilibrated, and then random snapshots were chosen as starting points for subsequent umbrella sampling simulations. Electronic structure calculations with the B3LYP DFT functional (54) and ccPVTZ basis set (55) were used for obtaining the partial charges of the AC lysine, which were incorporated in the standard AMBER ff99SB* protein force field. The DNA+protein system preparation involved the following steps. At first, each tail was placed in a box with a 20 bp DNA at a 25-Å center of mass separation (see Fig. S4 for more details), where the center of mass for the DNA was defined with respect to two base pairs in the middle of the DNA and for H4 tail it with respect to five residues centered at LYS-16. Afterward the whole system was solvated with TIP3P waters and sufficient amount of ions was added to reproduce 0.14 M NaCl concentration. Step-by-step energy minimization runs were carried out subsequently with the restrained and relaxed protein+DNA configurations. Next, the system was heated (Langevin NVT, approximately 500 ps) and equilibrated (Nose–Hoover NPT, approximately 1 ns), keeping the center of mass distance fixed.
Umbrella centers and spring constants were chosen via several trial simulations to ensure the significant overlap of parameter values between the adjacent windows. The two angles formed by the terminal bases of the DNA, the center of mass of the DNA, and the center of mass of the histone tails were restrained (with spring constants of approximately 90 kcal/mol) to facilitate the direct binding to the same specific spot on the DNA for both histone tails. In total, approximately 14 ns long simulations were carried out in each window and only the last 10 ns was used for the thermodynamic analyses. The WHAM (56) was employed for analyzing the umbrella sampling simulations. To check for the sufficient level of equilibration and to eliminate the possibility of bias due to specific initial condition, we have carried out additional and independent umbrella sampling runs (indicated as sim #2 in Fig. 5), with the same spring constants and restraining positions but using as starting configuration the H4 tail-DNA bound state. The quantitative (WT H4-DNA) and semiquantitative (AC H4-DNA) agreement between the corresponding PMFs are a strong indication of sufficient sampling along all the remaining directions orthogonal to the reaction coordinate (see Fig. 5). Prior to the umbrella sampling simulations, the bound configurations were equilibrated by running 2–3 nanoseconds long REMDs simulations. Afterwards, the most dissimilar (with respect to the starting structures) of the conformations were chosen for the subsequent PMF calculations. These are reported as simulation number: 1 and 2 on the Fig. 5 of the main text.
Both for postsimulation analyses and PMF curve calibrations, the electrostatic components of the free energy were separately estimated via the Debye–Huckel approximation for the spheres with finite radii, 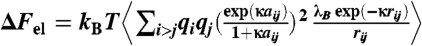 where the qis are standard AMBER charges (expressed in units of the elementary charge), rij is the interatomic distance, aij is the average radius of two interacting atoms, λB is the Bjerrum length (approximately 6.3 Å), κ-1 is the Debye length of the solution (approximately 7.1 Å), and the angular brackets indicate ensemble averaging. These estimates for the WT and AC form were used to calibrate the overall vertical offsets of each PMF curve by matching the rightmost points corresponding to the farthest distances between DNA and the tail (between approximately 0.5 kBT and 1 kBT). We have also estimated the electrostatic component of the interaction energy with periodic images using the DH approximation. In both cases the correction is significantly less than approximately 1 kBT and thus can be safely neglected.
where the qis are standard AMBER charges (expressed in units of the elementary charge), rij is the interatomic distance, aij is the average radius of two interacting atoms, λB is the Bjerrum length (approximately 6.3 Å), κ-1 is the Debye length of the solution (approximately 7.1 Å), and the angular brackets indicate ensemble averaging. These estimates for the WT and AC form were used to calibrate the overall vertical offsets of each PMF curve by matching the rightmost points corresponding to the farthest distances between DNA and the tail (between approximately 0.5 kBT and 1 kBT). We have also estimated the electrostatic component of the interaction energy with periodic images using the DH approximation. In both cases the correction is significantly less than approximately 1 kBT and thus can be safely neglected.
The simulations of the whole nucleosome, the snapshot of which is shown on Fig. 4, has been run for approximately 120 ns using explicit solvent and physiological ionic conditions. The starting configuration for the simulation was the X-ray structure from Richmond et. al (24) (Protein Data Base ID code 1KX5), with the complete structures of all histone tails.
Supplementary Material
ACKNOWLEDGMENTS.
All simulations were carried out with the help of supercomputers Deepthought’ at the University of Maryland and Topsail at the University of North Carolina. The help of Mr. Sean Doyle with the nucleosome simulations is gratefully acknowledged. This work was in part supported by the Beckman Young Investigator Award and Camille Dreyfus Teacher-Scholar Award.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1201805109/-/DCSupplemental.
References
- 1.Onuchic JN, Wolynes PG. Theory of protein folding. Curr Opin Struct Biol. 2004;14:70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Papoian GA. Proteins with weakly funneled energy landscapes challenge the classical structure-function paradigm. Proc Natl Acad Sci USA. 2008;105:14237–14238. doi: 10.1073/pnas.0807977105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCammon J, Gelin BR, Karplus M. Dynamics of folded proteins. Nature. 1977;267:585–590. doi: 10.1038/267585a0. [DOI] [PubMed] [Google Scholar]
- 4.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 5.Honeycutt JD, Thirumalai D. The nature of folded states of globular proteins. Biopolymers. 1992;32:695–709. doi: 10.1002/bip.360320610. [DOI] [PubMed] [Google Scholar]
- 6.Zhuravlev PI, Materese CK, Papoian GA. Deconstructing the native state: Energy landscapes, function, and dynamics of globular proteins. J Phys Chem B. 2009;113:8800–8812. doi: 10.1021/jp810659u. [DOI] [PubMed] [Google Scholar]
- 7.Chung HS, McHale K, Louis JM, Eaton WA. Single-molecule fluorescence experiments determine protein folding transition path times. Science. 2012;335:981–984. doi: 10.1126/science.1215768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dyson HJ. Expanding the proteome: Disordered and alternatively folded proteins. Q Rev Biophys. 2011;44:467–518. doi: 10.1017/S0033583511000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Potoyan DA, Papoian GA. Energy landscape analyses of disordered histone tails reveal special organization of their conformational dynamics. J Am Chem Soc. 2011;133:7405–7415. doi: 10.1021/ja1111964. [DOI] [PubMed] [Google Scholar]
- 11.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 12.Shogren-Knaak M, et al. Histone h4-k16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 13.Luo J, et al. Acetylation of p53 augments its site-specific dna binding both in vitro and in vivo. Proc Natl Acad Sci USA. 2004;101:2259–2264. doi: 10.1073/pnas.0308762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Angelov D, Vitolo J, Mutskov V, Dimitrov S. Preferential interaction of the core histone tail domains with linker DNA. Proc Natl Acad Sci USA. 2001;98:6599–6604. doi: 10.1073/pnas.121171498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: W. H. Freeman; 1998. [Google Scholar]
- 16.Papoian GA, Wolynes PG. The physics and bioinformatics of binding and folding-an energy landscape perspective. Biopolymers. 2003;68:333–349. doi: 10.1002/bip.10286. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Verkhivker GM. Energy landscape theory, funnels, specificity, and optimal criterion of biomolecular binding. Phys Rev Lett. 2003;90:188101–188105. doi: 10.1103/PhysRevLett.90.188101. [DOI] [PubMed] [Google Scholar]
- 18.Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc Natl Acad Sci USA. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levy Y, Papoian GA, Onuchic JN, Wolynes PG. Energy landscape analysis of protein dimers. Isr J Chem. 2004;44:281–297. [Google Scholar]
- 20.Levy Y, Onuchic JN, Wolynes PG. Fly-casting in protein-DNA binding: Frustration between protein folding and electrostatics facilitates target recognition. J Am Chem Soc. 2007;129:738–739. doi: 10.1021/ja065531n. [DOI] [PubMed] [Google Scholar]
- 21.Sigalov AB. Uncoupled binding and folding of immune signaling-related intrinsically disordered proteins. Prog Biophys Mol Biol. 2011;106:525–536. doi: 10.1016/j.pbiomolbio.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 22.Fuxreiter M. Fuzziness: Linking regulation to protein dynamics. Mol Biosyst. 2012;8:168–177. doi: 10.1039/c1mb05234a. [DOI] [PubMed] [Google Scholar]
- 23.Tompa P, Fuxreiter M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 24.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 A resolution. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 25.Alberts B, et al. Molecular Biology of the Cell. 5th Ed. New York: Garland Science; 2007. [Google Scholar]
- 26.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Woodcock CL, Ghosh RP. Chromatin higher-order structure and dynamics. Cold Spring Harb Perspect Biol. 2010;2:1–25. doi: 10.1101/cshperspect.a000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bannister A, Kozarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 30.Dorigo B, Schalch T, Bystricky K, Richmond TJ. Chromatin fiber folding: Requirement for the histone H4 N-terminal tail. J Mol Biol. 2003;327:85–96. doi: 10.1016/s0022-2836(03)00025-1. [DOI] [PubMed] [Google Scholar]
- 31.Allahverdi A, et al. The effects of histone h4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res. 2011;39:1680–1691. doi: 10.1093/nar/gkq900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee D, Hayes J, Pruss D, Wolffe AP. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72:73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- 33.Hansen JC. Conformational dynamics of the chromatin fiber in solution: Determinants, mechanisms, and functions. Annu Rev Biophys Biomol Struct. 2002;31:361–392. doi: 10.1146/annurev.biophys.31.101101.140858. [DOI] [PubMed] [Google Scholar]
- 34.Parisi G. Order parameter for spin-glasses. Phys Rev Lett. 1983;50:1946–1948. [Google Scholar]
- 35.Wang X, Moore SC, Laszckzak M, Ausio J. Acetylation increases the alpha-helical content of the histone tails of the nucleosome. J Biol Chem. 2000;275:35013–35020. doi: 10.1074/jbc.M004998200. [DOI] [PubMed] [Google Scholar]
- 36.Baneres JL, Martin A, Parello J. The n tails of histones h3 and h4 adopt a highly structured conformation in the nucleosome. J Mol Biol. 1997;273:503–508. doi: 10.1006/jmbi.1997.1297. [DOI] [PubMed] [Google Scholar]
- 37.Arbely E, et al. Acetylation of lysine 120 of p53 endows DNA-binding specificity at effective physiological salt concentration. Proc Natl Acad Sci USA. 2011;108:8251–8256. doi: 10.1073/pnas.1105028108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tafvizi A, Huang F, Fersht AR, Mirny LA, van Oijen AM. A single-molecule characterization of p53 search on DNA. Proc Natl Acad Sci USA. 2011;108:563–568. doi: 10.1073/pnas.1016020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cherstvy AG. Positively charged residues in DNA-binding domains of structural proteins follow sequence-specific positions of DNA phosphate groups. J Phys Chem B. 2009;113:4242–4247. doi: 10.1021/jp810009s. [DOI] [PubMed] [Google Scholar]
- 40.Spolar RS, Record MT. Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- 41.Sánchez IE, Ferreiro DU, Dellarole M, de Prat-Gay G. Experimental snapshots of a protein-DNA binding landscape. Proc Natl Acad Sci USA. 2010;107:7751–7756. doi: 10.1073/pnas.0911734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halford SE, Marko JF. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004;32:3040–3052. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Misra VK, Honig B. On the magnitude of the electrostatic contribution to ligand-DNA interactions. Proc Natl Acad Sci USA. 1995;92:4691–4695. doi: 10.1073/pnas.92.10.4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tóth K, Brun N, Langowski J. Chromatin compaction at the mononucleosome level. Biochemistry. 2006;45:1591–1598. doi: 10.1021/bi052110u. [DOI] [PubMed] [Google Scholar]
- 45.Gansen A, Tóth K, Schwarz N, Langowski J. Structural variability of nucleosomes detected by single-pair förster resonance energy transfer: Histone acetylation, sequence variation, and salt effects. J Phys Chem B. 2009;113:2604–2613. doi: 10.1021/jp7114737. [DOI] [PubMed] [Google Scholar]
- 46.Materese CK, Savelyev A, Papoian GA. Counterion atmosphere and hydration patterns near a nucleosome core particle. J Am Chem Soc. 2009;131:15005–15013. doi: 10.1021/ja905376q. [DOI] [PubMed] [Google Scholar]
- 47.Mühlbacher F, Holm C, Schiessel H. Controlled DNA compaction within chromatin: The tail-bridging effect. Europhys Lett. 2006;73:135–141. [Google Scholar]
- 48.Protacio RU, Li G, Lowary PT, Widom J. Effects of histone tail domains on the rate of transcriptional elongation through a nucleosome. Mol Cell Biol. 2000;20:8866–8878. doi: 10.1128/mcb.20.23.8866-8878.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anderson JD, Lowary PT, Widom J. Effects of histone acetylation on the equilibrium accessibility of nucleosomal DNA target sites. J Mol Biol. 2001;307:977–985. doi: 10.1006/jmbi.2001.4528. [DOI] [PubMed] [Google Scholar]
- 50.Best RB, Hummer G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J Phys Chem B. 2009;113:9004–9015. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pérez A, et al. Refinement of the amber force field for nucleic acids: Improving the description of [alpha]/[gamma] conformers. Biophys J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyamoto S, Kollman P. Settle: An analytical version of the shake and rattle algorithm for rigid water models. J Comput Chem. 1992;13:952–962. [Google Scholar]
- 53.Joung I, Cheatham T., III Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B. 2008;112:9020–9041. doi: 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Becke A. A new mixing of hartree-fock and local density-functional theories. J Chem Phys. 1993;98:1372. [Google Scholar]
- 55.Dunning T., Jr Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J Chem Phys. 1989;90:1007–1023. [Google Scholar]
- 56.Ferrenberg AM, Swendsen RH. Optimized monte carlo data analysis. Phys Rev Lett. 1989;63:1195–1198. doi: 10.1103/PhysRevLett.63.1195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.