

Abstract
The palladium-catalyzed cross-coupling reaction of potassium (4-methoxyphenyl)dimethylsilanolate (K+1−) with aryl bromides has been demonstrated using triphenylphosphine oxide as a stabilizing ligand. Unsymmetrical biaryls can be prepared from a variety of aryl bromides in good yield with short reaction times. Qualitative kinetics studies compared effects of different phosphine oxides on the rate of cross-coupling and established the beneficial effect of these ligands in the reaction of electron-rich arylsilanolates. The improved yield and reproducibility of the cross-coupling of several bromides was demonstrated by direct comparison of reactions performed with and without triphenylphosphine oxide under non-rigorous exclusion of oxygen.
Keywords: cross-coupling, biaryls, phosphine oxides, silanols
1. Introduction
The palladium-catalyzed cross-coupling reaction of organometallic nucleophiles is one of the premier methods for carbon-carbon bond formation.i The use of organostannanes,ii organoboranes,iii and organozinciv reagents has been comprehensively studied as the nucleophilic partner and has been elevated to name reaction status.v Although these reactions have been used extensively for myriad synthetic transformations, there are nevertheless inherent limitations that warrant the development of new methods. Organosilanes, initially investigated by Hiyama and coworkers, have become an attractive alternative to some of the more commonly used donors. Early implementation of organosilanes as donors with different transferable groups required the use of fluoride to promote the cross-coupling process (Hiyama-Hatanaka paradigm).vi Despite the broad applicability of this procedure for the cross-coupling of organosilanes, the requirement for fluoride activation is a significant disadvantage because of, inter alia, the incompatibility with silicon protecting groups and the aggressive nature of fluoride. Therefore, the development of fluoride-free, cross-coupling reactions of organosilanols that display the same substrate scope and rate as the fluoride-promoted processes would be valuable.vii This objective has been realized, in part, with the introduction of Ag2Oviii and KOSiMe3 as activators for the cross-coupling of alkenyl-viib and alkynylsilanols.ix
However, the formation of biaryls from cross-coupling of the corresponding arylsilanes (such as aryl(halo)silanes,x aryl(halo)silylcyclobutanes,viic aryl(triallyl)silanesxi and aryl(trialkoxy)silanesxii) has been dominated by the use of fluoride activation.xiii Although these reactions are effective, the coupling of the arylsilanes requires more forcing conditions. To address this concern, and remove the requirement for fluoride activation, we have recently demonstrated that electron-rich aryldimethylsilanols undergo cross-coupling reactions with aryl iodides and bromides under the action of Cs2CO3.xiv Although the scope of electrophile for the cross-coupling of (4-methoxyphenyl)dimethylsilanol (1) is good, the reactions are slow (3-24 h), additives are needed to suppress homocoupling, and other arylsilanols are poor substrates.xivb Because of our continued interest in the development of milder and more general cross-coupling reactions of organosilanes, we made recourse our to recent discovery that alkali metal silanolates are key reactive intermediates in these transformations.xv,xvi The ease of preparation, stability and competence of metal silanolates has been illustrated in the cross-coupling of both alkenylxvii and heteroarylxviii silanolates. The preformed silanolate can be directly charged into the reaction mixture and affords a significant rate increase compared to the use of TMSOK as the activator.xix Thus, we sought to develop cross-coupling conditions for the formation of biaryls with preformed silanolates to improve both the reaction time as well as the scope of the substrate.
2. Results
2.1. Orienting Experiments
Initial studies focused on the cross-coupling of preformed, alkali metal silanolates in the presence of various additives. Because the previous conditions for the coupling of silanols employed phosphine ligands in conjunction with the Cs2CO3 / H2O activator,xiv we were concerned about the in-situ formation of phosphine oxides through reduction of the palladium(II) precatalyst, allylpalladium(II)chloride dimer (APC) by the phosphine.xx The cross-coupling of potassium (4-methoxyphenyl)dimethylsilanolate K+1− with 4-bromobenzotrifluoride using APC (2.5 mol%) and dppb (5 mol%) in toluene at 90 °C reached 80% conversion in a 100 min, Figure 1 (compared to ca. 180 min with Cs2CO3 / H2O). To test the affect of phosphine oxides on the reaction rate and efficiency, the mono-oxide, dppb(O), was employed and a similar conversion was reached, but in a shorter time (70 min). Much to our surprise the use of dppb(O)2 dramatically increased the rate and the reaction went to completion in less than 15 min! Other phosphine oxides were then tested to establish if this phenomenon was unique to dppb(O)2. A more basic, chelating bis phosphine oxide, dppf(O)2 gave identical results. Remarkably either 1 and 2 equiv of triphenylphosphine oxide per palladium gave rates and conversions nearly indistinguishable from those with dppb(O)2. The beneficial effect of phosphine oxide ligands was clearly demonstrated in the “ligandless” experiment shown in Figure 1. Under identical conditions but without a phosphine oxide additive, the initial rate of the coupling is good, nearly matching the rate in the presence of dppp(O), however, the reaction stalled after 30 min with the obvious formation of palladium black.
Figure 1.
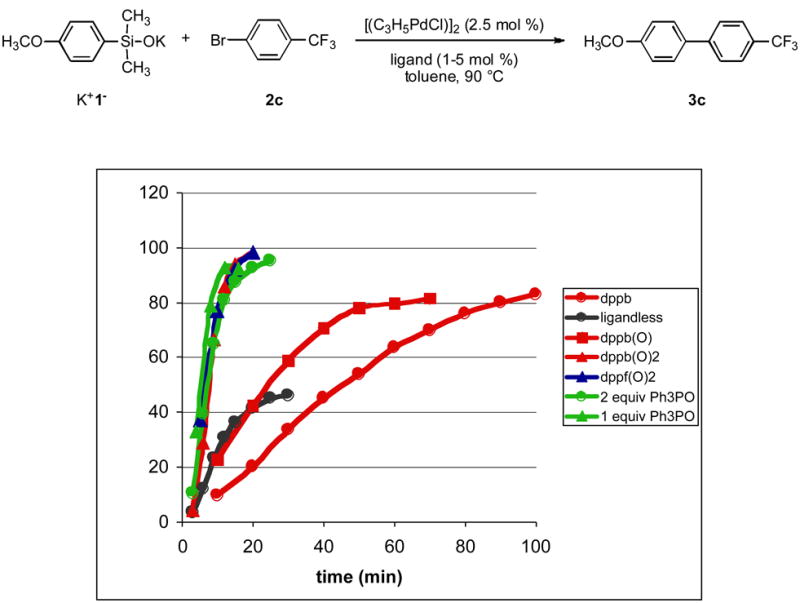
Rate of cross-coupling as a function of ligand.
Although control experiments have unambiguously established that phosphine oxides are not formed from the reduction of APC,xx our misconception has lead to the serendipitous discovery that phosphine oxides are useful ligands that allow for considerably faster cross-coupling reactions compared to the use of phosphine ligands. On the basis of these insights, we undertook a systematic study of the effect of triphenylphosphine oxide on the cross-coupling of K+1− with avariety of electronically and sterically demanding aryl bromides to expand the scope of biaryl synthesis with arylsilanolates.
2.2. Expansion of Scope of Aryl Bromide
To establish the utility of phosphine oxides in a preparative cross-coupling reaction, K+1− and 4-bromobenzotrifluoride 2c were combined in toluene at 90 °C with APC (2.5 mol%) and triphenylphosphine oxide (Ph3P(O), 5 mol%). The cross-coupling product was obtained in good yield (79%) after only 30 min. From this initial encouraging result, the cross-coupling with a variety of aryl bromides in the presence of Ph3P(O) was undertaken to fully establish reaction scope. The results are compiled in Table 1. Gratifyingly, aryl bromides of diverse structural and electronic nature underwent smooth cross-coupling to form unsymmetrical biaryl products rapidly and in good yield. Both electron-deficient (entry 1-3) and electron-rich (entry 5-7) 4-substituted aryl bromides reacted within 60 min to afford moderate to good yields. Unexpectedly, these reaction conditions were not compatible with certain sensitive functional groups. For example, aryl bromides containing a keto or cyano substituent led to poor conversions (< 10%) in contrast to their behavior in Cs2CO3-promoted cross-coupling reactions.xiv Further complications arose in the reaction of ethyl 4-bromobenzoate where unproductive consumption of the starting aryl bromide was observed. The loss of starting material was attributed to ester cleavage by the arylsilanolate at elevated temperatures. This problem could be easily remedied by making recourse to a tert-butyl ester. Compound 2a reacted smoothly to afford an 82% yield of the desired biaryl product (Table1, entry 1). In all cases, the symmetrical biaryl formed through homocoupling of the bromide was the primary side product (7-14% yield).
Table 1. Cross-Coupling of K+1− with Substituted Aryl Bromides Using Phosphine Oxidesa.
| |||||
|---|---|---|---|---|---|
| entry | aryl bromide | product | time, min | yield,%b | product ratioc (cross/homocoupling, 3:4) |
| 1 |
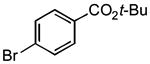
|
3a | 60 | 82 | 98:2 |
| 2 |
|
3b | 45 | 80 | 90:10 |
| 3 |
|
3c | 30 | 79 | - |
| 4 |
|
3d | 30 | 72 | 88:12 |
| 5 |
|
3e | 35 | 63 | 83:17 |
| 6 |
|
3f | 60 | 61 | 82:18 |
| 7 |
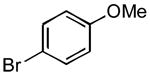
|
3g | 60 | 81 | - |
| 8 |
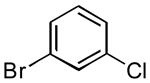
|
3h | 60 | 86 | 93:7 |
| 9 |
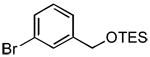
|
3i | 60 | 67d | - |
| 10 |
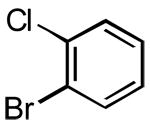
|
3j | 30 | 86 | - |
| 11 |
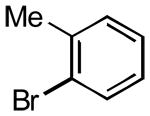
|
3k | 30 | 85 | - |
| 12 |
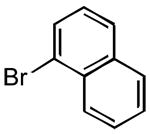
|
3l | 35 | 85 | - |
| 13e |
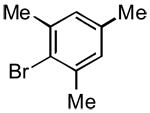
|
3m | 60 | 77 | - |
| 14e |
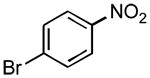
|
3n | 90 | 80f | 99:1g |
Reaction employed 1.3 equiv of K+1−.
Yields are based on chromatographically homogeneous products.
Ratios of cross/homocoupling are based on isolated yields unless otherwise mentioned.
Product was found to be 97% pure by GC analysis
Reaction employed 1.5 equiv of K+1− and dppbO (5 mol%).
Yield based on conversion as determined by 1H NMR.
Ratio of cross-/homocoupling determined by 1H NMR integration.
The rate of reaction was not sensitive to sterically demanding groups on the aryl bromide. Indeed, the 2-substituted aryl bromides reacted at a rate comparable to the corresponding 4-substituted isomers. Substrates bearing both electron-withdrawing (Table 1, entry 10) and electron-donating (Table 1, entry 11) substituents at the 2-position reacted to give the desired biaryl products in good yields. The successful coupling of the sterically hindered 2-bromomesitylene dramatically illustrated this point (Table 1, entry 13). The standard reaction conditions afforded only a 60% conversion with visible formation of Pd black after 10 min. Using 1.5 equiv of the arylsilanolate and the hemilabile ligand dppbO, the reaction was complete in 90 min and the biaryl product was isolated in good yield (77%). Cross-coupling reactions of aryl bromides bearing a heteroatom substituent at the 2-position were problematic and provided only 20% conversion in the best case. These groups containing Lewis basic atoms (e.g. O, N) most likely coordinate the metal center and thus inhibit productive cross-coupling from occurring.xxi On the other hand, reaction of K+1− with meta-substituted aryl bromides proceeded smoothly as both the 3-chloro as well the labile 3-triethylsilyloxy methyl substituents were compatible (Table 1, entries 8-9). The stability of the sensitive silicon-protecting group illustrates the mildness of the reaction conditions even at elevated temperatures.
Although the effectiveness of Ph3P(O) as a ligand had been established for the cross-coupling of K+1−, we nonetheless needed to perform the preparative reactions in the absence of a ligand for direct comparisons. Therefore, a representative set of substrates were subjected to the cross-coupling reaction conditions in the absence of Ph3P(O). Surprisingly, the preparative cross-coupling of K+1− with 4-bromobenzotrifluoride using APC (2.5 mol%) in toluene at 90 °C went to completion in 30 min without Ph3P(O). This result was contrary to the results obtained during the kinetic studies of this system. Interestingly, the cross-coupling in the absence of the ligand as rapid and general for the cases tested (Table 2). Both ortho and para-substituted aryl bromides reacted quickly all reaching >99% conversion. The reaction with the 4-bromobenzotrifluoride was repeated on a preparative scale and a 76% isolated yield was achieved with 7% undesired homocoupling product. These results contradicted those obtained during the kinetic analysis of this system. Thus, to reconcile the disparate behavior, it was necessary to revisit the previous examples to more precisely understand the effect of added phosphine oxide.
Table 2. Ligandless Cross-Coupling of K+1−with Substituted Aryl Bromidesa.
| ||||
|---|---|---|---|---|
| entry | aryl bromide | product | time, min | conversion,%b |
| 1 |
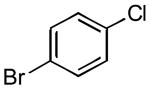
|
3b | 60 | 100 |
| 2 |
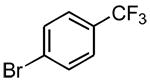
|
3c | 30 | 100 (76%)c,d |
| 3 |
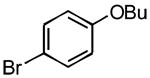
|
3f | 60 | > 99 |
| 4 |
|
3j | 30 | 100 |
| 5 |
|
3k | 30 | > 95 |
Reaction employed 1.3 equiv of K+1−.
Conversion determined by 1H NMR integration of the corresponding aryl bromide
Yield in parentheses based on pure, isolated material
7% homocoupling product isolated
2.3 The Role of Phosphine Oxide
The principle difference between the kinetic runs illustrated in Figure 1, and the results in Table 2 was the frequent sampling of the former reactions that could have compromised the inert atmosphere. To identify the impact of phosphine oxide additives on the rate and reproducibility of the cross-coupling under non-optimal reaction conditions, several representative bromides (2b, 2c, 2f, 2k) were reexamined. For operational simplicity and to assay the robustness of the procedure, all reagents (except the hygroscopic silanolate) were weighed open to the atmosphere and added to the reaction flask without taking the usual precautions to remove oxygen. Each reaction (0.5 mmol) was performed in triplicate in side-by-side experiments both with and without Ph3P(O) and the progress was monitored by GC analysis. The graphs in Figure 2 show the results for each of the four substrates. The blue lines represent the reactions run with Ph3P(O) and the red lines represent those without. As indicated in the legend, pairs of experiments executed side-by-side are represented by the same icons (squares, circles, diamonds, etc.). In addition, the effect of scale was examined and is illustrated by the dashed lines (2.0 mmol scale) on the charts for 2c and 2f. The beneficial effect of Ph3P(O) is immediately apparent in the coupling with 2b (Figure 2). In the presence of this ligand, the reaction proceeded smoothly and with excellent reproducibility (conversions 59–67%). In contrast, in the absence of Ph3P(O) the reaction was irreproducible and afforded consistently lower yields (18-57%). A similar irreproducibility was seen with 2c. In the presence of Ph3P(O) the reactions tracked nearly identically affording 76-78% conversion in 35 min, whereas the ligandless runs afforded 34-62% conversion in the same time. When the scale was increased four-fold, the yields for both reaction improved, nonetheless, the yield in the presence of Ph3P(O) remained higher (89 vs. 74%). Similar results were seen with bromides 2f and 2k. In both cases the presence of PPh3(O) resulted in improved conversion and reproducibility of the reaction. The striking behavior of 2-bromotoluene (2k) deserves comment. Despite the steric encumbrance, this substrate reacted faster than any other in the group reaching completion in less than 10 min in the presence of the ligand. As clearly seen in the graph, the ligandless reactions were slower and less reproducible.
Figure 2.
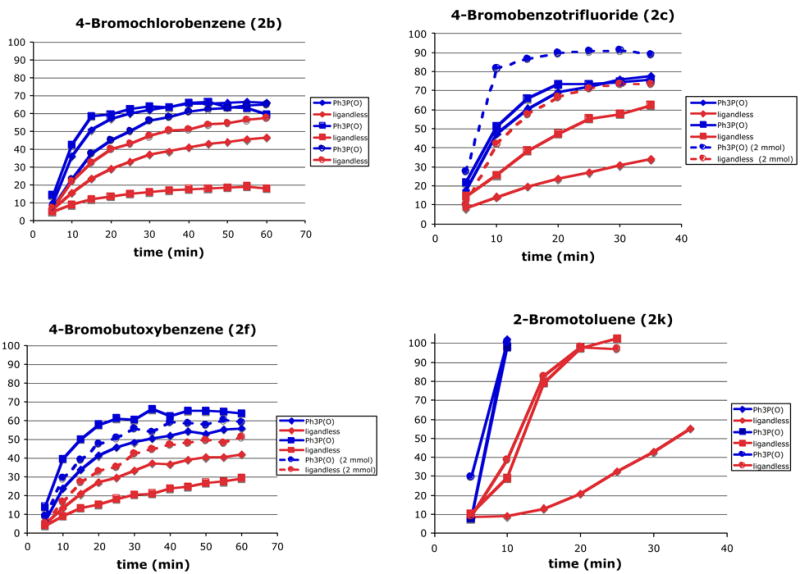
Comparison of reaction profiles.
3. Discussion
The results in Table 1 and Figure 2 clearly show that the phosphine oxide additive has a salutary effect in the cross-coupling reaction. However, the origin of this effect is not well understood, but may arise from a weak stabilizing interaction with the palladium catalyst. Because these reactions are performed in a non-coordinating solvent (toluene), stabilization from the solvent is not likely. Therefore, the phosphine oxide could be serving as a labile ligand that can prevent catalyst decomposition by a number of mechanisms including oxidation or agglomeration.
Although phosphine oxides are not generally recognized as good ligands for late transition metalsxxii a dppf(O)2palladium(II)Cl2 complex has been reported.xxiii The existence of this complex demonstrates the ability of phosphine oxides to bind directly to Pd(II), but no discrete coordination chemistry to Pd(0) has been reported and no synthetic transformation of these species has been described. On the other hand, there is a wealth of knowledge (both structural and synthetic) on the use of bisphosphine monooxides as “hemilabile” ligands for palladium. Hemilabile ligands, which contain both soft and hard coordinating atoms are a promising new class of ligands for a variety of synthetic transformations.xxiv The ability for these ligands to stabilize late transition metals while forming weak metal complexes affording more active metal centers is of interest. The bisphosphine monoxides are effective in reactions such as carbonylation,xxv cycloaddition,xxvi and oligomerization of olefins.xxvii
How then does Ph3P(O) influence the coupling reactions by itself? An intriguing hypothesis is that the phosphine oxide stabilizes the Pd(0) atoms/particles formed from direct reduction of APC by the silanolate.xx The reduction of APC leads to the formation of highly active Pd nanoparticles that can readily catalyze reactions, but that can also form inactive clusters.xxviii Several methods have been developed to stabilize Pd nanoparticles, such as the use of aminomethylated TentaGel in a resin form.xxix In addition, Li has described the use of secondary phosphine oxides (P-bound) in the palladium-catalyzed coupling of aryl chlorides with phenylboronic acid.xxx One of the more creative uses of phosphine oxides as labile ligands was reported by Bawendi and coworkers who prepared a poly(ethylene glycol) phosphine oxide polymer to stabilize water-soluble palladium nanoparticles.xxxi Interestingly, an example of a Suzuki cross-coupling was provided, clearly demonstrating the ability of phosphine oxides to intervene productively in catalytic processes. In addition, phosphine oxides have been used extensively to stabilize nanoparticles of other metals.xxxii
The weak coordination of the phosphine oxide to the palladium catalyst prevents the precipitation of palladium black. Therefore, a constant catalyst loading is maintained throughout the reaction resulting in good reaction rates and reproducible yields. In the absence of this additive, the reactions proceed, but with varying rates and conversions. The irreproducibility might arise from variable degrees of oxygen contamination of the reaction apparatus resulting in inconsistent rates of catalyst decomposition. Thus, under rigorously controlled anaerobic conditions, the effect of the phosphine oxide is negligible and reactions proceed rapidly and with excellent conversion as was seen in the control experiments in Table 2. However, the reactions become more robust and do no require special precautions if Ph3P(O) is added. In this sense, the phosphine oxide can be seen as a “buffer” or scavenger that allows reactions to proceed, even under less than ideal conditions.
4. Conclusions
In summary, the cross-coupling of an electron-rich arylsilanolate with a variety of aryl bromides proceeds smoothly with Ph3P(O) as a ligand. The use of a preformed arylsilanolate in combination with an extremely labile ligand to provide unsymmetrical biaryls compares very favorably with the use of Cs2CO3 and arylsilanols. This method has the additional advantage that the silanolate is easily prepared and stored, and can be charged directly into the reaction vessel. Although the cross-coupling reactions also function in the absence of ligands, control studies showed that this process has a sensitivity to oxygen or moisture and the Ph3P(O) can serve as a “buffering” ligand that helps to stabilize highly active palladium nanoparticles throughout the catalytic cycle. Further studies to extend this process to other arylsilanolates along with investigations into the mechanism of the homocoupling pathway are ongoing and will be reported in due course.
5. Experimental Section
5.1. General Experimental Methods
See the Supporting Information
5.2. General Procedure I: Analysis and Kinetic Measurements for Cross-coupling of K+1−with 4-Bromobenzotrifluoride Using Different Phosphines (Oxides)
5.2.1. Kinetic measurement using 1,3-bis(diphenylphosphino)butane dioxide (dppb(O)2)
In a dry box, naphthalene (48 mg), APC (4.6 mg, 0.0125 mmol, 0.025 equiv), dppb(O)2 (11.0 mg, 0.025 mmol, 0.05 equiv), and 4-bromobenzotrifluoride (70 μL, 0.50 mmol, 1.0 equiv) were dissolved in dry toluene (1.0 mL) at room temperature in a one-neck, 5-mL round-bottom flask with a magnetic stir bar and fitted with a reflux condenser and three-way stop-cock (fitted with an argon inlet and a rubber septum). To this solution was added K+1− (165 mg, 0.75 mmol, 1.5 equiv). The flask was removed from the dry box, the stopcock purged with argon, and was placed in a 90 °C oil bath. Reaction progress was monitored by GC analysis. Sampling of the reaction was performed by removing 30-μL aliquots of the mixture by syringe and quenching the aliquots at regular intervals. The quench was performed as follows: the withdrawn aliquot of the reaction mixture was injected into 100 μL of a 10% aqueous solution of 2-dimethylaminoethanethiol hydrochloride. This yellow solution was diluted with 1.5 mL of ether and the organic layer was filtered through a 0.5 × 1.0 cm plug of silica gel. The filtrate was analysis by GC. Naphthalene, tR 1.53 min; 3c, tR 5.72 min (HP-1, 170 °C (5 min), 50 °C/min, 225 °C (2 min), 16 psi).
5.3. General Procedure II: Cross-Coupling of Potassium Dimethyl(4-methoxyphenyl)silanolate with Aryl Bromides (Table 1)
5.3.1. Butyl 4′-methoxy-4-biphenylcarboxylate (3a) (Table 1, entry 1)
In a dry box, an oven-dried, 5-mL, 1-necked, round-bottomed flask containing a magnetic stir bar equipped with a reflux condenser and a three-way argon inlet capped with a septum, APC (9 mg, 0.025 mmol, 2.5 mol %) and triphenylphosphine oxide (Ph3P(O), 14 mg, 0.05 mmol, 5 mol %) were combined. Dry, deoxygenated toluene (2.0 mL) and 4-bromo(tert-butyl)benzoate (257 mg, 1.0 mmol, 1.0 equiv) were added followed by potassium dimethyl(4-methoxyphenyl)silanolate K+1− (286 mg, 1.3 mmol, 1.3 equiv). The flask was closed-off to the atmosphere and transferred to a hood. The reaction mixture was heated to 90 °C under argon. After TLC analysis showed complete consumption of the aryl bromide, the mixture was cooled to rt and subjected to an aqueous work-up. The reaction mixture was poured onto H2O (10 mL) and was extracted with ethyl acetate (3 × 10 mL). The organics were washed with brine (10 mL) and dried (Mg2SO4). The combined organics were concentrated in vacuo and the residue was purified by column chromatography (SiO2, 30 × 200 mm SiO2, CH2Cl2/hexanes, 1/1) followed by recrystallization from ethanol to afford 234 mg of 3a as analytically pure colorless plates. mp 107-108 °C (EtOH); 1H NMR (500 MHz, CDCl3) δ 8.04 (d, 2 H, J = 8.8 Hz, HC(3)), 7.60 (d, 2 H, J = 8.8 Hz, HC(2)), 7.57 (d, 2 H, J = 8.8 Hz, HC(2′)), 7.00 (d, 2 H, J = 8.8 Hz, HC(3′)), 3.86 (s, 3 H, H3CO(3″)), 1.62 (s, 9 H, H3C(2″)); 13C NMR (126 MHz, CDCl3) δ 165.7 C(5), 159.7 C(4′), 144.7 C(2′), 132.5 C(1), 130.1 C(1′), 129.9 C(3), 128.3 C(2′), 126.3 C(2), 114.3 C(3′), 80.8 C(1″), 55.3 C(3″), 28.2 C(2″); IR (KBr) 2979 (m), 2925 (m), 2827 (w), 1938 (w), 1892 (w), 1871 (w), 1709 (s), 1601 (s), 1531 (m), 1497 (m), 1452 (m), 1372 (m), 1301 (s), 1170 (s), 1112 (s), 1037 (s), 1015 (m), 833 (s), 774 (s), 698 (w) cm−1; MS (EI, 70eV) m/z 284 (M+, 4), 228 (100), 213 (21), 185 (21), 139 (12), 117 (9), 91 (6); Rf 0.30 (hexanes/CH2Cl2, 2/1) [silica gel, UV (256, 366)]; Anal. Calcd for C18H20O3: C, 76.03; H, 7.09. Found: C, 75.86; H, 7.22.
5.3.2. 4-Butoxy-4′-methoxybiphenyl (3f) (Table 1, entry 6)
Following General Procedure II, APC (15 mg, 0.025 mmol, 2.5 mol %), Ph3P(O) (23 mg, 0.05 mmol, 5 mol %), toluene (3.3 mL), 1-bromo-4-butoxybenzene (174 μL, 1.0 mmol, 1.0 equiv) and K+1c− (286 mg, 1.3 mmol, 1.3 equiv) were combined and heated to 90 °C. After 60 min, the mixture was cooled to rt and subjected to an aqueous work-up. Purification by column chromatography (SiO2, 40 mm × 200 mm, hexanes/CH2Cl2, gradient 4/1 to 2/1) followed by recrystallization afforded 155 mg (61%) of 3f as colorless plates. mp 132-133 °C (EtOH); 1H NMR (500 MHz, CDCl3) δ 7.49 (d, 2 H, J = 8.6 Hz, HC(2)), 7.48 (d, 2 H, J = 8.8 Hz, HC(2′)), 6.97 (d, 2 H, J = 8.6 Hz, HC(3)), 6.96 (d, 2 H, J = 8.8 Hz, HC(3′)), 4.01 (t, 2 H, J = 6.5 Hz, HC(1″)), 3.85 (s, 3 H, HC(1″)), 1.80 (tt, 2 H, J1 = 6.5 Hz, J2 = 7.5 Hz, HC(2″)), 1.52 (tq, 2 H, J1 = J2 = 7.5 Hz, HC(3″)), 1.00 (t, 3 H, J = 7.5Hz, HC(4″)); 13C NMR (126 MHz, CDCl3) δ 158.6 C(4), 158.3 C(4′), 133.5 C(1), 133.2 C(1′), 127.7 C(2), 127.7 C(2′), 114.7 C(3), 114.1 C(3′), 67.7 C(1″), 55.3 C(1″), 31.3 C(2″), 19.2 C(3″), 13.8 C(4″); IR (KBr) 2950 (s), 2925 (s), 2873 (m), 2372 (w), 2056 (w), 1891 (w), 1602 (s), 1501 (s), 1470 (m), 1398 (m), 1323 (m), 1274 (s), 1263 (s), 1178 (s), 1137 (m), 1121 (m), 1075 (w), 1044 (s), 1005 (m), 969 (w), 819 (s), 806 (s) cm−1; MS (EI, 70eV) m/z 256 (M+, 91), 200 (100), 185 (49), 157 (15), 128 (12), 102 (3); Rf 0.27 (hexanes/CH2Cl2, 2/1) [silica gel, UV]; Anal. Calcd for C17H20O2: C, 79.65; H, 7.86. Found: C, 79.85; H, 7.90.
5.3.3. 3-Chloro-4′methoxybiphenyl (3h) (Table 7, entry 8)
Following General Procedure II, APC (9 mg, 0.025 mmol, 2.5 mol %), Ph3P(O) (14 mg, 0.05 mmol, 5 mol %), toluene (2.0 mL), 3-chlorobromobenzene (117 μL, 1.0 mmol, 1.0 equiv) and K+1c− (286 mg, 1.3 mmol, 1.3 equiv) were combined and heated to 90 °C. After 60 min, the mixture was cooled to rt and subjected to an aqueous work-up. Purification by column chromatography (SiO2, 30 mm × 200 mm, hexanes/CH2Cl2, gradient 7/1 to 6/1) followed by recrystallization afforded 189 mg (86%) of 3h as analytically pure colorless plates. mp 56-57 °C (EtOH); 1H NMR (500 MHz, CDCl3) δ 7.55 (t, 1 H, J1=J2 = 1.9 Hz, HC(2)), 7.51 (d, 2 H, J = 8.8 Hz, HC(2′)), 7.43 (dt, 1 H, J1=J2 = 1.3 Hz, J3=7.7 Hz, HC(4)), 7.34 (t, 1 H, J = 7.7 Hz, HC(5)), 7.28 (dq, 1 H, J1 = 1.3 Hz, J2 = 2.1 Hz, J3 =7.7 Hz, HC(6)), 6.99 (d, 2 H, J = 8.8 Hz, HC(3′)), 3.86 (s, 3 H, H3CO(1″)); 13C NMR (126 MHz, CDCl3) δ 159.5 C(4′), 142.6 C(1), 134.6 C(3), 132.2 C(1′), 129.9 C(5), 128.1 C(2′), 126.8 C(2), 126.6 C(6), 124.8 C(4), 114.3 C(3′), 55.3 C(1″); IR (KBr) 3059 (w), 2956 (m), 2935 (w), 2904 (w), 2832 (w), 2362 (m), 2341 (w), 1894 (w), 1605 (m), 1576 (m), 1561 (m), 1512 (m), 1470 (m), 1390 (w), 1290 (m), 1279 (m), 1248 (s), 1184 (m)l, 1101 (m), 1021 (s), 884 (w), 832 (s), 786 (s), 687 (s), 667 (m) cm1; MS (EI, 70eV) m/z 220 (33), 218 (M+, 100), 205 (12), 203 (38), 177 (13), 175 (41), 168 (3), 149 (15), 139 (15), 113 (9), 98 (5), 87 (13), 75 (19), 63 (27); Rf 0.28 (hexanes/CH2Cl2, 6/1) [silica gel, UV]; Anal. Calcd for C13H11ClO: C, 71.40; H, 5.07. Found: C, 71.25; H, 4.99.
5.3.4. 4′-Methoxy-3-(triethylsiloxymethyl)biphenyl (3i) (Table 1, entry 9)
Following General Procedure II, APC (9 mg, 0.025 mmol, 2.5 mol %), Ph3P(O) (14 mg, 0.05 mmol, 5 mol %), toluene (2.0 mL), 1-bromo-3-(triethylsiloxymethyl)benzene (301 mg, 1.0 mmol, 1.0 equiv) and K+1c− (286 mg, 1.3 mmol, 1.3 equiv) were combined and heated to 90 °C. After 60 min, the mixture was cooled to rt and subjected to an aqueous work-up. Purification by column chromatography (SiO2, 30 mm × 220 mm, hexanes/CH2Cl2, 6/1) followed by reverse phase chromatography (reverse phase silica, 20 mm & times; 150 mm, MeOH/H2O, 12/1) afforded 220 mg (67 %) of 3i as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.57 (m, 1 H, HC(2)), 7.56 (d, 2 H, J = 8.8 Hz, HC(2′)), 7.46 (d, 1 H, J = 7.6 Hz, HC(6)), 7.40 (t, 1 H, J = 7.6 Hz, HC(5)), 7.31 (d, 1 H, J = 7.6 Hz, HC(4)), 7.00 (d, 2 H, J = 8.8 Hz, HC(3′)), 4.82 (s, 2 H, HC(2″)), 3.87 (s, 3 H, HC(1″)), 1.03 (t, 9 H, J= 7.8 Hz, HC(4″)), 0.70 (q, 6 H, J = 7.8 Hz, HC(3″)); 13C NMR (126 MHz, CDCl3) δ 159.1 C(4), 141.7 C(3), 140.7 C(1′), 133.8 C(1), 128.6 C(5), 128.1 C(2), C(2′), 125.4 C(6), 124.6 C(4), 114.1 C(3′), 64.8 C(7), 55.3 C(1″), 6.8 C(3″), 4.5 C(2″); IR (neat) 3023 (w), 2950 (s), 2909 (s), 2863 (s), 2361 (w), 2046 (w), 1886 (w), 1697 (w), 1607 (s), 1519 (s), 1483 (s), 1460 (s), 1439 (s), 1411 (m), 1372 (m), 1284 (s), 1248 (s), 1181 (s), 1108 (s), 1074 (s), 1008 (s), 971 (m), 858 (m), 829 (s), 783 (s), 744 (s) cm−1; MS (EI, 70eV) m/z 328 (M+, 56), 299 (70), 269 (26), 197 (100), 165 (9), 154 (7), 99 (9), 59 (4); HRMS (EI, 70eV) m/z calcd for C20H28O2Si (M+): 328.1859; found: 328.1862; Rf 0.38 (hexanes/CH2Cl2, 1/1) [silica gel, UV, CAN]; GC tR = 9.00 min (HP-1, 150 °C, 2 min, 50 °C/min, 250 °C, 6.5 min, 16 psi).
5.3.5. 2-Chloro-4′-methoxybiphenyl (3j) (Table 1, entry 10)
Following General Procedure II, APC (9 mg, 0.025 mmol, 2.5 mol %), Ph3P(O) (14 mg, 0.05 mmol, 5 mol %), toluene (2.0 mL), 2-chlorobromobenzene (116 μL, 1.0 mmol, 1.0 equiv) and K+1c− (286 mg, 1.3 mmol, 1.3 equiv) were combined and heated to 90 °C. After 30 min, the mixture was cooled to rt and subjected to an aqueous work-up. Purification by column chromatography (SiO2, 30 mm × 220 mm, hexanes/CH2Cl2, 6/1) followed by Kugelrohr distillation afforded 188 mg (86%) of 3j as ananalytically pure, colorless oil. Bp 130 °C (ABT, 0.2 mmHg); 1H NMR (500 MHz, CDCl3) δ 7.49 (dd, 1 H, J1 = 1.3 Hz, J2 = 7.9 Hz, HC(3)), 7.42 (d, 2 H, J = 8.8 Hz, HC(2′)), 7.36 (dd, 1 H, J1 = 1.9 Hz, J2 = 7.3 Hz, HC(6)), 7.32 (ddd, 1 H, J1 = 1.5 Hz, J2 = J3 = 7.9 Hz, HC(5)), 7.28 (ddd, 1 H, J1 = 1.9 Hz, J2 =J3 = 7.9 Hz, HC(4)), 7.01 (d, 2 H, J = 8.8 Hz, HC(6)), 3.88 (s, 3 H,H3CO(1″)); 13C NMR (126 MHz, CDCl3) δ 159.1 C(4′), 140.1 C(1), 132.6 C(2), 131.7 C(1′), 131.4 C(6), 130.6 C(2′), 129.9 C(3), 128.2 C(4), 126.8 C(5), 113.4 C(3′), 55.2 C(1″); IR (neat) 3062 (m), 3002 (m), 2957 (m), 2935 (m), 2836 (m), 2546 (w), 2359 (w), 2047 (w), 1888 (w), 1613 (w), 1579 (m), 1516 (s), 1469 (s), 1441 (s), 1408 (m), 1296 (s), 1250 (s), 1179 (s), 1126 (m), 1109 (m), 1074 (m), 1037 (s), 1018 (m), 1002 (m), 946 (w), 831 (s), 805 (m), 757 (s), 733 (s), 665 (m) cm−1; MS (EI, 70eV) m/z 220 (36), 218 (M+, 100), 205 (5), 203 (15), 177 (5), 175 (13), 149 (4), 139 (10), 115 (3), 75 (2), 63 (2); TLC Rf 0.43 (hexanes/CH2Cl2, 6/1) [silica gel, UV]; Anal. Calcd for C13H11ClO: C, 71.40; H, 5.07. Found: C, 71.21; H, 4.99.
5.4. General Procedure III: Cross-Coupling of Potassium Dimethyl(4-methoxyphenyl)silanolate with Aryl Bromides Without a Ligand (Table 2)
5.4.1. 4′-Methoxy-4-(trifluoromethyl)biphenyl (3c) (Table 2, entry 2)
In an oven-dried, 5-mL, 1-necked, round-bottomed flask containing a magnetic stir bar equipped with a reflux condenser and an argon inlet capped with a septum, was charged APC (9 mg, 0.025 mmol, 2.5 mol %). The flask was sequentially evacuated and purged with argon twice. Toluene (1.0 mL), and 4-bromobenzotrifluoride (138 μL, 1.0 mmol, 1.0 equiv) were added followed by dimethyl(4- methoxyphenyl)silanolate K+1c− (286 mg, 1.3 mmol, 1.3 equiv). The reaction mixture was heated to 90 °C under argon. After TLC analysis showed complete consumption of the aryl bromide, the mixture was cooled to rt and subjected to an aqueous work-up. . The reaction mixture was poured onto H2O (10 mL) and was extracted with ethyl acetate (3 × 10 mL). The organics were washed with brine (10 mL) and dried (Mg2SO4). The combined organics were concentrated in vacuo and the residue was purified by column chromatography (SiO2, 30 × 200 mm SiO2, hexanes/CH2Cl2, 5/1) followed by recrystallization from ethanol to afford 191 mg of 3c as colorless flake crystals. The spectroscopic data matched those from the literature.xiv mp121-122 °C (EtOH); 1H NMR (500 MHz, CDCl3) δ 7.68-7.64 (m, 4 H, HC(2), HC(3)), 7.55 (d,2 H, J = 8.8 Hz, HC(2′)), 7.01 (d, 2 H, J = 8.8 Hz, HC(3′)), 3.87 (s, 3 H, H3CO(1″)); 13C NMR (126 MHz, CDCl3) δ 159.9 C(4′), 144.3 C(1), 132.2 C(1′), 128.7 (q, JCF = 32.2 Hz, C(4)), 128.3 C(2′), 126.9 C(2), 125.7 (q, JCF = 3.7 Hz, C(3)), 124.4 (q, JCF = 271.6 Hz, C(5)), 114.4 C(3′), 55.3 C(1″); 19F NMR (376 MHz, CDCl3) δ -62.6 CF3(5); TLC Rf 0.63 (hexanes/CH2Cl2, 4/1) [silica gel, UV]; GC tR = 6.55 min (HP-1, 150 °C (5 min), 50 °C/min, 250 °C (2 min), 16 psi).
5.5. General Procedure IV: Comparison of Cross-Coupling of Potassium Dimethyl(4-methoxyphenyl)silanolate with Aryl Bromides with and without Triphenylphosphine Oxide
5.5.1. 4-Bromobenzotrifluoride using Ph3P(O)
In a dry box, K+1− (143 mg, 0 50 mmol, 13 equiv) was charged into an oven-dried vial and sealed. The vial was transferred from the dry box into the hood. Naphthalene (31.7 mg), APC (4.6 mg, 0.0125 mmol, 0.025 equiv), Ph3P(O) (7.0 mg, 0.025 mmol, 0.05 equiv) were charged into a one-neck, 5-mL round-bottom flask with a magnetic stir bar and fitted with a reflux condenser (during weighing, the round-bottom flask was kept open to an ambient atmosphere). The condenser capped with an argon adapter and the solids were dissolved in dry toluene (1.0 mL) resulting in a yellow solution. 4-Bromobenzotriflouride (69 μL, 0.5 mmol, 1.0 equiv) was then added by removing the argon adapter and adding the material via syringe. To this solution was added the solid K+1− by again removing the argon adapter and adding the material as a solid. The flask was opened to an argon atmosphere, and was placed in a 90-°C oil bath. Reaction progress was monitored by GC analysis. The reaction was sampled by removing 30-μL aliquots of the mixture syringe and quenching the aliquots at regular intervals (5 min). The quench was performed as follows: the withdrawn aliquot of the reaction mixture was injected into 100 μL of a 10% aqueous solution of 2-dimethylaminoethanethiol hydrochloride. This yellow solution was diluted with 1.5 mL of ether and the organic layer was filtered through a 0.5 × 1.0 cm plug of silica gel. The filtrate was subjected to GC analysis. Naphthalene, tR 1.53 min; 3c, tR 5.72 min (HP-1, 170 °C (5 min), 50 °C/min, 225 °C (2 min), 16 psi).
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health for generous financial support (GM 63167). R.C.S acknowledges the University of Illinois for a graduate fellowship. S.A.T. thanks Johnson and Johnson PRI for a graduate fellowship.
Footnotes
Supporting Information Available: Complete experimental details for all preparative procedures along with full characterization of all starting materials and products as well as raw data for kinetic analyses (46 pages) are provided.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (i).de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- (ii).(a) Farina V, Krishnamurthy V, Scott WJ. The Stille Reaction. Wiley-Interscience; New York: 1998. [Google Scholar]; (b) Stille J. Angew Chem Int Ed Engl. 1986;25:508–524. [Google Scholar]; (c) Mitchell TN. In: Metal-Catalyzed Cross-Coupling Reactions. de Meijere A, Diederich F, editors. Vol. 1. Wiley-VCH; Weinheim, Germany: 2004. Chapter 3. [Google Scholar]
- (iii).(a) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483. [Google Scholar]; (b) Suzuki AJ. Organomet Chem. 1999;576:147–168. [Google Scholar]; (c) Miyaura N. In: Metal-Catalyzed Cross-Coupling Reactions. de Meijere A, Diederich F, editors. Vol. 2. Wiley-VCH; Weinheim, Germany: 2004. Chapter 2. [Google Scholar]
- (iv).Knochel P, Calaz MI, Hupe E. In: Metal-Catalyzed Cross-Coupling Reactions. de Meijere A, Diederich F, editors. Vol. 1. Wiley-VCH; Weinheim, Germany: 2004. Chapter 11. [Google Scholar]
- (v).Kürti L, Czakó B. Strategic Application of Named Reaction in Organic Syntheses. Elsevier; Burlington: 2005. pp. 310–438.pp. 448 [Google Scholar]
- (vi).(a) Hiyama TJ. Organomet Chem. 2002;653:58–61. [Google Scholar]; (b) Hiyama T, Hatanka Y. Pure and Appl Chem. 1994;66:1471–1478. [Google Scholar]; (c) Hiyama T. In: Metal-Catalyzed, Cross-Coupling Reactions. Diederich F, Stang PJ, editors. Chapter. 10 Wiley-VCH; Weinheim, Germany: 1998. [Google Scholar]
- (vii).(a) Denmark SE, Sweis RF. Acc Chem Res. 2002;35:835–846. doi: 10.1021/ar020001r. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Sweis RF. J Am Chem Soc. 2001;123:6439–6440. doi: 10.1021/ja016021q. [DOI] [PubMed] [Google Scholar]; (c) Denmark SE, Sweis RF. In: Metal-Catalyzed Cross-Coupling Reactions. de Meijere A, Diederich F, editors. Vol. 1. Wiley-VCH; Weinheim, Germany: 2004. Chapter 4. [Google Scholar]
- (viii).Hirabayashi K, Mori A, Kawashima J, Sugoro M, Nishihara Y, Hisyama T. J Org Chem. 2000;65:5342–5349. doi: 10.1021/jo000679p. [DOI] [PubMed] [Google Scholar]
- (ix).Denmark SE, Tymonko SA. J Org Chem. 2003;68:9151–9154. doi: 10.1021/jo0351771. [DOI] [PubMed] [Google Scholar]
- (x).(a) Hatanaka Y, Hiyama T. Tetrahedron Lett. 1990;31:2719–2722. [Google Scholar]; (b) Hatanaka Y, Fukushima S, Hiyama T. Chem Lett. 1989:1711–1714. [Google Scholar]; (c) Homsi F, Hosoi K, Nozaki K, Hiyama T. J Organomet Chem. 2001;624:208–216. [Google Scholar]; (d) Hagiwara E, Gouda K, Hatanaka Y, Hiyama T. Tetrahedron Lett. 1997;38:439–442. [Google Scholar]; (e) Hatanaka Y, Goda K, Okahara Y, Hiyama T. Tetrahedron. 1994;50:8301–8316. [Google Scholar]
- (xi).(a) Nakao Y, Oda T, Sahoo AK, Hiyama T. J Organomet Chem. 2003;687:570–573. [Google Scholar]; (b) Sahoo AK, Nakao Y, Hiyama T. Chem Lett. 2004;33:632–633. [Google Scholar]
- (xii).(a) Mowery ME, DeShong P. Org Lett. 1999;1:2137–2140. doi: 10.1021/ol991186d. [DOI] [PubMed] [Google Scholar]; (b) Lee HM, Nolan SP. Org Lett. 2000;2:2053–2055. doi: 10.1021/ol005956t. [DOI] [PubMed] [Google Scholar]; (c) Mowery ME, DeShong P. J Org Chem. 1999;64:1684–1688. doi: 10.1021/jo982463h. [DOI] [PubMed] [Google Scholar]
- (xiii).A clever, internally activated, non-fluoride coupling of aryl[(2-hydroxymethyl)phenyl]dimethylsilanes has also been reported. Nakao Y, Imanaka H, Sahoo AK, Yada A, Hiyama T. J Am Chem Soc. 2005;127:6952–6953. doi: 10.1021/ja051281j.
- (xiv).(a) Denmark SE, Ober MH. Org Lett. 2003;5:1357–1360. doi: 10.1021/ol034328j. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Ober MH. Adv Synth Cat. 2004;346:1703–1714. [Google Scholar]
- (xv).Denmark SE, Sweis RF. J Am Chem Soc. 2004;126:4876–4882. doi: 10.1021/ja0372356. [DOI] [PubMed] [Google Scholar]
- (xvi).For a discussion of cross-coupling reactions using silanolates see: Denmark SE, Baird JD. Chem Eur J. 2006;12:4954–4963. doi: 10.1002/chem.200600034.
- (xvii).Denmark SE, Kallemeyn JM. J Am Chem Soc. 2006 doi: 10.1021/ja065988x. ASAP. [DOI] [PubMed] [Google Scholar]
- (xviii).Denmark SE, Baird JD. Org Lett. 2006;8:793–795. doi: 10.1021/ol053165r. [DOI] [PubMed] [Google Scholar]
- (xix).For comparison see: Denmark SE, Baird JD. Org Lett. 2004;6:3649–3652. doi: 10.1021/ol048328a.
- (xx).We have subsequently found that the reduction of APC does not involve phosphines, but rather silanolates, see: Denmark SE, Smith RC. Synlett. 2006:2921–2928.
- (xxi).Basu B, Freijd T. Acta Chem Scand. 1996;50:316–322. [Google Scholar]
- (xxii).(a) Slone CS, Weinberger DA, Mirkin CA. Prog Inorg Chem. 1999;48:233–350. [Google Scholar]; (b) Parr J. In: Comprehensive Coordination Chemistry II. McCleverty JA, Meyer TJ, editors. Vol. 1. Elsevier; Amsterdam: 2004. p. 280. [Google Scholar]; (c) Burford N. Coord Chem Rev. 1992;112:1–18. [Google Scholar]
- (xxiii).Yeo JSL, Vittal JJ, Hor TSA. Chem Commun. 1999:1477–1478. [Google Scholar]
- (xxiv).Grushin VV. Chem Rev. 2004;104:1629–1662. doi: 10.1021/cr030026j. [DOI] [PubMed] [Google Scholar]
- (xxv).(a) Wegman RW, Abatjoglou AG, Harrison AM. J Chem Soc Chem Commun. 1987:1891–1892. [Google Scholar]; (b) Wegman RW, Schrek DJ. Eur Pat Appl EP 173170, 1986. 1986 [Google Scholar]; (c) Wegman RW, Abatjoglou AG. U.S. Patent 4670570. PCT Int Appl 8600888 1986. 1987
- (xxvi).(a) Faller JW, Liu X, Parr J. Chirality. 2000;12:325–337. doi: 10.1002/(SICI)1520-636X(2000)12:5/6<325::AID-CHIR5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]; (b) Faller JW, Parr J. Organometallics. 2000;19:1829–1832. [Google Scholar]; (c) Faller JW, Grimmond BJ, Curtis M. Organometallics. 2000;19:5174–5181. [Google Scholar]; (d) Faller JW, Lavoie AR, Grimmond BJ. Organometallics. 2002;21:1662–1666. [Google Scholar]; (e) Faller JW, Parr J. Organometallics. 2000;19:3556–3561. [Google Scholar]; (f) Faller JW, Parr J. Organometallics. 2001;20:697–699. [Google Scholar]
- (xxvii).Brassat I, Keim W, Killat S, Mothrath M, Mastrorilli P, Nobile CF, Suranna GP. J Mol Catal A. 2000;157:41–58. [Google Scholar]
- (xxviii).(a) Moreno-Mañas M, Pleixats R. Acc Chem Res. 2003;36:638–643. doi: 10.1021/ar020267y. [DOI] [PubMed] [Google Scholar]; (b) Hu J, Liu Y. Langmuir. 2005;21:2121–2123. doi: 10.1021/la0471902. [DOI] [PubMed] [Google Scholar]
- (xxix).Cho JK, Najman R, Dean TW, Ichihara O, Muller C, Bradley MJ. Am Chem Soc. 2006;128:6276–6277. doi: 10.1021/ja057480k. [DOI] [PubMed] [Google Scholar]
- (xxx).(a) Li GY. Angew Chem Int Ed. 2001;40:1513–1516. doi: 10.1002/1521-3773(20010417)40:8<1513::aid-anie1513>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]; (b) Li GY E.I. du Pont de Nemours and Co. US Patent 6124462. 2000
- (xxxi).Kim SW, Kim S, Tracy JB, Jasanoff A, Bawendi MG. J Am Chem Soc. 2005;127:4556–4557. doi: 10.1021/ja043577f. [DOI] [PubMed] [Google Scholar]
- (xxxii).Masala O, Seshadri R. Annu Rev Mater Res. 2004;34:41–81. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.