

Abstract
Although superoxide dismutase 1 (SOD1) stands out as a relatively soluble protein in vitro, it can be made to fibrillate by mechanical agitation. The mechanism of this fibrillation process is yet poorly understood, but attains considerable interest due to SOD1’s involvement in the neurodegenerative disease amyotrophic lateral sclerosis (ALS). In this study, we map out the apoSOD1 fibrillation process from how it competes with the global folding events at increasing concentrations of urea: We determine how the fibrillation lag time (τlag) and maximum growth rate (νmax) depend on gradual titration of the folding equilibrium, from the native to the unfolded state. The results show that the agitation-induced fibrillation of apoSOD1 uses globally unfolded precursors and relies on fragmentation-assisted growth. Mutational screening and fibrillation m-values (∂ log τlag/∂[urea] and ∂ log νmax/∂[urea]) indicate moreover that the fibrillation pathway proceeds via a diffusely bound transient complex that responds to the global physiochemical properties of the SOD1 sequence. Fibrillation of apoSOD1, as it bifurcates from the denatured ensemble, seems thus mechanistically analogous to that of disordered peptides, save the competing folding transition to the native state. Finally, we examine by comparison with in vivo data to what extent this mode of fibrillation, originating from selective amplification of mechanically brittle aggregates by sample agitation, captures the mechanism of pathological SOD1 aggregation in ALS.
Keywords: protein aggregation, protein binding, protein folding
Intracellular aggregation of the homodimeric enzyme superoxide dismutase (SOD1) is a key pathological hallmark of amyotrophic lateral sclerosis (ALS). Despite this coupling with protein-aggregation disease, SOD1 stands out as a relatively soluble protein in vitro: Mutations that are destabilized to the extent that they fail to fold can be left for weeks in physiological buffer without aggregating. Studies of SOD1 aggregation need then to involve certain stimuli that prompts the process to occur (1). One such stimulus is mechanical agitation. Like many other proteins, SOD1 can be driven to form amyloid-like fibrils by shaking or stirring the sample (2–4). Although these experiments may at first seem artificial, they have come to play a key role in our understanding of the aggregation mechanism: Protein aggregation is a high-dimensional process and, as such, its predicted outcome relies on knowledge about all competing pathways (5). In the case of SOD1, this high dimensionality is underlined by the observation that the structures of the fibrillar aggregates are polymorphic and vary with mutation (6). Competing aggregation pathways are also indicated at macroscopic level where the morphology of the SOD1 aggregates can be changed from filamentous to branched protofibrillar or amorphous by replacing the mechanical agitation with incubation in organic solvent (7), addition of lipids (8), or allowing oxidative cross-linking of the protein’s solvent accessible cysteines (9). Non-amyloid aggregates can also be promoted without agitation in physiological buffer by simply increasing the protein concentration to 95–950 μM and extending the incubation times (10). More detailed insight into the SOD1 aggregation mechanism has so far been precluded by difficulty to identify at structural level the monomeric precursor material: Native SOD1 is a metallated dimer with a complex maturation and degradation pathway, including a plethora of intermediate states (11, 12). As observed for β2-microglobulin (13) and TTR (14); however, it is often assumed that the precursor for SOD1 aggregation is a ruptured apo species, which either exposes sticky material through local unfolding of the central β-barrel (15, 16) cross-links oxidatively via C6/C111 (9), polymerizes via the long active site loops (17), or promotes aggregation by a mixture of mechanisms (10). There is also alternative evidence, based on the congruent effects of ALS-associated mutations on protein stability, which implicate the globally unfolded state as the disease precursor in vivo (18). To distinguish between these possibilities, we examine here the agitation-induced aggregation of apoSOD1 with the toolbox from protein folding: We use linear free-energy relations and urea titration to determine how the fibrillation lag time (τlag) and maximum growth rate (νmax) correlates with the occupancy of states in the unimolecular folding equilibrium. Occupancies of states are determined from chevron kinetics and NMR, and the experiments were done at pH 6.3 and 37 °C to match as far as possible physiological conditions. The results show that the fibrillation process of SOD1 relies on globally unfolded monomers (D), and linear correlations between log τlag, log νmax and log [D] indicate that the fibril growth is assisted by mechanical fragmentation and holds no information about primary nucleation events. Systematic E-scanning of the SOD1 primary sequence suggests moreover that the globally unfolded monomers associate first diffusely to the fibrillar ends, in analogy with the general two-step association pathway for protein–protein binding.
Results and Discussion
Proteins.
Three commonly implicated driving forces for SOD1 aggregation are oxidative cross-linking by the protein’s cysteines (9), assembly via the structurally flexible active site loops (19, 20), and exposure of sticky sequence regions by rupture of the protein’s central β-barrel (6, 21). To determine the effects of these factors on agitated fibrillation in vitro, we examined three different SOD1 variants (Fig. 1): The apoSOD1 monomer containing the native disulphide bond C57–C146 but with the free cysteines C6 and C111 substituted for A to assure folding reversibility (apoSOD1C57–C146) (22), the reduced apoSOD1 monomer lacking the disulphide bond (apoSOD1SH SH), and a variant in which the active site loops IV and VII have been removed by protein engineering (apoSOD1ΔIV ΔVII) (23). Upon incubation at 25 μM under agitation by a magnetic stirrer at pH 6.3 and 37 °C, all three proteins aggregate into fibrils of indistinguishable morphology (Fig. 1). Accordingly, we conclude that the C57–C146 disulphide bond and loops IV/VII are not required for fibrillation, and that the fibrillar core is composed from segments of the β-barrel, which is the only common denominator of the three structures. The results are in good agreement with mass-spectroscopy data from Furukawa and co-workers, which identify a fibrillar core of β-strand segments, coated by loops IV and VII protruding loosely into the solvent (6).
Fig. 1.

Crystal structures and fibril morphologies of the apoSOD1 variants examined. The apoSOD1 monomer with intact C57–C146 crosslink (apoSOD1C57–C146), the corresponding protein in TCEP with reduced C57 and C146 moieties (apoSOD1SH SH), and the variant with loops IV (green) and VII (blue) removed by protein engineering (apoSOD1ΔIV ΔVII). Structures constructed from PDB entry 2XJK. The EM micrographs of the respective protein fibrils, produced by agitation in pure pH 6.3 buffer at 37 °C. Scale bars are 200 nm.
Tuning of the Folding Equilibrium.
The question is then to what extent the SOD1 barrel needs to rupture—or unfold—to become fibrillation competent. To find out, we gradually destabilized the SOD1 structure by urea titration while measuring the response on the aggregation kinetics. Determination of the concentrations of native (N) and unfolded species (D) at each urea concentration was by standard two-state analysis (12, 21, 22, 24, 25).
![]() Scheme 1 where D and N interconvert via a single transition state (‡), kf, and ku are the refolding- and unfolding-rate constants, respectively, and kdown is the kinetic pre-factor (26). The equilibrium constant for folding is here:
Scheme 1 where D and N interconvert via a single transition state (‡), kf, and ku are the refolding- and unfolding-rate constants, respectively, and kdown is the kinetic pre-factor (26). The equilibrium constant for folding is here:
| [1] |
and
 |
[2] |
where  ,
,  , and
, and  are the values at 0 M urea, and mD–N = mu - mf = ∂ log KD–N/∂[urea] = ∂ log ku/∂[urea]-∂ log kf/∂[urea] define the urea dependence. Consistent with the linear relations in Eqs. 1 and 2, apoSOD1C57–C146, apoSOD1SH SH and apoSOD1ΔIV ΔVII yield all characteristically V-shaped chevron plots under the conditions of the fibrillation assays at 37 °C (Fig. 2 and Table 1). We investigate here how these linear changes of log KD-N correlate with changes of the fibrillation kinetics; i.e., we vary the occupancy of putative fibrillation-prone material by urea titration (Fig. 2 and Table 1). A benefit of this approach is that it allows larger variation of protein concentration than conventional aggregation assays. With a fixed concentration of 25 μM apoSOD1ΔIV ΔVII, which is the most stable construct in this study, urea titration permits screening of [N] = 25 μM to 100 pM and [D] = 35 nM to 25 μM. The less-stable variants apoSOD1C57–C146 and apoSOD1SH SH yield correspondingly spans of [D] = 0.4 μM to 25 μM and [D] = 8.5 μM to 25 μM, respectively. Thus, to optimize the dynamic range of the fibrillation assays, we choose apoSOD1ΔIV ΔVII for further experiments as described next.
are the values at 0 M urea, and mD–N = mu - mf = ∂ log KD–N/∂[urea] = ∂ log ku/∂[urea]-∂ log kf/∂[urea] define the urea dependence. Consistent with the linear relations in Eqs. 1 and 2, apoSOD1C57–C146, apoSOD1SH SH and apoSOD1ΔIV ΔVII yield all characteristically V-shaped chevron plots under the conditions of the fibrillation assays at 37 °C (Fig. 2 and Table 1). We investigate here how these linear changes of log KD-N correlate with changes of the fibrillation kinetics; i.e., we vary the occupancy of putative fibrillation-prone material by urea titration (Fig. 2 and Table 1). A benefit of this approach is that it allows larger variation of protein concentration than conventional aggregation assays. With a fixed concentration of 25 μM apoSOD1ΔIV ΔVII, which is the most stable construct in this study, urea titration permits screening of [N] = 25 μM to 100 pM and [D] = 35 nM to 25 μM. The less-stable variants apoSOD1C57–C146 and apoSOD1SH SH yield correspondingly spans of [D] = 0.4 μM to 25 μM and [D] = 8.5 μM to 25 μM, respectively. Thus, to optimize the dynamic range of the fibrillation assays, we choose apoSOD1ΔIV ΔVII for further experiments as described next.
Fig. 2.

Folding and fibrillation data. (A) Chevron plots of apoSOD1C57–C146, apoSOD1SH SH, and apoSOD1ΔIV ΔVII showing fits of log kf and log ku [Eq. 8]. The curvatures in the unfolding limbs at high [urea] are due to transition-state shifts or native-state fraying and were excluded from the fits. The dotted line indicates the urea concentration where D reaches full occupancy. Units are in s-1. (B) Concentration of D in the fibrillation assays as calculated from the chevron data of apoSOD1ΔIV ΔVII. Total protein concentration is 25 μM, and units are in M. (C) Fibrillation lag times (τlag) of apoSOD1ΔIV ΔVII vs. [urea] (open circles), derived from fibrillation time courses as described in SI Text, Analysis of Fibrillation Time Courses. The intrinsic [urea] dependence of log τlag was measured at full occupancy of the globally unfolded state (D) above 4 M urea. Subtraction of this [urea] dependence yields the direct relation between log τlag and log [D] (closed circles). Under these conditions, ∂ log τlag/∂[urea] = mτlag = 0.13 ± 0.06. Units are in s. (D) Corresponding data and normalisation procedure for the elongation rate constants (νmax) of apoSOD1ΔIV ΔVII. ∂ log νmax/∂[urea] = mν max = -0.17 ± 0.05. Units are in s-1.
Table 1.
Kinetic parameters and protein stabilities. Rate constants are in units of s-1
| apoSOD1C57–C146 | apoSOD1SH SH | apoSOD1ΔIVΔVII | |
 * *
|
−0.89 ± 0.03 | −1.26 ± 0.08 | 0.13 ± 0.03 |
| mf* (M-1) | −0.92 ± 0.03 | −0.92 ± 0.03 ∥ | −0.82 ± 0.02 |
 * *
|
−2.68 ± 0.01 | −1.54 ± 0.02 | −2.72 ± 0.04 |
| mu* (M-1) | 0.44 ± 0.01 | 0.36 ± 0.01 | 0.22 ± 0.01 |
| MP † (M) | 1.32 ± 0.01 | 0.22 ± 0.07 | 2.74 ± 0.05 |
| mD-N ‡ (M-1) | 1.36 ± 0.03 | 1.28 ± 0.07 | 1.04 ± 0.02 |
 § §
|
−1.79 ± 0.04 | −0.28 ± 0.08 | −2.85 ± 0.05 |
 ¶ (kcal/mol) ¶ (kcal/mol) |
2.54 ± 0.07 | 0.40 ± 0.05 | 4.04 ± 0.07 |
| MP ** (M) | n.d. | n.d. | 2.67 ± 0.04/2.75 ± 0.04 |
| mD-N ** (M-1) | n.d. | n.d. | 0.89 ± 0.07/0.98 ± 0.07 |
| MP †† (M) | 1.53 ± 0.06 | ‡‡ | 2.88 ± 0.06 |
| mD-N †† (M-1) | 1.18 ± 0.23 | ‡‡ | 0.93 ± 0.09 |
*Derived from chevron data according to Eq. 8.
†Transition midpoint derived from the intersection between log kf and logku.
‡mD-N = mu - mf [Eq. 2].
§Calculated from Eq. 2.
¶ .
.
∥Locked to mf of apoSOD1C57–C146.
**Derived from NMR data in SI Text, NMR and Equilibrium Unfolding Analysis.
††Derived from equilibrium unfolding data in SI Text, NMR and Equilibrium Unfolding Analysis.
‡‡Lack of folded-state baseline prevents reliable estimate.
Verification of Two-State Behavior by Heteronuclear Single Quantum Correlation (HSQC) NMR.
As an independent control of the two-state folding behaviour of apoSOD1ΔIV ΔVII, we monitored at equilibrium the urea unfolding with HSQC NMR at 37 °C. The results show that the unfolding of apoSOD1ΔIV ΔVII is a clean first-order transition: The HSQC spectra obtained in the transition region are in all details linear combinations of the native and denatured cross peaks, with no trace of intermediate species (SI Text, NMR and Equilibrium Unfolding Analysis and Fig. S1). Moreover, plots of the levels of N and D vs. [urea] yield coinciding midpoints at MP ≈ 2.7 M urea and equilibrium mD-N values that match within experimental errors the kinetic mD-N from chevron data [Eqs. 1 and 2] (SI Text, NMR and Equilibrium Unfolding Analysis, Figs. S2 and S3, and Table 1). The locally unfolded and weakly populated intermediate (< 1%) revealed previously by relaxation-dispersion NMR (16) and hydrogen deuterium-exchange analysis (23), seems thus to remain at high-energy throughout the urea-unfolding transition at 37 °C.
Agitated SOD1 Fibrillation.
Like several other disease-associated proteins and peptides (5), the fibrillation process of apoSOD1 displays a characteristic lag phase followed by a sigmoidal increase of Thioflavin T (ThT) fluorescence (4) (SI Text, Analysis of Fibrillation Time Courses and Fig. S4). Determination of the fibrillation lag times (τlag) and maximum growth rates (νmax) of apoSOD1ΔIV ΔVII was by standard procedures (SI Text, Analysis of Fibrillation Time Courses), and the resulting plots of τlag and νmax vs. [urea] are shown in Fig. 2. Data shows that the fibrillation kinetics is, on the whole, accelerated by urea addition and that the apoSOD1ΔIV ΔVII construct maintains its basic fibrillation behaviour also under severely denaturing conditions at 8 M urea (Fig. 2). We find also that all of the SOD1 monomers are trapped in fibrillar form when the reaction has come to completion (SI Text, Analysis of Fibrillation Time Courses) and that the morphologies of the fibrils produced at 8 M urea are indistinguishable from those observed in pure buffer (SI Text, Analysis of Fibrillation Time Courses and Fig. S5). Taken together, this shows that agitated fibrillation is a remarkably robust process, which even persists under solvent conditions that strongly bias the folding equilibrium toward D (Scheme 1). At 8 M urea, the value of KD–N = [D]/[N] is 0.3·106 (Table 1), corresponding to nearly full occupancy of 25 μM D and merely pM levels of N. Similarly, low levels are expected for the transient intermediate visible by NMR in the absence of urea (16, 23), as this is close to N on the folding coordinate. On this basis, we conclude that the precursor for apoSOD1 fibrillation is not the native protein or the transient intermediate, but the globally unfolded state. Analogous fibrillation from globally unfolded material has been observed for the WW domain (27) and β2-microglobulin at low pH (28). An attractive, reductionist feature of this interpretation is that it renders apoSOD1 the same fibrillation mechanism as disordered peptides. It also explains why the apoSOD1 fibrils are polymorphic and so readily swap barrel segments in their spines upon point mutation (6): Globally unfolded precursors allow larger malleability than structurally restricted intermediates.
Accounting for the Urea-Dependence of the Fibrillation Kinetics.
From the urea titrations of apoSOD1ΔIV ΔVII in Fig. 2, it is evident that νmax reaches its highest values near, or just after, the transition midpoint, and then slowly decreases with increasing [urea]. A matching, opposite change is described by τlag. Similar trends are seen for apoSOD1C57–C146 and apoSOD1SH SH, albeit that their low-transition midpoints make data less amenable to detailed analysis (SI Text, Normalization of τlag and νmax to log KD-N and Fig. S6). One explanation for the νmax and τlag maxima would be that the protein populates an aggregation-prone intermediate in the unfolding transition region, which then gradually disappears at higher denaturant concentrations (29). Since there is no trace of intermediates in the NMR analysis, however, it is more likely that the maxima of νmax result from a trade-off between (i) increased occupancy of D and (ii) intrinsic suppression of fibrillation by the added urea itself. Such suppression by urea is expected for all processes involving burial of solvent-exposed surface area, and there is no reason to believe that folding and fibrillation should be different in this respect. To compensate for the urea dependence, linear functions were fitted the log νmax and log τlag values above [urea] = 4 M where D has full occupancy (Fig. 2). These baselines were subsequently subtracted from the data to produce graphs of log νmax and log τlag vs. [urea] that are linked directly to the occupancy of species in the folding equilibrium (Fig. 2).
Evidence for Fragmentation-Assisted Fibrillation.
The urea-compensated values of log νmax and log τlag show overall linear dependencies on log[D] (Fig. 3), consistent with observations from conventional fibrillation assays where protein concentration is varied by mass (10, 30, 31). A striking feature of the plots in Fig. 3 is that the dependence on protein concentration is very weak with slopes of ∂ log νmax/∂ log[D] = 0.56 ± 0.03 and ∂ log τlag/∂ log[D] = -0.47 ± 0.03. Similarly, low values of ∂ log τlag/∂ log[D] are observed for other proteins and peptides with globally unfolded precursors (30). These weak protein-concentration dependences are formally inconsistent with a primary nucleation mechanism where the formation of primary nuclei from soluble monomers is slower than their elongation (32): In the case of apoSOD1ΔIV ΔVII, it would suggest primary nuclei with an aggregation number of n = 0.5 monomers. Rather, the behavior indicates the involvement of secondary fibrillation pathways like heterogeneous nucleation on fibril surfaces and fragmentation (32). Of particular interest is the role of fragmentation, since the apoSOD1 fibrillation depends critically on mechanical agitation. To examine this further, we use the analytical expressions for νmax and τlag presented by Knowles et al. (30), which, in this case, translate to:
| [3] |
| [4] |
where C+ ∝ kn[D]n-1, where kn is the rate constant for forming a primary nucleus size n (30), and κ is the rate of fibril multiplication according to:
| [5] |
where k+ and k- are the rate constants for elongation and fragmentation, respectively. Notably, νmax is entirely controlled by κ and independent of the primary nucleation events when fragmentation is operative (k- > 0). Under such conditions, ∂ log νmax/∂ log[D] converges to 0.5 ( ). This is in remarkable agreement with the values of ∂ log νmax/∂ log[D] = 0.56 ± 0.03 measured in this study (Fig. 3). Correspondingly, we observe that ∂ log τlag/∂ log[D] = -0.47 ± 0.03, and ∂ log νmax/∂ log τlag = -1.11 ± 0.02 (Fig. 3). Similarly, weak concentration dependencies of νmax and τlag are found by Radford and co-workers in numeric simulations of secondary fragmentation processes (31). Taken together, these observations suggest that the fibrillation process of apoSOD1ΔIV ΔVII is dominated by mechanical fragmentation, and that the resulting fibrillar fragments elongate by incorporating globally unfolded monomers. The lag phase contains thus no information about the primary nucleation events, as is often assumed in protein-aggregation kinetics, but is the sole consequence of an exponential increase of growing ends. It remains to establish what gives rise to the first fragmentable species (33). Is it ordering by specific or colloidal association of unfolded species, and to what extent does this primary nucleation rely on external support from contaminant seeds or cuvette/gas interfaces?
). This is in remarkable agreement with the values of ∂ log νmax/∂ log[D] = 0.56 ± 0.03 measured in this study (Fig. 3). Correspondingly, we observe that ∂ log τlag/∂ log[D] = -0.47 ± 0.03, and ∂ log νmax/∂ log τlag = -1.11 ± 0.02 (Fig. 3). Similarly, weak concentration dependencies of νmax and τlag are found by Radford and co-workers in numeric simulations of secondary fragmentation processes (31). Taken together, these observations suggest that the fibrillation process of apoSOD1ΔIV ΔVII is dominated by mechanical fragmentation, and that the resulting fibrillar fragments elongate by incorporating globally unfolded monomers. The lag phase contains thus no information about the primary nucleation events, as is often assumed in protein-aggregation kinetics, but is the sole consequence of an exponential increase of growing ends. It remains to establish what gives rise to the first fragmentable species (33). Is it ordering by specific or colloidal association of unfolded species, and to what extent does this primary nucleation rely on external support from contaminant seeds or cuvette/gas interfaces?
Fig. 3.
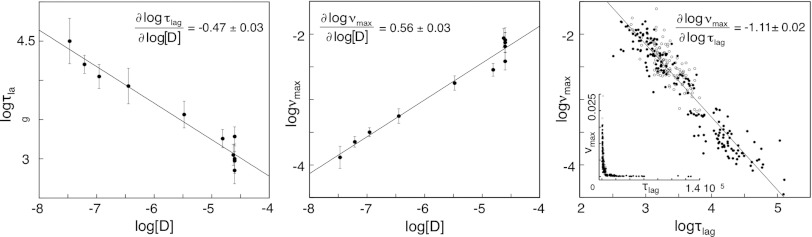
The fibrillation lag time (log τlag) and elongation rate constant (log νmax) show linear dependencies on the logarithmized concentration of globally unfolded apoSOD1ΔIV ΔVII (log[D]). The matching slopes of ∂ log τlag/∂ log[D] = -0.47 ± 0.03 and ∂ log νmax/∂ log[D] = 0.56 ± 0.03 suggest a fragmentation-assisted mechanism according to Eqs. 3 and 5. Plots of log τlag vs. log νmax, and τlag vs. νmax (Inset). Data from urea titration (closed circles) and mutagenesis (open circles).
Urea Dependence and Fibrillation m-values.
To shed further light on the apoSOD1ΔIV ΔVII fibrillation process, we used the urea dependencies of νmax and τlag, which reflect the changes in solvent accessible surface area of the underlying microscopic reaction steps. From the denatured baselines in Fig. 2, we obtain ∂ log νmax/∂[urea] = mν max = -0.17 ± 0.05 and ∂ log τlag/∂[urea] = mτlag = 0.13 ± 0.06 (Fig. 2). The matching absolute values of mνmax and mτlag lend further support to the existence of a common scaling factor κ, as inferred in Eqs. 4 and 5. Importantly, this scaling factor allows us to examine the microscopic rate constants k+ and k- within the same conceptual framework as the folding kinetics (Fig. 4). The results from such examination are described in SI Text, Fibrillation m-values and suggest that 30–50% of the SOD1ΔIV ΔVII sequence (i.e., 2 to 4 β-strands) buries in the fibril structure, depending on the position of the elongation barrier (‡elongation) along m-/+ (Fig. 4). Interestingly, this estimate is in good agreement with previous data from mass spectroscopy, which indicate fibrillar segments of 3 to 5 β-strands (6). To this end, the relatively low values of mν max and mτlag show that the persistent growth of SOD1 fibrils at high [urea] does not need to imply exceptional stability of the product, but could also stem from low sensitivity to denaturant.
Fig. 4.

Schematic illustration of the bifurcated folding and fibrillation free-energy profiles of apoSOD1ΔIV ΔVII. The reaction coordinate is solvent-accessible surface area, measured by the m-values in Table 1 and SI Text, Fibrillation m-values. The profiles indicate the case where the folding and fibrillation transition states (‡folding and ‡elongation) are placed symmetrically. For simplicity, the stabilities of the fibrillar state (A), the unfolded monomer (D) and the folded protein (N) are normalized, and barrier heights are not to scale. I* is the high-energy intermediate observed by NMR at 0 M urea (16, 23).
Mutational Analysis by E-Scanning.
Association of proteins is generally described as a multistep process that starts by random collision, leading first to a loosely bound transient complex that subsequently rearranges into the close-packed, final complex (34). In treatment of association data, the latter step is often assumed to be irreversible, which in this study yields:![]() Scheme 2 where D is the globally unfolded monomer, An, a fibril length n, and A′*An, the transient complex. The rate constant kc describes either folding-after-binding (35), binding by fly-casting (36), or conformational steering by hard-body rearrangement (34), depending on the structural nature of the transient complex A′*An. The overall rate constant of fibril elongation then becomes:
Scheme 2 where D is the globally unfolded monomer, An, a fibril length n, and A′*An, the transient complex. The rate constant kc describes either folding-after-binding (35), binding by fly-casting (36), or conformational steering by hard-body rearrangement (34), depending on the structural nature of the transient complex A′*An. The overall rate constant of fibril elongation then becomes:
| [6] |
which reduces to k+ ≈ kD when kc≫k-D, and to k+ ≈ (kD/k-D)kc when kc ≪ k-D (34). Mutational studies of association of rigid proteins have shown that the transient complex, as a rule, is dominated by long-range electrostatic contacts with close-range hydrophobic contacts playing a minor role (34). Accordingly, the effect of mutation on the association kinetics is found to be largest for substitution of charged side chains, whereas truncation of hydrophobic side chains show little effect (34). Under the assumption that similar principles apply to association of disordered precursors to fibrils (Scheme 2), we examined in this study the fibrillation kinetics by E-scanning: the SOD1ΔIV ΔVII sequence was screened by insertion of negatively charged glutamic-acid moieties (Fig. 5 and SI Text, Mutational Analysis of the Fibrillation Kinetics by E-Scanning, Fig. S7 and Table S1). The measurements were done under fully denaturing conditions at 5 M urea to probe specifically the globally unfolded state (Fig. 2). The results show, somewhat unexpectedly, that the response to mutation is not limited to a few fibrillation hot spots but is relatively uniform across the apoSOD1ΔIV ΔVII sequence (Fig. 5 and Table 2). Moreover, the effect on τlag displays an apparent correlation with global charge that extrapolates linearly to the values of apoSOD1SH SH and apoSOD1C57–C146 (Fig. 6). The latter correlation resembles data from other protein-protein interactions (34) and indicates that the reaction involves a loosely bound transient complex, as depicted in Scheme 2. Data implicates also that the decrease of τlag upon loop removal is fully accounted for by loss of loop charges: The associated effect of loop-entropy reduction seems subordinate. This weak influence of coil entropy is also evident from the similar τlag values of globally unfolded apoSOD1SH SH and apoSOD1C57–C146 that differ by the cross-link between C57 and C146 (Table 2). Looking at the individual positions, however, there is one notable outlier in the data set: The mutation V5E/V7E in β1 has no discernible effect on the fibrillation kinetics, despite introducing two new negative charges (Fig. 5 and Table 2). It is also clear that several of the other mutations show variations in τlag that go beyond their global charge. Additional factors seem to be at play. To examine possible contributions from sequence hydrophobicity, we selected the apoSOD1ΔIV ΔVII mutations with a common net charge of -5 (Table 2) and plotted these against local change in Kyte-Dolittle index (Fig. 6). Although data show some scatter, the results point at a basic correlation between decreased hydrophobicity and increased τlag; the β1 mutation V5E/V7E is, again, deviant because of its lacking impact on τlag. A seemingly analogous influence of loosely packed hydrophobic side chains has been observed in the recognition between the unfolded S peptide and ribonuclease S (37). Finally, we note that none of the apoSOD1ΔIV ΔVII mutations lead to an acceleration of the fibrillation kinetics as expected from a selective increase of the fragmentation rate constant k- [Eq. 5]. This indicates that the mutations affect mainly k+ in the κ term, consistent with the two-step association mechanism in Scheme 2. One explanation for this bias would be that k- is mainly controlled by close-range contacts in the final fibrillar product, and these are not probed by the current E mutations: Insertion of E side chains in this study yields binding ϕ-values of 1, as has been observed previously for charge mutations outside the binding sites of the TEM1-BLIP, barnase-barstar, and Ral-Ral complexes (34). Substitution of surface charges seems to yield similar effect on the ground state and transition state for breaking up the fibrillar core.
Fig. 5.
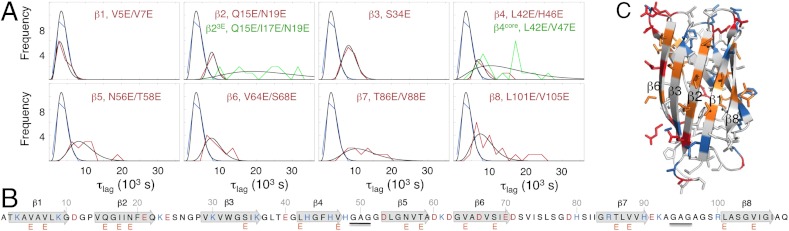
Examination of the apoSOD1ΔIV ΔVII fibrillation kinetics by E-scan under fully denaturing conditions at 5 M urea. (A) Histograms of mutant lag times (τlag) fitted by non-normalized gamma distributions (Table 2). Pseudo-wild type (black) and mutants (red and green). (B) Sequence positions of E mutations. (C) Structural positions of E mutations (orange). Positively and negatively charged side chains are shown in blue and red, respectively.
Table 2.
Parameters for fibrillation kinetics at 5 M urea
| τlag* (103 s) | δτlag† (103 s) | νmax* (10-3 s-1) | δνmax† (10-3 s-1) | Charge (e) | Hydrophobicity‡ | |
| apoSOD1ΔIVΔVII | 3.57 | 1.30 | 0.98 | 0.49 | −3 | 9.87 |
| β1, V5E/V7E | 3.93 | 1.50 | 1.40 | 0.45 | −5 | −2.83 |
| β2, Q15E/N19E | 7.82 | 1.74 | 0.57 | 0.22 | −5 | 9.87 |
| β3, S34E | 8.39 | 1.66 | 1.15 | 0.86 | −4 | 7.17 |
| β4, L42E/H46E | 11.42 | 4.35 | 0.74 | 0.34 | −5 | 2.27 |
| β5, N56E/T58E | 9.50 | 3.65 | 0.55 | 0.24 | −5 | 7.07 |
| β6, V64E/S68E | 9.22 | 2.99 | 0.46 | 0.19 | −5 | −0.63 |
| β7, T86E/V88E | 11.93 | 4.15 | 0.44 | 0.08 | −5 | −0.73 |
| β8, L101E/V105E | 11.22 | 5.71 | 0.47 | 0.42 | −5 | −5.41 |
| β2, Q15E/I17E/N19E | 20.86 | 6.91 | 0.19 | 0.08 | −6 | 1.87 |
| β4, L42E/V47E | 13.70 | 6.10 | 0.44 | 0.16 | −5 | −5.23 |
| apoSOD1C57–C146 | 40.30 § | 8.27 § | 0.10§ | 0.01§ | −8 | −65.33 |
| apoSOD1SH SH | 29.14 | 5.64 | 0.10 | 0.03 | −8 | −65.33 |
*Derived from ThT data according to SI Text, Analysis of Fibrillation Time Courses).
†Standard deviation from 7 to 20 measurements.
‡Calculated from the integrated Kyte-Doolittle plot with a running average of 5 residues (SI Text, Mutational Analysis of the Fibrillation Kinetics by E-Scanning).
§Estimated by interpolation of data at 4 and 6 M urea (SI Text, Analysis of Fibrillation Time Courses).
Fig. 6.

Correlations between fibrillation lag times (τlag) and the parameters net charge and hydrophobicity change. (A) τlag shows an overall linear dependence on net charge, consistent with the general behaviour of protein-protein interactions, according to Scheme 2 (34). ApoSOD1ΔIV ΔVII (-3), E-scan mutants (-4 to -6), and apoSOD1C57–C146/apoSOD1SH SH (-8). (B) Plot of τlag for the apoSOD1ΔIV ΔVII mutants with normalized net charge of -5 against change in local hydrophobicity (Table 2). The outlier is the β1 mutation V5E/V7E, which is unique by having no effect on τlag. The R-value of 0.85 is without the β1 measurement.
Summary and Conclusions.
The diffusely distributed effect of mutations along the apoSOD1ΔIV ΔVII sequence can be interpreted in two ways. One possibility is that all probed segments, save β1, are part of the fibrillar structure. Although this interpretation may, at first, seem inconsistent with the size estimate from the relatively low fibrillation m-values (Fig. 2), it can be accommodated with data if the fibrillar products are heterogeneous. The monitored reaction would then represent the average of several pathways, each of which involve different subsets of smaller sequence portions, consistent with the previously seen polymorphic nature of the SOD1 fibrils (6). In light of the global dependence on net change and sequence hydrophobicity (Fig. 6), an alternative scenario would be that the unfolded protein is first captured nonspecifically in a loosely bound transient complex and is thereafter guided into its specific, tightly bound state by the fibrillar surface (Scheme 2). A key feature of this two-step process is that the initial and final complexes need not to rely on the same interactions. In the case of apoSOD1ΔIV ΔVII, the diffusely bound coil would rearrange into a close-packed segment involving less than half of the sequence material (Fig. 4). Interestingly, such two-step binding would explain why protein solubility is generally found to depend on both global (38) and position-specific (39, 40) physiochemical properties: The diffuse association of the transient complex is to some extent global, whereas the close-packed architecture of the filamentous product relies on specific sequence motifs. Even so, we are not yet in position to identify the sequence regions that compose the fibrillar core of apoSOD1ΔIV ΔVII: The global correlations with net charge and hydrophobicity indicate that our mutational analysis targets the transient complex alone. The question is then to what extent these in vitro studies reflect the pathologic aggregation of SOD1 in ALS? Of particular concern is the effect of mechanical fragmentation, which biases the process by selecting for aggregates that are structurally brittle: Agitation of β2-microglobulin is seen to promote straight amyloid-like fibrils under conditions where resting samples would otherwise form curly aggregates (28). Further fuel to this concern is provided by the pathological SOD1 inclusions being not clearly fibrillar but resembling more the Lewy bodies observed in prion diseases (41). It has been hypothesized, however, that fragmentation represents a key component of the pathological fibrillation process, since this is expected to happen naturally, due to the ever present mechanical stress in living tissue (42). Such fragmentation of larger aggregates into smaller segments would not only amplify the precipitation but could also facilitate cell-to-cell transmission and hastening the disease progression (42). Sample agitation would then reproduce innate in vivo fluctuations, rather than biasing the experiments in nonphysiological direction. In this context, it is interesting that the congruent effects of ALS-associated SOD1 mutations point at the globally unfolded state as disease precursor. Either by directly shifting the folding equilibrium of the apo protein toward D (18) or indirectly by decreasing the affinity for the stabilizing Cu and Zn ions (12). Equally notable, bioinformatic analysis has shown that ALS-associated SOD1 mutations preferentially decrease the proteins net negative charge (43), in full correspondence with data in Fig. 6. Taken together, these consistencies indicate that that simplified studies of SOD1 fibrillation in vitro could, after all, capture the essence of the pathological aggregation mechanism: an inherent competition between folding and aggregation that bifurcates from the globally unfolded state.
Materials
Mutagenesis, Expression, Purification, and Experimental Conditions.
Mutagenesis, expression, and purification were as in (22, 23), and all experiments were performed at 37 °C in 10 mM Bis-Tris at pH 6.3, unless otherwise stated.
Electron Microscopy.
Carbon-coated 200-mesh copper grids were applied on top of 20 μL droplets of fibril preparation and blotted for 5 min. Staining was with 1% (weight/volume) uranyl acetate. Images of 18,000× magnification were recorded in a Tecnai G2 Spirit BioTWIN microscope with a tungsten filament operating at 80 kV.
Folding Kinetics.
Final protein concentration was 4 μM and the urea grade was ultrapure (MP Biomedicals, Inc.). Kinetic measurements were on a PiStar-180 stopped-flow apparatus (Applied Photophysics, Leatherhead, UK) or by manual mixing on a Varian Cary Eclipse spectrometer (Santa Clara, CA), with excitation at 280 nm and emission collected with a 305 nm long-pass filter or with monochromator at 360 nm. Reduction was by addition of 2.0 mM Tris (2-carboxyethyl) phosphine (TCEP) to all buffers and overnight incubation. Chevron data was fitted with
 |
[8] |
where kobs is the observed rate constant, and the parameters as defined in Eqs. 1 and 2. Data analysis was with KaleidaGraph (Synergy Software, Reading, PA).
Fibrillation Kinetics.
Fibrillation was measured at 37 °C in standard 3.5 ml quarts cuvettes in a Varian Cary Eclipse spectrometer (Santa Clara, CA) with Teflon-coated magnetic stirrers at 1,800 rpm. Sample volume was 2 mL and contained 25 μM protein and 20 μM ThT in 10 mM Bis-Tris at pH 6.3 and varying [urea]. Reduction was by 0.5 mM TCEP. ThT excitation was at 444 nm and emission at 485 nm and measured at every 300 s of incubation. Data treatment was by standard procedures as described in SI Text, Analysis of Fibrillation Time Courses.
Supplementary Material
ACKNOWLEDGMENTS.
This work was supported by grants from Swedish Research Council, the Knut and Alice Wallenberg Foundation, the Bertil Hållsten Foundation and Hjärnfonden. This work was also supported by the Access to Research Infrastructures Activity in the 7th Framework Program of the EC (Project no. 261863, Bio-NMR) for conducting the research at CERM. We thank Daniel Schaal and Ingrid Calone for early contributions to this project.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1201795109/-/DCSupplemental.
References
- 1.Rousseau F, Schymkowitz J, Oliveberg M. ALS precursor finally shaken into fibrils. Proc Natl Acad Sci USA. 2008;105:18649–18650. doi: 10.1073/pnas.0810568106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stathopulos PB, et al. Sonication of proteins causes formation of aggregates that resemble amyloid. Protein Sci. 2004;13:3017–3027. doi: 10.1110/ps.04831804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Furukawa Y, Kaneko K, Yamanaka K, O'Halloran TV, Nukina N. Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in familial form of ALS. J Biol Chem. 2008;283:24167–24176. doi: 10.1074/jbc.M802083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chattopadhyay M, et al. Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc Natl Acad Sci USA. 2008;105:18663–18668. doi: 10.1073/pnas.0807058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eichner T, Radford SE. A diversity of assembly mechanisms of a generic amyloid fold. Mol Cell. 2011;43:8–18. doi: 10.1016/j.molcel.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 6.Furukawa Y, Kaneko K, Yamanaka K, Nukina N. Mutation-dependent polymorphism of Cu,Zn-superoxide dismutase aggregates in the familial form of amyotrophic lateral sclerosis. J Biol Chem. 2010;285:22221–22231. doi: 10.1074/jbc.M110.113597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stathopulos PB, et al. Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc Natl Acad Sci USA. 2003;100:7021–7026. doi: 10.1073/pnas.1237797100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi I, et al. Lipid molecules induce the cytotoxic aggregation of Cu/Zn superoxide dismutase with structurally disordered regions. Biochim Biophys Acta. 2011;1812:41–48. doi: 10.1016/j.bbadis.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 9.Banci L, et al. Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: A possible general mechanism for familial ALS. Proc Natl Acad Sci USA. 2007;104:11263–11267. doi: 10.1073/pnas.0704307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hwang YM, et al. Nonamyloid aggregates arising from mature copper/zinc superoxide dismutases resemble those observed in amyotrophic lateral sclerosis. J Biol Chem. 2010;285:41701–41711. doi: 10.1074/jbc.M110.113696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw BF, Valentine JS. How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? Trends Biochem Sci. 2007;32:78–85. doi: 10.1016/j.tibs.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Leinartaite L, Saraboji K, Nordlund A, Logan DT, Oliveberg M. Folding catalysis by transient coordination of Zn2+ to the Cu ligands of the ALS-associated enzyme Cu/Zn superoxide dismutase 1. J Am Chem Soc. 2010;132:13495–13504. doi: 10.1021/ja1057136. [DOI] [PubMed] [Google Scholar]
- 13.Eichner T, Radford SE. Understanding the complex mechanisms of beta2-microglobulin amyloid assembly. FEBS J. 2011;278:3868–3883. doi: 10.1111/j.1742-4658.2011.08186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci USA. 2002;4(99 Suppl.):16427–16432. doi: 10.1073/pnas.202495199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DiDonato M, et al. ALS mutants of human superoxide dismutase form fibrous aggregates via framework destabilization. J Mol Biol. 2003;332:601–615. doi: 10.1016/s0022-2836(03)00889-1. [DOI] [PubMed] [Google Scholar]
- 16.Teilum K, et al. Transient structural distortion of metal-free Cu/Zn superoxide dismutase triggers aberrant oligomerization. Proc Natl Acad Sci USA. 2009;106:18273–18278. doi: 10.1073/pnas.0907387106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elam JS, et al. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol. 2003;10:461–467. doi: 10.1038/nsb935. [DOI] [PubMed] [Google Scholar]
- 18.Lindberg MJ, Bystrom R, Boknas N, Andersen PM, Oliveberg M. Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proc Natl Acad Sci USA. 2005;102:9754–9759. doi: 10.1073/pnas.0501957102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nordlund A, et al. Functional features cause misfolding of the ALS-provoking enzyme SOD1. Proc Natl Acad Sci USA. 2009;106:9667–9672. doi: 10.1073/pnas.0812046106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karch CM, Borchelt DR. Aggregation modulating elements in mutant human superoxide dismutase 1. Arch Biochem Biophys. 2010;503:175–182. doi: 10.1016/j.abb.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nordlund A, Oliveberg M. Folding of Cu/Zn superoxide dismutase suggests structural hotspots for gain of neurotoxic function in ALS: Parallels to precursors in amyloid disease. Proc Natl Acad Sci USA. 2006;103:10218–10223. doi: 10.1073/pnas.0601696103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindberg MJ, Normark J, Holmgren A, Oliveberg M. Folding of human superoxide dismutase: Disulfide reduction prevents dimerization and produces marginally stable monomers. Proc Natl Acad Sci USA. 2004;101:15893–15898. doi: 10.1073/pnas.0403979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Danielsson J, Kurnik M, Lang L, Oliveberg M. Cutting off functional loops from homodimeric enzyme superoxide dismutase 1 (SOD1) leaves monomeric beta-barrels. J Biol Chem. 2011;286:33070–33083. doi: 10.1074/jbc.M111.251223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fersht AR. Optimization of rates of protein folding: The nucleation-condensation mechanism and its implications. Proc Natl Acad Sci USA. 1995;92:10869–10873. doi: 10.1073/pnas.92.24.10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Svensson AK, Bilsel O, Kondrashkina E, Zitzewitz JA, Matthews CR. Mapping the folding free energy surface for metal-free human Cu,Zn superoxide dismutase. J Mol Biol. 2006;364:1084–1102. doi: 10.1016/j.jmb.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 26.Kubelka J, Hofrichter J, Eaton WA. The protein folding ‘speed limit’. Curr Opin Struct Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 27.Ferguson N, et al. General structural motifs of amyloid protofilaments. Proc Natl Acad Sci USA. 2006;103:16248–16253. doi: 10.1073/pnas.0607815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gosal WS, et al. Competing pathways determine fibril morphology in the self-assembly of beta2-microglobulin into amyloid. J Mol Biol. 2005;351:850–864. doi: 10.1016/j.jmb.2005.06.040. [DOI] [PubMed] [Google Scholar]
- 29.Hamada D, Dobson CM. A kinetic study of beta-lactoglobulin amyloid fibril formation promoted by urea. Protein Sci. 2002;11:2417–2426. doi: 10.1110/ps.0217702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knowles TP, et al. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326:1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 31.Xue WF, Homans SW, Radford SE. Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc Natl Acad Sci USA. 2008;105:8926–8931. doi: 10.1073/pnas.0711664105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferrone F. Analysis of protein aggregation kinetics. Methods Enzymol. 1999;309:256–274. doi: 10.1016/s0076-6879(99)09019-9. [DOI] [PubMed] [Google Scholar]
- 33.Thirumalai D, Klimov DK, Dima RI. Emerging ideas on the molecular basis of protein and peptide aggregation. Curr Opin Struct Biol. 2003;13:146–159. doi: 10.1016/s0959-440x(03)00032-0. [DOI] [PubMed] [Google Scholar]
- 34.Schreiber G, Haran G, Zhou HX. Fundamental aspects of protein–protein association kinetics. Chem Rev. 2009;109:839–860. doi: 10.1021/cr800373w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiefhaber T, Bachmann A, Jensen KS. Dynamics and mechanisms of coupled protein folding and binding reactions. Curr Opin Struct Biol. 2012;22:21–29. doi: 10.1016/j.sbi.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 36.Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc Natl Acad Sci USA. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bachmann A, Wildemann D, Praetorius F, Fischer G, Kiefhaber T. Mapping backbone and side-chain interactions in the transition state of a coupled protein folding and binding reaction. Proc Natl Acad Sci USA. 2011;108:3952–3957. doi: 10.1073/pnas.1012668108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- 39.Otzen DE, Kristensen O, Oliveberg M. Designed protein tetramer zipped together with a hydrophobic Alzheimer homology: A structural clue to amyloid assembly. Proc Natl Acad Sci USA. 2000;97:9907–9912. doi: 10.1073/pnas.160086297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buell AK, et al. Position-dependent electrostatic protection against protein aggregation. Chembiochem. 2009;10:1309–1312. doi: 10.1002/cbic.200900144. [DOI] [PubMed] [Google Scholar]
- 41.Forsberg K, et al. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One. 2010;5:e11552. doi: 10.1371/journal.pone.0011552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue WF, Hellewell AL, Hewitt EW, Radford SE. Fibril fragmentation in amyloid assembly and cytotoxicity: When size matters. Prion. 2010;4:20–25. doi: 10.4161/pri.4.1.11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sandelin E, Nordlund A, Andersen PM, Marklund SS, Oliveberg M. Amyotrophic lateral sclerosis-associated copper/zinc superoxide dismutase mutations preferentially reduce the repulsive charge of the proteins. J Biol Chem. 2007;282:21230–21236. doi: 10.1074/jbc.M700765200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.