

Abstract
In Nature, chiral natural products are usually produced in optically pure form; however, on occasion Nature is known to produce enantiomerically opposite metabolites. These enantiomeric natural products can arise in Nature from a single species, or from different genera and/or species. Extensive research has been carried out over the years in an attempt to understand the biogenesis of naturally occurring enantiomers, however, many fascinating puzzles and stereochemical anomalies still remain.
1. Introduction
Terrestrial and marine plants, animals, fungi, and bacteria (among others) are known to produce a multitude of secondary metabolites, often referred to as “natural products.”[1] Contrary to the required production of primary metabolites in order to sustain life, organisms can generally survive without the production of secondary metabolites; however, these metabolites often aid in the reproductive and/or defensive efforts of the species that produce them.[2,3] From a medicinal standpoint, many natural products also provide a rich source of bioactive agents, such as antitumoral, antibacterial, anti-insecticidal, anthelmintic, antinematodal, immunosuppressives, among other clinically relevant activities which have been widely exploited for both synthetic and semi-synthetic drug discovery and development efforts.[4,5]
In the vast majority of cases, chiral natural products are produced in Nature in optically pure form, where only one enantiomer is biosynthesized in the producing organism.[1,6] For example, only the biologically active (−)-isomer of morphine is produced by Nature, specifically by the opium poppy plant Papaver somniferum.[7] On the other hand, the production and isolation of enantiomeric metabolites is known, but in a relative sense to the overall abundance of secondary metabolites, remains a rare occurrence. These enantiomerically opposite metabolites can be produced by different genera or species, where one enantiomer is isolated from one species while the other enantiomer is isolated from a different species or genera; or both enantiomers may be produced and isolated as either a racemic or scalemic mixture (where one enantiomer predominates) from a single species.[6a]
Efforts to elucidate the biosynthetic pathway of bioactive natural products have been an area of intense research for over seventy-five years to both organic chemists and biologists.[2,4] However, the biogenesis of enantiomeric metabolites is generally not well understood. This is due in part to the fact that oftentimes one enantiomer always predominates over the other in Nature, as is the case with (−)-nicotine,[8] and in many other instances, the other natural enantiomer may be discovered years or decades later. As a result, the biosynthesis of the major and sometimes more bioactive enantiomer is well-studied, while the biosynthesis of the minor enantiomer remains unknown.
This Review is intended to provide an overview of the occurrence of well-known enantiomeric natural products produced in Nature, and to present a discussion, when applicable, of how these rare enantiomerically opposite metabolites arise biosynthetically. Due to the overwhelming number of known secondary metabolites, and the often overlooked reporting of the optical rotation, or CD spectra of like substances obtained from different sources, not all enantiomeric natural products have been identified. Furthermore, despite decades of research, not all of the biosynthetic pathways for the formation of enantiomeric natural products are fully understood; therefore, biogenetic discussions will focus on those metabolites where substantial and relevant biosynthetic research has been carried out. The present review is organized into classes of secondary metabolites based on their main biosynthetic derivations: terpenes (isoprene), phenylpropanoids (shikimic acid), polyketides (acetate), and alkaloids (amino acids). In many cases, these partitions are superficial since oftentimes, many natural products are of mixed biosynthetic origins (for example, the terpenoid alkaloids or mixed polyketide-nonribosomal peptide metabolites).
2. Terpenes
The terpenes are a large group of structurally diverse natural products numbering well over 30,000 compounds.[9,10] Typically isolated from a wide variety of plant species, these secondary metabolites display myriad biological activities ranging from pollinator attractants and chemical defenses for plants to essential oils and anti-cancer drugs for human clinical use.[10] All terpenoids are constructed from the head-to-tail condensation of repeating C5 isoprene units and are further subdivided into families based on the number of isoprenoid residues. The monoterpenes (C10) are the smallest structural type, followed by the sesquiterpenes (C15), diterpenes (C20), sesterterpenes (C25), triterpenes (C30), tetraterpenes (C40), and polyterpenes (>C40).
Enantiomeric terpenoids are (in a relative sense) rather common; however, they are generally limited to monoterpenes, sesquiterpenes, and on the rare occasion, diterpenes. Currently, (+)- and (−)-wistarin are the only examples to date of enantiomeric sesterterpenes (Figure 1), and the biosynthetic formation of these enantiomers is yet to be investigated.[11] Extensive research has been put forth toward determining the biosynthesis of enantiomeric monoterpenes, and while the biosynthesis of sesquiterpenes and diterpenes is understood, there are several unanswered questions about the formation of enantiomerically opposite secondary metabolites.
Figure 1.

(+)- and (−)-wistarin; the only known enantiomeric sesterterpenes.
2.1-Monoterpenes
The C10 monoterpene family of secondary metabolites are mainly isolated from higher plants and make up the flavor and aroma components of many essential oils of herbs, spices, citrus, and conifers.[12] The biosynthesis of monoterpenes has been thoroughly investigated via isotope studies with enzyme preparations and the isolation of cDNAs encoding monoterpene synthases;[13] however not all of the biosynthetic routes leading to the monoterpenes are fully understood.
Over the years, considerable attention has been placed specifically on the stereochemistry and mechanism of the cyclization reactions.[10,14,13a,15] Decades of research have established that monoterpene synthases are responsible for the formation of acyclic, monocyclic, and bicyclic monoterpenes, and that each synthase is capable of generating multiple products at the same active site. Since the co-occurrence of both monoterpene enantiomers in the same species is fairly common, interest has focused on determining how these enantiomers arise biosynthetically. With the isolation and characterization of numerous cyclases from Salvia, Mentha, Tanacetum, Foeniculum, Pinus, and Citrus species, including (+)-limonene synthase from Mentha piperita (peppermint), (−)-limonene synthase from Carum carvi L. (caraway seeds), and (+)- and (−)-α-pinene synthases from Salvia officinalis (sage), it was determined that monoterpene enantiomers can arise independently via stereochemically distinct routes; however, not all monoterpene synthases are completely stereospecific, as observed in the (−)-limonene synthase isolated from caraway seeds (discussed below).[10,13a]
The sizeable amount of research that has been carried out on the monoterpenes has led to the formation of a widely accepted mechanism for the monoterpene cyclization reaction (Scheme 1).[10,13a] To start, GPP stereoselectively binds in the active site as either a right-handed or left-handed helical conformer. GPP then undergoes ionization and isomerization to afford either (3R)- or (3S)-linalyl diphosphate (LPP), respectively. The enzyme-bound (3R)- or (3S)-LPP then undergoes a second ionization that initiates the C6-C1 cyclization to afford the α-terpinyl cation in either the (4R)- or (4S)-form, respectively. From this universal monocyclic intermediate a plethora of mechanisms (further electrophilic cyclization, hydride shifts, or Wagner-Meerwein rearrangements) are possible for the formation of the various monoterpene skeletons, which is finally terminated by deprotonation or nucleophilic capture.
Scheme 1.

Enantiomeric biogenesis of select monoterpenes.[15a]
As shown in Table 1, many of the chiral monoterpenes are produced in both enantiomeric forms, often by the same plant species. Additionally, many of the enantiomeric monoterpenes display unique biological activity, oftentimes with each enantiomer exhibiting distinct biological properties. Efforts to elucidate the biosynthetic formation of enantiomeric monoterpenes have been greatly aided by the isolation and characterization of several monoterpene synthases. Discussed below are three well-studied enantiomeric monoterpene biosyntheses (limonene, carvone, and α-pinene) for which distinct stereospecific enzymes have been identified that catalyze the cyclization of GPP to the corresponding monoterpene olefins of opposite configurations.
Table 1.
Occurrence and biological activity of enantiomeric monoterpenes.
| Monoterpene | Species | Biological Activity |
|---|---|---|
 (−)-(4S)-limonene |
Mentha piperita (peppermint),[17] Mentha spicata (spearmint),[17] Mentha pulegium (European pennyroyal),[17] Perilla frutescens (Chinese basil),[18,19] Perilla citriodora,[26] Abies grandis (grand fir),[20] Anethum graveolens L. (dill),[27] Semiardistomis puncticollis (carabid beetle),[28] Mentha cardiaca (Scotch spearmint),[21] Salvia officinalis (sage),[18] Pinus sylvestris (Scots pine),[29] pine needle oil,[9] Oleum cinae,[9] Pistacia vera L. (pistachio),[30] Angelica archangelica L. (wild celery)[31] | Turpentine odor[16a] Lemon odor[16b] |
| (+)-(4R)-limonene | Mentha piperita,[17] Mentha spicata,[17] Schizonepeta tenuifolia (Japanese catnip),[22] Citrus unshiu (mandarin orange),[21] Anethum graveolens L.,[27] Carum carvi L. (caraway fruit),[23] Ardistomis schaumii (carabid beetle),[28] Mentha cardiaca,[21] Salvia officinalis,[18] Pinus sylvestris,[29] Citrus limon (lemon),[24] Citrus sinensis (Valencia orange),[18] oil of orange rind,[9] dill oil,[9] oil of cumin, neroli, bergamot caraway and lemon (Citrus, Antethum, Juniperus, Peucedanum spp.),[9] Oleum cinae,[9] Pistacia vera L.,[30] Angelica archangelica L.[31] | Orange odor[16b] Shows insecticidal properties[9] |
 (−)-(4R)-carvone |
Mentha spicata,[32] Mentha cardiaca,[21] Tanacetum balsamita (balsam herb)[16] | Spearmint odor[16a] |
| (+)-(4S)-carvone | Anethum graveolens L.,[27] Carum carvi L.[23] | Caraway odor[16a] |
 (−)-α-pinene |
Abies grandis,[20] Pinus contorta(lodgepole pine),[33] Pinus taeda (loblolly pine),[34] Salvia officinalis,[18,35] Pinus sylvestris,[29] Pistacia vera L.,[30] Angelica archangelica L.,[31] Eucalyptus spp.[9] | Pine odor[16a] |
| (+)-α-pinene | Pinus contorta,[33] Pinus taeda,[34] Salvia officinalis,[18,35] Pinus sylvestris,[29] Pistacia vera L.,[30] Angelica archangelica L.,[31] Eucalyptus spp.[9] | Pine odor[16a] |
 (−)-β-pinene |
Abies grandis,[20] Pinus contorta,[33] Pinus taeda,[34] Salvia officinalis,[18,35] Pinus sylvestris,[29] Citrus limon, [24] Pistacia vera L.,[30] Angelica archangelica L.[31] | |
| (+)-β-pinene | Pinus contorta,[33] Pistacia vera L.,[30] Angelica archangelica L.[31] | |
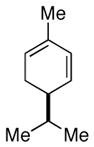 (−)-α-phellandrene |
Pinus contorta[36] | |
| (+)-α-phellandrene | Anethum graveolens L.[27] | |
 (−)-β-phellandrene |
Pinus contorta,[36] Pinus sylvestris,[29] Angelica archangelica L.,[31] Juniperus spp. (Juniper evergreen),[9] Pinus spp. (pine)[9] | |
| (+)-β-phellandrene | Anethum graveolens L.,[27] Angelica archangelica L.,[31] Bupleurum fruticosum (Shrubby Hare’s Ear),[9] Juniperus spp.[9] | |
 (−)-camphene |
Salvia officinalis,[37] Picea abies,[29] Pinus sylvestris,[29] Angelica archangelica L.[31] | |
| (+)-camphene | Salvia officinalis,[37] Picea abies,[29] Pinus sylvestris,[29] Angelica archangelica L.[31] | |
 (−)-camphor |
Picea pungens glauca (Colorado blue spruce),[38] Salvia officinalis,[38] Picea mariana nana (dwarf black spruce),[38] Thuja occidentalis (Northern Whitecedar),[38] Pinus sylvestris,[38] Tanacetum vulgare L. (tansy),[39] Chrysanthemum parthenium L. (feverfew),[40] Artemisia cana L. (silver sagebrush),[40] Chrysanthemum balsamita L. (costmary),[40] Matricaria parthenium (wild camomile),[9] Chrysanthemum sinense (mum),[9] Chrysanthemum indicum (mum)[9] | Camphoraceous odor[16a] |
| (+)-camphor | Picea mariana nana,[38] Picea albertiana conica (dwarf white spruce),[38] Picea sitchensis (Sitka spruce),[38] Artemesia californica (coastal sagebrush),[38] Chamaecyparis lawsoniana (Lawson’s cypress),[38] Pinus sylvestris,[38] Salvia officinalis,[38] Salvia leucophylla L. (San Luis purple sage),[40] Chrysanthemum sinese,[9] Chrysanthemum indicum,[9] Cinnamomum camphora (camphor tree)[9] | Camphoraceous odor[16a] Analeptic[9] Respiratory stimulant[9] Topical analgesic[9] Antipruritic[9] Antirheumatic[9] |
 (−)-borneol |
Thuja orientalis (Chinese Arborvitae),[38] Thuja standishii (Japanese Thuja),[38] Pinus sylvestris[29] | Camphoraceous odor with woody undertones[16b] |
| (+)-borneol | Picea sitchensis,[38] Chamaecyparis lawsoniana,[38] Pinus sylvestris,[29] Salvia officinalis[38] | Camphoraceous odor with earthy-peppery undertones[16b] |
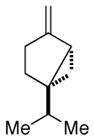 (−)-sabinene |
Pinus sylvestris,[29] Angelica archangelica L.[31] | |
| (+)-sabinene | Pinus sylvestris,[29] Salvia officinalis,[41] Angelica archangelica L.[31] |
2.1.1-Limonene and Carvone
Limonene is a widely distributed cyclic monoterpene and is a common precursor to the p-menthane family of natural products, as well as to the known monoterpene carvone. Both limonene and carvone are unique from a bioactivity perspective, in that each enantiomer exhibits a distint scent. Perhaps the most well known example of this is that (+)-carvone smells of caraway, while the enantiomer produces a spearmint odor.[16a]
The two limonene enantiomers known to occur in Nature are produced as either a single enantiomer or as a mixture of enantiomers, depending on the species. Limonene is derived from GPP, and through cell-free extracts and enzyme preparations two distinct limonene cyclases (synthases) have been identified from several species.[4,15d,17–24] As shown in Scheme 2, when tritium-labeled GPP was reacted with limonene synthase isolated from Mentha piperita (peppermint) and Mentha spicata (spearmint), (−)-3H-limonene was obtained.[17] On the other hand, a soluble limonene cyclase preparation was obtained from Citrus sinensis (Valencia oranges), which when reacted with tritium labeled GPP, enantiomerically pure (+)-limonene was obtained.[18] Likewise, when tritium-labeled GPP was reacted with a limonene synthase isolated from Carum carvi L. (caraway seeds), a 98:2 mixture of (+)- and (−)-limonene was observed, thus indicating that the limonene synthase from caraway seeds favors formation of (+)-limonene.[24]
Scheme 1.

Enantioselective biogenesis of limonene.
Carvone is also a cyclic monoterpene in which both enantiomers have been isolated. Through precursor incorporation studies and enzyme preparations, limonene has been established as a biosynthetic precursor to carvone.[17a,23,25] Two sets of enantioselective enzymes responsible for the conversion of (+)- or (−)-limonene to (+)- or (−)-carvone, respectively, have been isolated and characterized. As shown in Scheme 3, (−)-limonene-6-hydroxylase and (−)-trans-carveol dehydrogenase have been identified in Mentha spicata (spearmint), which catalyze the enantioselective conversion of (−)-limonene to (−)-trans-carveol to (−)-carvone.[25] While these enzymes are highly stereospecific, the corresponding enantiomeric enzymes found in Carum carvi L. were not completely stereo- or substrate-specific. Both (+)- and (−)-limonene served as substrates for (+)-limonene-6-hydroxylase, and when (+)-limonene was reacted with the enzyme, only 97% if the expected (+)-trans-carveol was isolated. The other 3% was made up of a mixture of (−)-trans-carveol and (−)-cis-carveol. These unexpected metabolites also displayed significant enzyme activity with (+)-trans-carveol dehydrogenase.
Scheme 3.

Enantiomeric carvone biosynthesis.
2.1.2-Pinene
α-Pinene and β-pinene are widely distributed bicyclic monoterpenes and serve as the major constituents in the volatile oil from Salvia officinalis (common sage).[18,35] Both enantiomers of α-pinene are natural products and can co-occur with either enantiomer predominanting. Contrary to this, β-pinene is almost always isolated as the optically pure (−)-isoform. The isolation of the (+)-β-pinene isomer is known, albeit the production of this metabolite is rare.[30,31,33] Biosynthetic studies carried out by Croteau and co-workers in the 1980s revealed that two enantiomeric pinene cyclases exist within Salvia officinalis, (+)-pinene cyclase (cyclase I) and (−)-pinene cyclase (cyclase II).[42] Croteau demonstrated that when 3H-GPP was reacted with each individual cyclase, the corresponding α-pinene enantiomers were formed (Scheme 4). In addition, (−)-β-pinene was formed from the reaction with cyclase II, whereas (+)-β-pinene was not detected from either cyclase reaction. From these reactions, minor amounts of (+)- and (−)-camphene and limonene were also formed as scalemic mixtures, (80% (+)-camphene isomer and 55% (−)-limonene isomer).[35,42,43]
Scheme 4.

Enantioselective pinene cyclases.
2.2-Sesquiterpenes
The sesquiterpenes make up a diverse group of acyclic and cyclic C15 terpenes isolated from a wide variety of plant, fungal, bacterial, marine and insect species. Like the monoterpenes, sesquiterpenes are often found as components of essential oils, such as vetiver oil and cubeb oil, and display a wide range of pharmacological activity.[44,45] Unfortunately, in most instances, the optical rotation of the sesquiterpenes is not reported, and as such, critical information regarding biological activity remains unknown.
Over the past two decades numerous sesquiterpene synthases have been isolated and characterized, including 5-epiaristolochene,[46] epiubenol,[47] pentalene,[48] germacrene C,[49] γ-humulene,[49] and δ-selinene,[50] and the mechanisms of these enzymes have also been investigated;[11,45,51] however, the biosynthetic formation of enantiomeric sesquiterpenes has, for the most part, remained obscure. Recently, König et al. isolated and characterized two enantioselective germacrene D synthases from S. canadensis.[44,45] The presence of these two cyclases within S. canadensis helps to explain the production of both enantiomers of germacrene D within the species, as well as support the possibility that the biosynthetic pathway of other enantiomeric sesquiterpenes arise from multiple enantioselective enzymes within different species of the same genera.
Most sesquiterpenes are chiral, and some members of this family of natural products have been found to be produced in both enantiomeric forms.[44] Generally, enantiomeric sesquiterpenes are produced by different species within the same genera as shown in Table 2; however, there are notable exceptions, such as the isolation of both enantiomers of germacrene D from Solidago canadensis and S. altissima.[45,52] Another unique example is the isolation of both enantiomers of furodysinin from Dysidea herbaceae. The (+)-isoform was isolated from D. herbaceae collected from Australia,[53] whereas the (−)-isoform is produced by the same species collected in Fiji.[54] Another observable trend in the isolation of enantiomeric sesquiterpenes is that terrestrial and marine sources have sometimes been observed to produce opposite enantiomers. An example of this is seen in the isolation of several sesquiterpenes from the soft coral Sinularia mayi. Seven of the major metabolites isolated from S. mayi were the opposite enantiomers of the more common forms found in terrestrial sources.[55] This is most likely a common occurrence, however, the stereochemical investigation of marine sesquiterpenoids is frequently disregarded.[56]
Table 2.
Enantiomeric sesquiterpenes.
| Sesquiterpene | Species | Biological Activity |
|---|---|---|
 (+)-germacrene D |
Ceroplastes rubens (scale insect),[57] Dendropanax trifidus M. (ivy tree),[58] Sinularia mayi (soft coral),[55] Preissia quadrata (liverwort),[59] Solidago altissima (late goldenrod),[52] Solidago canadensis (Canada goldenrod),[60] Podocarpus spicatus (black pine),[61] Zingiber officinale (ginger)[60] | |
| (−)-germacrene D | Ceroplastes ceriferus (scale insect),[57] Solidago altissima,[52] Solidago canadensis,[60] Pogostemon cablin (patchouli),[60,62] Pseudotsuga japonica (Japanese Douglas-fir),[9] Araucaria bidwillii (bunya pine),[63] Vitis vinifera (common grape vine),[60] Populus trichocarpa x deltoides (California poplar)[60] | |
 (+)-δ-cadinene |
Araucaria bidwillii,[63] Carum carvi L.,[64] Gossypium arboreum (tree cotton),[60] Gossypium hirsutum (upland cotton),[60] Mentha piperata[9] | |
| (−)-δ-cadinene | Ceroplastes ceriferus,[57] Sinularia mayi,[55] Araucaria araucana (Monkey-puzzle tree),[63] Araucaria bidwillii,[63] Carum carvi L.,[64] Heteroscyphus planus[65] | |
 (+)-epicubenol |
Streptomyces sp. LL-B7 (bacteria),[66] Heteroscyphus planus,[65] Scapania undulata (liverwort),[64] Juniperus rigida (temple juniper),[67] Streptomyces sp. B-7 (bacteria)[68] | |
| (−)-epicubenol | Cubeb oil,[69] Juniperus rigida,[67] Cedrus atlantica (Atlas Cedar)[9] | |
 (+)-δ-amorphene |
Leptospermum scoparium (Tea tree)[70] | |
| (−)-δ-amorphene | Vetiveria zizanioides (L.) Nash ex Small (vetiver oil)[71] | |
 (+)-cadina-3,5-diene |
Conocephalum conicum (scented liverwort)[70] | |
| (−)-cadina-3,5-diene | Leptospermum scoparium,[70] Piper cubeba oil[70] | |
 (+)-calamenene (trans) |
Ceroplastes ceriferus,[57] Conocephalum conicum[70] | |
| (−)-calamenene (trans) | Leptospermum scoparium,[70] Piper cubeba (tailed pepper)[70] | |
 (+)-furodysinin |
Dysidea sp. (marine sponge),[72] Dysidea herbaceae (marine sponge, Australia)[53] | |
| (−)-furodysinin | Dysidea herbaceae (marine sponge, Fiji),[54] Dysidea tupha,[73] Ceratosoma trilobatum (Sea Slug)[74] | Feeding deterent,[74] Ichthyotoxic[74] |
 (+)-chromazonarol |
Disidea pallescens (Black Sea sponge)[75] | |
| (−)-chromazonarol | Dictyopteris undulata (brown algae)[76] | Antimicrobial[77] |
 (+)-zonarene |
Dictyopteris zonarioides (brown seaweed),[64,78] Conocephalum conicum[70] | |
| (−)-zonarene | Dictyopteris zonarioides,[64,78] Leptospermum scoparium,[70] Piper cubeba[70] | |
 (+)-β-selinene |
Ceroplastes rubens[57] | |
| (−)-β-selinene | Ceroplastes ceriferus[57] | |
 (+)-β-bourbonene |
Ceroplastes rubens[57] | |
| (−)-β-bourbonene | Ceroplastes ceriferus[57] | |
 (+)-β-elemene |
Ceroplastes ceriferus[57] | |
| (−)-β-elemene | Ceroplastes rubens[57] | |
 (+)-pallescensin A |
Disidea pallescens[79] | |
| (−)-pallescensin A | Doriopsilla areolata (sea slug)[80] | |
 (±)-lucidene |
Uvaria lucida spp. lucida (African shrub)[81] |
2.3-Diterpenes
Diterpenes are isolated from numerous plants and fungi and are generally found in resins and essential oils. The structurally diverse family of diterpenes contains a C20 skeleton and is derived from the condensation of three equivalents of IPP with DMAPP to afford the acyclic geranylgeranyl diphosphate (GGPP) precursor.[10]
Similar to the monoterpenes and sesquiterpenes, enantiomeric diterpenes have been isolated, although their occurrence is rather rare (Table 3). The production of both enantiomers of various diterpenes can occur within the same species, or more commonly, within different species of the same genera. Unfortunately, biosynthetic studies on the formation of enantiomeric diterpenes have not been reported. Additionally, no biological activity has been reported for any of the individual diterpene enantiomers. As observed with the sesquiterpenes, optical rotation is generally not reported upon isolation of the diterpenes, and therefore, information concerning the occurance and biological activity of enantiomeric diterpenes is lacking.
Table 3.
Diterpenes.
| Diterpene | Species |
|---|---|
 (+)-beyerene |
Dacrydium intermedium (mountain pine),[82] Araucaria araucana[63] |
| (−)-beyerene | Podocarpus spicatus[61] |
 (±)-labda-8(20),13-diene-15-oic acid |
Eperua purpurea (Wapa tree),[83] Oxystigma oxyphyllum (African tree)[84] |
 (+)-rosadiene |
Podocarpus spicatus[61] |
| (−)-rosadiene | Dacrydium intermedium[82] |
 (+)-16-kaurene |
Podocarpus spicatus[61] |
| (−)-16-kaurene | Dacrydium intermedium T. Kirk,[82] Araucaria bidwillii,[63] Araucaria araucana,[63] Araucaria heterophylla[63] |
 (+)-sclarene |
Dacrydium intermedium,[82] Araucaria bidwillii,[63] Araucaria heterophylla[63] |
| (−)-sclarene | Podocarpus spicatus,[61] Dacrydium intermedium,[82] Araucaria araucana,[63] Araucaria heterophylla[63] |
3. Phenylpropanoids
Phenylpropanoids are found in numerous plant species and contribute greatly to plant defenses, structure, pigments, and reproduction.[85] Derived from the shikimate pathway, the phenylpropanoids are a class of natural products comprised of a vast array of diverse secondary metabolites formed from L-phenylalanine and/or L-tyrosine.[3] Lignins, lignans, flavonoids, coumarins, quinones, stilbenes, catechin, aurones and neoflavonoids are just a few of the many different types of phenylpropanoids derived from the enzymatic conversion of phenylalanine to the key intermediate p-coumaroyl-CoA, by way of the general phenylpropanoid pathway.[86]
3.1-Lignans
Lignans are phenylpropanoid dimers linked by the central C8 carbons of two phenylpropane units and make up an abundant class of phenylpropanoids.[87,88] Lignans are isolated from a wide range of plant species, specifically trees, and are believed to help prevent heart rot in trees.[88] They also display a plethora of biological activity, such as antitumor, antimitotic, and antiviral properties.[89] Some of the lignans produced early in the biosynthetic pathway also serve as lead compounds in the development of new drugs for the use in cancer therapies, such as the well-known podophyllotoxin-derived semisynthetic drug, etoposide.[89,90]
Enantiomerically, lignans can occur as mixtures of enantiomers with various enantiomeric compositions dependent upon the specific plant species. One extensively examined type of lignan is the early 9(9′) oxygenated lignans (pinoresinol, lariciresinol, secoisolariciresinol, and matairesinol). These lignans exists as either enantiomerically pure compounds, or as enantiomeric mixtures with various enantiomeric constitution.[91] As shown in Table 4, several trends are noticeable about these naturally occurring lignans: furofuran and furan lignans have never been isolated in optically pure form, while all dibenzylbutyrolactone lignans analyzed by chiral HPLC have been found to be optically pure. Furthermore, the predominant enantiomer of furofuran, furan, and dibenzylbutane lignans vary with plant species.[91] The optical rotation of the enantiomerically pure dibenzylbutyrolactone lignans were also found to vary between plant species.[92]
Table 4.
Enantiomeric lignans.
| Lignans | Species | Biological Activity |
|---|---|---|
 (+)-pinoresinol |
Forsythia koreana (Korean flowering plant, 82% e.e),[91] Linum flavum var. compactum (Dwarf Golden Flax, 65% e.e.),[91] Larix leptolepis (Japanese Larch, 92% e.e.),[91,92] Wikstroemia viridiflora,[92] Stellera chamaejasme (Tibetan Flowers),[92] Forsythia suspensa (Asian flowering plant),[92] Forsythia spp.,[92] Fraxinus spp.,[92] Helianthus annuus (common sunflower)[98] | Phytotoxic[98] |
| (−)-pinoresinol | Wikstroemia sikokiana (deciduous shrub, 74% e.e.),[91] Daphne odora (Winter Daphne, 95% e.e.),[91] Daphne genkwa (92% e.e.),[91] Daphne tangutica,[92] Zanthoxylum ailanthoides (Japanese Prickly-ash),[92] Zanthoxylum kellermanii,[92] Senecio scandens (wild daisy)[99] | Antioxidant[99] |
 (+)-syringaresinol |
Wikstroemia elliptica,[92] Daphne tangutica,[92] Passerina vulgaris,[92] Dirca occidentalis (Western Leatherwood)[93] | |
| (−)-syringaresinol | Daphne tangutica,[92] Zanthoxylum acanthopodium,[92] Daphne genkwa[100] | Anticancer[100] |
 (+)-sesamin |
Zanthoxylum acanthapodium,[92] Zanthoxylum valens,[92] Zanthoxylum setulosum[92] | Antihypertensive[101] |
| (−)-sesamin | Zanthoxylum piperitum (Japanese Pepper tree)[92] | |
 (+)-lariciresinol |
Forsythia koreana (35% e.e.),[91] Linum flavum var. compactum (70% e.e.),[91] Wikstroemia elliptica,[92] Larix leptolepis,[92] Abies sachalinensis (Sakhalin fir),[92] Araucaria angustifolia[92] | Phytotoxic (Inhibits lettuce germination)[102] |
| (−)-lariciresinol | Wikstroemia sikokiana (39% e.e.),92 Daphne odora (89% e.e.),92 Daphne genkwa (88% e.e.),92 Wikstroemia elliptica,93 Daphne tangutica,93 Dirca occidentalis93 | Phytotoxic (Inhibits root growth of Italian ryegrass)[102] |
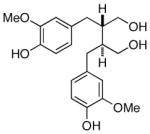 (+)-secoisolariciresinol |
Arctium lappa (Burdock, petiole) (81% e.e.),[91] Phyllanthus sp. (98% e.e.),[91] Daphne odora (>99% e.e.),[91] Daphne genkwa (97% e.e.)[91] | Antioxidant[103] |
| (−)-secoisolariciresinol | Arctium lappa (seeds) (65% e.e.),[91] Forsythia koreana (>99% e.e.),[91] Forsythia intermedia (>99% e.e.),[91] Wikstroemia sikokiana (45% e.e.),[91] Zanthoxylum ailanthoides,[92] Larix leptolepis,[92] Larix decidua (European larch),[92] Araucaria angustifolia,[92] Podocarpus spicatus[92] | Antioxidant[103] |
 (+)-arctigenin |
Wikstroemia indica,[92] Daphne genkwa[100] | Anticancer[100] |
| (−)-arctigenin | Arctium lappa (seeds) (>99% e.e.),[91] Forsythia koreana (>99% e.e.),[91] Forsythia intermedia (>99% e.e.),[91] Trachelospermum asiaticum var. intermedium (Yellow Star-jasmine),[92] Centaurea pamphylica[104] | Antitumoral,[105] Anticancer,[105] Antioxidant[104] |
 (+)-matairesinol |
Wikstroemia sikokiana (>99% e.e.),[91] Daphne odora (>99% e.e.),[91] Daphne genkwa (>99% e.e.),[91] Centaurea pamphylica[104] | |
| (−)-matairesinol | Arctium lappa (seeds) (>99% e.e.),[91] Forsythia koreana (>99% e.e.),[91] Forsythia intermedia (>99% e.e.),[91] Stellera chamaejasme,[92] Forsythia spp.,[92] Trachelospermum asiaticum var. intermedium,[92] Zanthoxylum kellermanii,[92] Picea excelsa (Norway spruce),[92] Tsuga mertensiana (Mountain Hemlock),[92] Thuja occidentalis (>99% e.e.)[91] | Antioxidant[104] |
 (+)-wikstromol |
Wikstroemia viridiflora,[92] Wikstroemia foetida,[92] Wikstroemia sikokiana (>99% e.e.),[91] Wikstroemia indica,[92] Daphne odora,[92] Passerina vulgaris[92] | Anticancer[106] |
| (−)-wikstromol | Thuja occidentalis (>99% e.e.),[91] Trachelospermum asiaticum var. intermedium,[92] Trachelospermum axillare[92] | Anticancer[107] |
 (+)-licarin A |
Leucas aspera (Common Leucas),[108] Machilus thunbergii (Japanese bay tree)[109] | Neuroprotective[109] |
| (−)-licarin A | Leucas aspera[108] | |
 (+)-chicanine |
Schisandra sp.[110] | |
| (−)-chicanine | Leucas aspera[108] | Antioxidant[108b] |
Like all phenylpropanoids, the lignans are derived via the cinnamate pathway. The biosynthetic pathway of 9(9′) oxygenated lignans is one of the more well-studied lignan pathways. The first five steps of this pathway have been extensively investigated and most of the enzymes responsible for the transformations and enantiomeric diversity seen in these types of lignans have been isolated and characterized. To date, several enantioselective lignan producing enzymes have been isolated and characterized. As shown in Scheme 5, the 9(9′) oxygenated lignans are formed by enantioselective dimerization of two coniferyl alcohol residues via an oxidase in the presences of a dirigent protein to afford pinoresinol in enantiomeric excess. The dirigent protein aids in controlling the stereospecificity of the bimolecular phenoxy radical coupling reactions of the two coniferyl alcohol units.[93]
Scheme 5.

Enantioselective biosynthesis of (+)-pinoresinol.
Next, pinoresinol is stereoselectively reduced to lariciresinol, which is subsequently reduced stereospecifically to secoisolariciresinol via pinoresinol/lariciresinol reductase. Two isoforms of this enzyme have been isolated, each displaying opposite enantioselectivity (Scheme 6).
Scheme 6.

Enantioselective conversion of pinoresinol to secoisolariciresinol via pinoresinol/lariciresinol reductase (PLR).
(+)-Pinoresinol/(+)-lariciresinol reductase has been isolated from Forsythia intermedia and Thuja plicata, whereas the (−)-pinoresinol/(−)-lariciresinol reductase was isolated from Thuja plicata.[94,95] Through incorporation studies it was determined that (+)-pinoresinol/lariciresinol reductases converts (+)-pinoresinol into (−)-secoisolariciresinol, and the opposite reductase converts (−)-pinoresinol into (+)-secoisolariciresinol.[95]
The final enzymatic conversion of secoisolariciresinol into enantiomerically pure matairesinol is not yet fully understood. (−)-Matairesinol is biosynthetically formed in various plant species (i.e. Forsythia intermedia, Arctium lappa, Thuja occidentalis, etc.); however, in Thymelaeaceae plants (Wikstroemia sikokiana and Daphne odora) the optically pure dextrorotatory enantiomer of matairesinol is produced. From Forsythia intermedia, secoisolariciresinol dehydrogenase was isolated and found to catalyze the enantioselective conversion of (−)-secoisolariciresinol into (−)-matairesinol (Scheme 7).[96] Secoisolariciresinol dehydrogenase preparation was also obtained from Daphne odora and Daphne genkwa, both known producers of the (+)-enantiomer of matairesinol; however, the in vitro reactions with enzyme preparation of both Daphne species resulted in the preferential formation of (−)-matairesinol.[97] To date, the biosynthesis of (+)-matairesinol remains unknown.
Scheme 7.

(−)-Matairesinol biosynthesis via secoisolariciresinol dehydrogenase (SIRD).
3.2-Flavonoids
Flavonoids make up a large, diverse family of aromatic secondary metabolites that are largely noted by the red, blue, and purple pigments found in plants.[111] Due to their colorful pigmentation, flavonoids are believed to act as an aid in plant reproduction by recruiting pollinators and seed dispersers. More recently, flavonoids have become an area of interest due to their association with the health benefits of wine, chocolate, fruits, and vegetables.
As shown in Table 5, the enantiomeric flavonoids occur mostly within three structural groups of flavonoids: the flavanones, flavonols, and isoflavonoids. Elucidation of the flavonoid biosynthetic pathway has been an area of growing research, with much of the attention recently being directed at the molecular genetics of the pathway.[71,86,111] Many of the enzymes responsible for the biosynthesis of the different subgroups of flavonoids have been isolated and characterized; however, the biosynthesis of enantiomeric flavonoids remains largely unresolved.
Table 5.
Enantiomeric flavonoids.
| Flavonoid | Species | Biological Activity |
|---|---|---|
 (+)-maackiain |
Dalbergia spruceana (Amazon rosewood),[114] Dalbergia stevensonii (Honduras rosewood),[114] ophora japonica (Pagoda Tree)[114] | Antimicrobial115] Phytoalexin[115] |
| (−)-maackiain | Dalbergia stevensonii,[114] Sophora japonica,[115] Trifolium pratense L. (red clover),[116] Pisum sativum L. (garden pea)[116] | Antimicrobial[115] Phytoalexin[115] |
 (+)-medicarpin |
Dalbergia decipularis Rizz. et Matt. (Tulipwood),[114] Dalbergia riparia,[114] Dalbergia variabilis,[114] Machaerium kuhlmannii Hoehne,[114] Machaerium nictitans,[114] Machaerium vestitum,[114] Arachis hypogea (peanut),[117] Sophora japonica[115] | Antimicrobial[115] Phytoalexin[115] |
| (−)-medicarpin | Dalbergia stevensonii,[114] Trigonella foenum-graecum (Fenugreek),[115] Medicago sativa (alfalfa)[118] | Antimicrobial[115] Phytoalexin[115] |
 (+)-catechin |
Acacia mearnsii (Black Wattle),[119] Acacia decurrens (Green Wattle),[119] Acacia dealbata (Silver Wattle),[119] Acacia pycnantha (Golden Wattle),[119] Chamaerops humilis (Mediterranean Dwarf Palm),[109] Phoenix canariensis (Canary Island Date Palm),[120] Butia capitata (Jelly Palm),[120] Howea forsteriana (Thatch Palm),[120] | |
| (−)-catechin | Chamaebatia foliolosa Benth (mountain misery), [121] chocolate[122] | |
 (+)-epicatechin |
Chamaerops humilis,[120] Livistona chinensis (Fountain Palm)[120] | |
| (−)-epicatechin | Acacia dealbata,[119] Acacia pycnantha[119] | |
 (+)-eriodictyol |
Arachis hypogaea (peanut hulls),[123] Hemizonia increscens (grassland tarweed)[123] | |
| (−)-eriodictyol | Arachis hypogaea,[123] Hemizonia increscens,[123] Thymus vulgaris[123] | |
 (+)-homoeriodictyol |
Eriodictyon glutinosum (Mountain Balm)[123] | |
| (−)-homoeriodictyol | Eriodictyon glutinosum[123] |
The biosynthesis of enantiomeric medicarpin has been investigated in both Medicago sativa L. (alfalfa) and Arachis hypogea (peanut), which are known producers of (−)- and (+)-medicarpin, respectively. The complete biosynthetic pathway of (−)-medicarpin has been determined by biochemical techniques and confirmed by gene cloning and expression experiments.[112,113] As shown in Scheme 8, the advanced achiral precursor 2′-hydroxyformononetin is converted to (R)-vestitone via isoflavone reductase, which subsequently reacts with pterocarpan synthase to yield (−)-medicarpin in alfalfa.[112] Contrary to what is known regarding the biosynthesis of (−)-medicarpin, there are several unanswered questions concerning the biosynthesis of (+)-medicarpin in peanut. Surprisingly, peanut isoflavone reductase in peanuts produces the same (R)-vestitone intermediate produced in alfalfa. This compound has the opposite substrate and product stereospecificity necessary for the pterocarpan synthase, thus indicating the possibility of an epimerase in peanut.
Scheme 8.

Enantiomeric medicarpin biosynthesis.[112]
3.3-Coumarins
Coumarins are generally produced by higher plants and are also derived from the general phenylpropanoid pathway. Coumarins play an important role for plants by acting as a defense against phytopathogens.[124] They also display myriad bioactivities for human therapeutics, including antibiotics, anticoagulants, and analgesic properties.[125] Unlike the lignans and flavonoids, the formation of enantiomeric coumarins is not as common (Table 6), and therefore the biosynthesis of these enantiomeric secondary metabolites has not been investigated.
Table 6.
Enantiomeric coumarins
| Coumarins | Species | Biological Activity |
|---|---|---|
 (+)-decursinol |
Angelica gigas (aerial),[128] Angelica gigas Nakai (roots)[129] | Anticancer,[130] Antihelicobacterpyloric,[131] Antinociceptive,[132] Inhibitor of acetyl cholinesterase[133] |
| (−)-aegelinol (decursinol enantiomer) | Angelica gigas (aerial),[128] Aegle marmelos,[134] Ferulago campestris (aegelinol benzolate),[135] Eryngium campestre (benzoyl aegelinol)[136] | Antibacterial[135] |
|
(S)-decursin |
Angelica gigas (roots),[137] Angelica gigas (aerial),[128] Angelica sinensis (female ginseng),[138] Angelica acutiloba[136] | Antibacterial,[137] Sedative[137] |
| (R)-grandivittin (decursin enantiomer) | Ferulago campestris,[135] Eryngium campestre[136] | |
 (S)-agasyllin |
Angelica gigas (aerial),[128] Angelica gigas (roots),[137] Angelica sinensis,[138] Angelica acutiloba[138] | Antibacterial[137] |
| (R)-agasyllin | Angelica gigas (aerial),[128] Ferulago campestris,[135] Eryngium campestre[136] | Antibacterial,[135] Antihelicobacterpyloric[135] |
 (+)-praeruptorin A |
Peucedanum praeruptorum Dunn.[139] | Reduces blood pressure[139] |
| (−)-praeruptorin A | Peucedanum praeruptorum Dunn.[139] | Reduces blood pressure[139] |
3.4-Neoflavonoids
The neoflavonoids are a group of secondary metabolites containing a C6-C3-C6 skeleton and are closely related both structurally and biogenetically to the flavonoids, isoflavonoids, coumarins and quinones.[3,126] The neoflavonoids are found in a wide variety of plant families, including the Guttiferae, the Leguminosae, the Rubiaceae, the Passifloraceae, the Polypodiaceae, and the Compositae.
4-Methoxydalbergione is an open-chained neoflavonoid that contains a stereogenic center at the C-7 position, and is naturally produced as the (R)- or (S)-isoform from various species of the genera Dalbergia.[127] As shown in Table 7, the occurrence of enantiomeric open-chain neoflavonoids in Nature is limited; therefore, the biogenesis of enantiomeric neoflavonoids is unknown.
Table 7.
Neoflavonoid enantiomers.
| Neoflavonoid | Species | Biological Activity |
|---|---|---|
 (S)-methoxydalbergione |
Dalbergia violacea,[127] Dalbergia baroni Baker (Madagascar Rosewood),[140] Dalbergia cultrate (Khamphi Rosewood),[141] Dalbergia melanoxylon (African Blackwood),[142] Dalbergia inundata,[143] Dalbergia nitidula,[144] Dalbergia miscolobium[145] | |
| (R)-methoxydalbergione | Dalbergia niger Fr. Allem. (Bahia Rosewood),[127] Dalbergia latifolia Roxb. (Indian Rosewood),[127c,146] Dalbergia parviflora,[147] Dalbergia cochinchinensis (Thiland Rosewood),[148] Dalbergia retusa (Cocobolo)[149] | Antiplasmodial[150] |
3.5-Quinones
Through numerous feeding experiments, it has been determined that quinones are also derived from phenylalanine, which is converted to the known intermediate p-hydroxybenzoic acid (PHB).[151] Subsequent prenylation at the C3 position affords m-geranyl-p-hydroxybenzoic acid, which is further converted to a key intermediate (geranylhydroquinone) in the biosynthesis of enantiomeric shikonin and alkannin (Scheme 9). Currently, the early steps in the biosynthesis of shikonin and alkannin are far more understood than the later steps. As shown in Table 8, these enantiomeric quinones and their derivatives display a plethora of biological activity, including anti-inflammatory, antitumor, and antimicrobial activity. For a more in depth review of the chemistry and biology of alkannin, shikonin, and their quinone derivatives, please refer to the review by Nicolaou et al.[151]
Scheme 9.

Proposed biosynthesis of shikonin and alkannin.
Table 8.
Quinone natural enantiomers[a]
| Quinone | Species | Biological Activity |
|---|---|---|
 alkannin |
Alkanna tinctoria (Alkanet), Arnebia hispidissima, Arnebia nobilis, Arnebia tinctoria, Macrotomia cephalotes, Macrotomia euchroma (Syrian Alkanet), Onosma echioides, Onosma paniculata, Plagiobotrys arizonicus | Wound healing, Antiinflammatory, Antibacterial, Inhibition of topoisomerase-I, Antithrombotic |
| shikonin (alkannin enantiomer) | Arnebia euchroma, Arnebia hispidissima, Arnebia guttata, Arnebia tibetiana, Cynoglossum officinale (Gypsyflower), Echium lycopsis, Echium rubrum, Echium vulgare (Blueweed), Eritrichium incanum, Eritrichium sichotenze, Jatropha glandulifera, Lappula consanguinea, Lappula echinata (Blue-bur), Lithospermum erythrorhizon (Purple Gromwell), Lithospermum officinale (European Stoneseed), Macrotomia echioides, Macrotomia ugamensis, Macrotomia euchroma, Mertensia maritima (Sea Lungwort), Onosma caucasicum, Onosma conferitum, Onosma hookeri, Onosma livanovii, Onosma polyphyllum, Onosma tauricum, Onosma sericium, Onosma setosum, Onosma visianii, Onosma zerizaminium | Antitumor, Antiamebic, Antipyretic and analgesic, Antifungal, Antibacterial, Wound healing, Chemopreventive, Antiinflammatory, Inhibition of topoisomerase-II, Inhibition of microsomal monooxygenase, Stimulation of peroxidase, Protection from uv-radiation, Inhibition of testosterone-α-reductase, Induction and secretion of nerve growth factors |
 acetylalkannin |
Alkanna tinctoria, Arnebia euchroma, Arnebia hispidissima, Arnebia nobilis, Macrotomia cephalotes | Antimicrobial, inhibition of topoisomerase-I, Antithrombotic, Antitumor |
| acetylshikonin | Arnebia decumbens, Arnebia euchroma, Arnebia guttata, Cynglossum officinale, Echium vulgare, Eritrichium incanum, Eritrichium sichotenze, Jatropha glandulifera, Lappula consanguinea, Lappul echinata, Lithospermum arvense (Field Gromwell), Lithospermum erythrorhizon, Mertensia maritima, Onosma confertum, Onosma hookeri, Onosma paniculatum | |
 isobutyrlalkannin |
Alkanna tinctoria | |
| isobutyrylshikonin | Cynoglossum officinale, Echium vulgare, Eritrichium sichotenze, Lappula consanguinea, Lappula echinata, Lithospermum arvense, Lithospermum erythrorhizon, Macrotomia euchroma, Mertensia maritima | |
 isovalerylalkannin |
Alkanna tinctoria, Arnebia hispidissima, Arnebia tinctoria, Macrotomia cephalotes, Onosma heterophylla | Inhibition of topoisomerase-I |
| isovalerylshikonin | Arnebia decumbens, Cynoglossum officinale, Echium vulgare, Lappula consanguinea, Lappula echinata, Lithospermum arvense, Lithospermum erythrorhizon, Macrotomia euchroma | |
 α-methylbutyrylalkannin |
Alkanna tinctoria, Macrotomia cephalotes | Antimicrobial |
| α-methylbutyrylshikonin | Cynoglossum officinale, Echium vulgare, Eritrichium incanum, Eritrichium sichotenze, Lappula consanguinea, Lappula echinata, Lappula erythrorhizon, Mertensia maritima | Antimicrobial |
 β,β-dimethylacrylalkannin |
Alkanna tinctoria, Arnebia euchroma, Arnebia gutatta, Arnebia nobilis, Lithospermum erythrorhizon, Macrotomia cephalotes, Onosma heterophylla, Onosma hookeri, Onosma paniculata | Inhibition of topoisomerase-I and anticancer, Antimicrobial, Antithrombotic, Antiinflammatory |
| β,β-dimethylacrylshikonin | Alkanna hirsutissima, Arnebia euchroma, Arnebia guttata, Arnebia tibetiana, Cynoglossum officinale, Echium vulgare, Eritrichium incanum, Eritrichium sichotenze, Jatropha glandulifera, Lappula consanguinea, Lappula echinata, Echium spp., Lithospermum erythrorhizon, Macrotomia ugamensis, Mertensia maritima, Moltkiopsis ciliata, Onosma confertum, Onosma paniculatum, Onosma hookeri, Onosma zerizaminum | |
 teracrylalkannin |
Arnebia densiflora | Antimicrobial |
| teracrylshikonin | Arnebia euchroma, Arnebia guttata, Lithospermum erythrorhizon, Lithospermum euchromum | Antimicrobial |
 angelylalkannin |
Alkanna tinctoria | |
| angelylshikonin | Alkanna hirsutissima | |
 β-hydroxyisovalerylalkannin |
Arnebia euchroma, Arnebia hispidissima, Macrotomia cephalotes | Antimicrobial |
| β-hydroxyisovalerylshikonin | Arnebia euchroma, Arnebia guttata, Lithospermum arvense, Lithospermum erythrorhizon, Lithospermum euchromum | Antimicrobial |
 β-acetoxyisovalerylalkannin |
Alkanna tinctoria, Arnebia euchroma, Moltkiopsis ciliata, Onosma heterophylla | Antimicrobial |
| β-acetoxyisovalerylshikonin | Macrotomia euchroma | |
 (+)-dunnione |
Streptocarpus dunnii (Cape Primrose),[152] Calceolaria integrifolia[153] | |
| (−)-dunnione | Streptocarpus dunnii,[152] Calceolaria integrifolia[153] | |
 (+)-α-dunnione |
Streptocarpus dunni[152] | |
| (−)-α-dunnione | Streptocarpus dunni[152] | |
 (±)-8-hydroxydunnione |
Streptocarpus dunni[152] |
For species and biological activity that do not have a reference, please refer to the Review by Nicolau et al. (reference 151)
4. Polyketides
Derived from acetate, polyketides represent a structurally diverse family of secondary metabolites produced by a wide variety of plants, fungi, bacteria, and insects.[3,154] The exact role of polyketides in producing organisms is not known, however, it appears as though several serve as either chemical defense agents or aid in the growth and development for plants. Polyketides also display important medicinal activity, such as antibiotic, anticancer and immunosuppressant properties.
There are several examples of enantiomeric polyketides biosynthesized throughout the plant kingdom (Table 9), however, not much is known about the enantioselective biosynthesis of many of these secondary metabolites. To date, extensive research has been carried out at an enzymatic level on the enantiomeric formation of macrotetrolide antibiotics (nactins) and benzylisochromanequinone antibiotics.
Table 9.
Enantiomeric polyketides.
| Polyketide | Species | Biological Activity |
|---|---|---|
 (+)-nonactic acid (and homologues) |
Streptomyces griseus (bacteria),[155] Streptomyces spec. JA 5909-1[156] | |
| (−)-nonactic acid (and homologues) | Streptomyces griseus,[155] Streptomyces spec. JA 5909-1[156] | |
 kalafungin |
Streptomyces tanashiensis strain Kala,[157] Streptomyces coelicolor A3(2)[158] | Antibiotic[159] |
| nanaomycin D (kalafungin enantiomer) | Streptomyces rosa var. notoensis OS3966[160] | Antibiotic[160] |
 (+)-nanaomycin A |
Nocardia sp. (bacteria)[161] | Antibiotic[161] |
| (−)-nanaomycin A | Streptomyces rosa var. notoensis OS3966[162] | Antibiotic, [162] Antifungal, [162] Antimycoplasma activity[162] |
 (+)-mellein |
Cercospora taiwanensis,[163] Fusarium larvarum,[163] Grignardia laricina,[163] Gyrostroma missouriense (fungus),[163] Helicascus kanaloanus (marine fungus),[164] unidentified fungus[163] | |
| (−)-mellein | Aspergillus melleus (fungus),[163] Aspergillus ochraceus (fungus),[163] Aspergillus oniki (fungus),[163] Camponotus spp.,[163] Cornitermes spp.,[163] Grapholithia molesta (Oriental Fruit Moth),[163] Hypoxylon spp.,[163] Lasiodiplodia theobromae (fungus),[163] Marasmiellus ramealis (Twig Parachute Mushroom),[163] Pestalotia ramulosa (fungus),[163] Rhytidoponera metallica (Green-head Ant),[163] Septoria nodorum (fungus)[163] | Hepatitis C Inhibitor,[165a] Antibacterial,[165b] Antiviral, [165b] Phytotoxic[165b] |
 (+)-dermolactone |
Dermocybe kula (fungus)[166] | Major orange-red fungal pigment[166] |
| (−)-dermolactone | Dermocybe kula[166] | Minor orange-red fungal pigment[166] |
 (+)-scytalone |
Verticillium dahliae (fungus),[167] Phialophora lagerbergii (fungus),[168] Scytalidium sp.[169] | |
| (−)-scytalone | Phialophora lagerbergii,[168] Scytalidium sp. [169] | |
 (1S,3S)-austrocortilutein |
Dermocybe splendida (Splendid Red Skinhead, fungus)[170] | Antibiotic,[171] Fungal pigment (yellow)[172] |
| (1R,3R)- austrocortilutein | Dermocybe sp. WAT 20934[172] | |
 (1S,3R)- austrocortilutein |
Dermocybe splendida,[170] Dermocybe sp. WAT 20934,[172] Dermocybe sp. WAT 21568[172] | Antibiotic,[171] Fungal pigment (yellow-D. splendida)[172] |
| (1R,3S)- austrocortilutein | Dermocybe sp. WAT 21567,[172] Dermocybe sp. WAT 20934[172] |
4.1-Macrotetrolides
The macrotetrolide antibiotics (nactins) are mainly produced by Streptomyces species and are biosynthetically formed from four monomeric units of nonactic acid (NA) or its homologs, homononactic acid and/or bishomonanactic acid (Figure 2).[155,156] Of the five known homologs (nonactin, monactic, dinactin, trinactin, and tetranactin), biosynthetic studies mostly focus on nonactin, a 32-membered macrocycle composed of two alternating units of (+)-nonactic acid and (−)-nonactic acid, which in turn makes nonactin achiral.[155,156,173–175]
Figure 2.

Nactins and the monomeric units that make up the macrotetrolide antibiotics.
The biosynthesis of nonactin has been well-studied through in vivo feeding esperiments with 13C-, 2H-, and 18O- labeled precursors[173] and by the isolation of both nonactic acid enantiomers and its dimmer.[156] Recently, the biosynthetic research of nonactin has centered around the isolation and characterization of the genes and enzymes responsible for the biosynthesis of both enantiomers of nonactic acid.[155i] The biosynthesis of both NA enantiomers resulted in the proposal that the enantiomeric polyketide intermediates arise from a pair of enantiospecific pathways. The proposed biogenesis of the macrotetrolides, is supported through both feeding and enzymatic studies, which were carried out individually by Robinson, Priestley, and Shen.[155i,173–175] As shown in Scheme 10, the nactins are derived from malonyl-CoA, succinyl-CoA, and acetyl-CoA, which results in the formation of proposed intermediate 1. Biosynthetic studies performed by Robinson and co-workers using 14C-labeled compounds established that propionate also serves as a primary metabolic precursor, which most likely results in the formation of the proposed achiral intermediate 2.[173] Since 2 is achiral, it can serve as a common precursor to both nonactic acid enantiocomplementary pathways.
Scheme 10.

Proposed enantiocomplimentary pathways for the biogenesis of (+)- and (−)-nonactic acid.[155i]
Opposite stereospecific reductions of 2 would result in the generation of enantiomeric precursors, 3 and 4. Work carried out by Robinson and Spavold confirmed that the acyclic intermediates 3 and 4, as well as (6R,8R)- and (6S, 8S)-2-methyl-6,8-dihydroxynon-2E-enoic acids (NEA), were enantioselectively incorporated into nonactin via the respective (+)- and (−)-nonactate precursors.[173] However, the details of the conversion of the primary metabolites into 3 and 4 are still relatively unknown. Shen and co-workers garnered additional support for enantioelective pathways through enzymatic studies with NonS.[155i] They demonstrated that nonS governs only the enantioselective formation of (−)-NA and its homologus in S. griseus, however, the enzyme responsible for the biosynthesis of (+)-NA remains elusive.
4.2-Benzoisochromanequinones
Kalafungin, actinorhodin, medermycin, dihydrogranaticin and nanaomycin are all antifungal and antimycoplasmal antibiotics that possess a benzoisochromanequinone (BIQ) skeleton and are produced by various Streptomyces species.[176] Structurally, the BIQs all show a trans configuration in respect of the C-3 and C-15 chiral centers, thus these metabolites can be grouped into one of two categories: dihydrogranaticin (DHGRA), which displays the (3R,15S) configuration; or actinorhodin (ACT), which display the (3S,15R) stereochemistry (Figure 3).[177,178]
Figure 3.

Structural diversity of the BIQ antibiotics.
With the formation of enantiomerically opposite stereocenters in the ACT and DHGRA families, early stage enantiomeric intermediates have been isolated from different sources. Upon analysis of the numerous gene clusters (act, kal, nnm, gra, etc.) that have been identified, two enantioselective ketoreductases were identified; and as shown in Scheme 11, from a bicyclic intermediate, RED1/2 stereospecifically reduces the carbonyl functionality, which sets the C-3 stereochemistry.[178] In the ACT biosynthesis, the (S)-configuration is established via act-VI-ORF1 (RED1),[179] whereas the (R)-configuration is established in the dihydrogranaticin (DHGRA) biosynthesis by a completely unrelated gra-ORF6 ketoreductase, RED2.[177,180] These two enzymes show a remarkable difference in their substrate specificities as well as in the three-dimensional structures and catalytic mechanisms; however, both recognize the same substrate motif of the bicyclic intermediate.[178a] From this intermediate, subsequent cyclization and reduction results in the formation of the respective enantiomeric intermediates (S)-DNPA and (R)-DNPA. From these chiral intermediates, the advanced BIQ natural products, such as actinorhodin and DHGRA are derived.
Scheme 11.

Proposed enantioselective biosynthetic pathways of actinorhodin and dihydrogranaticin. (RED1/2 = stereospecific C-3 reductase)
5. Alkaloids
Alkaloids make up a vast and structurally diverse group of nitrogenous metabolites that are isolated from plants, bacteria, fungi, and animals.[3] This family of natural products can be further classified into subgroups, which are based on the handful of α-amino acids alkaloids are derived from, mainly lysine, ornithine, phenylalanine, tyrosine and tryptophan. In addition to these primary building blocks, mevalonate and acetate also serve as important starting points in the biosynthesis of alkaloids.
Pharmacologically, alkaloids display myriad bioactivities and are often used as medications, as recreational drugs, or in entheogenic rituals.[181] Morphine, caffeine, and psilocin (a mushroom hallucinogen) are common and well-known bioactive examples of alkaloids. Several lesser-known alkaloids are also biologically active and display anticancer, antibacterial, anthelmintic, anti-inflammatory activity.[182]
The occurrence of enantiomeric alkaloids in Nature is known, however, they are generally produced and isolated as racemic or scalemic mixtures. As observed in the lignans, many of the advanced alkaloid metabolites are produced in optically pure form, but the metabolites produced in the early stages of alkaloid biosynthesis are often isolated as enantiomeric mixtures. Select examples of these enantiomeric alkaloids are discussed below.
5.1-Manzamine Alkaloids
The manzamines are a growing class of β-carboline-containing cytotoxic marine sponge alkaloids that display an unusual polycyclic diamine system.[183] These natural products were first identified in the late 1980s and were found to have a diverse range of biological activity, including, but not limited to, antitumor, anti-inflammatory, insecticidal, and antiparasitic. Several of these natural products also display promising anti-infective activity against malaria and Mtb.[183]
The diversity in the location (Okinawa, the Philippines, Indonesia, the Red Sea, Italy, South Africa, and Papua New Guinea) and genera of sponges (Amphimedon sp. and Acanthostrongylophora) responsible for the production of manzamine alkaloids is widely believed to be a result of a symbiotic relationship between these sponges with common or closely related microorganism(s), which may account for the generation of manzamine enantiomers.[183] To date, only a few enantiomeric manzamine natural products have been isolated (Table 10) and the biosynthetic formation of these enantiomeric metabolites is currently under investigation.
Table 10.
Enantiomeric manzamine alkaloids.
| Manzamine | Species | Biological Activity |
|---|---|---|
 (+)-8-hydroxymanzamine A |
Indonesian sponge Pachypellina sp.,[187] Okinawan sponge Xestospongia sp.,[188] Okinawan sponge Amphimedon sp.[188] | Anticancer[183] |
| (−)-8-hydroxymanzamine A | Unidentified Indo-Pacific sponge (family Petrosiidae, order Haplosclerida)[189] | Anticancer,[183] Antimalarial[183] |
 (+)-manzamine F |
Xestospongia sp.[190] | Antimicrobial,[183] Anticancer[183] |
| (−)-manzamine F | Unidentified Indo-Pacific sponge (family Petrosiidae, order Haplosclerida)[189] | Activity against Mycobacterium tuberculosis[183] |
 (±) keramaphidin B |
Amphimedon sp.,[191] Xestospongia ingens [optically active, [α]D +29.8° (c 1.1; MeOH)][192] | Anticancer[183] |
Within this class of alkaloids, the isolation of both enantiomers of 8-hydroxymanzamine A, manzamine F, and keramaphidin B have been reported, along with the enantiomeric congers, ircinal A and B and ircinol A and B.[183] Interestingly, ircinols A and B are enantiomeric congeners of the alcoholic forms of ircinal A and B, respectively, and they also represent the first manzamine alkaloids to possess the opposite absolute configuration to that of manzamines A and B.[184] As shown in Figure 4, one enantiomer of keramaphidin B, ircinals A and B, and manzamines A and B all belong to one configurational series, while the other enantiomer of keramaphidin B and ircinols A and B, ingenamine, ingamine A contain the opposite absolute configuration, and thus make up a second enantiomeric series.
Figure 4.

The two enantiomeric series of manzamine alkaloids.
Since it is likely that sponge-associated microbes produce the manzamines, efforts to elucidate the biosynthetic pathway of these unique compounds is limited.[183] The identification of bacterial isolates from a manzamine producing sponge, as well as culturing the bacteria responsible for these transformations are limiting factors to completely understanding the biosynthesis of the manzamines. However, following the identification, isolation, and screening of numerous microbes from manzamine producing sponges, the biotransformation of both 8-hydroxymanzamine A to manzamine A[185] and ent-8-hydroxymanzamine A to the known metabolite ent-12,34-oxamazamine F[186] have been successfully carried out (Scheme 12).
Scheme 12.

Biocataytic conversion of enantiomeric 8-hydroxymanzamine A.
5.2-Indole Alkaloids
Indole alkaloids are natural products derived from tryptophan and make up one of the largest group of alkaloid secondary metabolites.[193] Biogenetically, this class of alkaloids can be divided into two structural categories, isoprenoid containing natural products and non-isoprenoid containing alkaloids. The latter group is comprised of simple indole derivatives, simple derivatives of β-carboline, and pyrroloindole alkaloids.[194] The isoprenoid alkaloids contain terpenoid structural elements derived from dimethylallyl pyrophosphate (DMAPP) and/or isopentenyl pyrophosphate (IPP).[195] The formation of enantiomeric indole alkaloids has been documented within the various subdivisions of the more complex isoprenoid alkaloids as outlined below.
5.2.1-Terpenoid Indole Alkaloids
Terpenoid alkaloids are often found in plant species belonging to the Apocynaceae, Loganiaceae, Rubiaceae, and Nyssaceae families.[196] Madagascar periwinkle (Catharanthus roseus), from the family Apocynaceae, is known to produce over one hundred structurally diverse terpenoid indole alkaloids; and elucidation of the terpenoid indole alkaloid biosynthetic pathway in Catharanthus roseus has been extensively studied. More than twenty enzymatic steps have been identified in this intricate biosynthetic pathway, which leads from the primary metabolites to the structurally complex antineoplastic agent, vinblastine. As is the case for many secondary metabolites, the advanced late stage intermediates, such as vinblastine and vincristine, are produced and isolated as a single enantiomer, whereas the early stage metabolites are sometimes produced as scalemic mixtures. As shown in Table 11, these enantiomeric metabolites oftentimes occur in separate species as a single enantiomer. While the overall biosynthesis of terpenoid indole alkaloids is fairly well understood, the biogenesis of enantiomeric metabolites is not currently known.
Table 11.
Enantiomeric indole alkaloid metabolites.[a]
| Indole Alkaloid | Species | Biological Activity |
|---|---|---|
 (+)-vincadifformine |
Amsonia tabernaemontana (Eastern Bluestar),[197] Amsonia angustifolia, Rhazya stricta, Tabernaemontana riedelii, Vinca difformis (Intermediate Periwinkle), Macoubea guianensis[198] | |
| (−)-vincadifformine | Vinca minor (Dwarf Periwinkle), Vinca difformis, Rhazya stricta, Tabernaemontana riedelii, Macoubea guianensis[198] | |
 (+)-vincadine |
Vinca minor, Amsonia tabernaemontana, Amsonia angustifolia, Macoubea guianensis[198] | |
| (−)-vincadine | Amsonia tabernaemontana, Amsonia angustifolia, Macoubea guianensis | |
 (+)-vincamine |
Vinca minor, Vinca major (Blue Periwinkle), Vinca erecta, Vinca difformis, Tabernaemontana rigida | Antihypertensive |
| (−)-vincamine | Tabernaemontana rigida | |
 (+)-quebrachamine |
Vinca erecta, Pleiocarpa tubicina, Pleiocarpa pycnantha var. pycnantha, Stemmadenia donnell-smithii | |
| (−)-quebrachamine | Aspidosperma quebracho-blanco (South American tree), Aspidosperma chakensis, other Aspidosperma spp., Gonioma kamassi, Hunteria elliotii, Rhazya stricta | |
 (+)-apparicine |
Aspidosperma dasycarpon | |
| (−)-apparicine | Aspidosperma olivaceum, other Aspidosperma spp., Catharanthus ovalis (Rosy Periwinkle), Catharanthus roseus (Madagascar Periwinkle), Pandaca ochrascens, Pandaca eusepala, Ervatamia heyneana, Tabernaemontana cumminsii, Schizzygia caffaeoides | Anticancer, Antibacterial, Antiviral |
 (+)-eburnamonine |
Hunteria eburnea, Amsonia tabernaemontana, Vinca minor | |
| (−)-eburnamonine | Vinca minor | Stimulates muscle activity |
 (+)-akuammicine |
Picralima nitida | |
| (−)-akuammicine | Picralima nitida, Alstonia scholaris (Blackboard tree), other Alstonia spp., several Vinca spp., Rauwolfia volkensii, Hunteria congolana, Catharanthus microphyllus, Cabucala erythrocarpa, Pandaca ochrascens, Catharanthus roseus[199] | |
 (+)-9-methoxymitralactonine |
Mitragyna speciosa (Kratom)[200] | |
| (−)-9-methoxymitralactonine | Mitragyna speciosa[200] | |
 (+)-mitralactonine |
Mitragyna speciosa[201] | |
| (−)-mitralactonine | Mitragyna speciosa[201] | |
 (±)-nitrarine |
Nitraria schoberi (Nitre Bush)[193,202] |
For species and biological activity that do not have a reference, please refer to the Dictionary of Alkaloids (reference 193)
5.2.2-Reverse Prenylated Indole Alkaloids
The unique and diverse family of reverse prenylated indole alkaloids containing a bicyclo[2.2.2]diazaoctane ring system has been the subject of extensive research due to their complex molecular structure and wide array of biological activity.[203] Members of this family have been isolated from both marine and terrestrial sources, most notably from the genera Aspergillus and Penicillium, and have been reported to display insecticidal, anthelmintic, calmodulin-inhibitory, antibacterial and antitumor properties. The recent identification of enantiomeric metabolites from related Aspergillus species has sparked interest in elucidating the biosynthetic pathway of the stephacidin and notoamide family of reverse prenylated indole alkaloids.
In 2009, Tsukamoto and co-workers isolated the known natural product (+)-stephacidin A[204] from marine-derived Aspergillus sp. MF297-2, along with several new metabolites later named the notoamides.[205] Shortly following the isolation of the stephacidins and notoamides from Aspergillus sp. MF297-2, Gloer and co-workers isolated the corresponding enantiomers from the terrestrial-derived fungus Aspergillus versicolor NRRL 35600.[206] These enantiomeric alkaloids (Table 12) are hypothesized to arise via a biosynthetic Diels-Alder reaction, which implies that each Aspergillus species possesses enantiomerically distinct Diels-Alderases. Furthermore, each fungal culture must also possess enantiomerically distinct oxidases responsible for the face-selective pinacol-type rearrangement to form the spiro-oxindole moiety observed in notoamide B and versicolamide B. Thus, Williams and co-workers proposed that the stephacidin and notoamide family shared a common biosynthetic pathway and that the enantiomeric formation of these alkaloids was due to a key enantiodivergent step in an otherwise common biogenetic pathway.[203] This work was further aided through the identification and characterization of the Aspergillus sp. MF297-2 and the Aspergillus versicolor NRRL35600 genome clusters,[207] as well as parallel precursor incorporation studies with both fungal cultures.[208]
Table 12.
Enantiomeric reverse prenylated indole alkaloids.
| Reverse Prenylated Indole Alkaloid | Species | Biological Activity |
|---|---|---|
 (+)-stephacidin A |
Aspergillus sp. MF297-2 (fungus),[205] Aspergillus ochraceus (fungus) [204] | Anticancer[204] |
| (−)-stephacidin A | Aspergillus versicolor (fungus) [206] | |
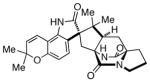 (+)-notoamide B |
Aspergillus versicolor[206] | |
| (−)-notoamide B | Aspergillus sp. MF297-2[205] | |
 (+)-versicolamide B |
Aspergillus versicolor[206] | |
| (−)-versicolamide B | Aspergillus sp. MF297-2[210] |
Based on results from genome mining and tracer studies, a biosynthetic pathway has been proposed.[207,208] As shown in Scheme 13, starting with the proposed pivotal intermediate notoamide S,[209] the pathway branches into at least two possible directions. Formation of the pyranoindole to yield notoamide E results in the biosynthesis of notoamide C, 3-epi-notoamide C, and notoamide D[208a,b] via the proposed enzyme NotB. However, notoamide S could also undergo a 2-electron oxidation by either NotD or NotH to give the achiral azadiene, which acts as the enantio-diverging point in the biosynthesis. The achiral azadiene can undergo a stereoselective [4+2] cycloaddition to yield either (+)-notoamide T in Aspergillus sp. MF297-2 or (−)-notoamide T in Aspergillus versicolor. From these putative intermediates, cyclization to form the pyranoindole ring system would furnish the enantiomeric pair of stephacidin A, respectively. Through precursor incorporation studies of 13C-labeled (±)-stephacidin A with both A. versicolor and Aspergillus sp. MF297-2, it was ascertained that face-selective oxidative enzymes (currently presumed to be flavo-enzymes) are present in both fungal cultures as evident by the enantioselective conversion of stephacidin A into notoamide B.[208c] Specifically in A. versicolor NRRL 35600, this oxidase is responsible for the biosynthetic conversion of (−)-stephacidin A into (+)-notoamide B, while a stereochemically complementary oxidase in the marine-derived Aspergillus sp. MF297-2 converts (+)-stephacidin A into (−)-notoamide B.
Scheme 13.

Putative enantio-divergent biosynthesis of stephacidin A and the notoamides.
It is significant that in each of these respective oxidation reactions of the 2,3-disubstituted indole moiety of stephacidin A, the oxidation must occur from distinct enantiotopic faces of the indole ring system and we presently doubt that this is accomplished by identical enzymes. More precisely, the oxidation of (+)-stephacidin A into (−)-notoamide B, must occur exclusively from the pro-R face of the indole in Aspergillus sp. MF297-2 and the oxidation of (−)-stephacidin A into (+)-notoamide B in Aspergillus versicolor, must occur exclusively from the pro-S face of the indole. To date, the diastereomeric oxindoles that would result from a putatively non-face-selective oxidation have not been detected. Of further intrigue, was the observation that Aspergillus sp. MF297-2 produces (−)-versicolamide B and that Aspergillus versicolor produces the enantiomer, (+)-versicolamide B. The putative precursor to versicolamide B, C6-epi-stephacidin A, has not yet been detected as a natural metabolite but its existence in each fungus is anticipated. Synthetic samples of this substance have been prepared and are under interrogation.
In an effort to understand the enzymatic basis for the biosynthesis of enantiomeric alkaloid natural products, we have pursued total genome sequencing and mining of the stephacidin/notaomide pathways from two fungal strains. The marine Aspergillus sp. MF297-2 strain generates (−)-notoamide B, whereas the terrestrial Aspergillus versicolor strain generates enantiomeric (+)-notoamide B, with the key chiral determinant hypothesized to reside within the presumed intramolecular Diels-Alderase enzyme. We have found the molecular architecture (e.g. gene placement and directionality of transcription) of these pathways to be remarkably similar with >70% identity of nucleotide sequences across the 35 kb gene clusters. The corresponding high level of amino acid sequence similarity suggests that subtle active site sequence variation plays a critical role in controlling chirality, and in accommodating the corresponding enantiomeric substrates for downstream assembly and tailoring reactions.
5.3-Quinolizidine (Lupine) Alkaloids
Quinolizidine alkaloids, often referred to as lupine (or lupin) alkaloids, are secondary metabolites found in a wide variety of leguminous plant and tree species.[211] There are over 550 known quinolizidine alkaloids, with many of these secondary metabolites occurring in the subfamily Papilionoideae of the Fabaceae. They are especially abundant in the tribes Genisteae, Sophoreae and Thermopsideae. Biologically, the lupine alkaloids have been implicated in plant-herbivore interactions with many of these alkaloids displaying toxic and/or teratogenic properties to livestock.[212]
The first reported isolations of the lupine alkaloids revealed that in many cases both enantiomers of a given alkaloid occur in Nature;[213] however, after further examination, many of the proposed racemates were found to actually occur as optically pure isoforms.[214] This review will only focus on select major enantiomeric lupine metabolites that are known to occur in Nature. Several of these metabolites are shown in Table 13, along with a limited listing of the respective sources of isolation.
Table 13.
Lupine alkaloid enantiomers.[a]
| Lupine Alkaloid | Species | Biological Activity |
|---|---|---|
 (+)-sparteine |
Cytisus caucasicus, Lupinus pusillus (Rusty Lupine), Genista monosperma (Bridal Broom), Pelargonium acutifolia, Pelargonium longifolia, Sophora pachycarpa, Ammodendron spp., Baptisia spp. Chamaecytisus proliferus, Adenocarpus hispanicus, Hovea linearis (Common Hovea),[214] Lygos raetam var. sarcocarpa,[215] Lupinus albus (White Lupine),[216] Genista lydia (Hardy Dwarf Broom)[217] | Highly toxic |
| (−)-sparteine | Cytisus scoparius (Scotch Broom), Lupinus spp., Adenocarpus spp., Piptanthus nanus, Sarothamnus spp., Chamaecytisus proliferus, Corothamnus rectipilosus,[215] Chamaecytisus austriacus,[216] Genista lydia[217] | Oxytoxic, Antiarrhythmic |
 (+)-lupanine |
Lupinus albus, Lupinus termis, Podalyria buxifolia, Virgilia capensis, Cytisus scoparius, other Cytisus spp., Cadia purpurea, Ammopiptanthus mongolicus, Thermopsis chinensis (Chinese Bush Pea), Leontice spp., Genista spp., Templetonia spp. Chamaespartium sagittale,[215] Corothamnus rectipilosus,[215] Genista rumelica,[215] Genista sessilifolia,[215] Chamaecytisus austriacus,[216] Genista lydia[217] | Toxic to livestock |
| (−)-lupanine | Lupinus albus, Lupinus termis, Podalyria buxifolia, Virgilia capensis, Lupinus pusillus, Lupinus macounii, Baptisia versicolor, Podalyria calyptrata (Water Blossom Pea), Ammodendron spp., Leontice smirnovii, Leontice eversmannii, Lygos raetam var. sarcocarpa,[215] Genista lydia,[217] Clathrotropis glaucophylla[218] | Toxic to livestock |
 (+)-β-isosparteine |
Lupinus spp.,[211a] Lupinus pusillus[219] | |
| (−)-β-isosparteine | Lupinus pusillus,[211a,220] Lupinus sericeus (Silky Lupine),[221] Lupinus argenteus stenophyllus (Silvery Lupine), Lupinus solosericeus, Sophora secundiflora (Texas Mountain Laurel) | |
 (+)-thermopsine |
Lupinus caudatus (Tailcup Lupine), Lupinus corymbosus | |
| (−)-thermopsine | Thermopsis lanceolata (Golden Banner), Thermopsis rhombifolia (Buffalo Bean), Sophora secundiflora | |
 (+)-ormosanine |
Ormosia panamensis (Coronil), Piptanthus nanus | |
| (−)-ormosanine | Podopetalum ormondii, Ormosia semicastrata, Ormosia jamaicensis, Piptanthus nanus | |
 (+)-piptanthine |
Hovea linearis, Templetonia retusa (Cockies Tounge), Ormosia semicastrata, Ammopiptanthus mongolicus | |
| (−)-piptanthine | Hovea linearis, Templetonia retusa, Ormosia semicastrata, Ammopiptanthus mongolicus, Piptanthus nanus |
For species and biological activity that do not have a reference, please refer to the Dictionary of Alkaloids (reference 193)
Unfortunately, biosynthetic studies of enantiomeric lupine alkaloids are rather limited. Independently, Spenser, Robins, and Wink demonstrated that the quinolizidine alkaloids are biosynthesized from lysine via a symmetrical cadaverine intermediate. Enzymatically, a lysine decarboxylase that converts lysine into cadaverine was isolated from lupine cell cultures and intact plants; however, late stage biosynthetic conversions remain elusive.[222] Through feeding studies using labeled precursors, both lysine and cadaverine have been shown to incorporate into (−)-sparteine, (+)-sparteine, and (+)-lupanine (Scheme 14).[223]
Scheme 14.

Biogenesis of (−)-sparteine.
5.4-Piperidine and Pyridine Alkaloids
Both piperidine and pyridine alkaloids are secondary metabolites containing a 6-membered heterocyclic ring system with a nitrogen containing nucleus, in which this heterocycle is saturated in piperidine alkaloids and unsaturated in pyridine alkaloids. Simple piperidine and pyridine natural products are generally associated with toxic alkaloids, as observed in one of the most well known examples of a pyridine alkaloid, nicotine.[8] Similarly, several piperidine alkaloids are known poisons produced by poison hemlock, Conium maculatum.[224] Many of the piperidine and pyridine alkaloids are known teratogenic agents,[225] and the ingestion of plants that produce these natural products by pregnant livestock can result in newborns with multiple congenital contractures and/or cleft palates.[226] As shown in Table 14, enantiomeric metabolites of these alkaloids are particularly rare, but those that are known are produced by a variety of plant sources. Several of these metabolites, such as ammodendrine, are produced as a nearly racemic mixture by a single species, whereas other metabolites occur as partial racemates.[6b] For example, the S- or (−)-isoform of nicotine generally makes up more than 95% of the natural product produced by tobacco.[8]
Table 14.
Enantiomeric piperidine and pyridine alkaloids.[a]
| Piperidine and Pyridine Alkaloid | Species | Biological Activity |
|---|---|---|
 (+)-ammodendrine |
Genista sphaerocarpa, Ammodendron conollyi, Ammodendron spp., Sophora franchetiana, Sophora tomentosa (Yellow Necklace Pod),[228] Coelidium fourcadei, Lupinus formosus (Summer Lupine),[225a–b] Lupinus varius,[225a] Lupinus hirsutus[225a] | Teratogen[225] |
| (−)-ammodendrine | Ammodendron conollyi, Ammodendron spp., Sophora franchetiana, Sophora tomentosa,[228] Coelidium fourcadei, Lupinus formosus,[225a–b] Castilleja miniata (Giant Red Indian Paintbrush)[225a] | Teratogen[225] |
 (+)-anabasine |
Nicotiana glauca (wild tobacco),[225d] Aphaenogaster subterranea (ant),[229] Aphaenogaster miamiana (ant)[229] | Teratogen[225d] |
| (−)-anabasine | Nicotiana glauca,[225d] Anabasis aphylla,[230] Aphaenogaster subterranea,[229] Aphaenogaster miamiana,[229] Messor sanctus (ant)[229] | Teratogen[225d] |
 (+)-coniine |
Conium maculatum (poison hemlock)[225c] | Toxic to livestock[225c] |
| (−)-coniine | Conium maculatum[225c] | Toxic to livestock[225c] |
 (+)-nicotine |
Nicotiana tabacum (cultivated tobacco) | Weakly binds to nicotinic acetylcholine receptors[8] |
| (−)-nicotine | Nicotiana tabacum, other Nicotiana spp., Asclepias syriaca (Common Milkweed), Lycopodium spp., Equisetum arvense (Field Horsetail), Sedum acre (Golden Stonecrop) | Incredibly reactive at nicotinic acetylcholine receptors[8] |
For species and biological activity that do not have a reference, please refer to the Dictionary of Alkaloids (reference 183)
While significant effort has been put forth towards elucidating the biosynthetic pathway of these metabolites,[8,227] the enantiomeric biogenesis of the piperidine and pyridine alkaloids has not been investigated. As observed with nicotine, biosynthetic studies have focused on the biogenesis of the major enantiomer, (−)-nicotine, and as such, there are currently no known explanations for the formation of (+)-nicotine. Furthermore, the characterization of enzymes responsible for the biosynthesis of piperidine and pyridine alkaloids, such as coniine, demonstrate a high substrate- and stereo-specificity, and therefore, the biosynthesis of only one enantiomer is known.[182] To date, no enantiomerically opposite enzymes responsible for the biosynthesis of pyridine or piperidine alkaloids have been identified.
5.5-Benzylisoquinoline Alkaloids
Benzylisoquinoline alkaloids (BIA) are a structurally diverse group of nitrogen-containing plant secondary metabolites, consisting of more than 2,500 defined structures found mostly in five plant families: the Papaveraceae, Fumariaceae, Ranunculaceae, Berberidaceae, and Menispermaceae.[196c,231] Structurally, the benzylisoquinoline natural products can be further divided into numerous groups, such as the aporphines, phthalideisoquinolines, morphinans, protoberberines, and pavines.[232] Benzylisoquinoline alkaloids are widely known to be produced by opium poppy (Papaver somniferum), and are well known for their wide range of biological activity and pharmaceutical importance, including: morphine and codeine, two well-known analgesics; papaverine, a muscle relaxant; noscapine, an antitumor agent; and sanguinarine, an antibiotic.[231b,233] The biosynthesis of benzylisoquinoline alkaloids has been thoroughly studied, and as such most of the biogenesis is understood at an enzymatic level.[196c,231–233] Furthermore, as shown in Table 15, the occurrence of enantiomeric benzylisoquinolines is known, however the biosynthetic formation of all these enantiomers is not fully understood.
Table 15.
Enantiomeric benzylisoquinoline alkaloid secondary metabolites.[a]
| Benzylisoquinoline Alkaloid | Species | Biological Activity |
|---|---|---|
 (+)-reticuline |
Annona reticulata (Wild-sweetsop), Phylica rogersii, Papaver somniferum (Opium Poppy) | |
| (−)-reticuline | Romneya coulteri var. trichocalyx (Coulter’s Matilija Poppy) | |
 (+)-salutaridine |
Croton salutaris, Croton balsamifera, Papaver spp., Glaucium spp. | Antitumor |
| (−)-sinoacutine (salutaridine enantiomer) | Sinomenium acutum, Corydalis spp., Gacium spp., Nandina spp., Croton salutaris, Stephania yunnanensis[234] | |
 (+)-scoulerine |
Corydalis tuberosa, Corydalis cava | |
| (−)-scoulerine | Many Corydalis spp., Erythrina orientalis, Bocconia frutescens (Tree Poppy), Glaucium spp., Eschscholzia lobbii (Frying Pans), Fumaria officinalis (Common Fumitory) | Antiemetic and Antitussive activity |
 (+)-canadine |
Corydalis tuberosa | Analgesic, Hypotensive |
| (−)-canadine | Hydrastis canadensis (Goldenseal), several Corydalis spp. | Analgesic, Hypotensive |
For species and biological activity that do not have a reference, please refer to the Dictionary of Alkaloids (reference 193)
Biosynthetically, benzylisoquinoline alkaloids are all derived from L-tyrosine along a basic benzylisoquinoline pathway.[232,233] As shown in Scheme 15, the first committed step in the BIA biosynthesis is the asymmetric Pictet-Spengler condensation of tyrosine derived dopamine and para-hydroxyphenylacetaldehyde (4-HPAA) via norcoclaurine synthase (NCS) to yield optically pure (S)-norcoclaurine.[231] Through four enzymatic transformations (S)-norcoclaurine is converted to optically pure (S)-reticuline. This intermediate serves as a vital branching point to the various benzylisoquinoline alkaloids, many of which display the same stereochemistry as (S)-reticuline; however, the promorphinan and morphinan subgroup of BIAs contain the opposite (R) stereochemistry.[232] These alkaloids are derived from (R)-reticuline, which arises from the inversion of stereochemistry of (S)-reticuline by way of oxidation and reduction via 1,2-dehydroreticuline synthase (DRS) and 1,2-dehydroreticuline reductase (DRR), respectively.[235]
Scheme 15.

Early stages of the benzylisoquinoline alkaloid biogenesis.
Early on, the biogenesis of some of the (R)-configured benzylisoquinoline alkaloids had been proposed to arise via (R)-reticuline; however, tracer incorporation studies have shown that this is not the case.[236] Based on the lack of incorporation of (R)-reticuline into the more advanced (R)-configured BIAs, it has been suggested that the formation of the (R)-series of metabolites arise from a stereochemical inversion of the (S)-enantiomer via enzymatic oxidation and reduction. Similar to the formation of enantiomeric reticuline, other BIA enantiomers, such as (R)- and (S)-canadine are formed from an inversion of stereochemistry. In the case of enantiomeric canadine, (S)-canadine is first oxidized by (S)-tetrahydroxyprotoberberine oxidase (STOX) to form berberine, which is subsequently reduced by berberine reductase to yield (R)-canadine (Scheme 16).[236,237]
Scheme 16.

Biosynthesis of enantiomeric canadine.
Unfortunately, substantial information is lacking in understanding the biosynthesis of all of the enantiomeric BIAs. As seen in the elucidation of the enantiomeric nicotine biosynthetic pathway, the biogenesis of only one enantiomer of the BIA natural products is understood, as is the case with (S)-scoulerine and (+)-salutaridine.[233] To date, no enantiomerically opposite enzymes in the biosynthesis of the benzylisoquinoline alkaloids have been isolated.
6. Summary and Outlook
As demonstrated in this Review, the formation of enantiomeric natural products by Nature is not as uncommon as one might have initially expected. While the number of enantiomeric natural products that have been discovered to date represent a small fraction (less than 1%) of the biospheres’ metabolome, it is clear that biogenetic mechanisms to create distinct enantiomers are widely expressed. Many puzzles and stereochemical anomalies remain and provide for a fertile area of future inquiry and discovery. A substantial body of research has been carried out over the years to attempt to understand the biogenesis of some enantiomeric metabolites, but our level of understanding remains in its’ infancy.
The points at which an enantiodivergence may occur in a biosynthetic pathway vary. For example, in the terpene cyclases (pinene, limonene, see Scheme 2), a simple precursor, such as geranyl diphosphate, can give rise to the two enantiomeric forms in the first committed step of the pathway. In other instances, the enantiodivergent step occurs deeper into the pathway, as appears to be the case with the stephacidins and notoamides. With recent advances in whole genome sequencing, proteomics, metabolomics and genome mining, significant leaps in unraveling many of these intriguing biosynthetic pathways are expected to accelerate. In many instances, enantiomeric metabolites arise from two distinct enzymes that function in an enantiodivergent and distinct mechanistic manifold, such as the (+)- and (−)-limonene synthases. On the other hand, the accumulation of both enantiomers from a single enzyme may be due in part to a lack of enzyme substrate- and stereo-specificity, which was observed in the biosynthesis of (+)-carvone via (+)-limonene-6-hydroxylase and (+)-trans-carveol dehydrogenase. In several examples, the biosynthetic formation of one enantiomer is understood at an enzymatic level, while the biogenesis of the minor enantiomer remains unknown. Furthermore, the enantioselective catabolism of an initially produced racemic metabolite, remains an additionally valid paradigm that has been little investigated. As more and more natural metabolites are discovered, despite the recent, severe cutback in research funding for natural products isolation and structural elucidation, it is almost a certainty, that additional families of enantiomeric natural substances will be discovered. The myriad of genetic and biochemical mechanisms controlling and leading to the phenotypic expression of enantiomeric natural substances will continue to tantalize the imagination. The impact of next-generation sequencing and bioinformatic tools to mine natural product biosynthetic genes, and assemble pathways from diverse microbial and plant species will have an enormous impact on future investigations. Moreover, the continued development of molecular tools to engineer gene-disruption mutants, and for heterologous expression will enable new insights into the details of natural product assembly and functional group elaboration. Moreover, the ability to clone, overexpress and purify biosynthetic enzymes from diverse microbial and plant species will allow in vitro studies of the metabolic pathway components with natural and unnatural substrates. These powerful approaches will provide access to even more chemical diversity from which exciting biological activities and potential drug leads might be identified.
Finally, the genetic mechanisms resulting in the formation of enantiomeric natural products has yet to be analyzed from an evolutionary molecular genetics perspective.[238] Focusing on molecular evolution, key questions remain about the mechanism(s) by which an enzyme making one enantiomeric form evolves to produce the enantiomeric form. Does the process, for example, require a gene duplication event, where one of the paralogs can be freed from selective constraints, thus allowing it to evolve by genetic drift to acquire multiple substitutions? Or, alternatively, can a single ortholog evolve this ability independently from different ancestral forms of an enzymatic function? Based on the examples covered here in this review, it appears that both may occur. A related question in cases where the two stereoselective enzymes have evolved from a common ancestor is, what is the ancestral state - an enzyme that produces a (partially) racemic mixture or an enzyme that produces one pure enantiomer? It is interesting to note that instances in which an enzyme produces both enantiomers are known, and also cases in which one enantiomer is produced in great excess over the other. Can the latter be thought of as an example of an intermediate state in the evolution of an enzyme from a nonselective form to a purely selective form? This provocative question merits thought and investigation because of course for evolution to proceed from one form of the enzyme to the other (i.e., from a (−)-producing form to a (+)-producing form), if it does not happen in a single mutation (say a single amino acid substitution in an active site) but rather through a transition requiring multiple amino acid changes, then all of the intermediate states have to be both possible functionally and also not be deleterious to the producing organism. Thus, the ancestral state and the number of changes in a protein required for the transition from one antipodal product to the other are fundamental questions that have been little, if at all studied. Another equally interesting question concerns the adaptive significance of these stereoselective transitions and this field is probably wide open. Phylogenetic and bioinformatic analysis of enantiomeric enzymes holds the promise of revealing mechanistic signatures of their functional evolution, including the roles of gene duplication, exon shuffling, natural selection, and genetic drift. What is particular tantalizing about enzymes that generate opposite enantiomers of a particular structure, is the ability to study many independent instances of this evolutionary process, a rare opportunity in molecular evolution. Such investigation has the potential of revealing generalities about the evolution of protein structure/function for this subtlest of possible change in natural product chemistry.
Biographies

Robert M. Williams received a B.A. (Chemistry, 1975) from Syracuse University (Ei-ichi Negishi), the Ph.D. (1979) at MIT (W.H. Rastetter) and was a post-doc at Harvard (1979–80; R.B. Woodward/Yoshito Kishi). He joined Colorado State University in 1980 and named University Distinguished Professor in 2002. Significant awards include the ACS Cope Scholars Award (2002) and the ACS Ernest Guenther Award in the Chemistry of Natural Products (2011). His interdisciplinary research program (>280 publications) at the chemistry-biology interface concerns the synthesis, biosynthesis and the chemical biology of biomedically significant natural products.

Jennifer M. Finefield graduated from Illinois State University in 2004 with a B.S. in Chemistry. She received her Ph.D. in 2011 from Colorado State University under the guidance of Professor Robert M. Williams, wherein her work focused on elucidating the biosynthesis of reverse prenylated indole secondary metabolites from Aspergillus versicolor. She is currently working on the synthesis of a novel HDAC inhibitor as a post-doc for Professor Robert M. Williams at Colorado State University.

David H. Sherman is the Hans W. Vahlteich Professor of Medicinal Chemistry, Professor of Chemistry, and Professor of Microbiology & Immunology at the University of Michigan. He obtained a B.A. from UC Santa Cruz (1978), the Ph.D. from Columbia (1981) and was a post-doc at Yale (1981–82) and MIT (1982–84). Dr. Sherman’s research concerns molecular genetic, biochemical and bioorganic chemical studies of microbial natural product biosynthesis. His lab employs pathway engineering, synthetic biology and chemoenzymatic synthesis to harness the tremendous metabolic capabilities of diverse bacteria, fungi and unculturable microbial symbionts.

Martin Kreitman, Ph.D. is a Professor at the University of Chicago. He received a B.S. in Biology (Stony Brook University, 1975), the M.S. in Zoology (University of Florida, 1977), and a Ph.D. in Population Genetics (Harvard, 1983). Significant awards include the 1991 MacArthur Fellows Program and became a Fellow of the American Academy of Arts and Sciences (2010). His research interests center on understanding evolutionary forces governing molecular variation and evolution. His lab is currently investigating the functional and evolutionary biology of eukaryotic cis-regulatory sequences and is developing models of complex human genetic diseases.
Footnotes
We are grateful to the National Institutes of Health for financial support (RO1CA070375 to RMW & DHS)
References
- 1.Miller KA, Tsukamoto S, Williams RM. Nature Chem. 2009;1:63–68. doi: 10.1038/nchem.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Challis GL, Hopwood DA. Proc Nat Acad Sci USA. 2003;100:14555–14561. doi: 10.1073/pnas.1934677100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herbert RB. The Biosynthesis of Secondary Metabolites. 2. Chapman and Hall; New York: 1989. [Google Scholar]
- 4.Clardy J, Walsh C. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- 5.Gunatilaka AAL. J Nat Prod. 2006;69:509–526. doi: 10.1021/np058128n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Colegate SM, Molyneux RJ. Bioactive Natural Products: Detection, Isolation, and Structural Determination. 2. Taylor and Francis; Boca Raton: 2008. pp. 209–219. [Google Scholar]; b) Lee ST, Gardner DR, Chang CWT, Panter KE, Molyneux RJ. Phytochem Anal. 2008;19:395–402. doi: 10.1002/pca.1064. [DOI] [PubMed] [Google Scholar]
- 7.Somogyi A, Bochner F, Foster D. Aust Prescr. 2004;27:47–49. [Google Scholar]
- 8.a) Gorrod JW, Jacob P. Analytical Determination of Nicotine and Related Compounds and Their Metabolites. Elsevier; Amsterdam: 1999. [Google Scholar]; b) Pogocki D, Ruman T, Danilczuk M, Danilczuk M, Celuch M, Walajtys-Rode E. Eur J Pharmacol. 2007;563:18–39. doi: 10.1016/j.ejphar.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 9.Connolly JD, Hill RA. Dictionary of Terpenoids. Chapman and Hall; London: 1991. [Google Scholar]
- 10.Davis EM, Croteau R. Top Curr Chem. 2000;209:53–95. [Google Scholar]
- 11.a) Gregson RP, Ouvrier D. J Nat Prod. 1982;45:412–414. [Google Scholar]; b) Fontana A, Fakhr I, Mollo E, Cimino G. Tetrahedron: Asymmetry. 1999;10:3869–3872. [Google Scholar]
- 12.Wise ML. Phycologia. 2003;42:370–377. [Google Scholar]
- 13.a) Croteau R. Chem Rev. 1987;87:929–954. [Google Scholar]; b) Lange BM, Rujan T, Martin W, Croteau R. Proc Natl Acad Sci USA. 2000;97:13172–13177. doi: 10.1073/pnas.240454797. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McGarvey DJ, Croteau R. Plant Cell. 1995;7:1015–1026. doi: 10.1105/tpc.7.7.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hyatt DC, Youn B, Zhao Y, Santhamma B, Coates RM, Croteau RB, Kang C. Proc Nat Acad Sci USA. 2007;104:5360–5365. doi: 10.1073/pnas.0700915104. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Croteau R, Cane DE. Methods Enzymol. 1985;110:352–405. [Google Scholar]; b) Cane DE. Acc Chem Res. 1985;18:220–226. [Google Scholar]; c) Wheeler CJ, Croteau R. J Biol Chem. 1987;262:8213–8219. [PubMed] [Google Scholar]; d) Schwab W, Williams DC, Davis EM, Croteau R. Arch Biochem Biophys. 2001;392:123–136. doi: 10.1006/abbi.2001.2442. [DOI] [PubMed] [Google Scholar]
- 16.a) Laska M, Teubner P. Chem Senses. 1999;24:161–170. doi: 10.1093/chemse/24.2.161. [DOI] [PubMed] [Google Scholar]; b) Bentley R. Chem Rev. 2006;106:4099–4112. doi: 10.1021/cr050049t. [DOI] [PubMed] [Google Scholar]
- 17.a) Kjonaas R, Croteau R. Arch Biochem Biophys. 1983;220:79–89. doi: 10.1016/0003-9861(83)90389-2. [DOI] [PubMed] [Google Scholar]; b) Alonso WR, Rajaonarivony JIM, Gershenzon J, Croteau R. J Biol Chem. 1992;267:7582–7587. [PubMed] [Google Scholar]; c) Colby SM, Alonso WR, Katahira EJ, McGarvey DJ, Croteau R. J Biol Chem. 1993;268:23016–23024. [PubMed] [Google Scholar]
- 18.Pyun H-J, Coates RM, Wagschal KC, McGeady P, Croteau RB. J Org Chem. 1993;58:3998–4009. [Google Scholar]
- 19.Yuba A, Yazaki K, Tabata M, Honda G, Croteau R. Arch Biochem Biophys. 1996;332:280–287. doi: 10.1006/abbi.1996.0343. [DOI] [PubMed] [Google Scholar]
- 20.Bohlmann J, Steele CL, Croteau R. J Biol Chem. 1997;272:21784–21792. doi: 10.1074/jbc.272.35.21784. [DOI] [PubMed] [Google Scholar]
- 21.Rani K, Akhila A. Fitoterapia. 1998;69:337–348. [Google Scholar]
- 22.Maruyama T, Ito M, Kiuchi F, Honda G. Biol Pharm Bull. 2001;24:373–377. doi: 10.1248/bpb.24.373. [DOI] [PubMed] [Google Scholar]
- 23.Bouwmeester HJ, Gerschenzon J, Konings MCJM, Croteau R. Plant Physiol. 1998;117:901–912. doi: 10.1104/pp.117.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lücker J, El Tamer MK, Schwab W, Verstappen FWA, van der Plas LHW, Bouwmeester HJ, Verhoeven HA. Eur J Biochem. 2002;269:3160–3171. doi: 10.1046/j.1432-1033.2002.02985.x. [DOI] [PubMed] [Google Scholar]
- 25.Gershenzon J, Maffei M, Croteau R. Plant Physiol. 1989;89:1351–1357. doi: 10.1104/pp.89.4.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito M, Kiuchi F, Yang LL, Honda G. Biol Pharm Bull. 2000;23:359–36. doi: 10.1248/bpb.23.359. [DOI] [PubMed] [Google Scholar]
- 27.a) Bouwmeester HJ, Davies JAR, Toxopeus H. J Agric Food Chem. 1995;43:3057–3064. [Google Scholar]; b) Faber B, Bangert K, Mosandl A. Flavour Frag J. 1997;12:305–314. [Google Scholar]
- 28.Attygalle AB, Wu X, Maddison DR, Will KW. Naturwissenschaften. 2009;96:1443–1449. doi: 10.1007/s00114-009-0596-8. [DOI] [PubMed] [Google Scholar]
- 29.Sjödin K, Persson M, Fäldt J, Ekberg I, Borg-Karlson AK. J Chem Ecol. 2000;26:1701–1720. [Google Scholar]
- 30.Tsokou A, Georgopoulou K, Melliou E, Magiatis P, Tsitsa E. Molecules. 2007;12:1233–1239. doi: 10.3390/12061233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holm Y, Vuorela P, Hiltunen R. Flavour Frag J. 1997;12:397–400. [Google Scholar]
- 32.Akhila A, Banthorpe DV, Rowan MG. Phytochemistry. 1980;19:1433–1437. [Google Scholar]
- 33.Savage TJ, Ichii H, Hume SD, Little DB, Croteau R. Arch Biochem Biophys. 1995;320:257–265. doi: 10.1016/0003-9861(95)90008-x. [DOI] [PubMed] [Google Scholar]
- 34.Phillips MA, Wildung MR, Williams DC, Hyatt DC, Croteau R. Arch Biochem Biophys. 2003;411:267–276. doi: 10.1016/s0003-9861(02)00746-4. [DOI] [PubMed] [Google Scholar]
- 35.Gambliel H, Croteau R. J Biol Chem. 1982;257:2335–2342. [PubMed] [Google Scholar]
- 36.Wagschal K, Savage TJ, Croteau R. Tetrahedron. 1991;47:5933–5944. [Google Scholar]
- 37.Croteau R, Gershenzon J, Wheeler CJ, Satterwhite DM. Arch Biochem Biophys. 1990;277:374–381. doi: 10.1016/0003-9861(90)90593-n. [DOI] [PubMed] [Google Scholar]
- 38.Battersby AR, Laing DG, Ramage R. J Chem Soc, Perkin Trans 1. 1972;21:2743–2748. doi: 10.1039/p19720002743. [DOI] [PubMed] [Google Scholar]
- 39.Croteau R, Shaskus J. Arch Biochem Biophys. 1985;236:535–543. doi: 10.1016/0003-9861(85)90656-3. [DOI] [PubMed] [Google Scholar]
- 40.Banthorpe DV, Baxendale D. J Chem Soc. 1970:2694–2696. doi: 10.1039/j39700002694. [DOI] [PubMed] [Google Scholar]
- 41.Wise ML, Savage TJ, Katahira E, Croteau R. J Biol Chem. 1998;273:14891–14899. doi: 10.1074/jbc.273.24.14891. [DOI] [PubMed] [Google Scholar]
- 42.Gambliel H, Croteau R. J Biol Chem. 1984;259:740–748. [PubMed] [Google Scholar]
- 43.a) Croteau R, Satterwhite DM, Wheeler CJ, Felton NM. J Biol Chem. 1989;264:2075–2080. [PubMed] [Google Scholar]; b) Croteau R, Satterwhite DM. J Biol Chem. 1989;264:15309–15315. [PubMed] [Google Scholar]; c) McGeady P, Pyun HJ, Coates RM, Croteau R. Arch Biochem Biophys. 1992;299:63–72. doi: 10.1016/0003-9861(92)90244-q. [DOI] [PubMed] [Google Scholar]
- 44.Prosser I, Altug IG, Phillips AL, König WA, Bouwmeester HJ, Beale MH. Arch Biochem Biophys. 2004;432:136–144. doi: 10.1016/j.abb.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt CO, Bouwmeester HJ, Franke S, König WA. Chirality. 1999;11:353–362. [Google Scholar]
- 46.Stark CM, Back K, Chappell J, Noel JP. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 47.Cane DE, Tandon M, Prabhakaran PC. J Am Chem Soc. 1993;115:8103–8106. [Google Scholar]
- 48.Cane DE, Oliver JS, Harrison PMH, Abell C, Hubbard BR, Kane CT, Lattman R. J Am Chem Soc. 1990;112:4513–4524. [Google Scholar]
- 49.Colby SM, Crock J, Dowdle-Rizzo B, Lemaux PG, Croteau R. Biochemistry. 1998;95:2216–2221. doi: 10.1073/pnas.95.5.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steele CL, Crock J, Bohlmann J, Croteau R. J Biol Chem. 1998;273:2078–2089. doi: 10.1074/jbc.273.4.2078. [DOI] [PubMed] [Google Scholar]
- 51.Cane DE. Chem Rev. 1990;90:1089–1103. [Google Scholar]
- 52.Niwa M, Iguchi M, Yamamura S. Chem Pharm Bull. 1980;28:997–999. [Google Scholar]
- 53.Searle PA, Jamal NM, Lee GM, Molinski TF. Tetrahedron. 1994;50:3879–3888. [Google Scholar]
- 54.Horton P, Inman WD, Crews P. J Nat Prod. 1990;53:143–151. [Google Scholar]
- 55.Beechan CM, Djerassi C, Eggert H. Tetrahedron. 1978;34:2503–2508. [Google Scholar]
- 56.Yong KWL, Jankam A, Hooper JNA, Suksamrarn A, Garson MJ. Tetrahedron. 2008;64:6341–6348. [Google Scholar]
- 57.Naya Y, Miyamoto F, Takemoto T. Experientia. 1978;34:984–986. [Google Scholar]
- 58.Yoshihara K, Hirose Y. Proceedings of the 21st Symposium on the Chemistry of Terpenes, Essential Oils and Aromatics; The Chemical Society of Japan, Okayama. 1973. p. Abstract no. 1E704. [Google Scholar]
- 59.König WA, Bülow N, Fricke C, Melching S, Rieck A, Muhle H. Phytochemistry. 1996;43:629–633. [Google Scholar]
- 60.Degenhardt J, Köllner TG, Gershenzon J. Phytochemistry. 2009;70:1621–1637. doi: 10.1016/j.phytochem.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 61.Lorimer SD, Weavers RT. Phytochemistry. 1987;26:3207–3215. [Google Scholar]
- 62.Deguerry F, Pastore L, Wu S, Clark A, Chappell J, Schalk M. Arch Biochem Biophys. 2006;454:123–136. doi: 10.1016/j.abb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 63.Pietsch M, König WA. Phytochem Anal. 2000;11:99–105. [Google Scholar]
- 64.Bordoloi M, Shukla VS, Nath SC, Sharma RP. Phytochemistry. 1989;28:2007–2037. [Google Scholar]
- 65.a) Nabeta K, Katayama N, Nakagawara S, Katoh K. Phytochemistry. 1993;32:117–122. [Google Scholar]; b) Nabeta K, Fujita M, Komuro K, Katayama K, Takasawa T. J Chem Soc, Perkin Trans 1. 1997:2065–2070. [Google Scholar]
- 66.Cane DE, Tandon M. J Am Chem Soc. 1995;117:5602–5603. [Google Scholar]
- 67.Tomita B, Hirose Y. Phytochemistry. 1972;11:3355–3357. [Google Scholar]
- 68.Gerber NN. Phytochemistry. 1971;10:185–189. [Google Scholar]
- 69.Ohta Y, Hirose Y. Tetrahedron Lett. 1967:2073–2075. [Google Scholar]
- 70.Melching S, Bülow N, Wihstutz K, Jung S, König WA. Phytochemistry. 1997;44:1291–1296. [Google Scholar]
- 71.Grotewold E. The Science of Flavonoids. Springer; New York: 2006. [Google Scholar]
- 72.Kazlauskas R, Murphy PT, Wells RJ. Tetrahedron Lett. 1978:4951–4954. [Google Scholar]
- 73.Guella G, Mancini I, Guerriero A, Pietra F. Helv Chim Acta. 1985;68:1276–1282. [Google Scholar]
- 74.Mollo E, Gavagnin M, Carbone M, Guo Y-W, Cimino G. Chemoecology. 2005;15:31–36. [Google Scholar]
- 75.Cimino G, De Stefano S, Minale L. Experientia. 1975;31:1117–1118. [Google Scholar]
- 76.Fenical W, McConnell O. Experientia. 1975;31:1004–1005. [Google Scholar]
- 77.Koker MES. J Pharm Bioresour. 2010;7:77–92. [Google Scholar]
- 78.Fenical W, Sims JJ, Squatrito D, Wing RM, Radlick R. J Org Chem. 1973;38:2383–2386. doi: 10.1021/jo00953a022. [DOI] [PubMed] [Google Scholar]
- 79.Cimino G, De Stefano S, Guerriero A, Minale L. Tetrahedron Lett. 1975:1425–1428. [Google Scholar]
- 80.Spinella A, Alvarez LA, Avila C, Cimino G. Tetrahedron Lett. 1994;35:8665–8668. [Google Scholar]
- 81.Weenen H, Nkunya MHH, El-Fadl AA, Harkema S, Zwanenburg B. J Org Chem. 1990;55:5107–5109. [Google Scholar]
- 82.Perry NB, Weavers RT. Phytochemistry. 1985;24:2899–2904. [Google Scholar]
- 83.De Santis V, Medina JD. J Nat Prod. 1981;44:370–372. [Google Scholar]
- 84.a) Bevan CWL, Ekong DEU, Okogun JI. J Chem Soc C. 1968:1067–1070. [Google Scholar]; b) Ekong DEU, Okogun JI. Chem Commun. 1967:72–73. [Google Scholar]
- 85.Weisshaar B, Jenkins GI. Curr Opin Plant Biol. 1998;1:21–257. doi: 10.1016/s1369-5266(98)80113-1. [DOI] [PubMed] [Google Scholar]
- 86.Vogt T. Mol Plant. 2010;3:2–20. doi: 10.1093/mp/ssp106. [DOI] [PubMed] [Google Scholar]
- 87.Suzuki S, Umezawa T, Shimada M. J Chem Soc, Perkin Trans 1. 2001:3252–3257. [Google Scholar]
- 88.Suzuki S, Umezawa T. J Wood Sci. 2007;53:273–284. [Google Scholar]
- 89.Davin LB, Lewis NG. Curr Opin Plant Biol. 2005;16:398–406. doi: 10.1016/j.copbio.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 90.Meresse P, Dechaux E, Monneret C, Bertounesque E. Curr Med Chem. 2004;11:2443–2466. doi: 10.2174/0929867043364531. [DOI] [PubMed] [Google Scholar]
- 91.Umezawa T. Photochem Rev. 2003;2:371–390. and references therein. [Google Scholar]
- 92.Umezawa T, Okunishi T, Shimada M. Wood Research. 1997;84:62–75. and references therein. [Google Scholar]
- 93.Davin LD, Wang H-B, Crowell AL, Bedgar DL, Martin DM, Sarkanen S, Lewis NG. Science. 1997;275:362–366. doi: 10.1126/science.275.5298.362. [DOI] [PubMed] [Google Scholar]
- 94.Dinkova-Kostova AT, Gang DR, Davin LB, Bedgar DL, Chu A, Lewis NG. J Biol Chem. 1996;271:29473–29482. doi: 10.1074/jbc.271.46.29473. [DOI] [PubMed] [Google Scholar]
- 95.Fujita M, Gang DR, Davin LB, Lewis NG. J Biol Chem. 1999;274:618–627. doi: 10.1074/jbc.274.2.618. [DOI] [PubMed] [Google Scholar]
- 96.Xia Z-Q, Costa MA, Pélissier HC, Davin LB, Lewis NG. J Biol Chem. 2001;276:12614–12623. doi: 10.1074/jbc.M008622200. [DOI] [PubMed] [Google Scholar]
- 97.Okunishi T, Sakakibara N, Suzuki S, Umezawa T, Shimada M. J Wood Sci. 2004;50:77–81. [Google Scholar]
- 98.Macías FA, López A, Varela RM, Torres A, Molinillo JMG. J Argric Food Chem. 2004;52:6443–6447. doi: 10.1021/jf048945d. [DOI] [PubMed] [Google Scholar]
- 99.Leu Y-L, Lin C-L, Kuo P-C. Arch Pharm Res. 2011;34:377–382. doi: 10.1007/s12272-011-0304-z. [DOI] [PubMed] [Google Scholar]
- 100.Park B-Y, Oh S-R, Ahn K-S, Kwon O-K, Lee H-K. Int Immunopharmacol. 2008;8:967–973. doi: 10.1016/j.intimp.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 101.Kong X, Yang J-R, Guo L-Q, Xiong Y, Wu X-Q, Huang K, Zhou Y. Eur J Pharmacol. 2009;620:84–89. doi: 10.1016/j.ejphar.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 102.Nishiwaki H, Kumamoto M, Shuto Y, Yamauchi S. J Agric Food Chem. 2011 doi: 10.1021/jf203222w. Just Accepted Manuscript. [DOI] [PubMed] [Google Scholar]
- 103.Masuda T, Akiyama J, Fujimoto A, Yamauchi S, Maekawa T, Sone Y. Food Chem. 2010;123:442–450. [Google Scholar]
- 104.Shoeb M, Macmanus SM, Kong-Thoo-Lin P, Celik S, Jaspars M, Nahar L, Sarker SD. DARU J Pharm Sci. 2007;15:118–122. [Google Scholar]
- 105.Zhang S, Won Y-K, Ong C-N, Shen H-M. Curr Med Chem. 2005;5:239–249. doi: 10.2174/1568011053765976. [DOI] [PubMed] [Google Scholar]
- 106.Lee K-H. ACS Symposium Series. 1992;507:367–379. [Google Scholar]
- 107.Desai AG, Qazi GN, Ganju RK, El-Tamer M, Singh J, Saxena AK, Bedi YS, Taneja SC, Bhat HK. Curr Drug Metab. 2008;9:581–591. doi: 10.2174/138920008785821657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.a) Sadhu SK, Okuyama E, Fujimoto H, Ishibashi M. Chem Pharm Bull. 2003;51:595–598. doi: 10.1248/cpb.51.595. [DOI] [PubMed] [Google Scholar]; b) Ahmed F, Sadhu SK, Ishibashi M. J Nat Med. 2010;64:393–401. doi: 10.1007/s11418-010-0424-7. [DOI] [PubMed] [Google Scholar]
- 109.Ma CJ, Sung SH, Kim YC. Planta Med. 2004;70:79–80. doi: 10.1055/s-2004-815463. [DOI] [PubMed] [Google Scholar]
- 110.Liu J-S, Huang M-F, Gao Y-L, Findlay JA. Can J Chem. 1981;59:1680–1684. [Google Scholar]
- 111.Winkel-Shirley B. Plant Physiol. 2001;126:485–493. doi: 10.1104/pp.126.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Allen DJ, Gray JC, Paiva NL, Smith JT. Electrophoresis. 2000;21:2051–2057. doi: 10.1002/1522-2683(20000601)21:10<2051::AID-ELPS2051>3.0.CO;2-6. and references therein. [DOI] [PubMed] [Google Scholar]
- 113.a) Guo L, Dixon RA, Paiva NL. J Biol Chem. 1994;269:22372–22378. [PubMed] [Google Scholar]; b) Dixon RA, Dalkin AD, Edwards R, Fahrendorf T, Gowri G, Harison MJ, Lamb CJ, Loake GJ, Maxwell CA, Orr JD, Paiva NL. In: Phenolic Metabolismn in Plants. Stafford HA, Ibrahim RK, editors. Plenum Press; New York: 1992. pp. 91–138. [Google Scholar]; c) Paiva NL, Edwards R, Sun Y, Hrazdina G, Dixon RA. Plant Mol Biol. 1991;17:653–667. doi: 10.1007/BF00037051. [DOI] [PubMed] [Google Scholar]
- 114.Cook JT, Ollis WD, Sutherland IO, Gottlieb OR. Phytochemistry. 1978;17:1419–1422. and reference therein. [Google Scholar]
- 115.VanEtten HD, Matthews PS, Mercer EH. Phytochemistry. 1983;22:2291–2295. and references therein. [Google Scholar]
- 116.a) Ingham JL. In: Phytoalexins. Bailey JA, Mansfield JW, editors. Glasgow; 1982. p. 21. [Google Scholar]; b) Delserone LM, Matthews DE, VanEtten HD. Phytochemistry. 1992;31:3813–3819. [Google Scholar]
- 117.Strange RN, Ingham JR, Cole DL, Cavill ME, Edwards C, Cooksey CJ, Garratt P. Zeitschr Naturforsch. 1985;40c:313–316. [Google Scholar]
- 118.Paiva NL, Edwards R, Sun Y, Hrazdina G, Dixon RA. Plant Mol Biol. 1991;17:653–667. doi: 10.1007/BF00037051. [DOI] [PubMed] [Google Scholar]
- 119.Roux DG, Maihs EA, Paulus E. Biochem J. 1961;78:834–839. doi: 10.1042/bj0780834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Delle Monache F, Ferrari F, Poce-Tucci A, Marini-Bettolo GB. Phytochemistry. 1972;11:2333–2335. [Google Scholar]
- 121.Nahrstedt A, Proksch P, Conn EE. Phytochemistry. 1987;26:1546–1547. [Google Scholar]
- 122.Donovan JL, Crespy V, Oliveira M, Cooper KA, Gibson BB, Williamson G. Free Radic Res. 2006;40:1029–1034. doi: 10.1080/10715760600868545. [DOI] [PubMed] [Google Scholar]
- 123.Krause M, Galensa R. Chromatographia. 1991;32:69–72. [Google Scholar]
- 124.Bourgaud E, Hehn A, Larbat R, Doerper S, Gontier E, Kellner S, Matern U. Phytochem Rev. 2006;5:293–308. [Google Scholar]
- 125.Mueller RL. Best Pract Res Clin Haematol. 2004;17:23–53. doi: 10.1016/j.beha.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 126.Donnelly DMX, Boland G. The Flavonoids: Advances in Research Since 1986. Vol. 239. Chapman and Hall; London: 1986. [Google Scholar]
- 127.a) Ollis WD, Landgraf HJPEM, Gottlieb OR, Taveira Magalhaes M. Anais Acad Bras Cienc. 1963;36:31. [Google Scholar]; b) Ollis WD. Experientia. 1966;22:777–783. doi: 10.1007/BF01897407. [DOI] [PubMed] [Google Scholar]; c) Eyton WB, Ollis WD, Sutherland IO, Gottlieb OR, Taveira Magalhaes M, Jackman LM. Tetrahedron. 1966;21:2683–2696. [Google Scholar]
- 128.Jung DJ, Porzel A, Huneck S. Phytochemistry. 1991;30:710–712. [Google Scholar]
- 129.Hata K, Sano K. Tetrahedron Lett. 1966:1461–1465. [Google Scholar]
- 130.Ahn K-S, Sim W-S, Kim I-H. Planta Med. 1996;62:7–9. doi: 10.1055/s-2006-957785. [DOI] [PubMed] [Google Scholar]
- 131.Bae E-A, Han MJ, Kim N-J, Kim D-H. Bio Pharm Bull. 1998;21:990–992. doi: 10.1248/bpb.21.990. [DOI] [PubMed] [Google Scholar]
- 132.Choi S-S, Han K-J, Lee J-K, Lee H-K, Han E-J, Kim D-H, Suh HW. Life Sciences. 2003;73:471–485. doi: 10.1016/s0024-3205(03)00311-4. [DOI] [PubMed] [Google Scholar]
- 133.Kang SY, Lee KY, Sung SH, Park MJ, Kim YC. J Nat Prod. 2001;64:683–685. doi: 10.1021/np000441w. [DOI] [PubMed] [Google Scholar]
- 134.Chatterjee A, Sen R, Ganguly D. Phytochemistry. 1978;17:328–329. [Google Scholar]
- 135.Basile A, Sorbo S, Spadaro V, Bruno M, Maggio A, Faraone N, Rosselli S. Molecules. 2009;14:939–952. doi: 10.3390/molecules14030939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Erdelmeier CAJ, Sticher O. Planta Med. 1985;51:407–409. doi: 10.1055/s-2007-969533. [DOI] [PubMed] [Google Scholar]
- 137.Lee S, Shin D-S, Kim JS, Oh K-B, Kang SS. Arch Pharm Res. 2003;26:449–452. doi: 10.1007/BF02976860. [DOI] [PubMed] [Google Scholar]
- 138.Cho S-K, El-Aty AMA, Choi J-H, Kim MR, Shim JH. J Pharmaceut Biomed Anal. 2007;44:1154–1158. doi: 10.1016/j.jpba.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 139.Xu Z, Wang X, Dai Y, Kong L, Wang F, Xu H, Lu D, Song J, Hou Z. Chem-Biol Interact. 2010;186:239–246. doi: 10.1016/j.cbi.2010.04.024. and references therein. [DOI] [PubMed] [Google Scholar]
- 140.Donnelly BJ, Donnelly DMX, Sharkey CB. Phytochemistry. 1965;4:337–340. [Google Scholar]
- 141.Donnelly DMX, O’Reilly J, Thompson J. Phytochemistry. 1972;11:823–826. [Google Scholar]
- 142.Donnelly DMX, O’Reilly J, Whalley WB. Phytochemistry. 1975;14:2287–2290. [Google Scholar]
- 143.Leite de Almeida ME, Gottlieb OR. Phytochemistry. 1974;13:751–752. [Google Scholar]
- 144.Letcher RM, Shirley IM. Phytochemistry. 1976;15:353–354. [Google Scholar]
- 145.Gregson M, Ollis WD, Sutherland IO, Gottlieb OR, Magalhaes MT. Phytochemistry. 1978;17:1375–1377. [Google Scholar]
- 146.a) Dempsey CB, Donnelly DMX, Laidlaw RA. Chem & Ind. 1963:491. [Google Scholar]; b) Rao MM, Seshadri TR. Tetrahedon Lett. 1963;4:211–215. [Google Scholar]
- 147.Muangnoicharoen N, Frahm AW. Phytochemistry. 1982;21:767–772. [Google Scholar]
- 148.Pathak V, Shirota O, Sekita S, Hirayama Y, Hakamata Y, Hayashi T, Yanagawa T, Satake M. Phytochemistry. 1997;46:1219–1223. [PubMed] [Google Scholar]
- 149.a) Jurd L, Stevens K, Manners G. Phytochemistry. 1972;11:3287–3292. [Google Scholar]; b) Gregson M, Ollis WD, Redman BT, Sutherland IO, Dietrichs HH, Gottlieb OR. Phytochemistry. 1978;17:1395–1400. [Google Scholar]
- 150.Beldjoudi N, Mambu L, Labaïd M, Grellier P, Ramanitrahasimbola D, Rasoanaivo P, Martin MT, Frappier F. J Nat Prod. 2003;66:1447–1450. doi: 10.1021/np030008x. [DOI] [PubMed] [Google Scholar]
- 151.Papageorgiou VP, Assimopoulou AN, Couladouros EA, Hepworth D, Nicolaou KC. Angew Chem Int Ed. 1999;38:270–300. doi: 10.1002/(SICI)1521-3773(19990201)38:3<270::AID-ANIE270>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 152.Inoue K, Ueda S, Nayeshiro H, Inouye H. Phytochemistry. 1983;22:737–741. [Google Scholar]
- 153.Rüedi P, Eugster CH. Helv Chim Acta. 1977;60:945–947. doi: 10.1002/hlca.19770600529. [DOI] [PubMed] [Google Scholar]
- 154.Baerson SR, Rimando AM. ACS Symposium Series 995. 2005:2–14. [Google Scholar]
- 155.Nonactin and its homologs are derived from repeating (+)- and (−) monomer precursor acids, therefore many of the following references refer to the isolation of the macrotetrolide natural product as opposed to the isolation of the individual monomer precursors. Bennett RE, Brindle SA, Giuffre NA, Jackson PW, Kowald J, Pansy FE, Perlman D, Trejo WH. Antimicrob Agents Chemother. 1961:169–172.Corbaz R, Ettinger L, Gaumann E, Keller-Schlierlein W, Kradolfer F, Kyburz E, Neipp L, Prelog V, Zahner H. Helv Chim Acta. 1955;38:1445.Dobler M. Helv Chim Acta. 1972;55:1371–1384. doi: 10.1002/hlca.19720550504.Dutcher JD. Antimicrob Agents Chemother. 1961:173–177.Gerlach H, Hutter R, Keller-Schlierlein W, Seibl J, Zahner H. Helv Chim Acta. 1967;50:1782–1793. doi: 10.1002/hlca.19670500711.Keller-Schierlein W, Gerlach H. Fortschr Chem Org Naturst. 1968;26:161–189.Menshikov GP, Rubinsthein MM. J Gen Chem USSR. 1956;26:2267.Meyers E, Pansy FE, Perlman D, Smith DA, Weisenborn FL. J Antibiot. 1965;18:128.Smith WC, Xiang L, Shen B. Antimicrob Agents Chemother. 2000:1809–1817. doi: 10.1128/aac.44.7.1809-1817.2000. and reference therein.
- 156.Fleck WF, Ritzau M, Heinze S, Gräfe U. J Basic Microbiol. 1996;36:235–238. [Google Scholar]
- 157.a) Bergy ME. J Antibiot. 1968;21:454–457. doi: 10.7164/antibiotics.21.454. [DOI] [PubMed] [Google Scholar]; b) Hoeksema H, Krueger WC. J Antibiot. 1976;29:704–709. doi: 10.7164/antibiotics.29.704. [DOI] [PubMed] [Google Scholar]
- 158.Cole SP, Rudd BAM, Hopwood DA, Chang C, Floss HG. J Antibiot. 1987;40:340–347. doi: 10.7164/antibiotics.40.340. [DOI] [PubMed] [Google Scholar]
- 159.Bergy ME. J Antibiot. 1968;21:454–457. doi: 10.7164/antibiotics.21.454. [DOI] [PubMed] [Google Scholar]
- 160.Ömura S, Tanaka H, Okada Y, Marumo H. J Chem Soc, Chem Comm. 1976:320–321. [Google Scholar]
- 161.Imai H, Suzuki K, Kadota S, Iwanami M, Saito T. J Antibiot. 1989;42:1186–1188. doi: 10.7164/antibiotics.42.1186. [DOI] [PubMed] [Google Scholar]
- 162.a) Ömura S, Tanaka H, Koyama Y, Öiwa R, Katagiri M, Awaya J, Nagai T, Hata T. J Antibiot. 1974;27:363–365. doi: 10.7164/antibiotics.27.363. [DOI] [PubMed] [Google Scholar]; b) Tanaka H, Koyama Y, Awaya J, Marumo H, Öiwa R, Katagiri M, Nagai T, Ömura S. J Antibiot. 1975;28:860–867. doi: 10.7164/antibiotics.28.860. [DOI] [PubMed] [Google Scholar]; c) Tanaka H, Koyama Y, Nagai T, Marumo H, Ömura S. J Antibiot. 1975;28:868–875. doi: 10.7164/antibiotics.28.868. [DOI] [PubMed] [Google Scholar]
- 163.Hill RA. Prog Chem Org Nat Prods. 1986;49:1–78. [Google Scholar]
- 164.Poch GK, Gloer JB. J Nat Prod. 1989;52:257–260. doi: 10.1021/np50062a006. [DOI] [PubMed] [Google Scholar]
- 165.a) Dai JR, Carté BK, Sidebottom PJ, Yew ALS, Ng SB, Huang Y, Butler MS. J Nat Prod. 2001;64:125–126. doi: 10.1021/np000381u. [DOI] [PubMed] [Google Scholar]; b) Gunatilaka AAL. J Nat Prod. 2006;69:509–526. doi: 10.1021/np058128n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.a) Gill M, Giménez A. J Chem Soc, Perkin Trans 1. 1990:2585–2591. [Google Scholar]; b) Donner CD, Gill M, Tewierik LM. Molecules. 2004;9:498–512. doi: 10.3390/90600498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bell AA, Stipanovic RD, Puhalla JE. Tetrahedron. 1976;32:1353–1356. [Google Scholar]
- 168.Aldridge DC, Davies AB, Jackson MR, Turner WB. J Chem Soc, Perkin Trans 1. 1974:1540–1541. [Google Scholar]
- 169.Findlay JA, Kwan D. Can J Chem. 1973;51:1617–1619. [Google Scholar]
- 170.Gill M, Smrdel AF, Strauch RJ, Begley MJ. J Chem Soc, Perkin Trans 1. 1990:1583–1592. [Google Scholar]
- 171.Beatte KD, Rouf R, Gander L, May TW, Ratkowsky D, Donner CD, Gill M, Grice ID, Tiralongo E. Phytochemistry. 2010;71:948–955. doi: 10.1016/j.phytochem.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 172.Gill M, Giménez A, Jhingran AG, Qureshi A. Phytochemistry. 1992;31:947–951. [Google Scholar]
- 173.a) Ashworth DM, Clark CA, Robinson JA. J Chem Soc, Perkin Trans 1. 1989:1461–1467. [Google Scholar]; b) Spavold ZM, Robinson JA. J Chem Soc, Chem Comm. 1988:4–6. [Google Scholar]; c) Ashworth DM, Robinson JA, Turner DL. J Chem Soc, Perkin Trans. 11988:1719–1727. [Google Scholar]
- 174.a) Woo AJ, Strohl WR, Priestley ND. Antimicrob Agents Chemother. 1999:1662–1668. doi: 10.1128/aac.43.7.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nelson ME, Priestley ND. J Am Chem Soc. 2002;124:2894–2902. doi: 10.1021/ja016965f. [DOI] [PubMed] [Google Scholar]; c) Cox JE, Priestley ND. J Am Chem Soc. 2005;127:7976–7977. doi: 10.1021/ja050068k. [DOI] [PubMed] [Google Scholar]; d) Walczak RJ, Nelson ME, Priestley ND. J Am Chem Soc. 2001;123:10415–10416. doi: 10.1021/ja005749o. [DOI] [PubMed] [Google Scholar]
- 175.Kwon H-J, Smith WC, Xiang L, Shen B. J Am Chem Soc. 2001;123:3385–3386. doi: 10.1021/ja0100827. [DOI] [PubMed] [Google Scholar]
- 176.Kakinuma S, Ikeda H, Ömura S. Tetrahedron. 1991;47:6059–6068. [Google Scholar]
- 177.a) Taguchi T, Ebizuka Y, Hopwood DA, Ichinose K. J Am Chem Soc. 2001;123:11376–11380. doi: 10.1021/ja015981+. [DOI] [PubMed] [Google Scholar]; b) Ichinose K. Actinomycetol. 2003;17:71–75. [Google Scholar]; c) Ichinose K, Taguchi T, Ebizuka Y, Hopwood DA. Actinomycetol. 1998;12:99–109. [Google Scholar]
- 178.(a) Taguchi T, Kunieda K, Takeda-Shitaka M, Takaya D, Kawano N, Kimberley MR, Booker-Milburn KI, Stephenson GR, Umeyama H, Ebizuka Y, Ichinose K. Bioorg Med Chem. 2004;12:5917–5927. doi: 10.1016/j.bmc.2004.08.026. [DOI] [PubMed] [Google Scholar]; (b) Li A, Itoh T, Taguchi T, Xiang T, Ebizuka Y, Ichinose K. Bioorg Med Chem. 2005;13:6856–6863. doi: 10.1016/j.bmc.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 179.a) Fernández-Moreno MA, Martinez E, Caballero JL, Ichinose K, Hopwood DA, Malpartida F. J Biol Chem. 1994;269:24854–24863. [PubMed] [Google Scholar]; b) Ichinose K, Surti C, Taguchi T, Malpartida F, Booker-Milburn KI, Stephenson GR, Ebizuka Y, Hopwood DA. Bioorg Med Chem Lett. 1999;9:395–400. doi: 10.1016/s0960-894x(99)00011-6. [DOI] [PubMed] [Google Scholar]; c) Taguchi T, Itou K, Ebizuka Y, Malpartida F, Hopwood DA, Surti CM, Booker-Milburn KI, Stephenson GR, Ichinose K. J Antibiot. 2000;53:144–152. doi: 10.7164/antibiotics.53.144. [DOI] [PubMed] [Google Scholar]
- 180.Ichinose K, Taguchi T, Bedford DJ, Ebizuka Y, Hopwood DA. J Bacteriol. 2001;183:3247–3250. doi: 10.1128/JB.183.10.3247-3250.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Freye E, Levy JV. Pharmacology and Abuse of Cocaine, Amphetamines, Ecstasy and Related Designer Drugs. Springer; London: 2009. [Google Scholar]
- 182.Roberts MF, Wink M. Alkaloids: Biochemistry, Ecology, and Medicinal Applications. Plenum Press; New York: 1998. [Google Scholar]
- 183.Hu J-F, Hamann MT, Hill R, Kelly M. Alkaloids Chem Biol. 2003;60:207–285. doi: 10.1016/s0099-9598(03)60004-0. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.a) Tsuda M, Kawasaki N, Kobayashi J. Tetrahedron. 1994;50:7957–7960. [Google Scholar]; b) Kondo K, Shigemori H, Kikuchi Y, Ishibashi M, Sasaki T, Kobayashi J. J Org Chem. 1992;57:2480–2483. [Google Scholar]
- 185.Kasanah N, Rao KV, Yousaf M, Wedge DE, Hamann MT. Tetrahedron Lett. 2003;44:1291–1293. [Google Scholar]
- 186.Yousaf M, El Sayed KA, Rao KV, Lim CW, Hu J-F, Kelly M, Franzblau SG, Zhang F, Peraud O, Hill RT, Hamann MT. Tetrahedron. 2002;58:7397–7402. [Google Scholar]
- 187.Ichiba T, Corgiat JM, Scheuer PJ, Kelly-Borges M. J Nat Prod. 1994;57:168–170. doi: 10.1021/np50103a027. [DOI] [PubMed] [Google Scholar]
- 188.Tsuda M, Kobayashi J. Heterocycles. 1997;46:765–794. [Google Scholar]
- 189.El Sayed KA, Kelly M, Kara UAK, Ang KKH, Katsuyama I, Dunbar DC, Khan AA, Hamann MT. J Am Chem Soc. 2001;123:1804–1808. doi: 10.1021/ja002073o. [DOI] [PubMed] [Google Scholar]
- 190.Ichiba T, Sakai R, Kohmoto S, Saucy G, Higa T. Tetrahedron Lett. 1988;29:3083–3086. [Google Scholar]
- 191.a) Kobayashi J, Tsuda M, Kawasaki N, Matsumoto K, Adachi T. Tetrahedron Lett. 1994;35:4383–4386. [Google Scholar]; b) Tsuda M, Inaba K, Kawasaki N, Honma K, Kobayashi J. Tetrahedron. 1996;52:2319–2324. [Google Scholar]
- 192.Kong F, Andersen RJ. Tetrahedron. 1995;51:2895–2906. [Google Scholar]
- 193.Cordell GA, Saxton JE, Shamma M, Smith GF, Southon IW, Buckingham J. Dictionary of Alkaloids. Chapman and Hall; London: 1989. [Google Scholar]
- 194.Ishikura M, Yamada K, Abe T. Nat Prod Rep. 2010;27:1630–1680. doi: 10.1039/c005345g. [DOI] [PubMed] [Google Scholar]
- 195.a) Scott AI. Acc Chem Res. 1970;3:151–157. [Google Scholar]; b) Ramos-Valdivia AC, van der Heijden R, Verpoorte R. Nat Prod Rep. 1997;14:591–603. doi: 10.1039/np9971400591. [DOI] [PubMed] [Google Scholar]
- 196.a) El-Sayed M, Verpoorte R. Phytochem Rev. 2007;6:277–305. [Google Scholar]; b) O’Connor SE, Maresh JJ. Nat Prod Rep. 2006;23:532–547. doi: 10.1039/b512615k. [DOI] [PubMed] [Google Scholar]; c) Facchini PJ, De Luca V. Plant J. 2008;54:763–784. doi: 10.1111/j.1365-313X.2008.03438.x. [DOI] [PubMed] [Google Scholar]
- 197.Zsadon B, Kaposi P. Tetrahedron Lett. 1970:4615–4616. [Google Scholar]
- 198.Anderson LA, Bisset NG, Phillipson JD, Zarucchi JL. J Ethnopharmacol. 1985;14:187–192. doi: 10.1016/0378-8741(85)90086-8. [DOI] [PubMed] [Google Scholar]
- 199.Constabel F, Rambold S, Chatson KB, Kurz WGM, Kutney JP. Plant Cell Rep. 1981;1:3–5. doi: 10.1007/BF00267645. [DOI] [PubMed] [Google Scholar]
- 200.Takayama H, Kurihara M, Kitajima M, Said IM, Aimi N. Tetrahedron. 2000;56:3145–3151. [Google Scholar]
- 201.Takayama H, Kurihara M, Kitajima M, Said IM, Aimi N. J Org Chem. 1999;64:1772–1773. doi: 10.1021/jo9823644. [DOI] [PubMed] [Google Scholar]
- 202.Normatov M, Yunusov SY. Khim Prir Soedin. 1968;4:139. [Google Scholar]
- 203.a) Stocking EM, Williams RM. Angew Chem Int Ed. 2003;42:3078–3115. doi: 10.1002/anie.200200534. [DOI] [PubMed] [Google Scholar]; b) Williams RM, Cox RJ. Acc Chem Res. 2003;36:127–139. doi: 10.1021/ar020229e. [DOI] [PubMed] [Google Scholar]; c) Sunderhaus JD, Sherman DH, Williams RM. Isr J Chem. 2011;51:442–452. doi: 10.1002/ijch.201100016. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Williams RM. J Org Chem. 2011;76:4221–4259. doi: 10.1021/jo2003693. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Williams RM. Chem Pharm Bull. 2002;50:711–740. doi: 10.1248/cpb.50.711. [DOI] [PubMed] [Google Scholar]; f) Miller KA, Williams RM. Chem Soc Rev. 2009;38:3160–3174. doi: 10.1039/b816705m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Qian-Cutrone J, Krampitz KD, Shu YZ, Chang LP. U.S. Patent 6,291,461. 2000
- 205.Kato H, Yoshida T, Tokue T, Nojiri Y, Hirota H, Ohta T, Williams RM, Tsukamoto S. Angew Chem Int Ed. 2007;46:2254–2256. doi: 10.1002/anie.200604381. [DOI] [PubMed] [Google Scholar]
- 206.Greshock TJ, Grubbs AW, Jiao P, Wicklow DT, Gloer JB, Williams RM. Angew Chem Int Ed. 2008;47:3573–3577. doi: 10.1002/anie.200800106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Genome cluster of Aspergillus sp. MF297–2: Ding Y, de Wet JR, Cavalcoli J, Li S, Greshock TJ, Miller KA, Finefield JM, Sunderhaus JD, McAfoos TJ, Tsukamoto S, Williams RM, Sherman DH. J Am Chem Soc. 2010;132:12733–12740. doi: 10.1021/ja1049302.Genome cluster of A. versicolor NRRL 35600: Li S, Finefield JM, Tsukamoto S, Williams RM, Sherman DH. Unpublished results.
- 208.a) Tsukamoto S, Kato H, Greshock TJ, Hirota H, Ohta T, Williams RM. J Am Chem Soc. 2009;131:3834–3835. doi: 10.1021/ja810029b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Finefield JM, Greshock TJ, Sherman DH, Tsukamoto S, Williams RM. Tetrahedron Lett. 2011;52:1987–1989. doi: 10.1016/j.tetlet.2011.02.078. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Finefield JM, Kato H, Greshock TJ, Sherman DH, Tsukamoto S, Williams RM. Org Lett. 2011;13:3802–3805. doi: 10.1021/ol201284y. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Finefield JM, Sherman DH, Tsukamoto S, Williams RM. J Org Chem. 2011;76:5954–5958. doi: 10.1021/jo200218a. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kato H, Nakamura Y, Finefield JM, Umaoka H, Nakahara T, Williams RM. Tetrahedron Lett. 2011;52:6923–6926. doi: 10.1016/j.tetlet.2011.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.McAfoos TJ, Li S, Tsukamoto S, Sherman DH, Williams RM. Heterocycles. 2010;82:461–472. doi: 10.3987/COM-10-S(E)19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tsukamoto S, Kawabata T, Kato H, Greshock TJ, Hirota H, Ohta T, Williams RM. Org Lett. 2009;11:1297–1300. doi: 10.1021/ol900071c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.a) Norcross NR, Melbardis JP, Solera MF, Sephton MA, Kilner C, Zakharov LN, Astles PC, Warriner SL, Blakemore PR. J Org Chem. 2008;73:7939–3951. doi: 10.1021/jo8013512. [DOI] [PubMed] [Google Scholar]; b) Van Wyk BE, Verdoorn GH. Pl Syst Evol. 1995;198:267–274. [Google Scholar]; c) Wink M, Meibner C, Witte L. Phytochemistry. 1995;38:139–153. [Google Scholar]
- 212.Cook D, Lee ST, Gardner DR, Pfister JA, Welch KD, Green BT, Davis TZ, Panter KE. J Agric Food Chem. 2009;57:1646–1653. doi: 10.1021/jf803468q. [DOI] [PubMed] [Google Scholar]
- 213.Golebiewski WM, Spenser ID. J Am Chem Soc. 1976;98:6726–6728. [Google Scholar]
- 214.a) Grundon MF. Nat Prod Rep. 1984;1:349–353. [Google Scholar]; b) Lamberton JA, Morton TC, Suares H. Aust J Chem. 1982;35:2577–2582. [Google Scholar]
- 215.Michael JP. Nat Prod Rep. 1994;11:639–657. doi: 10.1039/np9941100639. [DOI] [PubMed] [Google Scholar]
- 216.Michael JP. Nat Prod Rep. 1997;14:619–636. doi: 10.1039/np9971400619. [DOI] [PubMed] [Google Scholar]
- 217.Michael JP. Nat Prod Rep. 2002;19:719–741. doi: 10.1039/b104969k. [DOI] [PubMed] [Google Scholar]
- 218.Michael JP. Nat Prod Rep. 2004;21:625–649. doi: 10.1039/b310689f. [DOI] [PubMed] [Google Scholar]
- 219.Van Wyk DW, Greinwald R, Witte L. Biochem Syst Ecol. 1995;23:533–537. [Google Scholar]
- 220.Marion L, Fenton SW. J Org Chem. 1948;13:780–781. doi: 10.1021/jo01163a025. [DOI] [PubMed] [Google Scholar]
- 221.Carmack M, Douglas B, Martin EW, Suss H. J Am Chem Soc. 1955;77:4435. [Google Scholar]
- 222.a) Wink M, Hartmann T. Plant Physiol. 1982;70:74–77. doi: 10.1104/pp.70.1.74. and reference therein. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wink M. Planta Med. 1987;47:509–514. doi: 10.1055/s-2006-962797. and references therein. [DOI] [PubMed] [Google Scholar]
- 223.a) Golebiewski WM, Spencer ID. Can J Chem. 1985;63:2707–2718. [Google Scholar]; b) Spencer ID. Pure Appl Chem. 1985;57:453–470. [Google Scholar]; c) Golebiewski WM, Spencer ID. Can J Chem. 1988;66:1734–1748. [Google Scholar]; d) Fraser AM, Robins DJ. J Chem Soc, Chem Commun. 1986:545–547. [Google Scholar]; e) Fraser AM, Robins DJ. J Chem Soc, Perkin Trans 1. 1987:105–109. [Google Scholar]
- 224.a) Vetter J. Food Chem Toxicol. 2004;42:1373–1382. doi: 10.1016/j.fct.2004.04.009. [DOI] [PubMed] [Google Scholar]; b) Reynolds T. Phytochemistry. 2005;66:1399–1406. doi: 10.1016/j.phytochem.2005.04.039. [DOI] [PubMed] [Google Scholar]
- 225.a) Lee ST, Panter KE, Molyneux RJ, Chang C-WT, Gardern DR, Pfister JA. In: Poisonous Plants: Global Research and Solutions. Panter KE, Wierenga TL, Pfister JA, editors. 2007. p. 469. [Google Scholar]; b) Lee ST, Molyneux RJ, Panter KE, Chang CWT, Gardner DR, Pfister JA, Garrossian M. J Nat Prod. 2005;68:681–685. doi: 10.1021/np0580199. [DOI] [PubMed] [Google Scholar]; c) Lee ST, Green BT, Welch KD, Pfister JA, Panter KE. Chem Res Toxicol. 2008;21:2061–2064. doi: 10.1021/tx800229w. [DOI] [PubMed] [Google Scholar]; d) Lee ST, Wildeboer K, Panter KE, Kem WR, Gardner DR, Molyneux RJ, Chang CWT, Soti F, Pfister JA. Neurotoxicol Teratol. 2006;28:220–228. doi: 10.1016/j.ntt.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 226.a) Keeler RF, Shupe JL, Crowe MW, Olson A, Balls LD. Am J Vet Res. 1981;42:1231–1234. [PubMed] [Google Scholar]; b) Keeler RF, Crowe MW, Lambert EA. Teratology. 1984;30:61–69. doi: 10.1002/tera.1420300109. [DOI] [PubMed] [Google Scholar]; c) Keeler RF, Crowe MW. Cornell Vet. 1984;74:50–59. [PubMed] [Google Scholar]; d) Panter KE, Bunch TD, Keeler RF, Sisson DV, Callan RJ. J Toxicol: Clin Toxicol. 1990;28:69–83. doi: 10.3109/15563659008993477. [DOI] [PubMed] [Google Scholar]
- 227.a) Mothes K, Schütte HR. Angew Chem Int Ed. 1963;2:341–357. [Google Scholar]; b) Leete E. Annu Rev Plant Physiol. 1967;18:179–196. [Google Scholar]; c) Leete E. J Am Chem Soc. 1964;86:2509–2513. [Google Scholar]; d) Leete E. Acc Chem Res. 1971;4:100–107. [Google Scholar]; e) Alworth WL, Rapoport H. Arch Biochem Biophys. 1965;112:45–53. doi: 10.1016/0003-9861(65)90008-1. [DOI] [PubMed] [Google Scholar]; f) Solt ML, Dawson RF, Christman DR. Plant Physiol. 1960;35:887–894. doi: 10.1104/pp.35.6.887. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Leete E, Gros EG, Gilbertson TJ. J Am Chem Soc. 1964;86:3907–3908. [Google Scholar]; h) Leete E. J Nat Prod. 1982;45:197–205. [Google Scholar]; i) DeBoer KD, Lye JC, Aitken CD, Su AKK, Hamill JD. Plant Mol Biol. 2009;69:299–312. doi: 10.1007/s11103-008-9425-2. [DOI] [PubMed] [Google Scholar]; j) Katoh A, Ohki H, Inai K, Hashimoto T. Plant Biotechnology. 2005;22:389–392. [Google Scholar]
- 228.Murakoshi I, Kidoguchi E, Nakamura M, Haginiwa J, Ohmiya S, Higashiyama K, Otomasu H. Phytochemistry. 1981;20:1725–1730. [Google Scholar]
- 229.Leclercq S, Charles S, Daloze D, Braekman J-C, Aron S, Pasteels JM. J Chem Ecol. 2001;27:945–952. doi: 10.1023/a:1010335003297. [DOI] [PubMed] [Google Scholar]
- 230.Lukes R, Arojan AA, Kovar J, Blaha K. Collect Czech Chem Commun. 1962;27:751–756. [Google Scholar]
- 231.a) Bonamore A, Barba M, Botta B, Boffi A, Macone A. Molecules. 2010;15:2070–2078. doi: 10.3390/molecules15042070. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Samanani N, Facchini PJ. Planta. 2001;213:898–906. doi: 10.1007/s004250100581. [DOI] [PubMed] [Google Scholar]
- 232.Ziegler J, Facchini PJ, Geibler R, Schmidt J, Ammer C, Kramell R, Voigtländer S, Gesell A, Pienkny S, Brandt W. Phytochemistry. 2009;70:1696–1707. doi: 10.1016/j.phytochem.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 233.Desgagné-Penix I, Facchini PJ. In: Plant Metabolism and Biotechnology. Ashihara H, Crozier A, Komamine A, editors. Wiley; West Sussex: 2011. p. 241. [Google Scholar]
- 234.Zhang L-B, Rao G-X. Biochem Syst Ecol. 2009;37:622–625. [Google Scholar]
- 235.a) De-Eknamkul W, Zenk MH. Phytochemistry. 1992;31:813–821. [Google Scholar]; b) Hirata K, Poeaknapo C, Schmidt J, Zenk MH. Phytochemistry. 2004;65:1039–1046. doi: 10.1016/j.phytochem.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 236.Bauer W, Zenk MH. Tetrahedron Lett. 1991;32:487–490. [Google Scholar]
- 237.Mascavage LM, Jasmin S, Sonnet PE, Wilson M, Dalton DR. Ullmann’s Encyclopedia of Industrial Chemistry. Wiley; 2011. [Google Scholar]
- 238.a) Bridgham JT, Carroll SM, Thornton JW. Science. 2006;312:97–101. doi: 10.1681/01.asn.0000926836.46869.e5. [DOI] [PubMed] [Google Scholar]; b) Dean AM, Thornton JW. Nat Rev Genet. 2007;8:675–688. doi: 10.1038/nrg2160. [DOI] [PMC free article] [PubMed] [Google Scholar]