

Abstract

The classical geometry of the 6-endo transition state for nucleophilic additions into oxocarbenium ions can be perturbed by incorporating the reactive groups into medium sized rings, leading to the formation of 2,6-trans-dialkyl tetrahydropyrans. The bicyclic products exhibit inside-outside stereoisomerism, as seen in numerous macrolide natural products.
Intramolecular nucleophilic 6-endo additions into oxocarbenium ions provide cis-2,6-disubstituted tetrahydropyrans due to the favorable chair transition state in which the oxocarbenium ion has E-geometry and the substitutents occupy equatorial orientations.1 2,6-trans-Disubstituted tetrahydropyrans could, in principle, be accessed by switching the geometry of the oxocarbenium ion or forcing a substitutent into an axial orientation (Scheme 1). Accessing the trans-isomer as the major product is quite rare, however, with Loh’s reports2 of the axial preference for carboalkoxy groups being notable exceptions. The trans-stereochemical orientation is present in a number of biologically active macrolide natural products in which the tetrahydropyran is embedded in the macrocyclic core, including the cytotoxins apicularen A3 and leucascandrolide A.4 Notably, these compounds exhibit inside-outside stereoisomerism,5 in which one bridgehead hydrogen points into the macrocycle and one points outside. The biological activity of these structures and their structurally unique features provides ample justification for the development of new protocols for their synthesis.
Scheme 1.

Transition states for 6-endo cyclizations into oxocarbenium ions.
We have been engaged in the synthesis of tetrahydropyrans and related structures in which carbocation formation proceeds through oxidative carbon–hydrogen bond cleavage.6 Our application of this oxidative method to the formation of macrocyclic oxocarbenium ions6b is particularly relevant to the synthesis of 2,6-trans-disubstituted tetrahydropyrans. We hypothesized that we could exploit the geometric constraints of macrocyclic cores to direct the formation of these structures. This plan was predicated on the notion that, for certain tether lengths, oxocarbenium ion precursors to the trans-isomers would be thermodynamically favored over the corresponding precursors to the cis-isomers, particularly if the the macrocycle contained a strain-inducing element such as an alkyne or an E-alkene (Figure 1). Oxidative carbon–hydrogen bond cleavage is well-suited for this objective since the requisite strained oxocarbenium ion intermediates would be difficult to access through conventional ionization-based protocols.7 In this manuscript we report that propargyl ether-containing macrocycles undergo oxidative carbon–hydrogen bond cleavage to provide oxocarbenium ions that react with appended nucleophiles in a transannular manner to yield 2,6-trans-disubstituted tetrahydropyrans.8
Figure 1.

Macrocyclic precursor to 2,6-cis- and 2,6-trans-disubstituted tetrahydropyrans.
Success in this venture requires that the Z-oxocarbenium ion or the axial substituent not cause a prohibitive energetic penalty. We reasoned that alkynyl groups could satisfy this requirement based on their sterically-undemanding nature. We have reported6d that E- and Z-substituted oxocarbenium ions differ in energy by only ~0.4 kcal/mol, as shown by the conversion of 1 to a 1.6:1 mixture of 2 and 3 (Scheme 2). We prepared ether 4 as a test substrate to determine whether an axially-oriented alkynyl group would be energetically accessible in an oxidative cyclization reaction. Subjecting 4 to 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in 1,2-dichloroethane (DCE) resulted in the formation of tetrahydropyrones 5 and 6. Remarkably the 2,6-trans-diastereomer 5 was the major product in this reaction, demonstrating that the alkynyl group shows a slight preference for an axial orientation, as shown by transition state 7. Although this modestly contra steric result was initially surprising, the axial orientation is consistent with our recent postulate6e of an attractive interaction between π-electrons and the partial positive charge9 on the hydrogen of the oxocarbenium ion. The observation that the level of diastereocontrol was lower when the reaction was run in the polar solvent CH3NO2 is consistent with the electrostatic model. Additional support for this weak attractive interaction is provided by the increased selectivity in the cyclization of 8 to 9, in which the intermediate cation is destabilized relative to 7, and in the diminished selectivity in the cyclization of 10 to 11, in which the ester group reduces the ability of the alkynyl group to engage in an electrostatic interaction.
Scheme 2.

Alkynes in the synthesis of 2,6-trans-disubstituted tetrahydropyrans.
The successful demonstrations that alkynyl groups show a slight preference for an axial orientation and that alkynyl-substituted oxocarbenium ions can access a Z-configuration without accruing a significant energetic penalty provide two options for preparing bridged bicyclic tetrahydropyrans with inside-outside stereochemical relationships. In consideration of the lower reactivity of propargylic ethers toward DDQ relative to benzylic and allylic ethers6d we initially examined substrates in which the alkynyl group is designed to adapt an axial alignment in the cyclization transition state. The results of these studies are shown in Table 1.
Table 1.
Stereoselective formation of inside-outside bridged bicycles.a
| entry | substrateb | product | time | yield (%) (dr) |
|---|---|---|---|---|
| 1 | 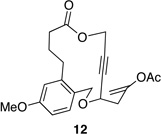 |
 |
4 h | 72 |
| 2 | 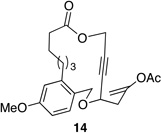 |
 |
1 h | 81 |
| 3 | 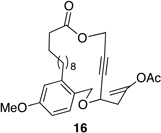 |
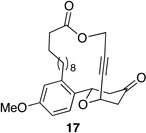 |
0.67 h | 65c (dr = 6:1)d |
| 4 | 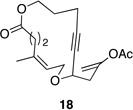 |
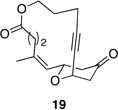 |
26 h | 54 |
| 5 | 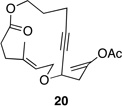 |
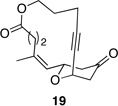 |
43 h | 35 |
| 6 | 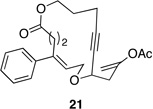 |
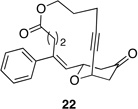 |
0.25 h | 72 |
Representative procedure: DDQ (2–3 equiv) was added to a 0.1 M solution of the substrate (1 equiv), LiClO4 (0.2 equiv), and 2,6-dichloropyridine (4 equiv) in DCE at rt. The reaction was stirred at rt for the indicated period for the indicated time. Please see the Supporting Information for details.
Please see the Supporting Information for details on substrate syntheses.
Combined yield of diastereomers.
Determined by integrating characteristic signals in the 1H NMR spectrum.
These reactions all proceed at room temperature and many provide good yields of the desired products. The reactions proceed more quickly when longer tether lengths are employed (entries 1–3). This most likely results from a reduction in the strain that results from the formation of the intermediate oxocarbenium ion, and is consistent with studies10 showing that cation stability is an important determinant in the rate of oxidative carbon–hydrogen bond cleavage. Stereochemical assignments were made based on coupling constants in 1H NMR spectra. The coupling constants also confirmed that the alkynyl groups occupy an axial orientation, as expected based on A-values. The formation of the 2,6-cis-disubstituted isomer was not observed until the cyclization substrate contained a 19-membered ring (entry 3), in accord with the analysis in Figure 1. Allylic ethers are also effective promoters of macrocyclic oxocarbenium ion formation, although the reactions are slower in comparison to the electron-rich benzylic ethers (entry 1 vs entry 4). Of note, E-alkene substrate 20 reacted quite slowly and changed configuration during the reaction (entry 5). This result can also be attributed to the formation of strained intermediates and shows that alkene rotational barriers of carbon–carbon π-bonds are significantly dimished in oxocarbenium ions. The addition of an aryl group to the alkene promotes a much faster cyclization reaction that proceeds quite efficiently.
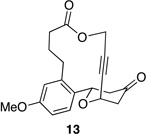
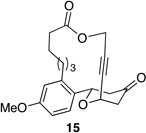
We also explored the possibility of preparing these bicylic structures through the intermediacy of alkynyl-substituted oxocarbenium ions. The results from this study are shown in Table 2. Propargylic ethers oxidize more slowly than allylic and benzylic ethers,5d resulting in lower yields of desired products and the need to heat these reactions for prolonged periods of time. The modest yields appear to result from non-specific decomposition of the strained oxocarbenium ions rather than overoxidation, although the reactions were not run with lower DDQ loadings because they were not kinetically viable under those conditions. All reactions produced the 2,6-trans-disubstituted stereoisomer as the major or exclusive product, with the cis-product only being observed for the cyclization of 16-membered ring substrate 27 (entry 3). Stereochemical assignments were based on 1H NMR coupling constants, and the structure of 24 was confirmed by crystallography.11 As noted in the reaction from Table 1, the reactions were marginally more efficient for substrates with larger rings (entry 1 vs entry 3). Stereoselectivity was also observed in the formation of tertiary ethers (entries 4 and 5). The difference in rates for substrates 29 and 31 can be attributed to the greater capacity of the proximal carbonyl group in 29 to destabilize the intermediate carbocation relative to the oxygen atom in 31. The reactions that proceed through the more highly-substituted oxocarbenium ions can be conducted at lower temperatures, though no improvement is seen in the yield. While the inductive effect that arises from the carbonyl group being proximal to the oxocarbenium ion has significant impact on reaction rates, increasing the distance between the carbocation intermediate and the oxygen of the lactone group has a negligible impact on cyclization efficiency (entry 1 vs entry 2). Although all the results in this discussion are consistent with reasonable thermodynamic analyses of the intermediate oxocarbenium ions, kinetic effects might also play a role in the oxidation rates since macrocyclic constraints could limit access to the proper geometry for carbon–hydrogen bond cleavage.12
Table 2.
Reactions through alkynyl-stabilized oxocarbenium ions.a
| entry | substrateb | product | time | temp | yield (%) (dr) |
|---|---|---|---|---|---|
| 1 | 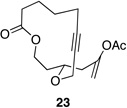 |
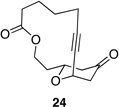 |
62 h | 50 °C | 36 |
| 2 | 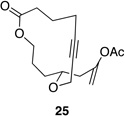 |
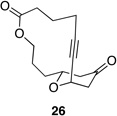 |
77 h | 40 °C | 36 |
| 3 | 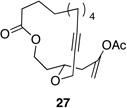 |
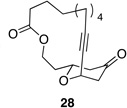 |
60 h | 40 °C | 46c (dr = 2:1)d |
| 4 | 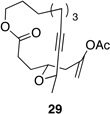 |
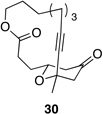 |
96 h | 30 °C | 40 |
| 5 | 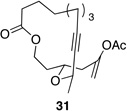 |
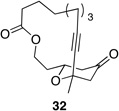 |
48 h | 30 °C | 39 |
Representative procedure: DDQ (2–3 equiv) was added to a 0.1 M solution of the substrate (1 equiv), LiClO4 (0.2 equiv), and 2,6-dichloropyridine (4 equiv) in DCE at rt. The reaction was stirred at the indicated period for the indicated time. Please see the Supporting Information for details.
Please see the Supporting Information for details on substrate syntheses.
Combined yield of diastereomers.
Determined by integrating characteristic signals in the 1H NMR spectrum.
Alkynes are not common structures in natural products or medicinal agents, but they are extremely versatile as precursors to numerous functional groups. To illustrate, alkyne 13 was subjected to the two step hydrosilylation/desilylation-based reduction that was reported by the Trost group13 to form E-alkene 33 (Scheme 3).14 This compound would be difficult to form directly due to the strong preference for the alkenyl group to occupy an equatorial orientation in the cyclization transition state, but the use of the alkynyl group for the cyclization followed by selective reduction makes this compound accessible. Moreover the vinylsilane intermediate can serve as a precursor to other functional groups through cross-coupling15 and oxidative16 transformations.
Scheme 3.

Selective alkyne functionalization.
We have shown that bridged ethers that show inside-outside stereoisomerism can be prepared through transannular cyclizations of macrocyclic propargylic ethers. Oxidative carbon–hydrogen bond cleavage was employed to generate the strained oxocarbenium ion intermediates in this study. These cyclizations are possible because the macrocycle constrains the geometry of the transition state and because of the sterically undemanding nature of alkynes. Two approaches to this objective were developed. One approach proceeds through the oxidation of a benzylic or allylic ether, where the alkynyl group adapts an axial orientation in the transition state, as is slightly preferred due to a possible electrostatic interaction. The other approach proceeds through the oxidation of propargylic ethers to form slighly disfavored but energetically accessible Z-oxocarbenium ion intermediates. The capacity of alkynes to act as versatile precursors to a number of functional groups makes the products of these reactions useful intermediates for the preparation of a range of compounds.
Supplementary Material
Acknowledgment
We thank the National Institutes of Health (GM062924) for generous support of this work. We thank Dr. Steve Geib (University of Pittsburgh) for crystallographic studies.
Footnotes
Supporting Information Available. Synthetic schemes for substrate syntheses, experimental procedures for coupling reactions, stereochemical determinations, crystallographic information, and characterization data for all new compounds. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent reviews, see: Olier C, Kaafarani M, Gastaldi S, Bertrand MP. Tetrahedron. 2010;66:413. Clarke PA, Santos S. Eur. J. Org. Chem. 2006:2045.
- 2.(a) Luo H-Q, Hu X-H, Loh T-P. Tetrahedron Lett. 2010;51:1041. [Google Scholar]; (b) Hu X-H, Liu F, Loh T-P. Org. Lett. 2009;11:1741. doi: 10.1021/ol900196w. [DOI] [PubMed] [Google Scholar]
- 3.Kunze B, Jansen R, Sasse R, Höfle G, Reichenbach H. J. Antibiot. 1998;51:1075. doi: 10.7164/antibiotics.51.1075. [DOI] [PubMed] [Google Scholar]
- 4.D’Ambrosio M, Guerriero A, Debitus C, Pietra F. Helv. Chim. Acta. 1996;79:51. [Google Scholar]
- 5.Alder RW, East SP. Chem. Rev. 1996;96:2097. doi: 10.1021/cr940246k. [DOI] [PubMed] [Google Scholar]
- 6.(a) Tu W, Liu L, Floreancig PE. Angew. Chem., Int. Ed. 2008;47:4184. doi: 10.1002/anie.200706002. [DOI] [PubMed] [Google Scholar]; (b) Tu W, Floreancig PE. Angew. Chem., Int. Ed. 2009;48:4567. doi: 10.1002/anie.200901489. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu L, Floreancig PE. Org. Lett. 2009;11:3152. doi: 10.1021/ol901188q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu L, Floreancig PE. Angew. Chem., Int. Ed. 2010;49:3069. doi: 10.1002/anie.201000033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liu L, Floreancig PE. Angew. Chem., Int. Ed. 2010;49:5894. doi: 10.1002/anie.201002281. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Brizgys G, Jung HH, Floreancig PE. Chem. Sci. 2012;3:438. [Google Scholar]; (g) Cui Y, Floreancig PE. Org. Lett. 2012;14:1720. doi: 10.1021/ol3002877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For recent representative examples of 2,6-cis-disubstituted tetrahydropyran synthesis through macrocyclic oxocarbenium ion formation, see: Crane EA, Scheidt KA. Angew. Chem., Int. Ed. 2010;49:8316. doi: 10.1002/anie.201002809. Custar DW, Zabawa TP, Scheidt KA. J. Am. Chem. Soc. 2008;130:804. doi: 10.1021/ja710080q. Woo SK, Kwon MS, Lee E. Angew. Chem., Int. Ed. 2008;47:3242. doi: 10.1002/anie.200800386. Yadav JS, Kumar GGKSN. Tetrahedron. 2010;66:480. Bahnck KB, Rychnovsky SD. J. Am. Chem. Soc. 2008;130:13177. doi: 10.1021/ja805187p. Wender PA, Schrier AJ. J. Am. Chem. Soc. 2011;133:9228. doi: 10.1021/ja203034k.
- 8.For other examples of transannular cyclizations to form inside-outside bridged ethers, see: Kühnert SM, Maier ME. Org. Lett. 2002;4:643. doi: 10.1021/ol017261d. Hilli F, White JM, Rizzacasa MA. Org. Lett. 2004;6:1289. doi: 10.1021/ol0497943. Li M, O'Doherty GA. Org. Lett. 2006;8:6087. doi: 10.1021/ol062595u.
- 9.Corey EJ, Lee TW. Chem. Commun. 2001:1321. [Google Scholar]
- 10.Jung HH, Floreancig PE. Tetrahedron. 2009;65:10830. doi: 10.1016/j.tet.2009.10.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Please see the Supporting Information for details.
- 12.For discussions of the stereoelectronic requirements for efficient bond cleavage reactions of radical cations, see: Tolbert LM, Khanna RK, Popp AE, Gelbaum L, Bottomley LA. J. Am. Chem. Soc. 1990;112:2373. Perrott AL, de Lijser HJP, Arnold DR. Can. J. Chem. 1997;75:384. Freccero M, Pratt A, Albini A, Long C. J. Am. Chem. Soc. 1998;120:284.
- 13.Trost BM, Ball ZT, Jöge T. J. Am. Chem. Soc. 2002;124:7922. doi: 10.1021/ja026457l. [DOI] [PubMed] [Google Scholar]
- 14.For other examples of hydrosilylation-based alkyne reduction in macrocycles, see: Fürstner A, Radkowski K. Chem. Commun. 2002:2182. doi: 10.1039/b207169j. Fürstner A, Bonnekessel M, Blank JT, Radkowski K, Seidel G, Lacombe F, Gabor B, Mynott R. Chem. Eur. J. 2007;13:8762. doi: 10.1002/chem.200700926. Micoine K, Fürstner A. J. Am. Chem. Soc. 2010;132:14064. doi: 10.1021/ja107141p.
- 15.(a) Hatanaka Y, Hiyama T. Tetrahedron Lett. 1990;31:2719. [Google Scholar]; (b) Denmark SE, Sweiss RF. Acc. Chem. Res. 2002;35:835. doi: 10.1021/ar020001r. [DOI] [PubMed] [Google Scholar]
- 16.Jones GR, Landais Y. Tetrahedron. 1996;52:7599. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.